DNA-herstelkinetiek en therapierespons bij gecombineerde...
59
DNA-herstelkinetiek en therapierespons bij gecombineerde radio-cetuximab therapie Cedric BRACKENIER Verhandeling ingediend tot het verkrijgen van de graad van Master in de Biomedische Wetenschappen Promotor: Prof. Dr. Hubert Thierens Begeleider: Dr. Ir. Kim De Ruyck Vakgroep medische basiswetenschappen Academiejaar 2011-2012
Transcript of DNA-herstelkinetiek en therapierespons bij gecombineerde...

DNA-herstelkinetiek en therapierespons bij
gecombineerde radio-cetuximab therapie
Cedric BRACKENIER
Verhandeling ingediend tot
het verkrijgen van de graad van
Master in de Biomedische Wetenschappen
Promotor: Prof. Dr. Hubert Thierens
Begeleider: Dr. Ir. Kim De Ruyck
Vakgroep medische basiswetenschappen
Academiejaar 2011-2012


DNA-herstelkinetiek en therapierespons bij
gecombineerde radio-cetuximab therapie
Cedric BRACKENIER
Verhandeling ingediend tot
het verkrijgen van de graad van
Master in de Biomedische Wetenschappen
Promotor: Prof. Dr. Hubert Thierens
Begeleider: Dr. Ir. Kim De Ruyck
Vakgroep medische basiswetenschappen
Academiejaar 2011-2012

i
“De auteur en de promotor geven de toelating deze masterproef voor consultatie beschikbaar
te stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder
de beperkingen van het auteursrecht, in het bijzonder met betrekking tot de verplichting
uitdrukkelijk de bron te vermelden bij het aanhalen van resultaten uit deze masterproef.”
21 mei 2012
Cedric Brackenier Prof. Dr. Hubert Thierens

ii
Voorwoord
Deze masterproef was niet tot stand kunnen komen zonder de hulp van een aantal mensen. Ik
wens hen dan ook oprecht te bedanken.
In de eerste plaats gaat mijn dank uit naar mijn promotor, prof. dr. Hubert Thierens, en mijn
begeleidster, dr. ir. Kim De Ruyck. Bedankt voor de begeleiding bij mijn eerste stappen in de
wereld van het wetenschappelijk onderzoek. Dit nieuwe onderzoek vereiste arbeidsintensieve
optimalisatieprocedures alvorens er kon worden overgegaan tot de eigenlijke experimenten.
Het nieuwe karakter van deze studie liet me echter toe om bijzonder veel bij te leren tijdens
dit academiejaar. Bedankt!
Mijn dank gaat ook uit naar het voltallige personeel van de diensten medische fysica en
fysische controle voor de aangename sfeer op de werkvloer. Bedankt!
Verder gaat mijn dank uit naar prof. dr. Jan Philippé voor het ter beschikking stellen van de
apparatuur voor flow cytometrie, en naar prof. dr. Stéphanie Laurent voor het verkrijgen van
cetuximab. Ik wens ook dr. Nancy Van Damme te bedanken voor het verkrijgen van de HT-
29 cellijn, en Koen Mertens en Julie Depuydt voor het overleg in verband met
celcultuursystemen. Bedankt!
Ook bedank ik mijn vrienden, bij wie ik steeds mijn hart kon luchten over ‘de kanker’
wanneer de experimenten iets moeizamer verliepen. Bedankt!
Wie ongetwijfeld ook een fundamentele bijdrage geleverd hebben tot het succes van deze
masterproef, zijn mijn medestudenten van ‘de straling’. Dankzij hen werd er, tussen het
werken door, heel wat afgelachen. Wat mij betreft heb ik vier extra vrienden overgehouden
aan deze thesis. Ik hoop hen in de toekomst nog regelmatig te zullen zien. Bedankt Lore,
Pieter, Mattias en Eline!
Mijn grootste dank gaat uit naar mijn ouders. Zonder hen had ik deze studies nooit kunnen
aanvatten en succesvol afronden. Bedankt!

iii
Inhoud
Voorwoord ................................................................................................................................. ii
Inhoud ........................................................................................................................................ iii
Overzicht afkortingen ................................................................................................................ vi
Samenvatting .............................................................................................................................. 1
1. Inleiding ................................................................................................................................. 2
1.1. Kanker ............................................................................................................................. 2
1.2. Colorectale kanker ........................................................................................................... 2
1.2.1. Epidemiologie ........................................................................................................... 2
1.2.2. Oorzaken en risicofactoren ....................................................................................... 3
1.2.3. Screening en preventie .............................................................................................. 3
1.2.4. Pathogenese ............................................................................................................... 4
1.2.5. Diagnose ................................................................................................................... 5
1.2.6. Behandeling .............................................................................................................. 5
1.3. Moleculair gerichte therapieën ........................................................................................ 6
1.3.1. De epidermale groeifactor receptor........................................................................... 6
1.3.2. Anti-EGFR therapie .................................................................................................. 8
1.3.3. Predictieve merkers voor anti-EGFR therapie ........................................................ 10
1.3.4. Rationale voor gecombineerde radio-cetuximab therapie ...................................... 11
1.4. Radiotherapie ................................................................................................................. 12
1.5. Detectie en herstel van stralingsgeïnduceerde DNA-schade ......................................... 13
1.5.1. DNA-schade checkpoints ........................................................................................ 14
1.5.2. Herstel van DNA-dubbelstrengbreuken .................................................................. 15
1.5.3. De γ-H2AX foci assay ............................................................................................ 16
1.6. Doel van de studie ......................................................................................................... 16
2. Materialen en methoden ....................................................................................................... 18

iv
2.1. Celcultuur ...................................................................................................................... 18
2.1.1. Oorsprong van de HT-29 cellijn ............................................................................. 18
2.1.2. Kweekomstandigheden van de HT-29 cellijn ......................................................... 18
2.1.3. Onderhoud van de HT-29 cellijn ............................................................................ 18
2.1.4. Periodieke controle op mycoplasmacontaminatie................................................... 18
2.1.5. Invriezen van de HT-29 cellijn ............................................................................... 20
2.1.6. Ontdooien en opstarten van de HT-29 cellijn ......................................................... 20
2.1.7. Kwantificatie van de HT-29 cellen ......................................................................... 20
2.2. Constructie van een groeicurve ..................................................................................... 21
2.3. Synchronisatie in G0/G1 fase van de celcyclus ............................................................. 21
2.3.1. Optimalisatie van de synchronisatieprocedure ....................................................... 21
2.3.2. Controle van de synchronisatietoestand .................................................................. 21
2.3.3. Synchronisatieprocedure voorafgaand aan de experimenten .................................. 22
2.4. Dosisrespons experimenten ........................................................................................... 22
2.4.1. De γH2AX foci assay voor dosisrespons experimenten ......................................... 22
2.5. DNA-herstelkinetiek experimenten ............................................................................... 23
2.5.1. De γH2AX foci assay na bestraling ........................................................................ 23
2.5.2. De γH2AX foci assay na gecombineerde radio-cetuximab therapie ...................... 24
2.6. Analyse van de γH2AX foci .......................................................................................... 25
2.6.1. Automatische analyse ............................................................................................. 25
2.6.2. Manuele analyse ...................................................................................................... 25
2.7. Statistische analysen ...................................................................................................... 26
3. Resultaten ............................................................................................................................. 27
3.1. Constructie van een groeicurve ..................................................................................... 27
3.2. Optimalisatie van de synchronisatieprocedure .............................................................. 29
3.3. Dosisrespons experimenten ........................................................................................... 30
3.4. DNA-herstelkinetiek experimenten ............................................................................... 32

v
4. Discussie ............................................................................................................................... 38
4.1. De constructie van een groeicurve ................................................................................. 38
4.2. Optimalisatie van de synchronisatieprocedure .............................................................. 38
4.3. Dosisrespons experimenten ........................................................................................... 39
4.4. DNA-herstelkinetiek experimenten ............................................................................... 41
4.5. Besluit ............................................................................................................................ 44
5. Referenties ............................................................................................................................ 45

vi
Overzicht afkortingen
60Co cobalt-60
ADP adenosine diphosphate
Akt RAC-alpha serine/threonine-protein kinase
AMP adenosine monophosphate
APC adenomatous polyposis coli
ATM ataxia telangiectasia mutated
ATP adenosine triphosphate
ATR ataxia telangiectasia and Rad3 related
ATRIP ATR interacting protein
BER base excision repair
BRAF v-raf murine sarcoma viral oncogene homolog B1
BSA bovine serum albumin
Cav-1 caveoline 1
CIMP CpG island methylator phenotype
CIN chromosomale instabiliteit
CO2 koolstofdioxide
CRC colorectal cancer
CT computed tomography
CU colitis ulcerosa
D-PBS Dulbecco’s phosphate-buffered saline
DAPI 4’,6-diamidino-2-phenylindole
DMSO dimethylsulfoxide
DNA deoxyribonucleic acid
DNA-PK DNA-dependent protein kinase
DNA-PKcs DNA-dependent protein kinase catalytic subunit

vii
DSB double strand breaks
EDTA ethylenediaminetetraacetic acid
EGFR epidermal growth factor receptor
erbB erythroblastic leukemia viral oncogene homolog
EU Europese Unie
FAP familial adenomatous polyposis
FBS fetal bovine serum
Fcγ Immunoglobulin gamma Fc
FOLFIRI chemotherapeutisch regime bestaande uit combinatie van 5-
fluorouracil, leucovorine en irinotecan
FOLFOX chemotherapeutisch regime bestaande uit combinatie van 5-
fluorouracil, leucovorine en oxaliplatine
HER tyrosine kinase-type cell surface receptor HER
HNPCC hereditary nonpolyposis colorectal cancer
HR homologe recombinatie
HRAS v-Ha-ras Harvey rat sarcoma viral oncogene homolog
HUS1 Hus1 checkpoint protein
IR ionizing radiation
JAK janus kinase
KRAS v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
Ku70 x-ray repair cross-complementing protein 6
Ku80 x-ray repair cross-complementing protein 5
MAB monoclonal antibody
MAPK mitogen-activated protein kinase
mCRC metastatic colorectal cancer
MDC1 mediator of DNA damage checkpoint protein 1
Mg2+
tweewaardig magnesium ion
MMR mismatch repair

viii
MRI magnetic resonance imaging
MRN Mre11, Rad50 en NBS1
MSI microsatelliet instabiliteit
nEGFR nuclear epidermal growth factor receptor
NHEJ non-homologous end joining
NRAS neuroblastoma RAS viral (v-ras) oncogene homolog
O2 zuurstofgas
PCNA proliferating cell nuclear antigen
PE phyco-erythrin
PET positron emission tomography
PFA paraformaldehyde
PI3K phosphoinositide-3-kinase
PI3KCA phosphoinositide-3-kinase, catalytic, alpha polypeptide
PIKK phosphatidylinositol-3-kinase-related kinases
PKC protein kinase C
PLC-n1 phospholipase C
PNK polynucleotide kinase
PPi pyrophosphate
PTEN phosphatase and tensin homolog
RAD GTP binding protein RAD
Raf Raf proto-oncogene serine/threonine protein kinase
RAM-TRITC tetramethyl rhodamine isothiocyanate conjugated rabbit anti-mouse
antibody
RPA replication protein A
RPA2 replication protein A2, 32 kDa
RT radiation therapy
SMAD mothers against decapentaplegic

ix
SQEY serine, glutamine, glutamaat, tyrosine
Src proto-oncogene tyrosine-protein kinase Src
SSA sessile serrated adenoma
SSM serum starvation medium
STAT signal transducer and activator of transcription
TdT terminal deoxynucleotidyltransferase
TGFBR1 transforming growth factor beta receptor 1
TGFBR2 transforming growth factor beta receptor 2
TGF-β transforming growth factor beta
TK tyrosine kinase
TKI tyrosine kinase inhibitor
TSA traditional serrated adenoma
VEGF vascular endothelial growth factor
WRN Werner syndrome, RecQ helicase-like
XLF synoniem voor NHEJ1, non-homologous end-joining factor 1
XRCC4 x-ray repair complementing defective repair in Chinese hamster cells 4

1
Samenvatting
Ondanks grote vooruitgang in de behandeling van geavanceerde kankers zijn conventionele
behandelingsstrategieën niet steeds genezend. Moleculair gerichte therapieën verhogen, door
hun specifieke focus op ontregelde signaalpathways in tumoren, de therapeutische index.
Bovendien zijn ze combineerbaar met conventionele cytotoxische therapieën. De epidermale
groeifactor receptor (EGFR) is een plasmamembranair glycoproteïne dat een kritische rol
speelt bij regulatie van cellulaire proliferatie, overleving, groei, migratie, tumorcel invasie en
inhibitie van apoptose. Overexpressie van EGFR wordt frequent gezien in humane tumoren en
is geassocieerd met een slechte prognose. Naast ligandbinding kan ook blootstelling aan
ioniserende straling EGFR activeren. Dit leidt tot translocatie van EGFR naar de nucleus.
Nucleaire EGFR draagt bij tot DNA-herstel, waardoor de cellulaire radiosensitiviteit en dus
ook het effect van radiotherapie afneemt. Dit maakt de EGFR een zinvol therapeutisch target.
Er bestaan 2 klassen van anti-EGFR therapeutica: tyrosine kinase inhibitoren en monoclonale
antilichamen (MAB). Cetuximab is een MAB dat specifiek bindt aan het extracellulair
domein van EGFR en receptorinternalisatie veroorzaakt. Het wordt toegepast bij de
behandeling van patiënten met KRAS wildtype metastatische colorectale kanker. Er is echter
nood aan predictieve merkers voor identificatie van patiënten die met grote waarschijnlijkheid
baat zullen hebben bij gecombineerde radio-cetuximab therapie. Het doel van deze studie was
de bepaling van de DNA-herstelkinetiek van de HT29 colonadenocarcinoma cellijn na
gecombineerde radio-cetuximab therapie. Hiervoor werden het celcultuursysteem en de
synchronisatieprocedure geoptimaliseerd. Dosisrespons experimenten werden uitgevoerd om
de optimale dosis 60
Co γ-straling te bepalen voor de DNA-herstelkinetiek experimenten.
DNA-herstelkinetiek experimenten werden uitgevoerd na gecombineerde radio-cetuximab
behandeling van de cellen. Hiervoor werd gebruik gemaakt van de γH2AX foci assay. De
γH2AX foci werden manueel gescoord met behulp van fluorescentiemicroscopie.
HT29 cellen die blootgesteld werden aan zowel cetuximab als aan ioniserende straling bleken
stralingsgeïnduceerde DNA dubbelstrengbreuken trager te herstellen in vergelijking met
HT29 cellen die enkel aan ioniserende straling werden blootgesteld. In vervolgonderzoek
kunnen de gegevens bekomen in deze pilootstudie gecorreleerd worden met therapierespons,
om zo na te gaan of DNA-herstelkinetiek al dan niet bruikbaar is als predictieve merker voor
radio-cetuximab therapie bij patiënten met colorectale kanker.

2
1. Inleiding
1.1. Kanker
Kanker is een ziekte die geassocieerd is met dynamische veranderingen in het genoom [1]. Bij
de progressieve evolutie van normale cellen naar een neoplastische toestand verwerven de
cellen geleidelijk wijzigingen in hun cellulaire fysiologie. Hierdoor verkrijgen ze nieuwe
functionele eigenschappen zoals het zelfvoorzienend zijn in groeisignalen, ongevoeligheid
aan groei-inhiberende signalen, ontwijking van apoptose, onbeperkte replicatiecapaciteit,
ondersteuning van angiogenese, weefselinvasie, metastasering, herprogrammering van het
cellulaire energiemetabolisme en actieve ontwijking van de immunologische respons. Het
ontstaan van deze nieuwe eigenschappen wordt bevorderd door de ontwikkeling van
genoominstabiliteit en inflammatie. Samen laten deze verworven kenmerken de kankercellen
toe te overleven, te prolifereren en zich te verspreiden [2].
Het wereldwijde aantal nieuwe kankergevallen en de kankergerelateerde mortaliteit worden in
2008 geschat op respectievelijk 12.7 en 7.6 miljoen [3]. Momenteel krijgen 1 op 3 Belgische
mannen en 1 op 4 Belgische vrouwen kanker voor het bereiken van de 75-jarige leeftijd [4].
De ziekte heeft dan ook een grote financiële impact op de gezondheidszorg [5].
1.2. Colorectale kanker
1.2.1. Epidemiologie
Zowel wereldwijd als in België vormt kanker van het colon en rectum (colorectale kanker,
CRC) bij mannen en vrouwen respectievelijk de derde en tweede meest voorkomende vorm
van kanker [3,4]. Zo wordt het wereldwijde aantal nieuwe CRC gevallen in 2008 geschat op
1.2 miljoen. Er zijn grote regionale verschillen in incidentie. De hoogste incidenties worden
gezien in de Oost-Europese landen, Japan, Nieuw-Zeeland, Australië, Duitsland en bij Afro-
Amerikanen. De laagste incidenties worden waargenomen in Centraal- en Zuid-Amerika,
Afrika en Centraal- en Zuid-Azië. Wegens de overname van Westerse levensgewoonten en
diëten neemt de incidentie sterk toe in gebieden waar vroeger een laag risico bestond op de
ontwikkeling van CRC. Anderzijds daalt de incidentie in sommige ontwikkelde landen door
de detectie en verwijdering van laesies in een voorstadium van CRC. De CRC-gerelateerde
mortaliteit daalt in regio’s met een goede gezondheidsinfrastructuur, maar blijft stijgen in
gebieden die daar niet over beschikken [3]. Momenteel is CRC bij Belgische mannen en

3
vrouwen respectievelijk de tweede en derde meest frequente oorzaak van kankergerelateerde
sterfte [4].
1.2.2. Oorzaken en risicofactoren
Circa 90% van alle CRC-gevallen zijn sporadisch van oorsprong. Gekende risicofactoren zijn
hoge leeftijd, mannelijk geslacht, cholecystectomie, ureterocolische anastomosen, vroege
menopauze, hoge leeftijd bij eerste zwangerschap, nullipariteit, vlees- en vetrijk dieet, dieet
arm aan vezels, foliumzuur en calcium, sedentaire levensstijl, obesitas, diabetes mellitus,
roken, blootstelling aan ioniserende straling, occupationele blootstellingen, hoge
alcoholinname en een persoonlijke historiek van sporadische tumoren [6]. Regelmatig
gebruik van aspirine of niet-steroïdale anti-inflammatoire middelen zou, net als gebruik van
oestrogenen bij vrouwen, dan weer beschermend werken [7]. Circa 20% van de sporadische
patiënten hebben bovendien een familiale risicocomponent, maar voldoen niet aan de
gedefinieerde criteria voor erfelijke CRC. Slechts 5 tot 10% van alle colorectale kankers
ontwikkelen in de context van erfelijke kankersyndromen zoals FAP1 en HNPCC
2. Tot slot
liggen bij 1 tot 2 procent der CRC-gevallen inflammatoire darmziekten zoals CU3 of de ziekte
van Crohn aan de basis [6].
1.2.3. Screening en preventie
De mogelijkheid tot verwijdering van letsels in een voorstadium van CRC maakt dat
screening kan bijdragen tot verlaging van incidentie en CRC-gerelateerde mortaliteit [7].
Diverse methoden, waaronder sigmoïdoscopie, colonoscopie, dubbelcontrast barium klysma
en de feces occult bloed test, zijn beschikbaar [6]. Gezien 94% van de patiënten ouder zijn
dan 50 jaar, kan screening beperkt worden tot deze leeftijdsgroep [7].
De belangrijkste en tevens de goedkoopste preventiemanier is aanpassing van de levensstijl.
Door te zorgen voor voldoende fysische activiteit, tabakgebruik te vermijden, eetgewoonten
aan te passen en het lichaamsgewicht onder controle te houden kan het risico op CRC
verlaagd worden. Daarnaast kunnen ook geneesmiddelen en chirurgie bijdragen tot
risicoverlaging, zij het dan in het geval van FAP, CU en eventueel HNPCC [6].
1 FAP: Familial adenomatous polyposis
2 HNPCC: Hereditary nonpolyposis colorectal cancer
3 CU: Colitis ulcerosa

4
1.2.4. Pathogenese
CRC is een heterogene ziekte die zich ontwikkelt via een toenemende graad van dysplasie van
de normale colonmucosa. Diverse moleculaire pathways dragen bij tot maligne transformatie
in deze adenoma-carcinoma sequentie [8].
Een eerste pathway, chromosomale instabiliteit (CIN), vormt de meest frequente genetische
aberratie in CRC en is geassocieerd met mutatie van het APC4 tumorsuppressorgen. Deze
mutaties treden zeer vroeg op in de ontwikkeling van adenoma’s en zorgen voor cellulaire
accumulatie van β-catenine. Daarnaast leidt inactivatie van APC tot abnormale chromosomale
segregatie, wat bijdraagt tot chromosomale instabiliteit. Ook het KRAS5 proto-oncogen, dat
een rol speelt bij cellulaire proliferatie, kan betrokken zijn bij chromosomale instabiliteit.
Constitutieve activatie van KRAS draagt bij tot de ontwikkeling en progressie van poliepen
[8].
De CpG island methylator phenotype (CIMP) pathway vormt een tweede pathway. Hij wordt
gekenmerkt door epigenetische instabiliteit, ontstaan door hypermethylatie-geïnduceerde
silencing van tumorsuppressorgenen en door globale DNA-hypomethylatie [8].
Een derde pathway, microsatelliet instabiliteit (MSI), ontstaat bij het optreden van mutaties in
nucleotide herhalingssequenties in het DNA. Deze pathway is geassocieerd met het mismatch
repair (MMR) systeem, dat instaat voor herstel van polymerase-geïnduceerde fouten bij
DNA-replicatie. Daar waar deficiëntie van het MMR systeem bij HNPCC berust op germline
mutaties in de MMR genen, is deze bij sporadische MSI tumoren te wijten aan silencing van
de genen via promotor hypermethylatie [8].
Een vierde grote pathway is de serrated pathway. Sessile serrated adenomas (SSA) en
traditional serrated adenomas (TSA) kunnen op een verschillende manier aanleiding geven tot
maligne progressie. Hoewel MSI en CIMP daarbij een overlappende rol spelen, zijn er toch
specifieke mutaties geïdentificeerd voor elke pathway. Daar waar KRAS mutaties vooral
worden aangetroffen bij TSA, zijn mutaties in BRAF6, een gen waarvan het eiwitproduct een
rol speelt in cellulaire proliferatie en inhibitie van apoptose, karakteristiek voor SSA [8].
Naast deze vier grote pathways kan ook chronische inflammatie bijdragen tot maligne
transformatie. Dit is te wijten aan een foutieve respons van het immuunsysteem tegenover de
4 APC: Adenomatous polyposis coli
5 KRAS: v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
6 BRAF: v-raf murine sarcoma viral oncogene homolog B1

5
intraluminale bacteriën, gecombineerd met bepaalde voorbeschikkende genetische
wijzigingen [8].
Tot slot zijn er nog diverse biochemische pathways die, wanneer getroffen door mutaties,
kunnen bijdragen tot de ontwikkeling van maligne weefsel. Dit is van toepassing voor de
TGF-β7 pathway, wanneer getroffen door mutaties in TGFBR1
8, TGFBR2
9 en de SMAD
10
genen, alsook bij verlies van de lange arm van chromosoom 18. De tumorsuppressor p53
speelt een rol in diverse pathways en kan dan ook op verschillende manieren geïnactiveerd
worden. Mutaties in p53 worden gezien in de helft van alle CRC-gevallen. Ook verhoogde
expressie van de epidermale groeifactor receptor (EGFR), een transmembranaire receptor die
een rol speelt bij cellulaire proliferatie, angiogenese en apoptose, is geassocieerd met
geavanceerde ziekte [8].
1.2.5. Diagnose
De gouden standaard voor de diagnose van CRC is colonoscopie [6]. Diverse alternatieven,
zoals sigmoïdoscopie, flexibele sigmoïdoscopie, virtuele colonoscopie, dubbelcontrast
bariumklysma en pneumocolon computer tomografie (CT), zijn beschikbaar [7]. Naast fysisch
onderzoek worden ook abdominale echografie en thoraxradiografie routinematig uitgevoerd.
Goede beeldvorming van rectale kanker, noodzakelijk voor de behandelingsplanning, gebeurt
bij voorkeur door middel van magnetische resonantie beeldvorming (MRI). Positron emissie
tomografie (PET) wordt dan weer gebruikt voor detectie van CRC recidieven. Voor de
detectie van levermetastasen vormen CT en MRI de diagnostische standaard [6].
1.2.6. Behandeling
Chirurgie vormt de basisbehandeling voor CRC. Voor patiënten met stadium I of II
colonkanker volstaat chirurgische behandeling. Voor patiënten met stadium III of een
specifieke risicovolle stadium II colonkanker wordt chirurgie gecombineerd met adjuvante
fluorouracil gebaseerde therapie [6]. Adjuvante therapie is een systemische behandeling die
wordt toegediend na resectie van de primaire tumor [9].
Ook bij vroege stadia van rectale kanker volstaat chirurgische behandeling. In meer lokaal
geavanceerde kankers wordt preoperatieve radiotherapie toegepast. Bij de meest lokaal
7 TGF-β: Transforming growth factor beta
8 TGFBR1:Transforming growth factor beta receptor 1
9 TGFBR2: Transforming growth factor beta receptor 2
10 SMAD: Mothers against decapentaplegic

6
geavanceerde kankers wordt, voorafgaand aan radicale chirurgie, preoperatieve
radiochemotherapie toegediend gebaseerd op fluorouracil [10].
Bijna 50% van de CRC-patiënten ontwikkelen metastasen. Eerstelijns palliatieve
chemotherapie voor metastatische CRC (mCRC) is gebaseerd op fluoropyrimidines.
Combinaties van 5-fluorouracil, leucovorine en oxaliplatine (FOLFOX) of 5-fluorouracil,
leucovorine en irinotecan (FOLFIRI) leiden tot hogere overleving. Ook monoclonale
antilichamen gericht tegen de vasculaire endotheliale groeifactor (VEGF) en tegen de
epidermale groeifactor receptor (EGFR) kunnen, eventueel in combinatie met chemotherapie,
overwogen worden. In sommige gevallen kan chemotherapie leiden tot downstaging,
waardoor initieel onresecteerbare metastasen toch resecteerbaar worden [11].
1.3. Moleculair gerichte therapieën
Ondanks grote vooruitgang in de behandeling van geavanceerde kankers zijn conventionele
behandelingsstrategieën niet steeds genezend [12]. Dit is te wijten aan de ontwikkeling van
primaire en verworven resistentie aan conventionele chemo- en radiotherapie. Moleculair
gerichte therapieën verhogen, door hun specifieke focus op ontregelde signaalpathways in
tumoren, de therapeutische index. Bovendien maken de verschillende biologische effecten en
het andere toxiciteitsprofiel combinatie met conventionele cytotoxische therapieën mogelijk
[13].
1.3.1. De epidermale groeifactor receptor
Groeifactoren en hun receptoren spelen een fundamentele rol in de communicatie tussen
cellen en hun omgeving [14]. Cellulaire ontwikkeling is dan ook afhankelijk van de activiteit
van plasmamembraan receptoren die intracellulaire signaalbanen controleren die, op hun
beurt, instaan voor de regulatie van cellulaire responsen. De epidermale groeifactor receptor
(EGFR, erbB1, HER1) is een dergelijk transmembranair glycoproteïne dat bestaat uit een
extracellulair ligandbindend domein, een hydrofobe transmembranaire regio en een
intracellulair domein met tyrosine kinase activiteit. Hij vormt samen met erbB2 (HER2),
erbB3 (HER3) en erbB4 (HER4) de erbB familie van transmembranaire proteïnekinase
receptoren [12].
1.3.1.1. EGFR signalering na ligandbinding
Binding van specifieke liganden ter hoogte van het extracellulair domein van EGFR
veroorzaakt receptor homo- of heterodimerisatie, gevolgd door activatie van intrinsieke

7
proteïnekinase activiteit en tyrosine autofosforylatie. Dit leidt tot activatie van de Ras-Raf-
MAPK11
, PI3K12
-Akt13
, PKC14
, JAK15
/STAT16
, STAT3 en PLC-n117
signaalpathways [12].
Hierdoor speelt EGFR-gemedieerde intracellulaire signalering een kritische rol bij cellulaire
proliferatie, overleving, groei, migratie, tumorcel invasie en inhibitie van apoptose [15].
Figuur 1 geeft een overzicht van de cytoplasmatische signaalpathways van EGFR.
Figuur 1. Overzicht van de cytoplasmatische signaalpathways geïnduceerd door EGFR-activatie. Figuur
overgenomen uit Rodemann et al. [16].
Omdat signaleringsniveaus strikt gereguleerd dienen te zijn [17], is receptorinactivatie
noodzakelijk. Hiervoor is receptorinternalisatie essentieel [18]. Internalisatie van EGFR na
ligandbinding gebeurt via clathrine-gemedieerde endocytose [12]. Na fusie van de
endocytotische vesikels met vroege endosomen wordt EGFR afgebroken in lysosomen of
gerecycleerd naar de plasmamembraan [17].
De geïnternaliseerde EGFR molecule kan echter ook naar de nucleus getransporteerd worden.
Na endocytose is het onduidelijk welke pathway EGFR naar de nucleus brengt [17]. De
nucleaire EGFR (nEGFR) is betrokken bij transcriptionele activatie van diverse genen [18].
11
MAPK: mitogen activated protein kinase 12
PI3K: phosphoinositide 3-kinase 13
Akt: RAC-alpha serine/threonine-protein kinase 14
PKC: Protein kinase C 15
JAK: Janus kinase 16
STAT: Signal transducer and activator of transcription 17
PLC-n1: phospholipase C

8
Bovendien fosforyleert nEGFR chromatingebonden PCNA18
, wat leidt tot verhoogde
stabiliteit van actief PCNA en dus stimulatie van DNA-replicatie en DNA-herstel [17]. Ook
kan nEGFR rechtsreeks bijdragen tot DNA-herstel en cellulaire overleving door te interageren
met DNA-PK19
[18], een essentieel onderdeel van de NHEJ (non homologous end joining)
DNA-herstel pathway [19]. Aldus speelt nEGFR een belangrijke rol in verschillende
biologische functies zoals tumorprogressie, DNA-herstel, DNA-replicatie en weerstand aan
bepaalde kankertherapieën [17].
1.3.1.2. EGFR signalering na blootstelling aan ioniserende straling
Naast binding van specifieke liganden kan ook blootstelling aan ioniserende straling (IR) en
oxidatieve stress leiden tot activatie van EGFR. Deze manier van EGFR-activatie is niet
geassocieerd met een proliferatieve celrespons, maar met regulatie van de cellulaire
overleving en herstel van DNA-schade. Blootstelling aan IR resulteert in de snelle stabilisatie
en activatie van Src kinase20
, dat vervolgens caveoline-1 (Cav-1) en EGFR fosforyleert. Dit
induceert internalisatie van EGFR via caveolae, en is geassocieerd met perinucleaire
accumulatie van EGFR en persisterende kinase activiteit. Caveolae bestaan uit associaties van
caveolines met sfingolipiden en cholesterol ter hoogte van de plasmamembraan. Ze vormen
samen met geassocieerde eiwitten het caveosoom, dat kan fuseren met vroege endosomen.
Compartimentatie van EGFR in caveolae voorkomt degradatie en laat intracellulaire EGFR
kinase-gelinkte signalering toe. De stralingsgeïnduceerde Cav-1 en EGFR fosforylaties zijn
bovendien geassocieerd met transport van EGFR naar de nucleus [18]. In bestraalde cellen
wordt nEGFR dan ook in complex gevonden met DNA-PK [19].
1.3.2. Anti-EGFR therapie
Gezien de complexiteit van erbB-gerelateerde signaaltransductie en haar belang voor de
cellulaire groei en overleving, is het niet verwonderlijk dat wijzigingen hierin kunnen
bijdragen tot carcinogenese [14]. EGFR overexpressie wordt dan ook frequent gezien in
humane tumoren [17] en is geassocieerd met een slechte prognose [14]. De epidermale
groeifactor receptor kan daarom beschouwd worden als een zinvol moleculair therapeutisch
doelwit.
18
PCNA: Proliferating cell nuclear antigen 19
DNA-PK: DNA-dependent protein kinase 20
Src-kinase: Proto-oncogene tyrosine-protein kinase Src

9
1.3.2.1. Monoclonale antilichamen versus tyrosine kinase inhibitoren
Binnen de anti-EGFR therapeutica kan men 2 grote klassen onderscheiden. Tyrosine kinase
inhibitoren (TKI) blokkeren de binding van adenosine trifosfaat aan het tyrosine kinase (TK)
domein van EGFR, waardoor ze TK activiteit en intracellulaire signalering blokkeren.
Monoclonale antilichamen (MAB) daarentegen binden ter hoogte van het extracellulair
domein en verhinderen zo ligandgeïnduceerde receptoractivatie [14]. Hoewel beide
benaderingen interfereren met EGFR-gemedieerde signalering, vertonen ze toch een aantal
functionele verschillen. MAB leiden, door inductie van receptorinternalisatie en -degradatie,
tot langdurige neerregulatie van EGFR op de plasmamembraan. TKI binden reversibel ter
hoogte van de kinase actieve site en vertonen deze werking niet. Ook kunnen MAB, in
tegenstelling tot TKI, immuniteitsreacties induceren tegen de tumorcel. Doordat MAB
intraveneus worden toegediend, treden hierbij minder geneesmiddeleninteracties op dan bij
peroraal toegediende TKI. Bovendien gebeuren sommige EGFR functies onafhankelijk van de
TK activiteit. Al deze verschillen in acht genomen, lijken MAB meer geschikt voor
toediening in combinatie met cytotoxische therapieën [13].
1.3.2.2. Anti-EGFR monoclonale antilichamen gebruikt bij therapie voor CRC
Voor de behandeling van geavanceerde chemorefractaire CRC zijn twee anti-EGFR
monoclonale antilichamen goedgekeurd: cetuximab en panitumumab [20].
1.3.2.2.1. Cetuximab
Cetuximab is een recombinant, humaan-muis chimeer immunoglobuline G1 MAB dat
specifiek bindt aan het extracellulair domein van humaan EGFR. Het heeft een hogere
affiniteit voor de receptor dan de endogene liganden en veroorzaakt receptorinternalisatie
zonder stimulatie van receptorfosforylatie. De antitumorale efficaciteit van cetuximab berust
aldus op inhibitie van celcyclusprogressie, promotie van apoptose, antiangiogenese en
verhoging van de immunologische activiteit [12]. In een fase III klinische studie werd
aangetoond dat cetuximab, bij patiënten met geavanceerde EGFR-expresserende CRC die
geen baat bleken te hebben bij eerdere behandelingen met irinotecan, oxaliplatin of
fluoropyrimidines, zowel de totale als de progressievrije overleving kan verhogen. Ook kon
het de achteruitgang van de levenskwaliteit bij deze patiënten beperken [21]. Gezien
moleculair gerichte therapieën een ander toxiciteitsprofiel vertonen dan conventionele
cytotoxische therapieën, kunnen beide gecombineerd worden [13]. Een fase III klinische
studie toonde aan dat additie van cetuximab aan irinotecan leidt tot langere progressievrije

10
overleving, een hogere graad van respons en een betere levenskwaliteit dan irinotecan
monotherapie bij patiënten met EGFR-expresserende metastatische CRC (mCRC) die eerder
onsuccesvol behandeld werden met fluoropyrimidine en oxaliplatin [22]. Additie van
cetuximab aan FOLFIRI voor eerstelijnsbehandeling van mCRC blijkt het risico op
ziekteprogressie te verminderen. Dit voordeel wordt echter enkel gezien bij patiënten met
wildtype KRAS tumoren. Het KRAS-gen codeert voor een G-eiwit dat ligandafhankelijke
receptoractivatie linkt met intracellulaire pathways van de EGFR signaalcascade. Mutaties in
codons 12 en 13 veroorzaken constitutieve activatie van KRAS-geassocieerde signaalpathways
[23], wat de klinische respons op EGFR-inhibitie beperkt. In een fase II klinische studie is
cetuximab ook in combinatie met FOLFOX veilig en actief gebleken voor eerstelijnstherapie
van mCRC. Door verbetering van de resectiegraad kan het de overleving verhogen bij
geavanceerde ziekte [24]. Het gebruik van cetuximab is binnen de Europese Unie (EU)
goedgekeurd voor de behandeling van patiënten met EGFR-expresserende KRAS-wildtype
mCRC in combinatie met irinotecan-gebaseerde chemotherapie of FOLFOX4, of als
monotherapie voor patiënten waarbij eerdere oxaliplatin en irinotecan gebaseerde therapieën
faalden en die intolerant zijn voor irinotecan [25].
1.3.2.2.2. Panitumumab
Panitumumab is een volledig humaan immunoglobuline G2 MAB. Het bindt ter hoogte van
het extracellulaire domein van EGFR en inhibeert zo ligandgeïnduceerde receptoractivatie en
tyrosine kinase fosforylatie. Het vertoont minder immunogeniciteit en veroorzaakt minder
infusiereacties dan cetuximab [14]. Ook voor respons op panitumumab therapie is KRAS-
status van belang [12]. Panitumumab is in de EU goedgekeurd voor eerstelijnsbehandeling
van patiënten met KRAS-wildtype mCRC in combinatie met FOLFOX en voor
tweedelijnsbehandeling in combinatie met FOLFIRI in patiënten die reeds eerstelijns
fluoropyrimidine-gebaseerde chemotherapie kregen (exclusief irinotecan) of als monotherapie
na falen van fluoropyrimidine, oxaliplatin en irinotecan bevattende chemotherapieregimes
[25].
1.3.3. Predictieve merkers voor anti-EGFR therapie
Wegens biologische heterogeniteit zijn veel van de moleculair gerichte therapieën slechts
effectief bij een minderheid der patiënten [20]. Bovendien ontwikkelen initieel responsieve
patiënten na verloop van tijd resistentie. Mogelijke mechanismen voor deze resistentie zijn
activatie van alternatieve receptor tyrosine kinasen die de EGFR pathway omzeilen,

11
constitutieve activatie van signaalpathways downstream van EGFR, EGFR genamplificatie en
receptormutaties [14]. Predictieve merkers bieden de mogelijkheid om op voorhand patiënten
te identificeren die met hoge waarschijnlijkheid baat zullen hebben bij een specifieke
behandeling en zijn bijgevolg van grote klinische waarde. Hun beschikbaarheid leidt tot een
verhoogde therapeutische doeltreffendheid en daling van de toxiciteit, hetgeen zowel de
levenskwaliteit van de patiënten als de kosten voor de gezondheidszorg ten goede komt [20].
De hoge kostprijs en de potentiële toxiciteit van cetuximab maken predictieve merkers van
groot belang voor dit type therapie.
KRAS, een intracellulaire mediator van de signaaltransductie stroomafwaarts van EGFR, is
een predictieve merker voor cetuximab en panitumumab therapie bij CRC. Mutaties in codons
12, 13 of 61, die leiden tot constitutieve activatie van KRAS, maken KRAS signalering
onafhankelijk van EGFR functie [13]. Gezien in diverse studies werd aangetoond dat
patiënten met mutaties in codons 12 of 13 van KRAS geen baat hebben bij cetuximab of
panitumumab therapie, dient de KRAS mutatiestatus in deze codons bepaald te worden bij alle
mCRC patiënten die kandidaat zijn voor deze therapievorm. Indien deze mutaties worden
aangetroffen zal er niet gestart worden met anti-EGFR therapie. Toch zijn deze mutaties in
codons 12 en 13 slechts verantwoordelijk voor hoogstens 40 % van de onresponsieve gevallen
[20]. Er is dus nood aan bijkomende predictieve merkers. Mogelijke merkers voor lage
therapierespons zijn mutaties in codon 61 van KRAS, mutaties in HRAS21
of NRAS22
, mutaties
in genen die coderen voor eiwitten stroomafwaarts van KRAS zoals de BRAF, PI3KCA23
en
PTEN24
genen en verlies van PTEN expressie [20]. Ook het aantal kopijen van EGFR [14],
het tumorale expressieniveau van EGFR liganden en Fcγ25
receptorpolymorfismen van patiënt
immuuncellen zijn mogelijke predictieve merkers [13].
1.3.4. Rationale voor gecombineerde radio-cetuximab therapie
De epidermale groeifactor receptor kan op diverse manieren bijdragen tot de ontwikkeling
van radioresistentie. Drie temporele fasen kunnen worden onderscheiden [15]. Deze worden
geïllustreerd in figuur 2.
21
HRAS: v-Ha-ras Harvey rat sarcoma viral oncogene homolog 22
NRAS: Neuroblastoma RAS viral (v-ras) oncogene homolog 23
PI3KCA: Phosphoinositide-3-kinase, catalytic, alpha polypeptide 24
PTEN: Phosphatase and tensin homolog 25
Fcγ receptor: Immunoglobulin gamma Fc receptor

12
Figuur 2. De EGFR-gemedieerde radioprotectie bestaat uit 3 temporele fasen. In een eerste fase interageert
nEGFR met DNA-PK (A). In een tweede fase leidt snelle activatie van de PI3K-Akt signaalbaan tot
onderdrukking van apoptose (B). Tot slot dragen de Ras/Raf/Mek/Erk en STAT pathways bij tot tumor
repopulatie door stimulatie van cellulaire proliferatie (C). Overgenomen uit Chen et al. [15].
In een eerste vroege fase leidt interactie van de nucleaire EGFR met DNA-PKcs26
tot herstel
van DNA dubbelstrengbreuken. In een tweede fase zorgt stralingsgeïnduceerde activatie van
EGFR voor activering van de PI3K-Akt pathway, die leidt tot blokkering van apoptotische
signaalpathways. Tot slot biedt activatie van de Ras-MAPK en STAT signaalpathway de
tumorcellen een kritisch overlevingsvoordeel door ondersteuning van tumorrepopulatie en -
progressie [15]. Dit maakt dat combinatie van cetuximab, dat EGFR-signalering en transport
van EGFR naar de nucleus blokkeert, met radiotherapie een zinvolle benadering is.
1.4. Radiotherapie
Radiotherapie (RT) maakt gebruik van ioniserende straling (IR) en wordt frequent toegepast
als eerstelijnsbehandeling met curatieve intentie voor primaire kankers en als palliatieve
therapie voor patiënten met geavanceerde metastatische ziekte [15]. Ioniserende straling is
deeltjes- of elektromagnetische straling die voldoende energie bezit om 1 of meerdere orbitale
elektronen te verwijderen van de atomen of moleculen waarmee ze interageert. Via diverse
absorptiemechanismen wordt het merendeel van de energie van de fotonen omgezet in
kinetische energie van secundaire elektronen. IR kan op een directe manier interageren met
kritische cellulaire doelwitstructuren en deze ioniseren of exciteren. Gezien het hoge
26
DNA-PKcs: DNA-dependent protein kinase catalytic subunit

13
watergehalte van cellen zal het grootste deel van de neergezette energie echter geabsorbeerd
worden in water. Deze indirecte werking van IR leidt tot vorming van vrije radicalen, die
wegens de aanwezigheid van een ongepaard elektron zeer reactief zijn en op hun beurt de
kritische doelwitten kunnen beschadigen [26]. Figuur 3 geeft een overzicht van de directe en
indirecte werking van ioniserende straling.
Figuur 3. De directe en indirecte werking van ioniserende straling. Bij de directe werking interageert het
secundaire elektron rechtstreeks met de doelwitten. Bij de indirecte werking interageren de vrije radicalen
geproduceerd door radiolyse van water met de doelwitten. Figuur overgenomen uit [26].
Het hoofddoel van RT is de destructie van het reproductiepotentieel van tumorcellen, wat kan
gebeuren door inductie van celdood. Dit kan optreden door apoptotische mechanismen, maar
zal bij solide tumoren vooral geschieden door mitotische celdood, een vorm van celdood die
optreedt tijdens of als resultaat van foutieve mitose [27].
1.5. Detectie en herstel van stralingsgeïnduceerde DNA-schade
Gezien DNA haar meest kritische doelwit is [7], geeft ioniserende straling aanleiding tot
diverse vormen van DNA-schade [26]. DNA dubbelstrengbreuken (DSB) vormen zich als het
resultaat van 2 enkelstrengbreuken in tegenoverliggende DNA strengen in elkaars nabijheid,
gewoonlijk binnen de 10 tot 20 basenparen [28]. Hoewel ze weinig frequent voorkomen in
vergelijking met andere vormen van DNA-schade, vormen DSB een bedreiging voor de
genomische integriteit en de cellulaire overleving. Dit is te wijten aan hun moeilijke herstel
door gebrek aan een intacte templatestreng en aan de verstoring van de continuïteit van de
DNA-molecule [29]. Aldus vormen DSB de meest cytotoxische vorm van DNA-schade.
Cellen beschikken dan ook over diverse herstelmechanismen voor deze vorm van schade [30].

14
1.5.1. DNA-schade checkpoints
Zoogdiercellen beschikken over DNA-schade checkpoints die werkzaam zijn bij de overgang
van de G1- naar de S-fase, tijdens de S-fase en bij de overgang van de G2- naar de M-fase
[31]. Deze checkpoints zorgen, na detectie van onherstelde DSB, voor een vertraging van de
celcyclusprogressie, waardoor DNA-herstel mogelijk wordt [32]. Ze bestaan uit complexe
fosforylatiecascades [32] en de deelnemende eiwitten kunnen onderverdeeld worden als
schadesensoren, signaalmediatoren, signaaltransducers en effectoren [31].
Belangrijke sensoren zijn het MRN27
complex en het Ku7028
/Ku8029
heterodimeer [31]. Na
detectie van DSB recruteert het MRN complex het ATM30
transducerkinase, dat
checkpointactivatie triggert. Ook stimuleert ATM DSB-processing door nucleasen, die de
vorming van enkelstrengig DNA katalyseren. Vervolgens wordt dit enkelstrengig DNA
gebonden door RPA31
, waarna RPA via de cofactor ATRIP32
herkend wordt door het
transducerkinase ATR33
. Volledige activatie van ATR vereist ook RPA-gemedieerde
rekrutering van het RAD9-RAD1-HUS134
complex [32]. ATM en ATR vormen samen met
DNA-PKcs de PIKK35
kinasefamilie. Rekrutering van DNA-PKcs naar de DSB gebeurt door
zijn interactie met de sensor Ku70/Ku80 [31]. Uiteindelijk worden door de transducerkinasen
effectorkinasen geactiveerd die transcriptiefactoren, celcyclusregulerende eiwitten en DSB-
herstelfactoren als doelwitten hebben [32].
Daarnaast staan de drie transducerkinasen van de PIKK familie in voor fosforylatie van histon
H2AX ter hoogte van serine 139, wat dan wordt aangeduid als γH2AX [31,32]. γH2AX wordt
vervolgens gebonden door signaalmediatoren. MDC136
is een dergelijke mediator die door
constitutieve interactie met het MRN complex ATM activeert [31]. Dit veroorzaakt
amplificatie van het γH2AX signaal [33], wat leidt tot stabiele accumulatie van diverse
eiwitten betrokken bij DSB-herstel [32].
27
MRN: Complex bestaande uit de Mre11, Rad50 en NBS1 eiwitten 28
Ku70: X-ray repair cross-complementing protein 6 29
Ku80: X-ray repair cross-complementing protein 5 30
ATM: Ataxia telangiectasia mutated 31
RPA: Replication protein A 32
ATRIP: ATR interacting protein 33
ATR: Ataxia telangiectasia and Rad3 related 34
RAD9-RAD1-HUS1: Complex gevormd door de RAD1, RAD9 en HUS1eiwitten 35
PIKK: Phosphatidylinositol 3-kinase-related kinases 36
MDC1: Mediator of DNA damage checkpoint protein 1

15
1.5.2. Herstel van DNA-dubbelstrengbreuken
Hogere eukaryoten beschikken over twee belangrijke systemen om cellen te beschermen
tegen stralingsgeïnduceerde DNA-schade: ‘homologe recombinatie’ (HR) en ‘non-
homologous end joining’ (NHEJ) [15]. HR vereist de aanwezigheid van homologe sequenties
[33] en is beperkt tot de S/G2 fasen van de celcyclus [30,32]. NHEJ daarentegen werkt
onafhankelijk van homologie voor de hereniging van DSB-uiteinden [33] en is mogelijk
gedurende de volledige celcyclus [30,32]. Hoewel beide pathways samenwerken, vormt
NHEJ de dominante pathway voor herstel van stralingsgeïnduceerde DSB in eukaryoten [15].
Bijgevolg wordt hier enkel ingegaan op deze pathway. Het principe van NHEJ wordt
weergegeven in figuur 4.
Figuur 4. Principe van NHEJ. Associatie van Ku eiwitten faciliteert de recrutering van DNA-PKcs, wat
processing door Artemis en daaropvolgende hereniging van de DSB uiteinden door het DNA ligase IV-XRCC4-
XLF complex promoot. Dit leidt finaal tot herstel van de genomische integriteit zonder verzekering van
sequentiebehoud. Overgenomen uit Mladenov et al [29].
DSB-inductie veroorzaakt binding van de Ku70 en Ku80 eiwitten ter hoogte van de DSB
[29,30,34], welke alignering van beide uiteinden promoten [33]. Het MRN-complex speelt
hierin mogelijks ook een rol, maar lijkt niet essentieel bij hogere eukaryoten [32]. Ku
transloceert 1 helicale draai inwaarts, recruteert vervolgens DNA-PKcs, stabiliseert zijn
binding op DNA en activeert zijn kinase functie [15,33]. DNA-PKcs bindt en activeert het

16
endonuclease Artemis. Verder fosforyleert DNA-PKcs de XLF37
, DNA ligase IV, XRCC438
,
WRN39
en RPA240
eiwitten. Bovendien treedt er DNA-PKcs autofosforylatie op, wat leidt tot
een conformationele verandering waardoor processing van de DSB-uiteinden (door TdT41
en
PNK42
), polymerisatie (door Polymerasen µ en λ) en ligatie (door een complex gevormd door
XLF, XRCC4 en DNA ligase IV) mogelijk worden. Vermits IR-geïnduceerde DSB vaak
geassocieerd zijn met zowel schade aan de pentosefosfaatruggengraat als met basenschade ter
hoogte van de terminale nucleotiden, is processing meestal vereist alvorens ligatie kan
gebeuren. Modificaties van DNA uiteinden die op deze manier ontstaan, kunnen leiden tot
inserties en deleties ter hoogte van de gevormde junctie, wat de mogelijke inductie van
sequentiewijzigingen door NHEJ verklaart [29]. Hoewel de NHEJ-componenten
onafhankelijk van elkaar kunnen werken, werken ze synergistisch in elkaars nabijheid [34].
Dit maakt NHEJ tot een zeer dynamische reactie [32].
1.5.3. De γ-H2AX foci assay
Ioniserende straling geïnduceerde DSB leiden tot lokale fosforylatie ter hoogte van het sterk
geconserveerde SQEY-motief43
van H2AX, een substraat voor de PIKK eiwitfamilie.
Significante signaalamplificatie treedt op, waardoor het γH2AX signaal zich verspreidt naar
naburige chromatineregio’s [28]. De ontwikkeling van een antilichaam tegen γH2AX maakte
het mogelijk om H2AX fosforylatie te detecteren. Dit laat in situ detectie toe van DNA-
schade en –herstel in individuele cellen [35]. Bij klinisch relevante dosissen is DSB detectie
via de γH2AX foci assay minstens een factor 100 gevoeliger dan alternatieve methoden.
Doordat γH2AX de novo gevormd word, is het bovendien een meer betrouwbare merker van
DSB dan hersteleiwitten die ook aanwezig zijn in afwezigheid van beschadigd DNA [28].
1.6. Doel van de studie
Het doel van deze studie was om de DNA-herstelkinetiek van HT-29 colonadenocarcinoma
cellen na gecombineerde radio-cetuximab therapie te bepalen. In vervolgonderzoek kunnen de
gegevens bekomen in deze pilootstudie gecorreleerd worden met therapierespons, om zo na te
37
XLF: Synoniem voor NHEJ1, non-homologous end-joining factor 1 38
XRCC4: X-ray repair complementing defective repair in Chinese hamster cells 4 39
WRN: Werner syndrome, RecQ helicase-like 40
RPA2: Replication protein A2, 32kDa 41
TdT: Terminal deoxynucleotidyltransferase 42
PNK: Polynucleotide kinase 43
SQEY-motief: Aminozuursequentiemotief bestaande uit serine, glutamine, glutamaat en tyrosine

17
gaan of DNA-herstelkinetiek al dan niet bruikbaar is als predictieve merker voor radio-
cetuximab therapie bij patiënten met colorectale kanker.

18
2. Materialen en methoden
2.1. Celcultuur
2.1.1. Oorsprong van de HT-29 cellijn
De HT-29 cellijn werd verkregen van de dienst gastro-enterologie van het Universitair
Ziekenhuis Gent. Deze dienst kocht de cellijn aan bij de Deutsche Sammlung von
Mikroorganismen und Zellkulturen GmbH (DSMZ). De HT-29 cellijn is een adherente cellijn
afkomstig van een 44-jarige caucasische vrouw met colon adenocarcinoma.
2.1.2. Kweekomstandigheden van de HT-29 cellijn
De HT-29 cellijn werd gekweekt in polystyreen filtercap cultuurflessen met oppervlakten van
25 of 75 cm² (Thermo Scientific). De kweekomstandigheden waren 37°C, 5% CO2 en 100%
luchtvochtigheid. Het groeimedium bestond uit Mc Coy’s 5A medium, aangerijkt met 10%
FBS44
en 1% antibiotica-antimycotica 100X (allen Invitrogen).
2.1.3. Onderhoud van de HT-29 cellijn
Om de twee dagen werd het groeimedium ververst. Subculturen werden aangelegd bij het
bereiken van circa 95% confluentie. Hiervoor werden, na verwijdering van het groeimedium,
de cellen tweemaal gespoeld met D-PBS45
(Sigma Aldrich), waarna gedurende 10 seconden
trypsine-EDTA46
(Invitrogen) aan de cellen werd toegevoegd. Vervolgens werd de falcon
gedurende 5 minuten te rusten gelegd. Om dissociatie van de cellen te bevorderen werd een
aantal keer met de falcon op het tafelblad geklopt. Indien de cellen moeilijk loskwamen werd
gebruik gemaakt van een cell scraper (BD). Tot slot werd vers groeimedium aan de cellen
toegevoegd, waarna de celsuspensie in de gewenste verhouding werd overgebracht naar een
nieuwe falcon. Groeimedium, D-PBS en trypsine-EDTA werden telkens op 37°C gebracht
vóór toevoeging aan de culturen. De cellen werden nooit langer dan 30 passages in cultuur
gehouden.
2.1.4. Periodieke controle op mycoplasmacontaminatie
Mycoplasma is de veelgebruikte triviale naam voor micro-organismen van de klasse
Mollicutes. Het zijn de kleinste vrijlevende bacteriële vormen die wijdverspreid zijn in de
natuur. Gedurende de evolutie hebben mycoplasma’s zowel hun bacteriële celwand als enkele
metabolische pathways verloren. Door deze structurele en metabolische deficiënties is
44
FBS: Foetal bovine serum 45
D-PBS: Dulbecco’s phosphate buffered saline 46
EDTA: Ethylenediaminetetraacetic acid

19
replicatie van mycoplasma grotendeels afhankelijk van de aanwezigheid van voedingsstoffen
in de omgeving. Wegens hun brede verspreiding zijn mycoplasma’s een frequente oorzaak
van celcultuurcontaminatie [36]. Hun aanwezigheid uit zich niet steeds door macroscopische
veranderingen in de cellen of media. Ook met routinemicroscopie is detectie niet
vanzelfsprekend. Veel mycoplasmacontaminanten groeien traag zonder de gastheercel te
vernietigen [37]. Ze veroorzaken echter wel wijzigingen in de biologische eigenschappen van
de gecontamineerde cellen [36]. Vermits dit de betrouwbaarheid van experimentele resultaten
kan ondermijnen, werd maandelijks getest op de aanwezigheid van mycoplasmacontaminatie.
2.1.4.1. Principe van de test
Voor detectie van mycoplasma contaminatie werd gebruik gemaakt van de MycoAlert
detectiekit (Lonza). Mycoplasmatische enzymen reageren met MycoAlert substraat, wat leidt
tot katalysering van de omzetting van ADP47
tot ATP48
. Bepaling van de ATP-niveau’s
gebeurt door middel van een bioluminiscente reactie, weergegeven in figuur 5.
Luciferase, Mg2+
ATP + Luciferine + O2 Oxyluciferine + AMP + PPi + CO2 + Licht
Figuur 5. Bioluminiscente reactie gebruikt voor de bepaling van de ATP niveaus. De hoeveelheid uitgezonden
licht is lineair gerelateerd aan de ATP-concentratie. AMP en PPi staan respectievelijk voor adenosine
monophosphate en pyrophosphate. O2, Mg2+
en CO2 staan respectievelijk voor zuurstofgas, een tweewaardig
positief magnesium atoom en koolstofdioxide. Figuur overgenomen uit [38].
Bepaling van de ATP-niveaus voor en na toevoeging van het substraat geeft een ratio die
indicatief is voor de aan- of afwezigheid van mycoplasma in het staal [38].
2.1.4.2. Uitvoering van de test
Groeimedium dat minstens 3 dagen op de te testen cultuur had gestaan werd gedurende 5
minuten gecentrifugeerd aan 330 g. Van elk staal werd 100 µl supernatans overgebracht naar
een well in een witte FluoroNunc module met Maxisorp oppervlakte (Thermo Scientific). Er
werden ook 2 wells gevuld met een positief en negatief controlestaal van de MycoAlert Assay
Control Set (Lonza). Aan elk staal werd 100 µl MycoAlert Reagens (Lonza) toegevoegd. Na
resuspendering en incubatie gedurende 5 minuten werd de eerste meting uitgevoerd. De
meting gebeurde met de Microlumat LB96P luminometer (EG&G Berthold) en WinGlow
software (EG&G Berthold). Vervolgens werd aan elk staal 100 µl MycoAlert Substraat
47
ADP: Adenosine diphosphate 48
ATP: Adenosine triphosphate

20
(Lonza) toegevoegd. Na resuspendering en incubatie gedurende 10 minuten werd de meting
herhaald. De verhouding van de luminiscentie van de tweede meting tot die van de eerste
meting werd bepaald. Bij een verhouding groter dan 1 werd het staal positief beschouwd voor
mycoplasma.
2.1.5. Invriezen van de HT-29 cellijn
Voor het invriezen van de HT-29 cellen werd de procedure van de aanleg van subculturen
gevolgd tot het punt waarop er vers groeimedium in de falcon gebracht wordt (zie sectie 2.1.3,
‘Onderhoud van de HT-29 cellijn’). De bekomen celsuspensie werd vervolgens overgebracht
naar een tube en gecentrifugeerd aan 330 g gedurende 5 minuten. Na weggieten van het
supernatans werd de pellet opgelost in vriesmedium bestaande uit Mc Coy’s 5A medium
aangerijkt met 20% FBS en 10% DMSO49
(Sigma-Aldrich). De oplossing werd daarna
verdeeld over cryotubes (Thermo Scientific), welke dan gedurende minstens 4 uur in een
CoolCell (BioCision) bij -80°C geplaatst werden. Vervolgens werden de cryovials
overgebracht naar een vat met vloeibare stikstof voor lange termijn stockering.
2.1.6. Ontdooien en opstarten van de HT-29 cellijn
De bevroren HT-29 cellen werden ontdooid door druppelsgewijze toevoeging van tot 37°C
opgewarmd groeimedium. Vervolgens werd de celsuspensie gecentrifugeerd gedurende 5
minuten aan 330 g. Na afgieten van het supernatans werd de pellet opgelost in groeimedium
en verdeeld over twee 25 cm² cultuurfalcons in de verhoudingen 1:5 en 4:5.
2.1.7. Kwantificatie van de HT-29 cellen
Voor kwantificatie van de cellen werd 100µl celsuspensie in een epje gebracht. Hieraan werd
100 µl trypaanblauw 0.4% (Sigma-Aldrich) toegevoegd. Na resuspendering werd deze
oplossing in de Bürker telkamer (Marienfeld) gebracht. Per 50 vakjes werden alle levende
cellen geteld, gelegen volledig binnen het vakje of rakend aan de rechtse of onderste lijn van
het vakje. Deze telling werd in totaal viermaal herhaald en de gemiddelde waarde, n, werd
berekend. De gebruikte formule om deze waarde te herleiden tot het aantal cellen in de
celsuspensie wordt weergegeven in figuur 6.
49
DMSO: Dimethylsulfoxide

21
s
Figuur 6. Formule gebruikt ter omzetting van het gemiddelde getelde celaantal per 50 vakjes van de Bürker
telkamer, n, naar het aantal cellen in het totale volume celsuspensie (Vs). TB staat voor trypaanblauw.
2.2. Constructie van een groeicurve
Voor de constructie van een groeicurve werden de HT-29 cellen op diverse tijdstippen na
subcultuur geteld (zie secties 2.1.3, ‘onderhoud van de HT-29 cellijn’ en 2.1.7, ‘Kwantificatie
van de HT-29 cellen’). Met behulp van Excel 2010 (Microsoft) werden de bekomen
celaantallen op semilogaritmische schaal uitgezet in functie van het aantal uren na subcultuur.
Aan de hand van de punten gelegen binnen het exponentiële deel van de groeicurve werden de
vergelijking en de determinatiecoëfficiënt van de best passende exponentiële trendlijn
bepaald. In de bekomen vergelijking werden 2 celaantallen ingevuld die bereikt werden in het
exponentiële deel van de groeicurve, en waarvan de ene waarde het dubbele is van de andere.
Uit het verschil tussen beide bekomen tijdstippen werd de verdubbelingstijd verkregen. De
duur van de lagfase werd bekomen door invulling van de hoeveelheid cellen op tijdstip 0 in de
vergelijking van de trendlijn.
2.3. Synchronisatie in G0/G1 fase van de celcyclus
2.3.1. Optimalisatie van de synchronisatieprocedure
Synchronisatie van de cellen in de G0/G1 fase van de celcyclus werd bekomen via
serumdeprivatie. Diverse condities werden uitgetest om de optimale methode te ontdekken.
Voor de optimalisatie-experimenten werd de periode waarin de cellen in normaal
groeimedium groeiden gevarieerd tussen 24 of 48 uur. De serumconcentratie in het
vervangende medium werd gevarieerd tussen 0 (SSM 0%), 0.1 (SSM 0.1%) of 10 procent (N
medium) en de periode waarin de cellen geïncubeerd werden in dit vervangende medium
werd gevarieerd tussen 24 en 48 uur. Telkens werd de synchronisatietoestand bepaald.
2.3.2. Controle van de synchronisatietoestand
Voor de bepaling van de synchronisatietoestand werd gebruik gemaakt van de Coulter DNA
Prep Reagents Kit (Beckman Coulter). Aan 100 µl celsuspensie werd eenzelfde volume
lysebuffer toegevoegd. Na mengen werd hieraan 1 ml DNA-stain oplossing toegevoegd,
waarna de oplossing opnieuw werd gemengd. Vervolgens werd de oplossing in het donker en
op kamertemperatuur bewaard gedurende 15 minuten tot maximaal 3 uren. Na deze incubatie

22
werd de DNA-index bepaald met behulp van de FACSCanto II flow cytometer (BD) en
FACSDiva v.6.1.3 software (BD). De flowrate werd op laag ingesteld. Als controlestaal werd
een humaan bloedstaal gebruikt dat een identieke behandeling onderging met lysebuffer en
DNA-stain oplossing. Bij analyse van dit controlestaal werd de PE50
waarde aangepast tot de
singlet piek zich ter hoogte van de waarde 50 bevond. Vervolgens werd de analyse van de
HT-29 cellen uitgevoerd met dezelfde PE-waarde als het controlestaal. De flow cytometer en
de Coulter DNA Prep Reagents Kit werden ter beschikking gesteld door prof. dr. Jan Philippé
van het laboratorium voor klinische biologie.
2.3.3. Synchronisatieprocedure voorafgaand aan de experimenten
Voorafgaand aan elk experiment werden de cellen gesynchroniseerd. Hiervoor werden 5 tot
7.5 miljoen cellen uitgezaaid per 75 cm² falcon in normaal groeimedium (N medium)
bestaande uit Mc Coy’s 5A medium met 10% FBS en 1% antibiotica-antimycotica 100X. Na
24 uur werd het medium verwijderd en werden de cellen tweemaal gespoeld met D-PBS.
Hierna werd medium toegevoegd dat enkel bestaat uit Mc Coy’s 5A medium met 1 %
antibiotica-antimycotica 100X (SSM 0%). Bij de DNA-herstelkinetiek experimenten werd
voor de hersteltijden langer dan 1 uur SSM 2% (bestaande uit Mc Coy’s 5A medium met 2%
FBS en 1% antibiotica-antimycotica 100X) gebruikt omdat dit in het aangepaste
cultuursysteem aanleiding gaf tot betere cellulaire morfologie. Na 24 uur incubatie in het
verarmde medium werden de cellen getrypsiniseerd en heropgelost in SSM 0% medium (of in
SSM 2% voor de lange herstelpunten). Ten slotte werd de celcyclusdistributie geanalyseerd.
2.4. Dosisrespons experimenten
2.4.1. De γH2AX foci assay voor dosisrespons experimenten
De gesynchroniseerde HT-29 cellen werden geoogst door trypsinisatie en heropgelost in SSM
0% aan een concentratie van 1.2 x 106 cellen per 2 ml. De 2 ml culturen werden bestraald met
60Co γ-straling (Gammatron R, Siemens) aan een dosis van 0.2, 0.5, 1, 1.5 of 2 Gy in een
warmwaterbad op 37°C. Er werd ook een onbestraald controlestaal geïncorporeerd in het
experiment. Per conditie werden 3 preparaten gemaakt. Na de bestraling werden de culturen
gedurende 15 minuten op 37°C geplaatst om DNA-herstel toe te laten. Dit DNA-herstel werd
gestopt door de culturen gedurende 10 minuten op ijs te plaatsen. Vervolgens werd 0.5 ml van
de celsuspensie gecentrifugeerd op poly-l-lysine gecoate slides (VWR International) door
middel van cytospin centrifugatie (Sigma). Nadien werden de slides gedurende minstens 20
50
PE: Phyco-erythrin

23
minuten gefixeerd in 3% PFA51
(Sigma-Aldrich). Bewaring overnacht gebeurde in 0.5% PFA
op 4°C. De volgende dag werden de gefixeerde cellen gewassen met D-PBS gedurende 5
minuten, waarna ze gedurende 10 minuten geïncubeerd werden met ijskoude Triton X-100
(Fluka Analytical). Vervolgens werden de cellen driemaal gedurende 10 minuten gewassen
met D-PBS met 1% BSA52
(Roche). Na de cellen te hebben bedekt met 100 µl van een 1:500
verdunning van het anti-γ-H2AX antilichaam (Biolegend) in D-PBS met 1% BSA, werden de
slides gedurende 1 uur in een vochtige kamer bewaard. De vochtige kamer bestaat uit een
grote petri-plaat waarin enkele vochtige tissues gelegd worden. Na opnieuw drie wasstappen
van 10 minuten met D-PBS met 1% BSA, werden de cellen bedekt met 100 µl van een 1:1000
verdunning van RAM-TRITC53
antilichaam (dakoCytomation) in D-PBS met 1% BSA. Na 1
uur incubatie in een verduisterde vochtige kamer volgden drie wasstappen van 10 minuten
met D-PBS. Tot slot werd 35 µl van een 200 ng DAPI54
(Sigma-Aldrich) per ml fluoromount
(Sigma-Aldrich) oplossing op de slides gebracht, en werden de slides bedekt met een proper
dekglaasje (Knittel Glass). De slides werden bewaard in het donker bij 4°C. Om uitdroging te
voorkomen werden de randen van de dekglaasjes bedekt met nagellak.
2.5. DNA-herstelkinetiek experimenten
Alvorens de eigenlijke DNA-herstelkinetiek experimenten te starten werden 20 cm x 20 cm
dekglaasjes (Menzel-Gläser) gecoat met poly-l-lysine. Een oplossing van 50 µl poly-l-lysine
(Sigma-Aldrich) per ml steriel water werd gesteriliseerd door ze doorheen een 0.22µm filter
(Millipore) te spuiten. De door droge autoclavering gesteriliseerde dekglaasjes werden in de
gesteriliseerde poly-l-lysine oplossing gespoeld en in een 6-well plaat (BD) geplaatst.
Vervolgens werd per well 2 ml steriele poly-l-lysine oplossing toegevoegd. Na een incubatie
van 1 uur bij 37°C werd de poly-l-lysine oplossing uit de wells verwijderd en werden de wells
vijfmaal gespoeld met steriel water. De platen werden overnacht te drogen gelegd in een
droge incubator. Ten slotte werden de 6-well platen omwikkeld met parafilm en bewaard bij
4°C.
2.5.1. De γH2AX foci assay na bestraling
De gesynchroniseerde HT-29 cellen werden geoogst door trypsinisatie en heropgelost in SSM
2% aan een concentratie van 1.2 x 106 cellen per 2 ml. De 2 ml culturen werden bestraald met
51
PFA: Paraformaldehyde 52
BSA: Bovine serum albumin 53
RAM-TRITC: Tetramethyl rhodamine isothiocyanate conjugated rabbit anti-mouse antibody 54
DAPI: 4’,6-diamidino-2-phenylindole

24
60Co γ-straling aan een dosis van 0.5 Gy in een warmwaterbad op 37°C. Er werden ook
onbestraalde controlestalen geïncorporeerd in het experiment. Per conditie werden 3
preparaten gemaakt. Na de bestraling werden de culturen gedurende diverse perioden (15
minuten, 2 uur, 4 uur) op 37°C geplaatst om DNA-herstel toe te laten.
Voor de hersteltijden van 2 uur en 4 uur werden de 2 ml culturen op de vooraf met poly-l-
lysine gecoate dekglaasjes in een 6-well plaat gebracht.
DNA-herstel werd gestopt door de culturen gedurende 10 minuten op ijs te plaatsen.
Vervolgens werd voor het herstelpunt van 15 minuten 0.5 ml van de celsuspensie
gecentrifugeerd op poly-l-lysine gecoate slides door middel van cytospin centrifugatie. Bij de
lange herstelpunten werd het medium uit de wells verwijderd en werden de dekglaasjes
tweemaal gespoeld met D-PBS.
De cellen werden vervolgens gedurende 20 minuten gefixeerd in 3% PFA. Bewaring
overnacht gebeurde in 0.5% PFA bij 4°C. De volgende dag werden de gefixeerde cellen
gewassen met D-PBS gedurende 5 minuten, waarna ze gedurende 10 minuten geïncubeerd
werden met ijskoude Triton X-100. Vervolgens werden de cellen driemaal gedurende 10
minuten gewassen met D-PBS met 1% BSA (Roche). Na de cellen te hebben bedekt met 100
µl van een 1:500 verdunning van het anti-γ-H2AX antilichaam (Biolegend) in D-PBS met 1%
BSA, werden de slides gedurende 1 uur in een vochtige kamer bewaard. Na opnieuw drie
wasstappen van 10 minuten met D-PBS met 1% BSA, werden de cellen bedekt met 100 µl
van een 1:1000 verdunning van RAM-TRITC antilichaam (dakoCytomation) in D-PBS met
1% BSA. Na 1 uur incubatie in een verduisterde vochtige kamer volgden drie wasstappen van
10 minuten met D-PBS.
De slides van het 15 minuten herstelpunt werden bedekt met 35 µl van een 200 ng DAPI per
ml fluoromount oplossing, waarna ze bedekt werden met een proper dekglaasje. De
dekglaasjes van de lange herstelpunten werden na de laatste van de drie wasstappen met D-
PBS uit de 6-well plaat verwijderd. Er werd 35 µl van een 200 ng DAPI per ml fluoromount
oplossing op poly-l-lysine gecoate slides gebracht, waarna de dekglaasjes hier omgekeerd
werden opgelegd.
Bewaring van de slides gebeurde in het donker bij 4°C. Om uitdroging te voorkomen werden
de randen van de dekglaasjes bedekt met nagellak.
2.5.2. De γH2AX foci assay na gecombineerde radio-cetuximab therapie
Een eerste experiment gebeurde analoog aan het DNA-herstelkinetiek experiment waar enkel
bestraling werd toegepast. Nu werd 4 uur voor trypsinisatie van de cellen 1µM cetuximab

25
(Merck) toegevoegd per 1 x 106 cellen. Het aantal γH2AX foci werd enkel bepaald na 15
minuten en na 4 uur DNA-herstel. Cetuximab werd ter beschikking gesteld door prof. dr.
Stéphanie Laurent van de dienst gastro-enterologie van het UZ Gent.
Voor een volgend DNA-herstelkinetiek experiment werden de cellen uitgezaaid op vooraf met
poly-l-lysine gecoate dekglaasjes in 6-well platen in plaats van in 75 cm² cultuurfalcons. De
cellen werden in deze platen gesynchroniseerd door 24 uur incubatie in N medium, gevolgd
door 24 uur incubatie in SSM 2%. Om het celaantal te bepalen dat na de
synchronisatieprocedure aanleiding zou geven tot een geschikt aantal cellen voor het γH2AX
foci experiment, werd een deelexperiment opgezet. Hierbij werden diverse celaantallen
uitgezaaid op vooraf met poly-l-lysine gecoate dekglaasjes in een 6 well plaat. Na doorlopen
van de synchronisatieprocedure werden de culturen visueel geïnspecteerd. Er dienden
voldoende cellen aanwezig te zijn op de dekglaasjes zonder elkaar te overlappen. Op basis
van dit deelexperiment werd gekozen om 400 000 cellen uit te zaaien per well. Vier uur voor
bestraling met 0.5 Gy 60
Co γ-straling werd 1µM cetuximab toegevoegd per 1 x 106 cellen. In
het experiment werden ook controlestalen geïncorporeerd die niet werden blootgesteld aan
cetuximab of 60
Co γ-straling.
2.6. Analyse van de γH2AX foci
2.6.1. Automatische analyse
Automatische analyse werd uitgevoerd met de Metacyte module van het Metafer 4
scanningssysteem (Metasystems) in combinatie met een Zeiss Axio Imager Z.2 microscoop
(Carl Zeiss MicroImaging) met Plan-Neofluar 40x/0.75 M27 objectief (Carl Zeiss
International). Gezien dit systeem nog niet eerder gebruikt werd voor de kwantificatie van
γH2AX foci in HT-29 cellen, drong een optimalisatieprocedure specifiek voor dit celtype zich
op. Er werd hiermee gestart, maar volledige optimalisatie bleek te tijdrovend voor het
tijdsbestek van deze masterproef. Automatische analyse werd bijgevolg enkel uitgevoerd voor
het dosisrespons experiment. Andere experimenten werden manueel geanalyseerd.
2.6.2. Manuele analyse
Manuele analyse werd uitgevoerd met het ChromoScan systeem (Applied Imaging),
bestaande uit een Olympus BX60 fluoroscentiemicroscoop met UPIan FL 100x/1.30 oil
objectief (Olympus) en Cytovision v2.8 software (Applied Imaging). Per beeld werd het
TRITC-signaal opgenomen op 10 optische secties van 1.03 µm volgens de z-as. Het finale

26
beeld werd bekomen door alle secties te projecteren op 1 vlak en dit te combineren met de
DAPI kleuring, waarvoor geen focusvlakken werden gebruikt. Per preparaat werden 100
cellen geteld. Voor elk van deze 100 cellen werd het aantal γH2AX foci geteld. Met behulp
van Excel 2010 (Microsoft) werd, per preparaat, het gemiddeld aantal foci per cel bepaald en
uitgezet in functie van de dosis (voor dosisresponsrespons experimenten) of in functie van de
periode van DNA-herstel (voor DNA-herstelkinetiek experimenten).
2.7. Statistische analysen
De resultaten werden verwerkt met behulp van Office Excel 2010 (Microsoft). Voor elk
preparaat werd het gemiddeld aantal γH2AX foci per cel bepaald. Vervolgens werden de
gemiddelde waarde en de standaarddeviatie voor de verschillende preparaten van dezelfde
conditie bepaald. Dit gemiddelde werd voor elke conditie gecorrigeerd met de gemiddelde
waarde voor de controlestalen. De standaardfout werd bekomen door middel van
onderstaande formule.
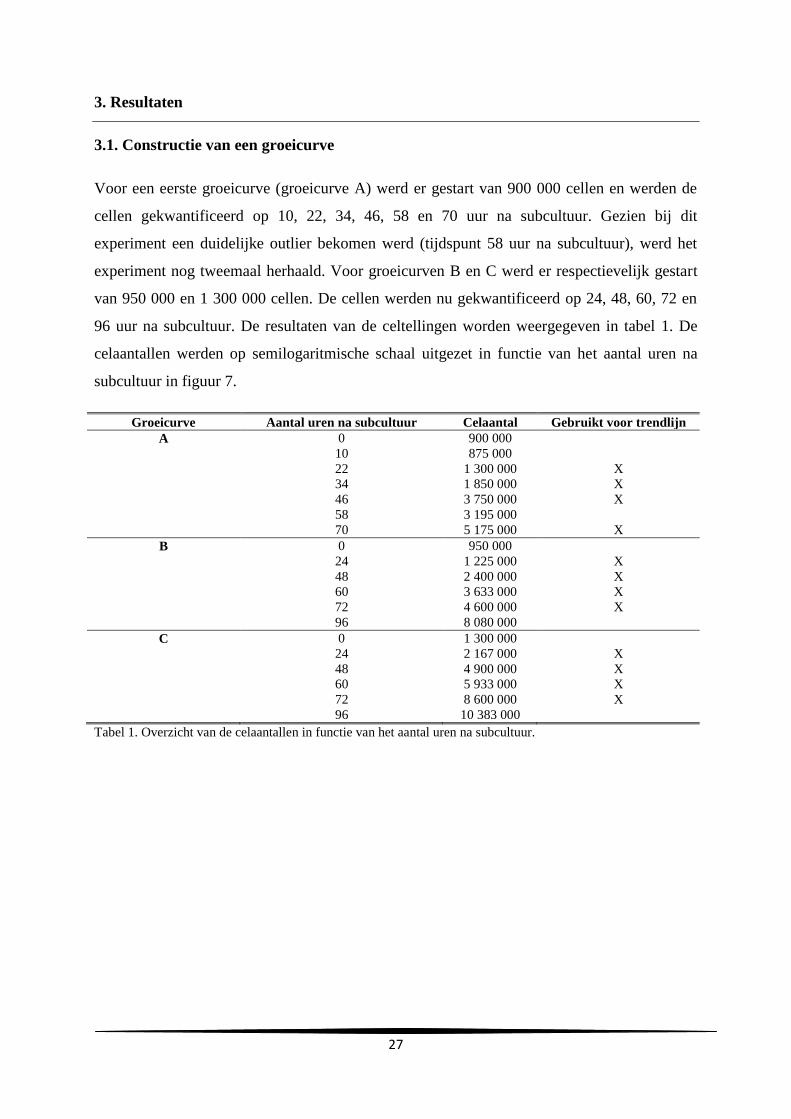
27
3. Resultaten
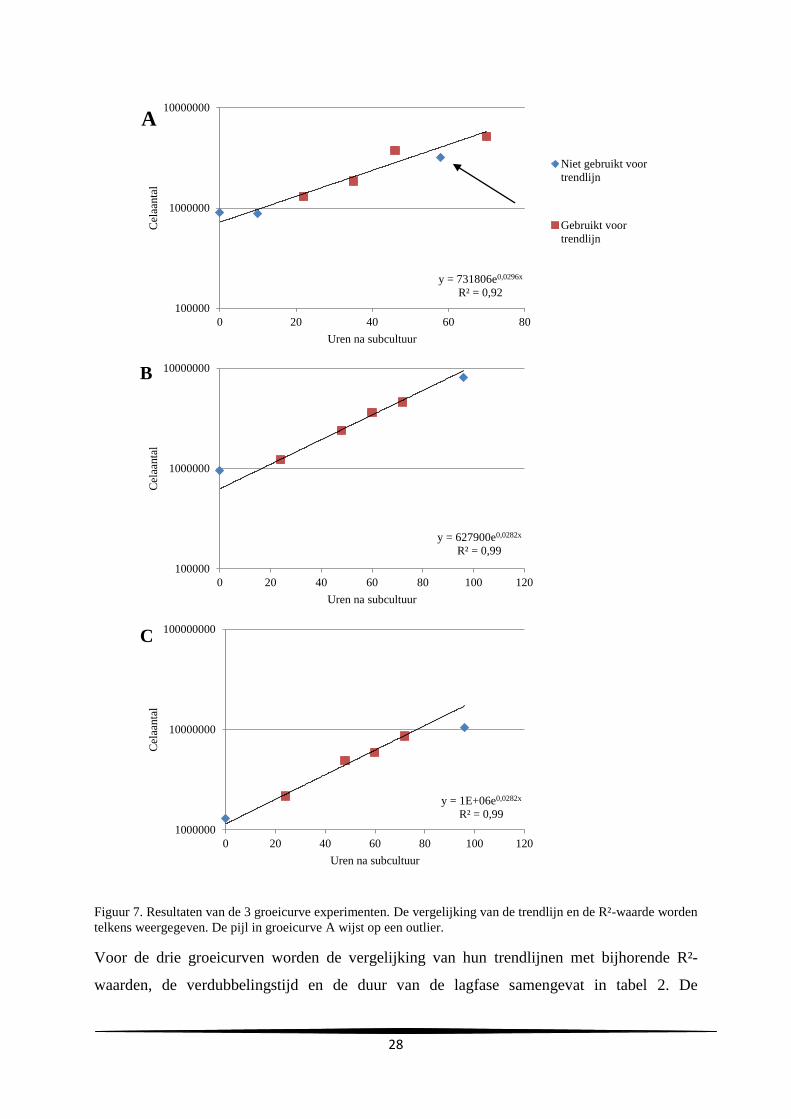
3.1. Constructie van een groeicurve
Voor een eerste groeicurve (groeicurve A) werd er gestart van 900 000 cellen en werden de
cellen gekwantificeerd op 10, 22, 34, 46, 58 en 70 uur na subcultuur. Gezien bij dit
experiment een duidelijke outlier bekomen werd (tijdspunt 58 uur na subcultuur), werd het
experiment nog tweemaal herhaald. Voor groeicurven B en C werd er respectievelijk gestart
van 950 000 en 1 300 000 cellen. De cellen werden nu gekwantificeerd op 24, 48, 60, 72 en
96 uur na subcultuur. De resultaten van de celtellingen worden weergegeven in tabel 1. De
celaantallen werden op semilogaritmische schaal uitgezet in functie van het aantal uren na
subcultuur in figuur 7.
Groeicurve Aantal uren na subcultuur Celaantal Gebruikt voor trendlijn
A 0 900 000
10 875 000
22 1 300 000 X
34 1 850 000 X
46 3 750 000 X
58 3 195 000
70 5 175 000 X
B 0 950 000
24 1 225 000 X
48 2 400 000 X
60 3 633 000 X
72 4 600 000 X
96 8 080 000
C 0 1 300 000
24 2 167 000 X
48 4 900 000 X
60 5 933 000 X
72 8 600 000 X
96 10 383 000
Tabel 1. Overzicht van de celaantallen in functie van het aantal uren na subcultuur.
28
Figuur 7. Resultaten van de 3 groeicurve experimenten. De vergelijking van de trendlijn en de R²-waarde worden
telkens weergegeven. De pijl in groeicurve A wijst op een outlier.
Voor de drie groeicurven worden de vergelijking van hun trendlijnen met bijhorende R²-
waarden, de verdubbelingstijd en de duur van de lagfase samengevat in tabel 2. De
y = 731806e0,0296x
R² = 0,92
100000
1000000
10000000
0 20 40 60 80
Cel
aanta
l
Uren na subcultuur
Niet gebruikt voor
trendlijn
Gebruikt voor
trendlijn
y = 627900e0,0282x
R² = 0,99
100000
1000000
10000000
0 20 40 60 80 100 120
Cel
aanta
l
Uren na subcultuur
B
y = 1E+06e0,0282x
R² = 0,99
1000000
10000000
100000000
0 20 40 60 80 100 120
Cel
aanta
l
Uren na subcultuur
C
A
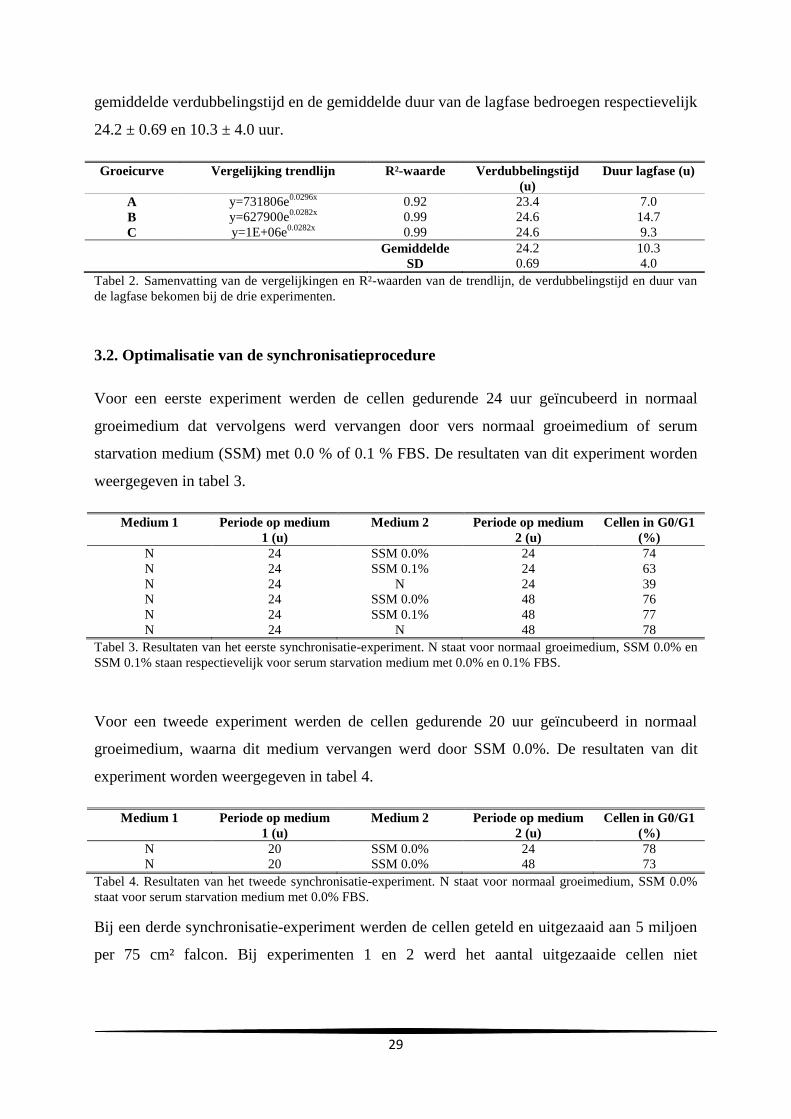
29
gemiddelde verdubbelingstijd en de gemiddelde duur van de lagfase bedroegen respectievelijk
24.2 ± 0.69 en 10.3 ± 4.0 uur.
Groeicurve Vergelijking trendlijn R²-waarde Verdubbelingstijd
(u)
Duur lagfase (u)
A y=731806e0.0296x
0.92 23.4 7.0
B y=627900e0.0282x
0.99 24.6 14.7
C y=1E+06e0.0282x
0.99 24.6 9.3
Gemiddelde
SD
24.2
0.69
10.3
4.0
Tabel 2. Samenvatting van de vergelijkingen en R²-waarden van de trendlijn, de verdubbelingstijd en duur van
de lagfase bekomen bij de drie experimenten.
3.2. Optimalisatie van de synchronisatieprocedure
Voor een eerste experiment werden de cellen gedurende 24 uur geïncubeerd in normaal
groeimedium dat vervolgens werd vervangen door vers normaal groeimedium of serum
starvation medium (SSM) met 0.0 % of 0.1 % FBS. De resultaten van dit experiment worden
weergegeven in tabel 3.
Medium 1 Periode op medium
1 (u)
Medium 2 Periode op medium
2 (u)
Cellen in G0/G1
(%)
N 24 SSM 0.0% 24 74
N 24 SSM 0.1% 24 63
N 24 N 24 39
N 24 SSM 0.0% 48 76
N 24 SSM 0.1% 48 77
N 24 N 48 78
Tabel 3. Resultaten van het eerste synchronisatie-experiment. N staat voor normaal groeimedium, SSM 0.0% en
SSM 0.1% staan respectievelijk voor serum starvation medium met 0.0% en 0.1% FBS.
Voor een tweede experiment werden de cellen gedurende 20 uur geïncubeerd in normaal
groeimedium, waarna dit medium vervangen werd door SSM 0.0%. De resultaten van dit
experiment worden weergegeven in tabel 4.
Medium 1 Periode op medium
1 (u)
Medium 2 Periode op medium
2 (u)
Cellen in G0/G1
(%)
N 20 SSM 0.0% 24 78
N 20 SSM 0.0% 48 73
Tabel 4. Resultaten van het tweede synchronisatie-experiment. N staat voor normaal groeimedium, SSM 0.0%
staat voor serum starvation medium met 0.0% FBS.
Bij een derde synchronisatie-experiment werden de cellen geteld en uitgezaaid aan 5 miljoen
per 75 cm² falcon. Bij experimenten 1 en 2 werd het aantal uitgezaaide cellen niet
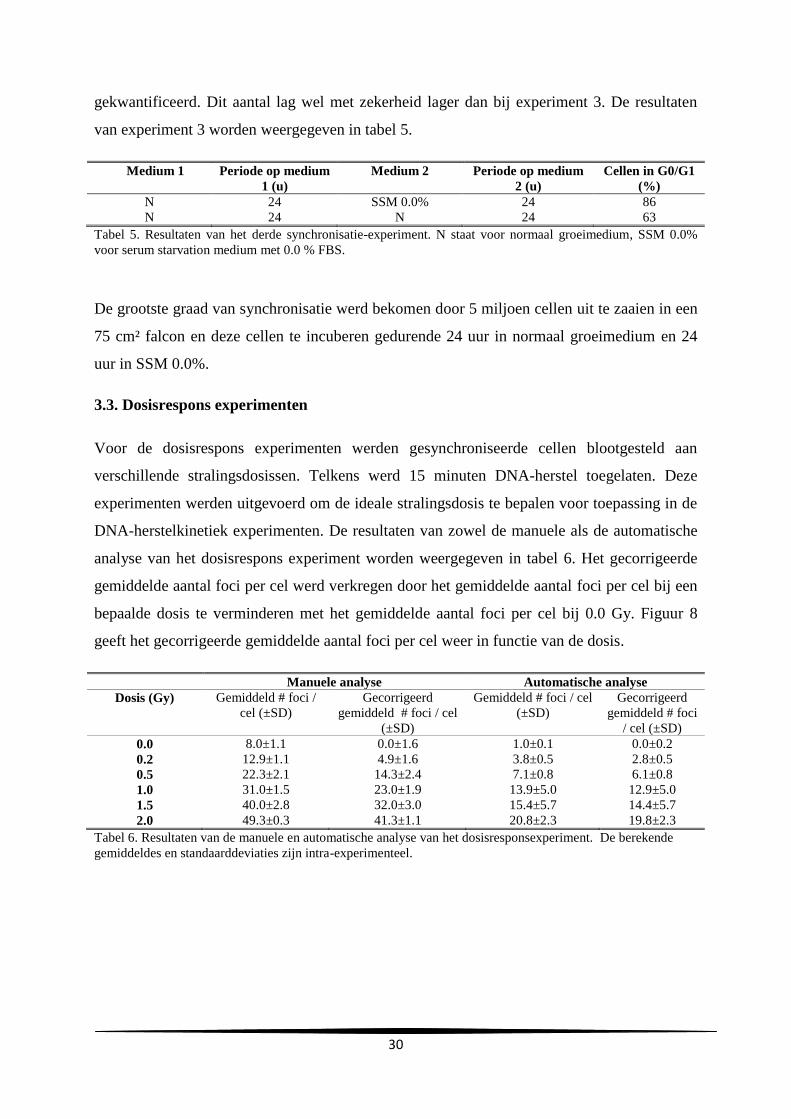
30
gekwantificeerd. Dit aantal lag wel met zekerheid lager dan bij experiment 3. De resultaten
van experiment 3 worden weergegeven in tabel 5.
Medium 1 Periode op medium
1 (u)
Medium 2 Periode op medium
2 (u)
Cellen in G0/G1
(%)
N 24 SSM 0.0% 24 86
N 24 N 24 63
Tabel 5. Resultaten van het derde synchronisatie-experiment. N staat voor normaal groeimedium, SSM 0.0%
voor serum starvation medium met 0.0 % FBS.
De grootste graad van synchronisatie werd bekomen door 5 miljoen cellen uit te zaaien in een
75 cm² falcon en deze cellen te incuberen gedurende 24 uur in normaal groeimedium en 24
uur in SSM 0.0%.
3.3. Dosisrespons experimenten
Voor de dosisrespons experimenten werden gesynchroniseerde cellen blootgesteld aan
verschillende stralingsdosissen. Telkens werd 15 minuten DNA-herstel toegelaten. Deze
experimenten werden uitgevoerd om de ideale stralingsdosis te bepalen voor toepassing in de
DNA-herstelkinetiek experimenten. De resultaten van zowel de manuele als de automatische
analyse van het dosisrespons experiment worden weergegeven in tabel 6. Het gecorrigeerde
gemiddelde aantal foci per cel werd verkregen door het gemiddelde aantal foci per cel bij een
bepaalde dosis te verminderen met het gemiddelde aantal foci per cel bij 0.0 Gy. Figuur 8
geeft het gecorrigeerde gemiddelde aantal foci per cel weer in functie van de dosis.
Manuele analyse Automatische analyse
Dosis (Gy) Gemiddeld # foci /
cel (±SD)
Gecorrigeerd
gemiddeld # foci / cel
(±SD)
Gemiddeld # foci / cel
(±SD)
Gecorrigeerd
gemiddeld # foci
/ cel (±SD)
0.0 8.0±1.1 0.0±1.6 1.0±0.1 0.0±0.2
0.2 12.9±1.1 4.9±1.6 3.8±0.5 2.8±0.5
0.5 22.3±2.1 14.3±2.4 7.1±0.8 6.1±0.8
1.0 31.0±1.5 23.0±1.9 13.9±5.0 12.9±5.0
1.5 40.0±2.8 32.0±3.0 15.4±5.7 14.4±5.7
2.0 49.3±0.3 41.3±1.1 20.8±2.3 19.8±2.3
Tabel 6. Resultaten van de manuele en automatische analyse van het dosisresponsexperiment. De berekende
gemiddeldes en standaarddeviaties zijn intra-experimenteel.
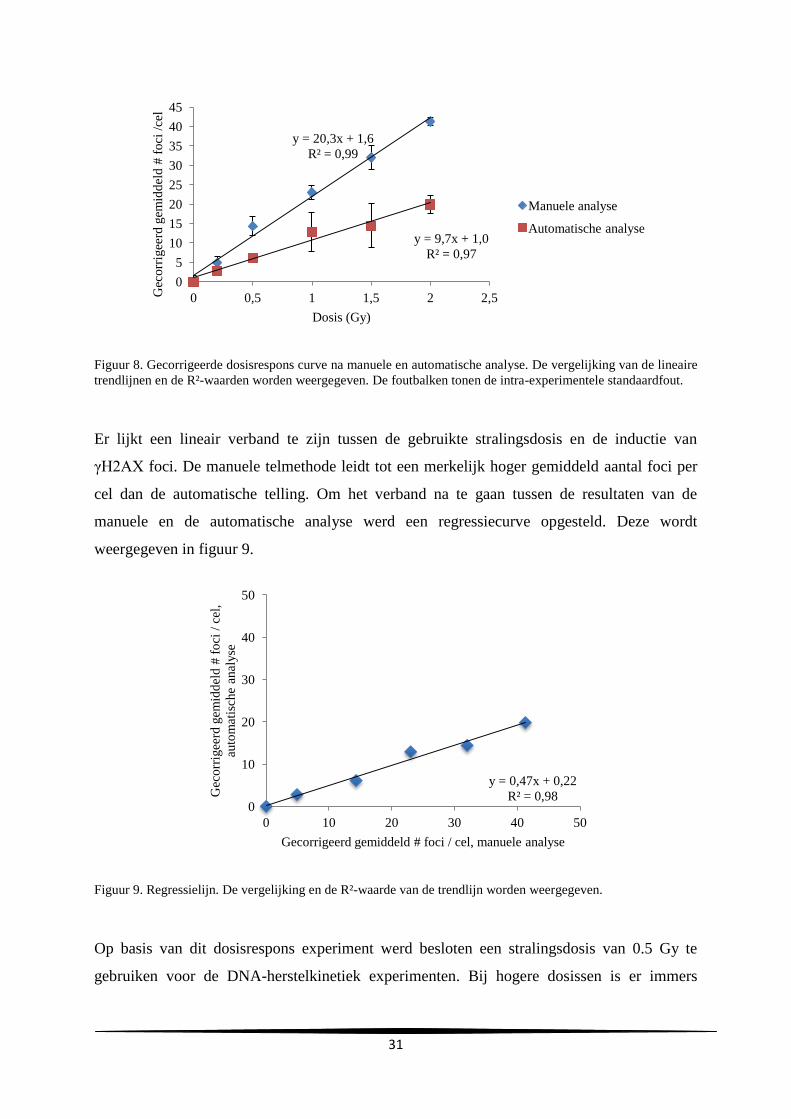
31
Figuur 8. Gecorrigeerde dosisrespons curve na manuele en automatische analyse. De vergelijking van de lineaire
trendlijnen en de R²-waarden worden weergegeven. De foutbalken tonen de intra-experimentele standaardfout.
Er lijkt een lineair verband te zijn tussen de gebruikte stralingsdosis en de inductie van
γH2AX foci. De manuele telmethode leidt tot een merkelijk hoger gemiddeld aantal foci per
cel dan de automatische telling. Om het verband na te gaan tussen de resultaten van de
manuele en de automatische analyse werd een regressiecurve opgesteld. Deze wordt
weergegeven in figuur 9.
Figuur 9. Regressielijn. De vergelijking en de R²-waarde van de trendlijn worden weergegeven.
Op basis van dit dosisrespons experiment werd besloten een stralingsdosis van 0.5 Gy te
gebruiken voor de DNA-herstelkinetiek experimenten. Bij hogere dosissen is er immers
y = 20,3x + 1,6
R² = 0,99
y = 9,7x + 1,0
R² = 0,97
0
5
10
15
20
25
30
35
40
45
0 0,5 1 1,5 2 2,5 Gec
orr
igee
rd g
emid
del
d #
fo
ci /
cel
Dosis (Gy)
Manuele analyse
Automatische analyse
y = 0,47x + 0,22
R² = 0,98 0
10
20
30
40
50
0 10 20 30 40 50
Gec
orr
igee
rd g
emid
del
d #
fo
ci /
cel
,
auto
mat
isch
e an
alyse
Gecorrigeerd gemiddeld # foci / cel, manuele analyse
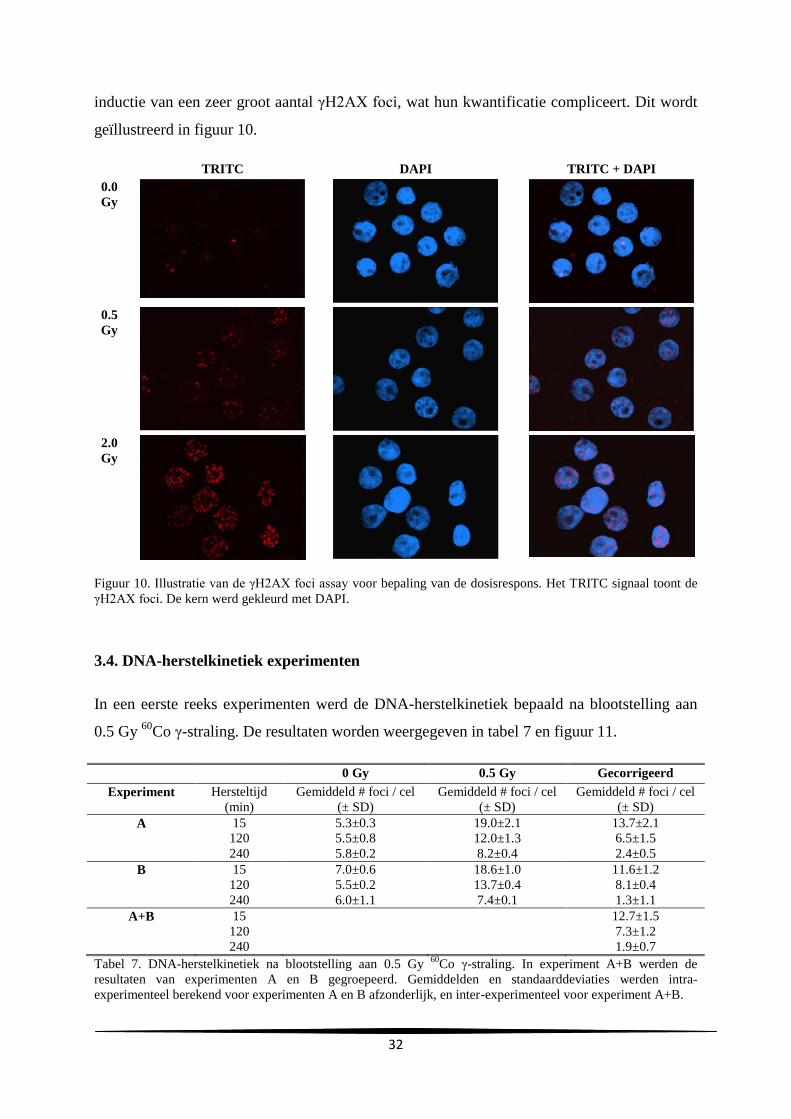
32
inductie van een zeer groot aantal γH2AX foci, wat hun kwantificatie compliceert. Dit wordt
geïllustreerd in figuur 10.
TRITC DAPI TRITC + DAPI
0.0
Gy
0.5
Gy
2.0
Gy
Figuur 10. Illustratie van de γH2AX foci assay voor bepaling van de dosisrespons. Het TRITC signaal toont de
γH2AX foci. De kern werd gekleurd met DAPI.
3.4. DNA-herstelkinetiek experimenten
In een eerste reeks experimenten werd de DNA-herstelkinetiek bepaald na blootstelling aan
0.5 Gy 60
Co γ-straling. De resultaten worden weergegeven in tabel 7 en figuur 11.
0 Gy 0.5 Gy Gecorrigeerd
Experiment Hersteltijd
(min)
Gemiddeld # foci / cel
(± SD)
Gemiddeld # foci / cel
(± SD)
Gemiddeld # foci / cel
(± SD)
A 15 5.3±0.3 19.0±2.1 13.7±2.1
120 5.5±0.8 12.0±1.3 6.5±1.5
240 5.8±0.2 8.2±0.4 2.4±0.5
B 15 7.0±0.6 18.6±1.0 11.6±1.2
120 5.5±0.2 13.7±0.4 8.1±0.4
240 6.0±1.1 7.4±0.1 1.3±1.1
A+B 15 12.7±1.5
120 7.3±1.2
240 1.9±0.7
Tabel 7. DNA-herstelkinetiek na blootstelling aan 0.5 Gy 60
Co γ-straling. In experiment A+B werden de
resultaten van experimenten A en B gegroepeerd. Gemiddelden en standaarddeviaties werden intra-
experimenteel berekend voor experimenten A en B afzonderlijk, en inter-experimenteel voor experiment A+B.
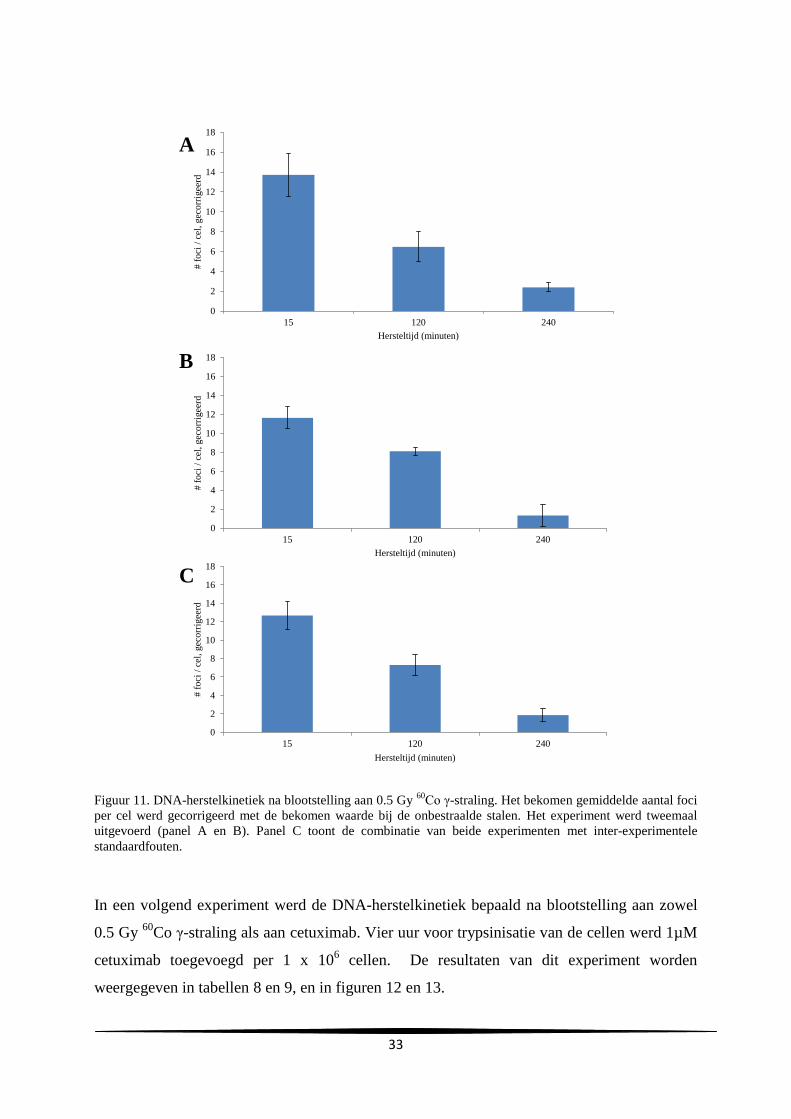
33
Figuur 11. DNA-herstelkinetiek na blootstelling aan 0.5 Gy 60
Co γ-straling. Het bekomen gemiddelde aantal foci
per cel werd gecorrigeerd met de bekomen waarde bij de onbestraalde stalen. Het experiment werd tweemaal
uitgevoerd (panel A en B). Panel C toont de combinatie van beide experimenten met inter-experimentele
standaardfouten.
In een volgend experiment werd de DNA-herstelkinetiek bepaald na blootstelling aan zowel
0.5 Gy 60
Co γ-straling als aan cetuximab. Vier uur voor trypsinisatie van de cellen werd 1µM
cetuximab toegevoegd per 1 x 106 cellen. De resultaten van dit experiment worden
weergegeven in tabellen 8 en 9, en in figuren 12 en 13.
0
2
4
6
8
10
12
14
16
18
15 120 240
# f
oci
/ c
el,
gec
orr
igee
rd
Hersteltijd (minuten)
0
2
4
6
8
10
12
14
16
18
15 120 240
# f
oci
/ c
el,
gec
orr
igee
rd
Hersteltijd (minuten)
0
2
4
6
8
10
12
14
16
18
15 120 240
# f
oci
/ c
el,
gec
orr
igee
rd
Hersteltijd (minuten)
A
B
C
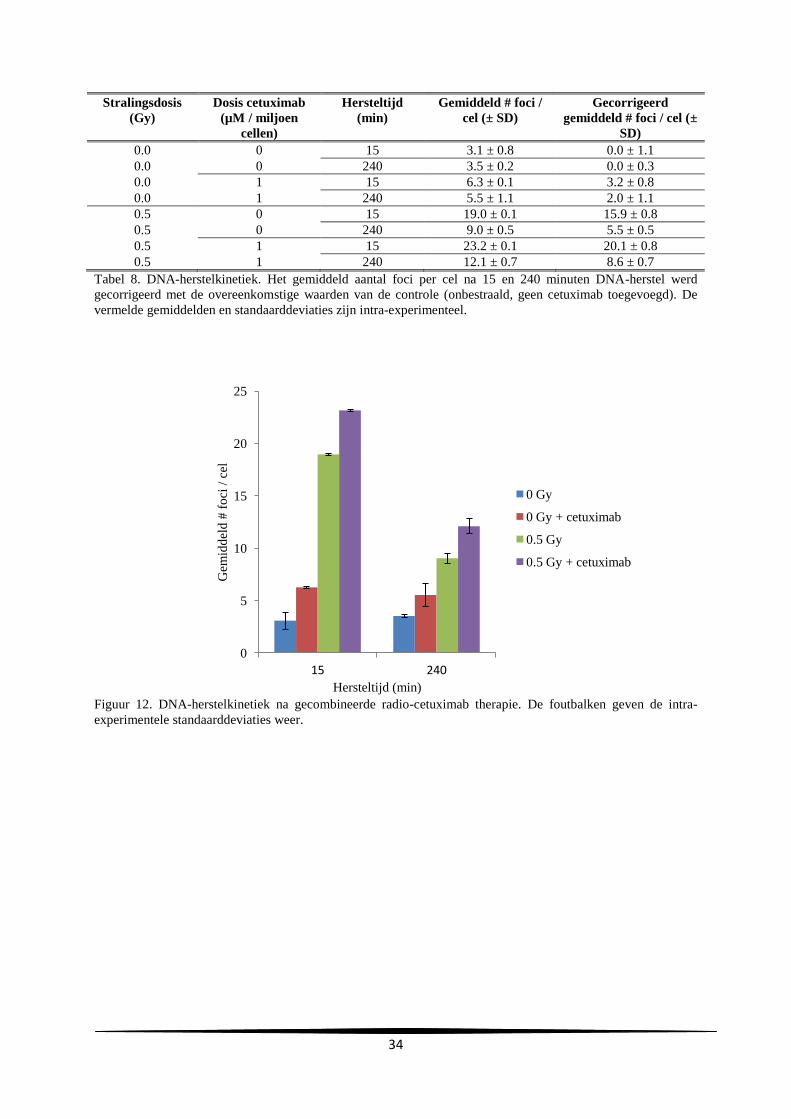
34
Stralingsdosis
(Gy)
Dosis cetuximab
(µM / miljoen
cellen)
Hersteltijd
(min)
Gemiddeld # foci /
cel (± SD)
Gecorrigeerd
gemiddeld # foci / cel (±
SD)
0.0 0 15 3.1 ± 0.8 0.0 ± 1.1
0.0 0 240 3.5 ± 0.2 0.0 ± 0.3
0.0 1 15 6.3 ± 0.1 3.2 ± 0.8
0.0 1 240 5.5 ± 1.1 2.0 ± 1.1
0.5 0 15 19.0 ± 0.1 15.9 ± 0.8
0.5 0 240 9.0 ± 0.5 5.5 ± 0.5
0.5 1 15 23.2 ± 0.1 20.1 ± 0.8
0.5 1 240 12.1 ± 0.7 8.6 ± 0.7
Tabel 8. DNA-herstelkinetiek. Het gemiddeld aantal foci per cel na 15 en 240 minuten DNA-herstel werd
gecorrigeerd met de overeenkomstige waarden van de controle (onbestraald, geen cetuximab toegevoegd). De
vermelde gemiddelden en standaarddeviaties zijn intra-experimenteel.
Figuur 12. DNA-herstelkinetiek na gecombineerde radio-cetuximab therapie. De foutbalken geven de intra-
experimentele standaarddeviaties weer.
0
5
10
15
20
25
15 240
Gem
idd
eld
# f
oci
/ c
el
Hersteltijd (min)
0 Gy
0 Gy + cetuximab
0.5 Gy
0.5 Gy + cetuximab
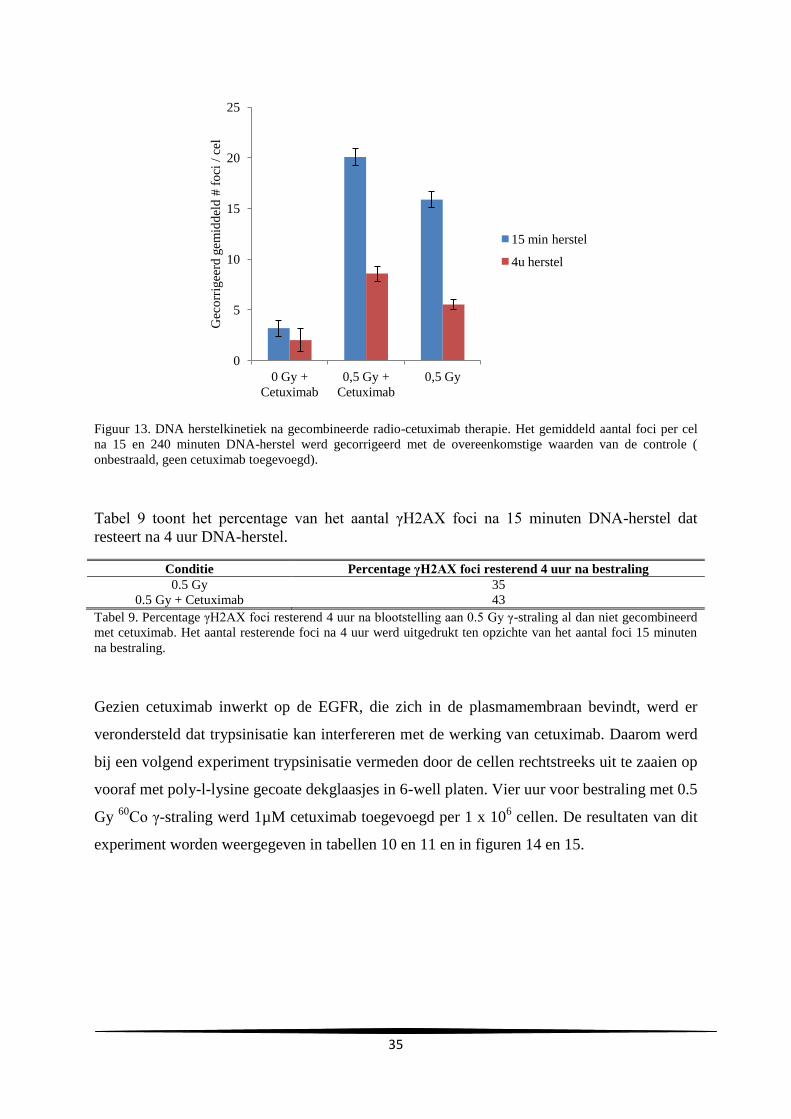
35
Figuur 13. DNA herstelkinetiek na gecombineerde radio-cetuximab therapie. Het gemiddeld aantal foci per cel
na 15 en 240 minuten DNA-herstel werd gecorrigeerd met de overeenkomstige waarden van de controle (
onbestraald, geen cetuximab toegevoegd).
Tabel 9 toont het percentage van het aantal γH2AX foci na 15 minuten DNA-herstel dat
resteert na 4 uur DNA-herstel.
Conditie Percentage γH2AX foci resterend 4 uur na bestraling
0.5 Gy 35
0.5 Gy + Cetuximab 43
Tabel 9. Percentage γH2AX foci resterend 4 uur na blootstelling aan 0.5 Gy γ-straling al dan niet gecombineerd
met cetuximab. Het aantal resterende foci na 4 uur werd uitgedrukt ten opzichte van het aantal foci 15 minuten
na bestraling.
Gezien cetuximab inwerkt op de EGFR, die zich in de plasmamembraan bevindt, werd er
verondersteld dat trypsinisatie kan interfereren met de werking van cetuximab. Daarom werd
bij een volgend experiment trypsinisatie vermeden door de cellen rechtstreeks uit te zaaien op
vooraf met poly-l-lysine gecoate dekglaasjes in 6-well platen. Vier uur voor bestraling met 0.5
Gy 60
Co γ-straling werd 1µM cetuximab toegevoegd per 1 x 106 cellen. De resultaten van dit
experiment worden weergegeven in tabellen 10 en 11 en in figuren 14 en 15.
0
5
10
15
20
25
0 Gy +
Cetuximab
0,5 Gy +
Cetuximab
0,5 Gy
Gec
orr
igee
rd g
emid
del
d #
fo
ci /
cel
15 min herstel
4u herstel
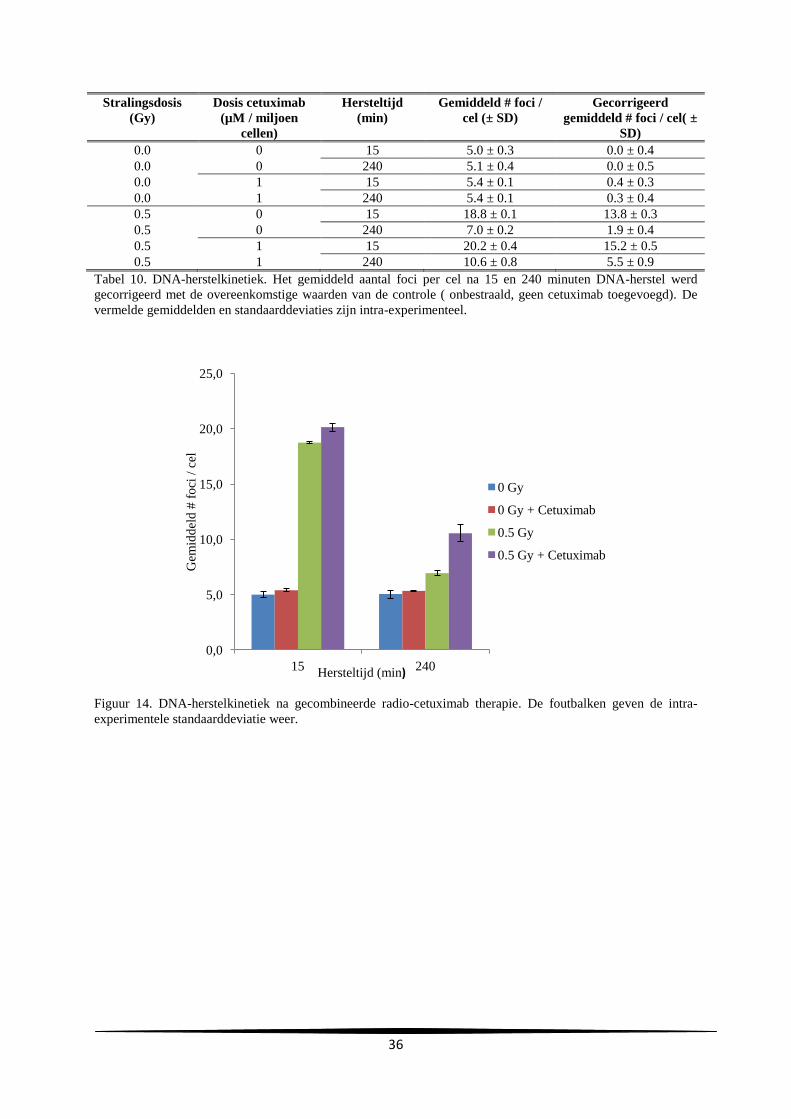
36
Stralingsdosis
(Gy)
Dosis cetuximab
(µM / miljoen
cellen)
Hersteltijd
(min)
Gemiddeld # foci /
cel (± SD)
Gecorrigeerd
gemiddeld # foci / cel( ±
SD)
0.0 0 15 5.0 ± 0.3 0.0 ± 0.4
0.0 0 240 5.1 ± 0.4 0.0 ± 0.5
0.0 1 15 5.4 ± 0.1 0.4 ± 0.3
0.0 1 240 5.4 ± 0.1 0.3 ± 0.4
0.5 0 15 18.8 ± 0.1 13.8 ± 0.3
0.5 0 240 7.0 ± 0.2 1.9 ± 0.4
0.5 1 15 20.2 ± 0.4 15.2 ± 0.5
0.5 1 240 10.6 ± 0.8 5.5 ± 0.9
Tabel 10. DNA-herstelkinetiek. Het gemiddeld aantal foci per cel na 15 en 240 minuten DNA-herstel werd
gecorrigeerd met de overeenkomstige waarden van de controle ( onbestraald, geen cetuximab toegevoegd). De
vermelde gemiddelden en standaarddeviaties zijn intra-experimenteel.
Figuur 14. DNA-herstelkinetiek na gecombineerde radio-cetuximab therapie. De foutbalken geven de intra-
experimentele standaarddeviatie weer.
0,0
5,0
10,0
15,0
20,0
25,0
15 240
Gem
idd
eld
# f
oci
/ c
el
Hersteltijd (min)
0 Gy
0 Gy + Cetuximab
0.5 Gy
0.5 Gy + Cetuximab
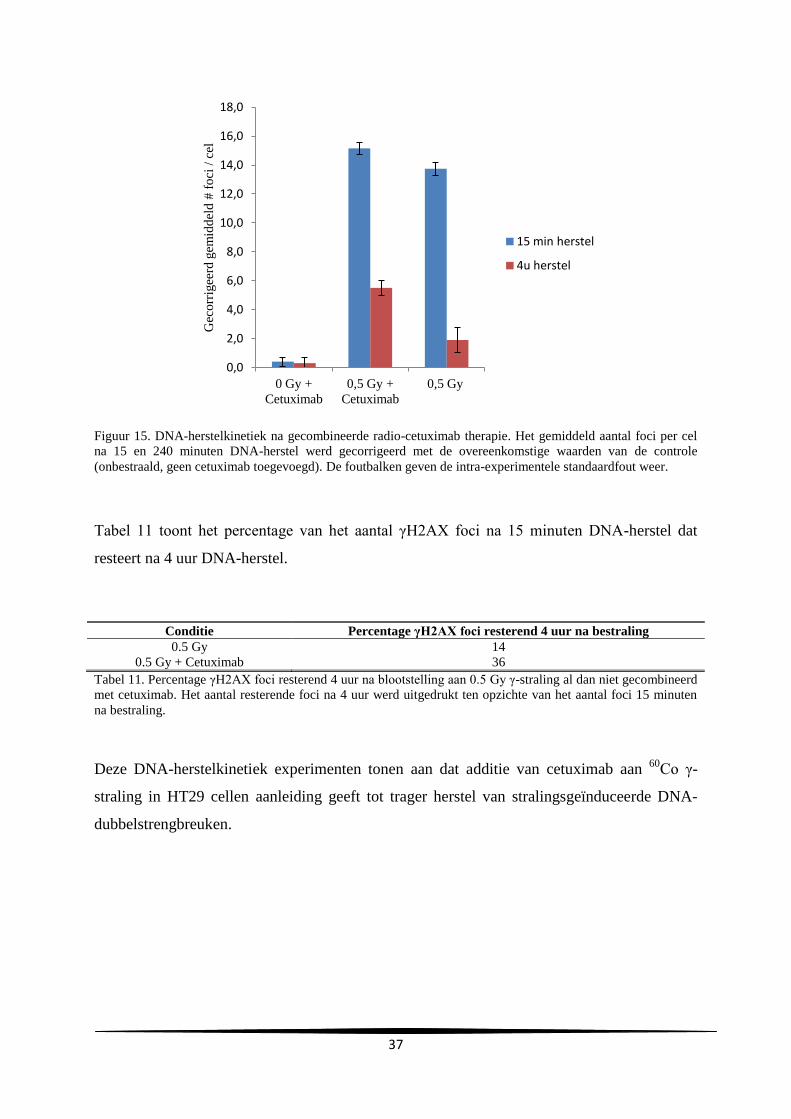
37
Figuur 15. DNA-herstelkinetiek na gecombineerde radio-cetuximab therapie. Het gemiddeld aantal foci per cel
na 15 en 240 minuten DNA-herstel werd gecorrigeerd met de overeenkomstige waarden van de controle
(onbestraald, geen cetuximab toegevoegd). De foutbalken geven de intra-experimentele standaardfout weer.
Tabel 11 toont het percentage van het aantal γH2AX foci na 15 minuten DNA-herstel dat
resteert na 4 uur DNA-herstel.
Conditie Percentage γH2AX foci resterend 4 uur na bestraling
0.5 Gy 14
0.5 Gy + Cetuximab 36
Tabel 11. Percentage γH2AX foci resterend 4 uur na blootstelling aan 0.5 Gy γ-straling al dan niet gecombineerd
met cetuximab. Het aantal resterende foci na 4 uur werd uitgedrukt ten opzichte van het aantal foci 15 minuten
na bestraling.
Deze DNA-herstelkinetiek experimenten tonen aan dat additie van cetuximab aan 60
Co γ-
straling in HT29 cellen aanleiding geeft tot trager herstel van stralingsgeïnduceerde DNA-
dubbelstrengbreuken.
0,0
2,0
4,0
6,0
8,0
10,0
12,0
14,0
16,0
18,0
0 Gy +
Cetuximab
0,5 Gy +
Cetuximab
0,5 Gy
Gec
orr
igee
rd g
emid
del
d #
fo
ci /
cel
15 min herstel
4u herstel

38
4. Discussie
4.1. De constructie van een groeicurve
De groeicurven werden opgesteld om een betere inschatting toe te laten van de cellulaire
proliferatie, en dus ook van de tijdstippen waarop subculturen moeten worden gemaakt.
De bekomen groeicurven (zie figuur 7) vertonen een sigmoïdaal verloop. De cellen nemen
niet toe in aantal tijdens de lagfase. Dit is een adaptatieperiode waarin de cellen aanhechten
aan het substraat en waarin ze componenten van de plasmamembraan en de extracellulaire
matrix vervangen die verloren gingen bij de trypsinisatie. Tijdens de daaropvolgende logfase
is er een exponentiële toename van het celaantal. Wanneer uiteindelijk de volledige
beschikbare oppervlakte door de cellen is ingenomen, vertoont de groeicurve een plateaufase
[37].
De drie groeicurven leverden vergelijkbare verdubbelingstijden op. De gemiddelde
verdubbelingstijd van 24.2 uur ligt tevens dicht bij deze beschreven in de literatuur
[39,40,41,42]. Tabel 12 toont de resultaten van een vergelijkende literatuurstudie.
Publicatie Gerapporteerde verdubbelingstijd HT-29 (u)
Forgue-Lafitte et al. [39]
Zhu et al. [40]
Potla et al. [41]
Yao et al. [42]
Eigen studie
24
22
26
25.9 ± 2.3
24.2 ± 0.7
Tabel 12. Vergelijkende literatuurstudie met betrekking tot de verdubbelingstijd van HT-29 cellen.
4.2. Optimalisatie van de synchronisatieprocedure
De cellen werden gesynchroniseerd om 2 redenen. De cellulaire radiosensitiviteit varieert
doorheen de celcyclus. Hij is het hoogst tijdens de M/G2 fasen, intermediair tijdens G1 en het
laagst tijdens de late S-fase [43]. Een eerste reden om te synchroniseren is het vermijden van
variatie in de resultaten omwille van een variabele celcyclusdistributie. DNA DSB worden
spontaan geïnduceerd wanneer de replicatievork tijdens de S-fase een beschadigde template
bereikt [33]. Een tweede reden om de cellen te synchroniseren is de beperking van de
hoeveelheid endogeen geïnduceerde DSB. Daarom werd geopteerd voor synchronisatie in de
G0/G1 fase. Hiertoe bestaan diverse methoden. Gezien farmacologische agentia cellulaire
bijwerkingen kunnen vertonen [44], werd in deze studie geopteerd voor synchronisatie door
serumdeprivatie. Hierbij resulteert het gebrek aan groeifactoren in het medium in arrest van

39
de celcyclus in de G1-fase of in overgang van de cellen naar de G0-fase [45]. Deze manier
van synchronisatie werd bovendien reeds eerder toegepast voor HT-29 cellen [46,47,48].
Het eerste synchronisatie-experiment (Tabel 3) toont dat 24 uur incubatie met normaal
groeimedium gevolgd door 24 uur incubatie met media met diverse hoeveelheden serum leidt
tot duidelijke verschillen in de celcyclusdistributie. Zoals verwacht steeg het procentuele
aantal cellen in G1/G0 bij daling van het serumgehalte van het medium. Bij 24 uur incubatie
met normaal groeimedium, gevolgd door 48 uur incubatie met media met diverse
hoeveelheden serum, zijn er geen duidelijke verschillen in synchronisatietoestand meer
merkbaar tussen de verschillende condities. Dit is mogelijk te wijten aan het optreden van
contactinhibitie, of aan de mogelijke uitputting van groeifactoren in de serumbevattende
media na 48 uur.
Uit een tweede experiment (tabel 4) kan worden afgeleid dat 48 uur incubatie op SSM 0%
medium geen meerwaarde biedt ten opzichte van 24 uur incubatie op dit medium.
Voor een derde experiment (tabel 5) werd er gestart van een welbepaald aantal cellen,
namelijk 5 miljoen per 75 cm² falcon. Deze werden 24 uur geïncubeerd in N medium,
gevolgd door 24 uur incubatie in SSM 0%. Op deze manier werden circa 85 % van de cellen
in G0/G1 aangetroffen. Dit werd als voldoende beschouwd en deze procedure werd dan ook
verkozen om de cellen te synchroniseren voorafgaand aan elk experiment. Telkens werd de
synchronisatietoestand gecontroleerd. Het procentueel celaantal in G0/G1 bedroeg steeds
minstens 85.
4.3. Dosisrespons experimenten
De dosisrespons experimenten werden uitgevoerd om de optimale stralingsdosis te bepalen
voor de DNA-herstelkinetiek experimenten. Te hoge dosissen zouden resulteren in niet meer
telbare hoeveelheden foci. Te lage dosissen zouden niet meer representatief zijn voor
radiotherapie-effecten. Op basis van de dosisrespons experimenten werd beslist de DNA-
herstelkinetiek experimenten uit te voeren met een stralingsdosis van 0.5 Gy.
In de literatuur werden geen studies gevonden die handelen over herstelkinetiek van
stralingsgeïnduceerde DNA-schade in de HT29 cellijn. Om toch een idee te krijgen over de
relatieve positie van de bekomen dosisrespons curve ten opzichte van reeds uitgevoerde
dosisrespons experimenten, wordt in figuur 16 de bekomen dosisrespons curve weergegeven
samen met een aantal dosisrespons curven die werden gevonden in de literatuur.
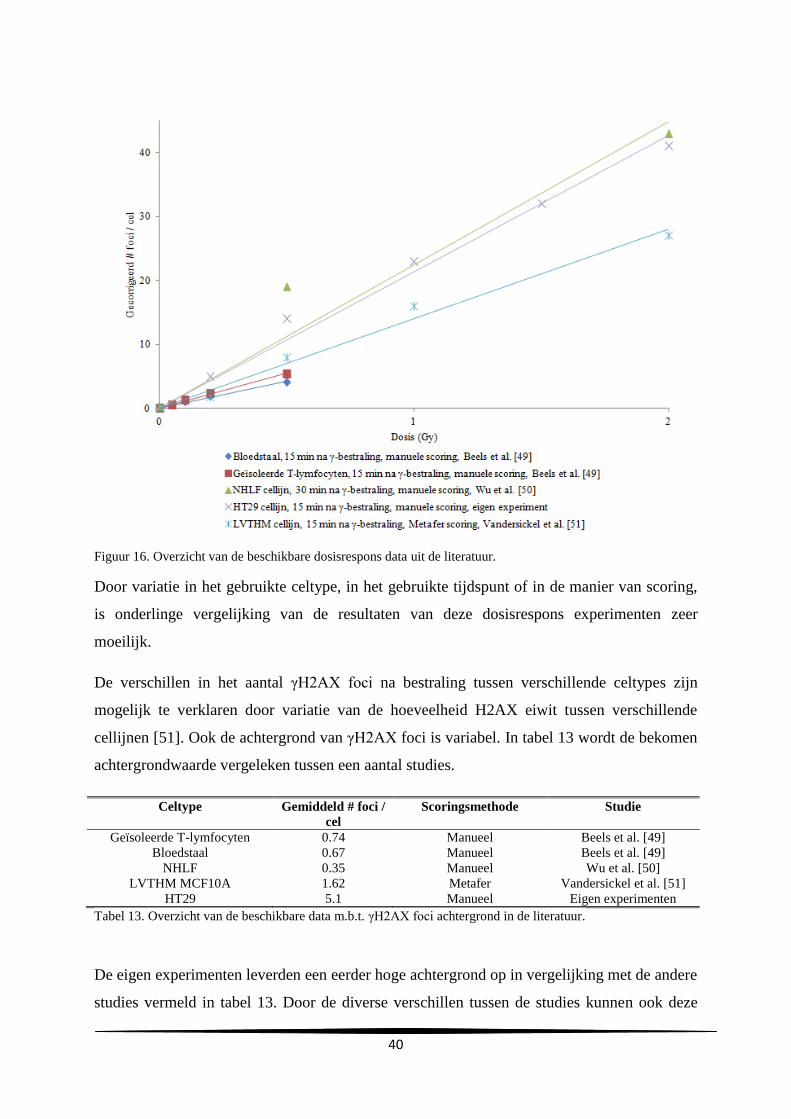
40
Figuur 16. Overzicht van de beschikbare dosisrespons data uit de literatuur.
Door variatie in het gebruikte celtype, in het gebruikte tijdspunt of in de manier van scoring,
is onderlinge vergelijking van de resultaten van deze dosisrespons experimenten zeer
moeilijk.
De verschillen in het aantal γH2AX foci na bestraling tussen verschillende celtypes zijn
mogelijk te verklaren door variatie van de hoeveelheid H2AX eiwit tussen verschillende
cellijnen [51]. Ook de achtergrond van γH2AX foci is variabel. In tabel 13 wordt de bekomen
achtergrondwaarde vergeleken tussen een aantal studies.
Celtype Gemiddeld # foci /
cel
Scoringsmethode Studie
Geïsoleerde T-lymfocyten 0.74 Manueel Beels et al. [49]
Bloedstaal 0.67 Manueel Beels et al. [49]
NHLF 0.35 Manueel Wu et al. [50]
LVTHM MCF10A 1.62 Metafer Vandersickel et al. [51]
HT29 5.1 Manueel Eigen experimenten
Tabel 13. Overzicht van de beschikbare data m.b.t. γH2AX foci achtergrond in de literatuur.
De eigen experimenten leverden een eerder hoge achtergrond op in vergelijking met de andere
studies vermeld in tabel 13. Door de diverse verschillen tussen de studies kunnen ook deze
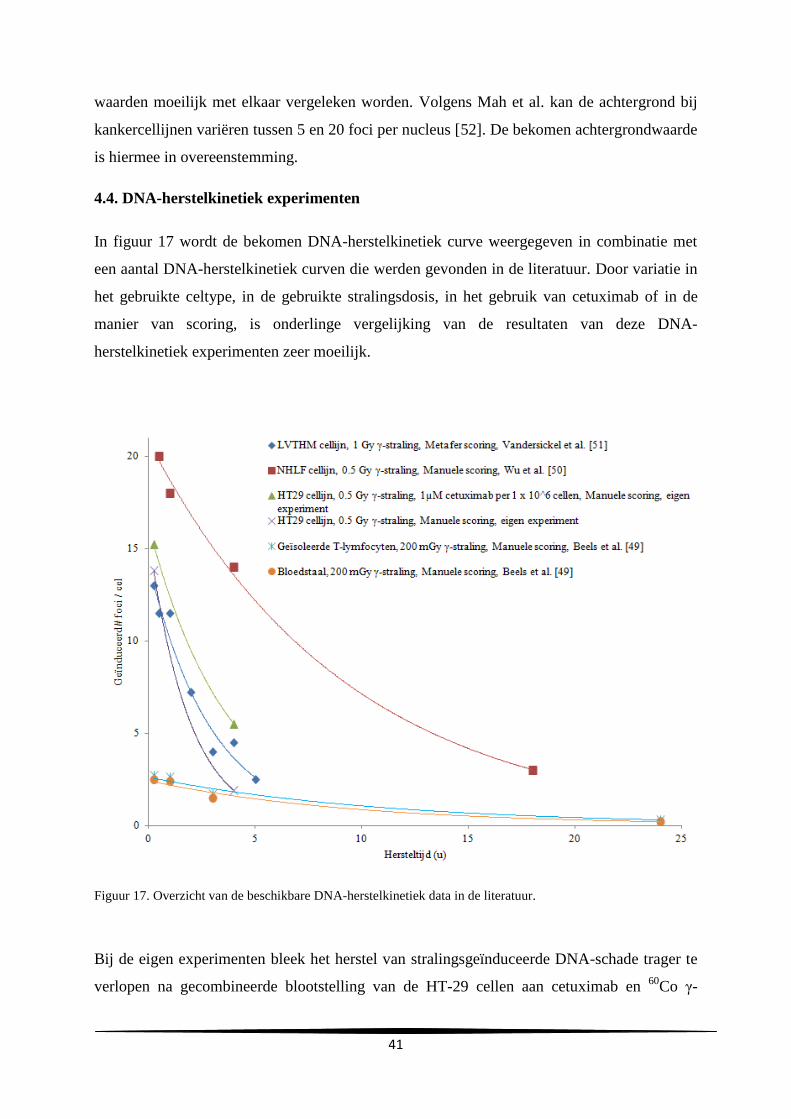
41
waarden moeilijk met elkaar vergeleken worden. Volgens Mah et al. kan de achtergrond bij
kankercellijnen variëren tussen 5 en 20 foci per nucleus [52]. De bekomen achtergrondwaarde
is hiermee in overeenstemming.
4.4. DNA-herstelkinetiek experimenten
In figuur 17 wordt de bekomen DNA-herstelkinetiek curve weergegeven in combinatie met
een aantal DNA-herstelkinetiek curven die werden gevonden in de literatuur. Door variatie in
het gebruikte celtype, in de gebruikte stralingsdosis, in het gebruik van cetuximab of in de
manier van scoring, is onderlinge vergelijking van de resultaten van deze DNA-
herstelkinetiek experimenten zeer moeilijk.
Figuur 17. Overzicht van de beschikbare DNA-herstelkinetiek data in de literatuur.
Bij de eigen experimenten bleek het herstel van stralingsgeïnduceerde DNA-schade trager te
verlopen na gecombineerde blootstelling van de HT-29 cellen aan cetuximab en 60
Co γ-

42
straling (Zie tabel 10 en grafiek 15). Vier uur na blootstelling van de HT-29 cellen aan 0.5 Gy
60Co γ-straling resteerde 14 % van het aantal aanwezige γH2AX foci 15 minuten na
bestraling. Indien echter 4 uur voor de bestraling 1µM cetuximab werd toegevoegd per 1 x
106 cellen, bleek nog 36 % van het oorspronkelijk aantal foci te resteren (Zie tabel 11).
González et al. [53] maakten ook gebruik van gecombineerde radio-cetuximab therapie. A431
cellen werden behandeld met verschillende concentraties cetuximab in combinatie met 4 Gy
137Cs straling. Cetuximab werd toegevoegd 1 uur voor de bestraling. Het aantal γH2AX foci
per nucleus werd bepaald 24 uur na bestraling. Een cetuximab concentratie van 50 µg/ml in
combinatie met een stralingsdosis van 4 Gy leverde na 24 uur DNA-herstel 13 foci op. Bij een
concentratie van 25 µg/ml resteerden 6.5 foci. 4 Gy 137
Cs straling in afwezigheid van
cetuximab leverde na 24 u DNA-herstel nog 3 γH2AX foci op [53]. Ook hier leidde additie
van cetuximab aan radiotherapie, net als bij de eigen experimenten, tot vertraagd herstel van
DNA dubbelstrengbreuken.
De functie van EGFR als regulator van DNA-herstel ligt mogelijk aan de basis van de
verklaring van het vertraagde herstel van stralingsgeïnduceerde DNA DSB na behandeling
met cetuximab.
Blootstelling aan ioniserende straling veroorzaakt translocatie van EGFR naar de nucleus,
waar EGFR associeert met DNA-PK. Dit leidt tot activatie van NHEJ DNA DSB herstel.
Blokkering van EGFR met cetuximab, een anti-EGFR monoclonaal antilichaam, verhindert
de stralingsgeïnduceerde translocatie van EGFR naar de nucleus [54]. Dit leidt tot
gereduceerde activiteit van DNA-PK, waardoor DNA-PK autofosforylatie op residu Thr2609,
wat essentieel is voor NHEJ, wordt gereduceerd [16]. Dittmann et al. konden in A549 cellen
inderdaad aantonen dat blokkering van nucleair EGFR transport door cetuximab DNA PK
activiteit deed afnemen. Dit had verhoogde residuele DNA schade en verminderde cellulaire
overleving tot gevolg [18].
Activatie van DNA-PKcs kan ook gebeuren door EGFR signalering via de PI3K-Akt
pathway. Hierbij interageert Akt rechtstreeks met DNA-PKcs. In cellen met gemuteerde Ras
eiwitten leidt constitutieve activatie van de MAPK pathway tot een autocriene lus van EGFR
ligandproductie en signalering, die vooral de PI3K-Akt pathway zal stimuleren [16]. Dit
mechanisme wordt geïllustreerd in figuur 18.

43
Figuur 18. De EGFR-PI3K-AKT pathway. Ioniserende straling kan rechtstreeks de EGFR activeren, die
vervolgens vooral aanleiding geeft tot activatie van de PI3K-AKT cascade. AKT signalering staat in voor anti-
apoptotische responsen en activeert herstel van DNA DSB door directe interactie met DNA-PKcs. In cellen met
een gemuteerd Ras eiwit, stimuleert de constitutief actieve MAPK pathway de productie en vrijstelling van
EGFR liganden. Deze stimuleren op hun beurt EGFR-afhankelijke PI3K-AKT signalering via een autocrien
mechanisme. Figuur overgenomen uit Rodemann et al. [16].
Anderzijds staat de nucleaire EGFR in voor fosforylatie van PCNA (proliferating cell nuclear
antigen) [18], een essentieel onderdeel van de ‘DNA sliding clamp’ [16]. Deze fosforylatie
verhoogt de stabiliteit van PCNA op chromatine [18], waardoor replicatie en herstel van DNA
gestimuleerd worden [16].
Daarnaast zorgt nEGFR voor opregulatie van de expressie van XRCC1, een eiwit uit de base
excision repair (BER) pathway dat instaat voor herstel van DNA enkelstrengbreuken.
Bijgevolg kan door inactivatie van EGFR de frequentie van geclusterde DNA-schade
toenemen, wat kan leiden tot een hoger aantal DNA DSB [16]. Dit verklaart het verhoogd
aantal γH2AX foci 15 minuten na bestraling bij radio-cetuximab combinatietherapie ten
opzichte van bestraling alleen (zie figuur 15).
Doordat cetuximab zowel nucleair transport [54], als de cytoplasmatische pathways [13] van
EGFR blokkeert, draagt het bij tot inhibitie van EGFR geïnduceerd DNA herstel. Dit verklaart
de geobserveerde radiosensitiserende werking van cetuximab.
Cetuximab monotherapie lijkt het aantal γH2AX foci licht te verhogen (Zie tabel 10 en figuur
15). In eerste instantie kan dit doen vermoeden dat cetuximab in beperkte mate DNA
dubbelstrengbreuken zou induceren. Er werd echter reeds aangetoond dat cetuximab geen

44
genotoxisch effect heeft [55]. De lichte verhoging van het aantal γH2AX foci kan mogelijk
verklaard worden door een hoger aantal cellen in apoptose na cetuximab behandeling, te
wijten aan inhibitie van de anti-apoptotische PI3K-AKT pathway. Om de statistische
significantie van deze lichte verhoging van het aantal γH2AX foci na te gaan, zijn er echter
meer herhalingen van het experiment vereist.
4.5. Besluit
Uit deze studie blijkt dat HT29 cellen die blootgesteld worden aan zowel cetuximab als aan
ioniserende straling, stralingsgeïnduceerde DNA dubbelstrengbreuken trager herstellen in
vergelijking met HT29 cellen die enkel aan ioniserende straling worden blootgesteld. Verder
onderzoek is nodig om deze resultaten te bevestigen. Door de resultaten te correleren met
therapierespons, kan worden nagegaan of de γH2AX foci assay bruikbaar is als predictieve
merker voor radio-cetuximab therapie bij patiënten met colorectale kanker. Op die manier
zouden patiënten die baat zullen hebben bij deze therapievorm op voorhand geïdentificeerd
kunnen worden, waardoor nodeloze bijwerkingen voor de patiënt alsook kosten voor de
gezondheidszorg vermeden kunnen worden. De resultaten van deze studie dienen nog
bevestigd te worden, zodat de statistische significantie ervan kan worden nagegaan. Eventueel
kan de procedure herhaald worden voor andere monoclonale antilichamen.

45
5. Referenties
1. Hanahan D, Weinberg RA (2000). The hallmarks of cancer. Cell. 100(1):57-70
2. Hanahan D, Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell. 144(5):646-74
3. Jemal A, Center MM, DeSantis C, Ward EM (2010). Global patterns of cancer incidence and mortality
rates and trends. Cancer Epidemiol Biomarkers Prev. 19(8):1893-907
4. Cancer incidence in Belgium (2008). Belgian cancer registry. Online beschikbaar op
www.kankerregister.be
5. Meropol NJ, Schrag D, Smith TJ, Mulvey TM, Langdon RM Jr, Blum D, Ubel PA, Schnipper LE;
American Society of Clinical Oncology (2009). American Society of Clinical Oncology guidance
statement: the cost of cancer care. J Clin Oncol. 27(23):3868-74
6. Weitz J, Koch M, Debus J, Höhler T, Galle PR, Büchler MW (2005). Colorectal cancer. Lancet.
14;365(9454):153-65
7. Souhami R, Tobias J (2007). Cancer and its management. 5th
ed. Blackwell publishing.
8. Harrison S, Benziger H (2011). The molecular biology of colorectal carcinoma and its implications: a
review. Surgeon. 9(4):200-10
9. Labianca R, Nordlinger B, Beretta GD, Brouquet A, Cervantes A; ESMO Guidelines Working Group
(2010). Primary colon cancer: ESMO Clinical Practice Guidelines for diagnosis, adjuvant treatment and
follow-up. Ann Oncol. 21 Suppl 5:v70-7
10. Glimelius B, Påhlman L, Cervantes A; ESMO Guidelines Working Group (2010). Rectal cancer: ESMO
Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 21 Suppl 5:v82-6
11. Van Cutsem E, Nordlinger B, Cervantes A; ESMO Guidelines Working Group (2010). Advanced
colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann Oncol. 21 Suppl 5:v93-7
12. Capdevila J, Elez E, Macarulla T, Ramos FJ, Ruiz-Echarri M, Tabernero J (2009). Anti-epidermal growth
factor receptor monoclonal antibodies in cancer treatment. Cancer Treat Rev. 35(4):354-63
13. Gerber DE, Choy H (2010). Cetuximab in combination therapy: from bench to clinic. Cancer Metastasis
Rev. 29(1):171-80
14. Lurje G, Lenz HJ (2009). EGFR signaling and drug discovery. Oncology. 77(6):400-10
15. Chen DJ, Nirodi CS (2007). The epidermal growth factor receptor: a role in repair of radiation-induced
DNA damage. Clin Cancer Res. 13(22 Pt 1):6555-60
16. Rodemann HP, Dittmann K, Toulany M (2007). Radiation-induced EGFR-signaling and control of DNA-
damage repair. Int J Radiat Biol. 83(11-12):781-91
17. Wang YN, Yamaguchi H, Hsu JM, Hung MC (2010). Nuclear trafficking of the epidermal growth factor
receptor family membrane proteins. Oncogene. 29(28):3997-4006
18. Dittmann K, Mayer C, Rodemann HP (2010). Nuclear EGFR as novel therapeutic target: insights into
nuclear translocation and function. Strahlenther Onkol. 186(1):1-6
19. Dittmann K, Mayer C, Kehlbach R, Rodemann HP (2008). Radiation-induced caveolin-1 associated
EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol Cancer.
7:69
20. Duffy MJ, O'Donovan N, Crown J (2011). Use of molecular markers for predicting therapy response in
cancer patients. Cancer Treat Rev. 37(2):151-9
21. Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes
RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ (2007). N Engl J Med. 357(20):2040-8
22. Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, Vega-Villegas ME, Eng
C, Steinhauer EU, Prausova J, Lenz HJ, Borg C, Middleton G, Kröning H, Luppi G, Kisker O, Zubel A,
Langer C, Kopit J, Burris HA 3rd
(2008). EPIC: phase III trial of cetuximab plus irinotecan after
fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol.
26(14):2311-9
23. Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim
R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P
(2009). Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med.
360(14):1408-17.
24. Tabernero J, Van Cutsem E, Díaz-Rubio E, Cervantes A, Humblet Y, André T, Van Laethem JL, Soulié
P, Casado E, Verslype C, Valera JS, Tortora G, Ciardiello F, Kisker O, de Gramont A (2007). Phase II
trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first-line treatment
of metastatic colorectal cancer. J Clin Oncol. 25(33):5225-32
25. www.ema.europa.eu
26. Lehnert S (2008). Biomolecular action of ionizing radiation. Taylor and Francis.

46
27. Eriksson D, Stigbrand T (2010). Radiation-induced cell death mechanisms. Tumor Biol. 31(4):363-72
28. Mah LJ, El-Osta A, Karagiannis TC (2010). gammaH2AX: a sensitive molecular marker of DNA damage
and repair. Leukemia. 24(4):679-86
29. Mladenov E, Iliakis G (2011). Induction and repair of DNA double strand breaks: the increasing spectrum
of non-homologous end joining pathways. Mutat Res. 711(1-2):61-72
30. Kass EM, Jasin M (2010). Collaboration and competition between DNA double-strand break repair
pathways. FEBS Lett. 584(17):3703-8
31. Langerak P, Russell P (2011). Regulatory networks integrating cell cycle control with DNA damage
checkpoints and double-strand break repair. Philos Trans R Soc Lond B Biol Sci. 366(1584):3562-71
32. Pardo B, Gómez-González B, Aguilera A (2009). DNA repair in mammalian cells: DNA double-strand
break repair: how to fix a broken relationship. Cell Mol Life Sci. 66(6):1039-56
33. Hartlerode AJ, Scully R (2009). Mechanisms of double-strand break repair in somatic mammalian cells.
Biochem J. 423(2):157-68
34. Lieber MR (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-
joining pathway. Annu Rev Biochem. 79:181-211
35. Podhorecka M, Skladanowski A, Bozko P (2010). H2AX Phosphorylation: Its Role in DNA Damage
Response and Cancer Therapy. J Nucleic Acids. pii: 920161
36. Volokhov DV, Graham LJ, Brorson KA, Chizhikov VE (2011). Mycoplasma testing of cell substrates and
biologics: Review of alternative non-microbiological techniques. Mol Cell Probes. 25(2-3):69-77
37. Freshney RI (2010). Culture of animal cells: a manual of basic technique and specialized applications. 6th
rev. ed. Wiley-Blackwell.
38. Mycoplasma detection assay - Instructions for use. Online beschikbaar op www.lonza.com
39. Forgue-Lafitte ME, Coudray AM, Bréant B, Mester J (1989). Proliferation of the human colon carcinoma
cell line HT29: autocrine growth and deregulated expression of the c-myc oncogene. Cancer Res.
49(23):6566-71
40. Zhu B, Vemavarapu L, Thompson WJ, Strada SJ (2005). Suppression of cyclic GMP-specific
phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J Cell Biochem. 94(2):336-50
41. Potla L, Boghaert ER, Armellino D, Frost P, Damle NK (2002). Reduced expression of EphrinA1
(EFNA1) inhibits three-dimensional growth of HT29 colon carcinoma cells. Cancer Lett. 175(2):187-95
42. Yao K, Gietema JA, Shida S, Selvakumaran M, Fonrose X, Haas NB, Testa J, O'Dwyer PJ (2005). In
vitro hypoxia-conditioned colon cancer cell lines derived from HCT116 and HT29 exhibit altered
apoptosis susceptibility and a more angiogenic profile in vivo. Br J Cancer. 93(12):1356-63
43. Pawlik TM, Keyomarsi K (2004). Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat
Oncol Biol Phys. 59(4):928-42
44. Davis PK, Ho A, Dowdy SF (2001). Biological methods for cell-cycle synchronization of mammalian
cells. Biotechniques. 30(6):1322-6, 1328, 1330-1
45. Uzbekov RE (2003). Analysis of the cell cycle and a method employing synchronized cells for study of
protein expression at various stages of the cell cycle. Biochemistry (Mosc). 69(5):485-96
46. Parnaud G, Corpet DE, Gamet-Payrastre L (2001). Cytostatic effect of polyethylene glycol on human
colonic adenocarcinoma cells. Int J Cancer. 92(1):63-9
47. Abal M, Bras-Goncalves R, Judde JG, Fsihi H, De Cremoux P, Louvard D, Magdelenat H, Robine S,
Poupon MF (2004). Enhanced sensitivity to irinotecan by Cdk1 inhibition in the p53-deficient HT29
human colon cancer cell line. Oncogene. 23(9):1737-44
48. Liu BP, Chong EY, Cheung FW, Duan JA, Che CT, Liu WK (2005). Tangutorine induces p21 expression
and abnormal mitosis in human colon cancer HT-29 cells. Biochem Pharmacol. 70(2):287-99
49. Beels L, Werbrouck J, Thierens H (2010). Dose response and repair kinetics of gamma-H2AX foci
induced by in vitro irradiation of whole blood and T-lymphocytes with X- and gamma-radiation. Int J
Radiat Biol. 86(9):760-8
50. Wu J, Fu Q, Quan Y, Wang W, Mei T, Li J, Yang G, Ren X, Xue J, Wang Y (2012). Repair rates of DNA
double-strand breaks under different doses of proton and γ-ray irradiation. Nucl Instrum Meth B. 276:1-6
51. Vandersickel V, Depuydt J, Van Bockstaele B, Perletti G, Philippe J, Thierens H, Vral A (2010). Early
increase of radiation-induced γH2AX foci in a human Ku70/80 knockdown cell line characterized by an
enhanced radiosensitivity. J Radiat Res. 51(6):633-41
52. Mah LJ, Orlowski C, Ververis K, Vasireddy RS, El-Osta A, Karagiannis TC (2011). Evaluation of the
efficacy of radiation-modifying compounds using γH2AX as a molecular marker of DNA double-strand
breaks. Genome Integr. 2(1):3
53. González JE, Barquinero JF, Lee M, García O, Casaco A (2012). Radiosensitization induced by the anti-
epidermal growth factor receptor monoclonal antibodies cetuximab and nimotuzumab in A431 cells.
Cancer Biol Ther. 13(2):71-6

47
54. Dittmann K, Mayer C, Rodemann HP (2005). Inhibition of radiation-induced EGFR nuclear import by
C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 76(2):157-61
55. Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann
HP (2005). Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of
DNA-dependent protein kinase. J Biol Chem. 280(35):31182-9