
Drie kinderen met algehele malaise, koorts, gewichtsverlies en...
5
Dames en Heren, Wij beschrijven u de ziektegeschiedenissen van 3 kinde- ren met algehele malaise, koorts, gewichtsverlies en cer- vicale lymfadenopathie. Dit is een symptomencombina- tie met een uitgebreide differentiaaldiagnose. De eerste patiënt leed aan de ziekte van Hodgkin, de tweede aan een gemengde bindweefselziekte (‘mixed connective tis- sue disease’) en de derde aan de ziekte van Hodgkin met lupus erythematodes disseminatus (SLE). De casussen vertonen veel overeenkomst, zoals u zult zien. Met behulp van specifieke anamnestische gege- vens, nauwkeurig lichamelijk onderzoek en aanvullende diagnostiek konden de juiste diagnosen uiteindelijk gesteld worden. Patiënt A, een 11-jarig creools meisje, had een pijnlijke zwelling rechts in de hals. Zij was al enige tijd progres- sief moe, met koorts, en werd wegens snelle toename van de zwelling en vermoeden van een lymfoprolifera- tieve maligniteit verwezen. Bij lichamelijk onderzoek zag men een niet-ziek, alert meisje, met een lichaamslengte van 153 cm (standaard- deviatiescore (SDS): 0), een lichaamsgewicht van 40 kg (SDS: 0). De huid toonde geen afwijkingen. Rechts supra- claviculair en in de rechter oksel bestonden vergrote, vast aanvoelende, verbakken lymfeklieren respectieve- lijk met afmetingen 6 × 8 cm en 5 cm diameter. Alge- meen intern-geneeskundig onderzoek toonde geen bij- zonderheden, met name geen hepatosplenomegalie. Patiënte werd opgenomen. Tijdens de opname nam de zwelling in geringe mate toe. Een maligne of infectieuze aandoening werd overwogen. Laboratoriumonderzoek toonde een anemie, geen af- wijkingen aan leukocytengetal en differentiatie, en een verhoogde bezinking (tabel 1). Verder onderzoek toon- de geen afwijkingen. Echografie van de hals en CT van de thorax bevestigden klierpakketten laag in de hals en toonden een mediastinaal klierpakket. Histologisch pas- te het excisiebiopt van de supraclaviculaire klierregio bij de ziekte van Hodgkin (nodulair scleroserend type), klinisch stadium IIA (volgens de Ann-Arbor-classifica- tie) (figuur). Patiënte kreeg tweewekelijks kuren van vincristine, prednison, procarbazine en doxorubicine (zogenaamd GPOH-95-protocol). Na 2 kuren was zij in partiële remissie, waarna zij radiotherapie kreeg. Patiënt B, een 13-jarig Curaçaos meisje, was sinds enke- le weken vermoeid met verminderde inspanningstole- rantie, hoofdpijn en 3 kg gewichtsverlies in 3 weken. Pijnklachten en verminderde spierkracht waren er voor- al in handen en benen. Ook had zij ochtendstijfheid. De hals en het achterhoofd toonden al enige tijd opgezette lymfeklieren. Bij lichamelijk onderzoek zagen wij een opvallend klein, prepuberaal meisje, niet bleek of acuut ziek. Haar lichaamslengte was 144 cm (SDS: –2,5) en haar gewicht 32 kg (SDS: –2). Er waren forse lymfeklierpakketten oc- cipitaal, submandibulair en beiderzijds ter hoogte van de M. sternocleidomastoideus. Tevens waren er niet-pijn- lijke, supraclaviculaire, axillaire en inguïnale lymfeklie- ren palpabel die elastisch aanvoelden en los lagen van onder- en bovenlaag. Er waren nagelriemafwijkingen alsmede een proximale myopathie met verminderde beweeglijkheid van de rechter elleboog. Op de handen werden peesnoduli van de extensorpezen gezien, zonder gezwollen gewrichten. Overig intern-geneeskundig on- derzoek leverde geen bijzonderheden op, met name geen hepatosplenomegalie. Bij aanvullend onderzoek waren er anemie, niet- afwijkende uitslagen voor leukocytengetal en normale differentiatie, met lichte trombocytose en verhoogde be- zinking (zie tabel 1). Er waren antistoffen aanwezig tegen extraheerbare kernantigenen (anti-ENA) en de uitslagen van anti-Sm-, anti-Sjögren-syndroom(SS)-A-, anti-SS-B- en anti-topo-isomerase-I-(Scl-70)-bepalingen waren eveneens positief. De serumactiviteit van creati- nekinase was verhoogd. Het urinesediment toonde geen afwijkingen. Klinisch beeld en aanvullend onderzoek pasten het best bij een gemengde bindweefselziekte. Wegens de algehele malaise, het gewichtsverlies, de koorts en de myopathie werd patiënte behandeld met naproxen, hy- droxychloroquine en een lage dosis prednison. Na een exacerbatie van myositis ontwikkelde zich glomerulonefritis (klasse II volgens de WHO-classifica- tie, hetgeen betekent immuundeposities en hypercellu- lariteit van het mesangium). Patiënte leed aan SLE met myositis en nefritis en was bij de laatste follow-up onder behandeling met steroïden, imuran, methotrexaat en NSAID’s. Ned Tijdschr Geneeskd 2004 6 maart;148(10) 453 Klinische lessen Drie kinderen met algehele malaise, koorts, gewichtsverlies en cervicale lymfadenopathie k.c.j.m.kraal, n.van paassen, l.m.ball, p.m.jansen en r.ten cate Leids Universitair Medisch Centrum, Postbus 9600, 2300 RC Leiden. Afd. Immunologie, Hematologie, Oncologie, Beenmergtransplantatie en Auto-immuunziekten: mw.K.C.J.M.Kraal, assistent-geneeskundige; mw.N.van Paassen, co-assistent; mw.L.M.Ball, kinderarts/hemato-onco- loog; mw.dr.R.ten Cate, kinderarts-reumatoloog. Afd. Pathologie: mw.P.M.Jansen, patholoog. Correspondentieadres: mw.R.ten Cate ([email protected]).
Transcript of Drie kinderen met algehele malaise, koorts, gewichtsverlies en...

Dames en Heren,Wij beschrijven u de ziektegeschiedenissen van 3 kinde-ren met algehele malaise, koorts, gewichtsverlies en cer-vicale lymfadenopathie. Dit is een symptomencombina-tie met een uitgebreide differentiaaldiagnose. De eerstepatiënt leed aan de ziekte van Hodgkin, de tweede aaneen gemengde bindweefselziekte (‘mixed connective tis-sue disease’) en de derde aan de ziekte van Hodgkin metlupus erythematodes disseminatus (SLE).
De casussen vertonen veel overeenkomst, zoals u zultzien. Met behulp van specifieke anamnestische gege-vens, nauwkeurig lichamelijk onderzoek en aanvullendediagnostiek konden de juiste diagnosen uiteindelijkgesteld worden.
Patiënt A, een 11-jarig creools meisje, had een pijnlijkezwelling rechts in de hals. Zij was al enige tijd progres-sief moe, met koorts, en werd wegens snelle toenamevan de zwelling en vermoeden van een lymfoprolifera-tieve maligniteit verwezen.
Bij lichamelijk onderzoek zag men een niet-ziek, alertmeisje, met een lichaamslengte van 153 cm (standaard-deviatiescore (SDS): 0), een lichaamsgewicht van 40 kg(SDS: 0). De huid toonde geen afwijkingen. Rechts supra-claviculair en in de rechter oksel bestonden vergrote,vast aanvoelende, verbakken lymfeklieren respectieve-lijk met afmetingen 6 × 8 cm en 5 cm diameter. Alge-meen intern-geneeskundig onderzoek toonde geen bij-zonderheden, met name geen hepatosplenomegalie.Patiënte werd opgenomen. Tijdens de opname nam dezwelling in geringe mate toe. Een maligne of infectieuzeaandoening werd overwogen.
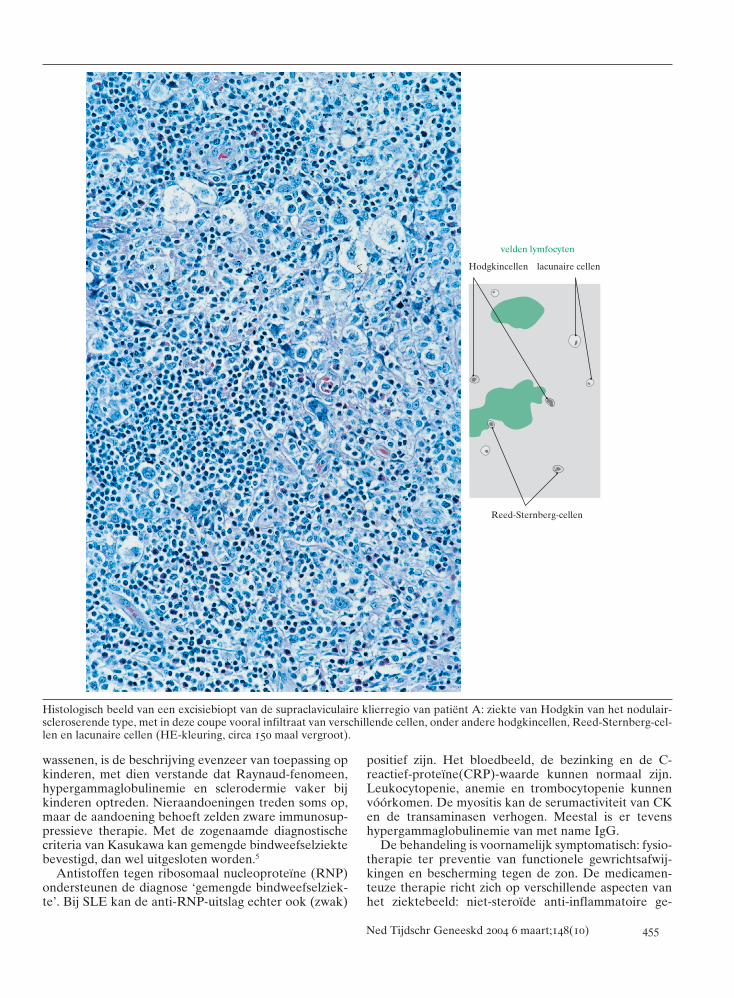
Laboratoriumonderzoek toonde een anemie, geen af-wijkingen aan leukocytengetal en differentiatie, en eenverhoogde bezinking (tabel 1). Verder onderzoek toon-de geen afwijkingen. Echografie van de hals en CT vande thorax bevestigden klierpakketten laag in de hals entoonden een mediastinaal klierpakket. Histologisch pas-te het excisiebiopt van de supraclaviculaire klierregio bijde ziekte van Hodgkin (nodulair scleroserend type),klinisch stadium IIA (volgens de Ann-Arbor-classifica-
tie) (figuur). Patiënte kreeg tweewekelijks kuren vanvincristine, prednison, procarbazine en doxorubicine(zogenaamd GPOH-95-protocol). Na 2 kuren was zij inpartiële remissie, waarna zij radiotherapie kreeg.
Patiënt B, een 13-jarig Curaçaos meisje, was sinds enke-le weken vermoeid met verminderde inspanningstole-rantie, hoofdpijn en 3 kg gewichtsverlies in 3 weken.Pijnklachten en verminderde spierkracht waren er voor-al in handen en benen. Ook had zij ochtendstijfheid. Dehals en het achterhoofd toonden al enige tijd opgezettelymfeklieren.
Bij lichamelijk onderzoek zagen wij een opvallendklein, prepuberaal meisje, niet bleek of acuut ziek. Haarlichaamslengte was 144 cm (SDS: –2,5) en haar gewicht32 kg (SDS: –2). Er waren forse lymfeklierpakketten oc-cipitaal, submandibulair en beiderzijds ter hoogte van deM. sternocleidomastoideus. Tevens waren er niet-pijn-lijke, supraclaviculaire, axillaire en inguïnale lymfeklie-ren palpabel die elastisch aanvoelden en los lagen vanonder- en bovenlaag. Er waren nagelriemafwijkingenalsmede een proximale myopathie met verminderdebeweeglijkheid van de rechter elleboog. Op de handenwerden peesnoduli van de extensorpezen gezien, zondergezwollen gewrichten. Overig intern-geneeskundig on-derzoek leverde geen bijzonderheden op, met namegeen hepatosplenomegalie.
Bij aanvullend onderzoek waren er anemie, niet-afwijkende uitslagen voor leukocytengetal en normaledifferentiatie, met lichte trombocytose en verhoogde be-zinking (zie tabel 1). Er waren antistoffen aanwezigtegen extraheerbare kernantigenen (anti-ENA) en deuitslagen van anti-Sm-, anti-Sjögren-syndroom(SS)-A-,anti-SS-B- en anti-topo-isomerase-I-(Scl-70)-bepalingenwaren eveneens positief. De serumactiviteit van creati-nekinase was verhoogd. Het urinesediment toonde geenafwijkingen.
Klinisch beeld en aanvullend onderzoek pasten hetbest bij een gemengde bindweefselziekte. Wegens dealgehele malaise, het gewichtsverlies, de koorts en demyopathie werd patiënte behandeld met naproxen, hy-droxychloroquine en een lage dosis prednison.
Na een exacerbatie van myositis ontwikkelde zichglomerulonefritis (klasse II volgens de WHO-classifica-tie, hetgeen betekent immuundeposities en hypercellu-lariteit van het mesangium). Patiënte leed aan SLE metmyositis en nefritis en was bij de laatste follow-up onderbehandeling met steroïden, imuran, methotrexaat enNSAID’s.
Ned Tijdschr Geneeskd 2004 6 maart;148(10) 453
Klinische lessen
Drie kinderen met algehele malaise, koorts, gewichtsverlies en cervicalelymfadenopathie
k.c.j.m.kraal, n.van paassen, l.m.ball, p.m.jansen en r.ten cate
Leids Universitair Medisch Centrum, Postbus 9600, 2300 RC Leiden.Afd. Immunologie, Hematologie, Oncologie, Beenmergtransplantatieen Auto-immuunziekten: mw.K.C.J.M.Kraal, assistent-geneeskundige;mw.N.van Paassen, co-assistent; mw.L.M.Ball, kinderarts/hemato-onco-loog; mw.dr.R.ten Cate, kinderarts-reumatoloog.Afd. Pathologie: mw.P.M.Jansen, patholoog.Correspondentieadres: mw.R.ten Cate ([email protected]).

Patiënt C was een 17-jarig creools-Aziatisch meisje metde ziekte van Hodgkin stadium IIB, bevestigd door his-topathologisch onderzoek van een lymfeklierbiopt. Zijpresenteerde zich met gegeneraliseerde lymfadenopa-thie, gewichtsverlies en algehele malaise. Volgens pro-tocol had zij een behandeling ondergaan en was zij eenjaar nadat de diagnose was gesteld in complete remissie.Zij maakte in Suriname een infectie door met hogekoorts, huiduitslag en hoesten. Er bestond sinds 3 wekenstekende, brandende pijn in de rechter schouder die nietrood, dik of warm was, met onderbroken pijnklachtenvan linker schouder en kuit. Zij had last van stijfheid,niet specifiek ’s ochtends, met gezwollen vingers en rodeplekken op de onderbenen; ook had zij verminderdeinspanningstolerantie.
Bij lichamelijk onderzoek zagen wij een meisje vangemengd-Aziatische afkomst in matige voedingstoe-stand. Haar lichaamslengte was 160,4 cm (SDS: –1,5) enhaar gewicht 52,8 kg (SDS: –2). Er waren geen tekenenvan vasculitis, de slijmvliezen waren bleek met een ulcusop het verhemelte. Er waren geen vergrote lymfeklierenof milt palpabel. De sclerae waren geel verkleurd doorhemolyse. Over de longen werd een piepend, verlengdexpirium gehoord alsmede een systolische souffle overhet hart, mogelijk op basis van een anemie. Hoewelpatiënte pijn aangaf in de linker schouder, was er geenartritis aantoonbaar en evenmin een myopathie.Laboratoriumonderzoek liet anemie, trombocytopenieen leukocytopenie zien (zie tabel 1). Bij aanvullendkinderreumatologisch onderzoek werden antinucleaireantistoffen (ANA) en antistoffen tegen dubbelstrengs-DNA aangetoond,1 waarmee het ziektebeeld voldeedaan de criteria van het American College of Rheuma-tology voor SLE. Na de succesvolle behandeling van de
hemolytische anemie, kreeg patiënte prednison in hogedosis.
Ziekte van Hodgkin. Onze eerste patiënte bleek een ma-ligne lymfoom te hebben, de derde maligniteit bij kin-deren na leukemie en hersentumoren. De ziekte vanHodgkin toont een bimodale leeftijdsdistributie piekendop 15-30 en 45-55 jaar en veroorzaakt 12% van de ma-ligniteiten onder de 18 jaar en 30% van de lymfomen opdeze leeftijd. De oorzaak is niet bekend, maar Epstein-Barr-virus speelt mogelijk een rol door de lymfocytenaan te zetten tot transformatie en ongeremde prolifera-tie. Daarnaast zijn verscheidene chromosomale veran-deringen beschreven.2 Het histologisch beeld wordtgekenmerkt door Reed-Sternberg-cellen, dat zijn ge-transformeerde lymfocyten.
Hodgkinlymfomen worden volgens de Rye-classifica-tie in 4 typen onderverdeeld; het nodulair-scleroserendetype komt het meest voor (45-60%). De meest voorko-mende presentatie is pijnloze zwelling van één klier ofvan een groep klieren: 80% bevindt zich cervicaal metbij 60% van de betreffende patiënten mediastinale uit-breiding. Soms is er tijdelijke regressie gevolgd doorlatere uitgroei. 20-40% van de kinderen heeft bij eerstepresentatie algemene symptomen, zogenaamde ‘B-symp-tomen’: vermoeidheid, koorts, gewichtsverlies (� 10%in 6 maanden), nachtzweten en jeuk.3
De diagnose wordt gesteld op basis van een uit-gebreide evaluatie, onder andere anamnese, lichamelijkonderzoek en laboratoriumonderzoek, met name ge-richt op BSE, volledig bloedbeeld, lever- en nierfunctie.Aanvullend onderzoek bestaat uit klierbiopsie, beeld-vormend onderzoek (echografie van de buik, thorax-foto, op indicatie CT van buik en thorax). De ziektewordt gestageerd volgens de Ann-Arbor-classificatie.
Hoewel alle kinderen met de ziekte van Hodgkin be-handeld worden in studieverband, met name om de toxi-citeit van de behandeling te verminderen, bestaan er,zelfs voor kinderen met de ziekte van Hodgkin in het-zelfde stadium, verschillende studieprotocollen die in-ternationaal naast elkaar worden gebruikt. De algeheleprognose is een overleving van circa 80%.
Gemengde bindweefselziekte. Onze tweede patiëntebleek een auto-immuunziekte te hebben. Dit zijn klini-sche syndromen met een complexe, multifactoriële etio-logie, gekenmerkt door ontsteking, waarbij vrijwel alleorganen en systemen in het lichaam betrokken kunnenzijn.
Bij gemengde bindweefselziekte worden klinischeverschijnselen van verschillende reumatische aandoe-ningen (juveniele idiopathische artritis, SLE, dermato-myositis) tezamen gezien (zogenaamd overlapbeeld).4
Klinische verschijnselen van gemengde bindweefsel-ziekte zijn onder andere: artritis, Raynaud-fenomeen,sclerodermieachtige huidafwijkingen, erythemen (zoalsgezien worden bij SLE), dermatomyositis, koorts, moti-liteitsstoornissen van de oesofagus, cardiale afwijkingen,restrictieve longfunctiestoornissen en hepatosplenome-galie.
Hoewel de ziekte voornamelijk is beschreven bij vol-
454 Ned Tijdschr Geneeskd 2004 6 maart;148(10)
tabel 1. Laboratoriumuitslagen bij initiële presentatie van patiëntenA, B en C
bepaling referentie patiënt
A B C
hemoglobine 6,6-9,6 mmol/l 5,9 5,0 3,4hematocriet 0,32-0,47 0,29 0,25 0,14leukocyten 4,0-12,5 × 109/l 12,2 6,2 5,1differentiatie
neutrofiele granulocyten 39-70% 67 53 11
trombocyten 150-450 × 109/l 347 424 21BSE 0-25 mm/1e h 41 67 120ANA negatief – sterk positief
positiefanti-SS-A-/-SS-B-
antistoffen negatief – sterk negatiefpositief
anti-RNP-antistoffen negatief – negatief negatiefanti-Sm-antistoffen negatief – sterk negatief
positiefanti-dubbelstrengs-DNA negatief – – positiefC-reactief proteïne 0 mg/ml – 0 8IgG 6-15 g/l – – 23,4
ANA = antinucleaire antistoffen; SS-A = Sjögren-syndroom-A-anti-geen; RNP = ribosomaal nucleoproteïne.
wassenen, is de beschrijving evenzeer van toepassing opkinderen, met dien verstande dat Raynaud-fenomeen,hypergammaglobulinemie en sclerodermie vaker bijkinderen optreden. Nieraandoeningen treden soms op,maar de aandoening behoeft zelden zware immunosup-pressieve therapie. Met de zogenaamde diagnostischecriteria van Kasukawa kan gemengde bindweefselziektebevestigd, dan wel uitgesloten worden.5
Antistoffen tegen ribosomaal nucleoproteïne (RNP)ondersteunen de diagnose ‘gemengde bindweefselziek-te’. Bij SLE kan de anti-RNP-uitslag echter ook (zwak)
positief zijn. Het bloedbeeld, de bezinking en de C-reactief-proteïne(CRP)-waarde kunnen normaal zijn.Leukocytopenie, anemie en trombocytopenie kunnenvóórkomen. De myositis kan de serumactiviteit van CKen de transaminasen verhogen. Meestal is er tevenshypergammaglobulinemie van met name IgG.
De behandeling is voornamelijk symptomatisch: fysio-therapie ter preventie van functionele gewrichtsafwij-kingen en bescherming tegen de zon. De medicamen-teuze therapie richt zich op verschillende aspecten vanhet ziektebeeld: niet-steroïde anti-inflammatoire ge-
Ned Tijdschr Geneeskd 2004 6 maart;148(10) 455
Histologisch beeld van een excisiebiopt van de supraclaviculaire klierregio van patiënt A: ziekte van Hodgkin van het nodulair-scleroserende type, met in deze coupe vooral infiltraat van verschillende cellen, onder andere hodgkincellen, Reed-Sternberg-cel-len en lacunaire cellen (HE-kleuring, circa 150 maal vergroot).
Hodgkincellen lacunaire cellen
velden lymfocyten
Reed-Sternberg-cellen

neesmiddelen (NSAID’s) ter bestrijding van artritis; hy-droxychloroquine kan een gunstig effect hebben ophuidsymptomen, artritis en klachten van malaise. Corti-costeroïden in hoge dosering zijn alleen geïndiceerd bijernstige cytopenie, myositis, hemodynamisch belangrij-ke pericarditis, glomerulonefritis en andere levensbe-dreigende complicaties. Eventueel kunnen andere im-muunsuppressiva gebruikt worden zoals methotrexaaten azathioprine.
SLE. De derde patiënte, met eerdere ziekte vanHodgkin, presenteerde zich één jaar na remissie metklachten die deden denken aan een recidief met para-neoplastische verschijnselen. Ondanks uitgebreid on-derzoek werden hiervoor geen aanwijzingen gevonden.Zij gebruikte al een jaar geen geneesmiddelen, zodat bijhaar uiteindelijk de diagnose ‘SLE’ werd gesteld.
SLE is het prototype van een auto-immuunziekte, bijgeen andere auto-immuunziekte komen zoveel immu-nologische stoornissen voor. Karakteristiek zijn de veleverschillende (auto)antilichamen. Van de patiënten pre-senteert 15-17% zich vóór het 16e levensjaar.6 De ziek-te komt vaker (4,5 maal zo vaak) voor bij meisjes, bijAziatische personen en in bevolkingsgroepen rond deMiddellandse Zee. De incidentie bij kinderen wordt ge-schat op 0,53-0,6 per 100.000/jaar.7
Behalve bij zogenaamde door geneesmiddelen ver-oorzaakte SLE is over de etiologie weinig bekend.Onder meer immuunontregeling, hormonale disbalansen omgevingsfactoren kunnen samen met een geneti-sche predispositie het beeld tot uiting doen komen.
De diagnose ‘SLE’ mag worden gesteld bij aanwezigzijn van tenminste 4 symptomen die voldoen aan de cri-teria volgens de American College of Rheumatology.1 2
Deze symptomen mogen tegelijkertijd of opeenvolgendaanwezig zijn. De klinische symptomen bij het begin vande ziekte zijn: moeheid, malaise, koorts, artritis, myopa-thie, huid- en slijmvliesafwijkingen, nefritis, longproble-men, cardiale afwijkingen, hematologische afwijkingenen verschillende neurologische beelden.8
Karakteristieke pathologische afwijkingen van SLEzijn immuuncomplex-gemedieerde vasculitiden met fi-brinoïde necrose. Dit proces kan op zichzelf echter nietalle klinische verschijnselen verklaren. Anemie (normo-cytaire of hypochrome) treedt bij ongeveer 50% van depatiënten op, typisch bij chronisch verlopende SLE.
Antinucleaire antilichamen bestaan bij vrijwel allekinderen met actieve SLE. Vooral antilichamen tegendsDNA (specifiek voor SLE), in hoge titers aanwezig bijkinderen met actieve nefritis, worden zelden gezien bijandere reumatische ziekten. De reumafactoren zijn bij10-30% van de patiënten verhoogd.
De medicatie wordt bepaald door de klinische symp-tomen. Huidafwijkingen, artritis en moeheid/malaisereageren meestal goed op antimalariamidelen in combi-natie met NSAID’s. Behandeling met corticosteroïdendient men te reserveren voor kinderen met levensbe-dreigende verschijnselen, zoals serositis, ernstige ane-mie, aantasting van lever, nieren of centraal zenuwstel-sel. Voordat gestart wordt met immuunsuppressie dientmen de belangrijke orgaansystemen te evalueren, om-
dat interpretatie van onderzoeksuitslagen moeilijk iswanneer patiënten reeds immunosuppressiva krijgen.Intensieve immuunsuppressie (corticosteroïden met aza-thioprine of cyclofosfamide) wordt toegepast bij WHO-klasse-IV-nefritis (dat wil zeggen: diffuse glomerulone-fritis met segmentale (klasse IV-S) of algehele (klasseIV-G) betrokkenheid, met verdere subdivisies voor actie-ve en sclerotische afwijkingen) en bij cerebrale SLE.
SLE heeft een aanzienlijke morbiditeit en sterfte. Hetis een chronische ziekte gekenmerkt door perioden vanexacerbatie en remissie. De prognose, die wordt bepaalddoor de mate van aantasting van het centrale zenuwstel-sel en de nieren, is sinds de toepassing van immuunsup-pressie verbeterd.
Recent werd de relatie tussen auto-immuunziektenzoals SLE en hematologische maligniteiten weer be-sproken.9 10 Overwogen worden onderliggende, gedeel-de afwijkingen van de B-celreeks en een gemeenschap-pelijke virale etiologie (bijvoorbeeld Epstein-Barr-virus). Veel van dergelijke patiënten worden echterlangdurig behandeld met immuunsuppressiva in hogedoseringen, wat de relatie vertroebelt.
Algehele malaise, koorts, gewichtsverlies en cervicalelymfadenopathie. De beschreven patiënten, allen tie-ners, hadden als initiële symptomen algehele malaise,koorts, gewichtsverlies en cervicale lymfadenopathie.De differentiaaldiagnose van lymfadenopathie beslaatverschillende ziektebeelden (tabel 2). De ziekte bij der-gelijke kinderen vormt een lastig diagnostisch probleemvoor de algemene kinderarts. Meestal betreft het echtereen tijdelijke, zelflimiterende aandoening. De overigekinderen behoeven echter uitvoeriger evaluatie.
456 Ned Tijdschr Geneeskd 2004 6 maart;148(10)
tabel 2. Differentiaaldiagnose van lymfadenopathie
infectieviralebacteriëleparasitairemycotische
auto-immuunziektejuveniele idiopathische artritislupus erythematodes disseminatusgemengde bindweefselziekte
maligniteitleukemielymfoomhistiocytoseneuroblastoom
medicamenteus-geïnduceerde aandoeningallopurinolatenololcaptopriltegretol
stapelingsziekteNiemann-Pick-ziekteziekte van Gaucher
andere aandoeningziekte van Kawasakisarcoïdosekattenkrabziekte

Met anamnestische gegevens kan men de differen-tiaaldiagnose verkleinen. Met name belangrijk zijn ge-detailleerde informatie over de koorts, eventuele bezoe-ken aan het buitenland, gewichtsverlies en de kenmer-ken van de lymfadenopathie. Als deel van de familie-anamnese moet men ook vragen naar het vóórkomenvan systeemziekten.
Bij lichamelijk onderzoek verdient de status localisvan een lymfeklierzwelling veel aandacht: hoe groot isde klier of het lymfeklierpakket, hoe is de consistentie,is er verkleuring van de huid erboven, is er verklevingaan de onderlaag? Gaat het om regionale of gegenerali-seerde lymfekliervergroting? Een indruk krijgen van degrootte van lever en milt is van belang, evenals de be-vindingen aan huid, spieren en gewrichten.
Aanvullend diagnostisch onderzoek leidt veelal tot deuiteindelijke diagnose. Veel informatie wordt verkregenuit het volledig bloedbeeld met een handmatige diffe-rentiatie, de bezinking, de CRP-waarde, de leverfunc-ties, de bloedkweek, de virusserologische uitslagen, demantouxtest en de thoraxröntgenfoto. Bij uitgebreidepijnloze, niet-rode, cervicale lymfadenopathie, zondergewrichtsklachten en het ontbreken van andere sympto-men passende bij een auto-immuunziekte, staat verderehematologische evaluatie op de voorgrond. Bij onzekerblijven van de diagnose en indien een maligniteit nietuitgesloten kan worden, moet men altijd een lymfeklier-biopt en een beenmergpunctie verrichten.10 Als men eer-der denkt aan een reumatologische aandoening, dientaanvullend onderzoek zich te richten op reumafactorenen immuunglobulinestatus. Men moet er echter reke-ning mee houden dat bij maligniteiten en infectieziektenjuist de uitslagen van onderzoek op autoantistoffen fout-positief kunnen zijn.
Dames en Heren, gezien de uitgebreide differentiaal-diagnose bij algehele malaise, koorts, gewichtsverlies encervicale lymfadenopathie dient men zoveel mogelijkgegevens te verkrijgen uit de anamnese en het lichame-lijk onderzoek. Nagelriemafwijkingen, tekenen van myo-pathie en peesnoduli kunnen wijzen op gemengde bind-weefselziekte en/of SLE, terwijl gegeneraliseerde of lo-kale lymfadenopathie in combinatie met recente extre-me vermoeidheid op de ziekte van Hodgkin wijzen.Gezien de complexiteit en het vaak atypische beloop isgedegen theoretische kennis van belang voor het stellenvan de diagnose.
Aanvullend onderzoek dient zoveel mogelijk op deindividuele patiënt afgestemd te worden. Hiermee kanmen vertraging bij het stellen van de diagnose tot eenminimum beperken, zodat de behandeling eerder inge-steld kan worden.
Belangenconflict: geen gemeld. Financiële ondersteuning: geengemeld.
abstractThree children with general malaise, fever, weight loss and cer-vical lymphadenopathy. – Combinations of symptoms such asgeneral malaise, fever, weight loss and cervical lymphadeno-pathy have extensive differential diagnoses. In three children,girls aged 11, 13 and 17 years who presented with these symp-toms, three different diagnoses were obtained. The first hadHodgkin’s disease, the second mixed connective tissue disease(MCTD), and the third Hodgkin’s disease in combination withsystemic lupus erythematosus (SLE). A systematic approach isnecessary for the diagnosis of such conditions. Careful historytaking can provide valuable information while a physical ex-amination provides essential clues for the final diagnosis. In par-ticular, nail-fold lesions, tendon nodules and signs of myopathyshould be looked for in patients suspected of MCTD and/orSLE. In Hodgkin’s disease, generalised or localised lymphade-nopathy combined with a short history of extreme fatigue arethe most important. Additional investigations should be indi-vidualised in order to minimise the diagnostic delay and makepossible early treatment.
literatuur1 Hochberg MC. Updating the American College of Rheumatology
revised criteria for the classification of systemic lupus erythema-tosus. Arthritis Rheum 1997;40:1725.
2 Nathan DG, Oski FA. Hematology of infancy and childhood. Vol 2.4th ed. Philadelphia: Saunders; 1992. p. 1334-48.
3 Voûte PA, Kraker J de, Caron HN. Kinderoncologie. Houten: BohnStafleu Van Loghum; 1997. p. 78-85.
4 Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed con-nective tissue disease – an apparently distinct rheumatic disease syn-drome associated with a specific antibody to an extractable nuclearantigen (ENA). Am J Med 1972;52:148-59.
5 Shen N, Chen S, Yang H, Fan L. Mixed connective tissue disease: adisease entity? Chin Med J 1998;111:214-7.
6 Cate RN ten, Fiselier ThJW, Kuis W. Werkboek kinderreumatolo-gie. Amsterdam: Nederlandse Vereniging voor Kinderreumatologie;2003.
7 Peschken CA, Esdaile JM. Systemic lupus erythematosus in NorthAmerican Indians: a population based study. J Rheumatol 2000;27:1884-91.
8 Rood MJ, Cate R ten, Suijlekom-Smit LWA van, Ouden EJ den,Ouwerkerk FE, Breedveld FC, et al. Childhood-onset systemiclupus erythematosus: clinical presentation and prognosis in 31patients. Scand J Rheumatol 1999;28:222-6.
9 Sultan SM, Ioannou Y, Isenberg DA. Is there an association ofmalignancy with systemic lupus erythematosus? An analysis of 276patients under long-term review. Rheumatology (Oxford) 2000;39:1147-52.
10 Abu-Shakra M, Gladman DD, Urowitz MB. Malignancy in systemiclupus erythematosus. Arthritis Rheum 1996;39:1050-4.
Aanvaard op 10 september 2003
Ned Tijdschr Geneeskd 2004 6 maart;148(10) 457


















