
Twee patiënten met een zeldzame manifestatie van ...ig.extras.bsl.nl/bestanden/file_1324.pdf · De...
6
2055 Ned Tijdschr Geneeskd. 2007 15 september;151(37) Nicolaas Fonteyn beschreef in 1639 een patiënt met een zo- genaamde ‘sagomilt’. 1 In retrospect betrof het waarschijn- lijk een geval van amyloïdose. Amyloïd dankt zijn naam aan het feit dat het proteïne blauw aankleurt door jodium. Ook zetmeel (Latijn: amylum) kleurt met jodium blauw aan. 2 Amyloïdneerslagen kunnen overal in het lichaam optreden. De klinische verschijnselen aan de tractus respiratorius door amyloïdose zijn divers. Ze worden echter maar zelden gerapporteerd. Dit kan komen doordat respiratoire amy- loïdose weinig voorkomt, maar ook doordat er niet over wordt geschreven. Wij beschrijven hieronder 2 casussen van patiënten met respiratoire amyloïdose. ziektegeschiedenissen Patiënt A, een 62-jarige man, werd opgenomen vanwege progressieve dyspneuklachten, hoestklachten en het opge- ven van wit, taai sputum. De voorgeschiedenis vermeldde matig ernstig obstructief longlijden in stadium II volgens de criteria van het Global Initiative for Chronic Obstructive Lung Disease (GOLD; www.goldcopd.org). Longfunctie- onderzoek 7 jaar tevoren toonde een geforceerde expiratoire éénsecondevolume(FEV 1 )-waarde van 2,18 l (60% van voor- speld). Patiënt had al 4 jaar een pacemaker wegens chrono- trope incompetentie, dat wil zeggen dyspneuklachten bij een te traag oplopende hartslag. 3 Hij was 11 jaar tevoren ge- stopt met roken. Lichamelijk onderzoek toonde cyanose, forse dyspneu, bij auscultatie zachte cortonen zonder souf- fles en over de longen zacht ademgeruis met een verlengd, piepend exspirium. Oriënterend bloedonderzoek toonde een verhoogde bezinking: 56 mm/1e uur, maar geen afwijkingen van het bloedbeeld, de elektrolyten of de nierfunctie. Arteriële bloedgasanalyse toonde ernstige hypoxemie met een Po 2 van 4,3 kPa bij ademen van kamerlucht. De thoraxfoto toonde een versterkte interstitiële fibrotische tekening (fi- guur 1). Deze tekening was toegenomen in vergelijking met die op eerdere beelden. De longfunctie was ernstig obstruc- tief gestoord. De FEV 1 was in de laatste 7 jaar sterk gedaald en was nu 0,79 l (24% van voorspeld). De koolmonoxidedif- fusiecapaciteit was ernstig gestoord: 2,5 mmol per kPa/min (26% van voorspeld). De werkhypothese was dat patiënt interstitiële longafwijkingen had bij een pre-existente COPD. In de differentiaaldiagnose overwoog men: idiopathi- sche pulmonale fibrose bij COPD, pulmonale fibrose bij col- lageenziekte, lymfangitis carcinomatosa, sarcoïdose, chro- nische extrinsieke allergische alveolitis, asbestose, systemi- sche amyloïdose met longlokalisatie, hartfalen, en een com- binatie van deze ziekten. Om meer informatie te krijgen omtrent de aard van het interstitiële longbeeld werd hoge- resolutie-CT van de thorax verricht. Deze liet verspreid gele- gen, intrapulmonale noduli zien, alsook bulleus gedegene- reerd longweefsel en met name dorsobasaal subpleurale fi- casuïstische mededelingen Twee patiënten met een zeldzame manifestatie van amyloïdose in het respiratoire systeem W.Jacobs, A.Vonk Noordegraaf, T.G.Sutedja, K.Grünberg en P.E.Postmus Zie ook de artikelen op bl. 2021 en 2032. Bij 2 patiënten werd amyloïdose van de tractus respiratorius vastgesteld. Het betrof mannen van res- pectievelijk 62 en 55 jaar. De eerste patiënt kwam met dyspneu en dubbelzijdige longparenchymafwij- kingen. Er was een snel progressieve obstructieve longfunctiestoornis met een ernstige diffusiestoornis. Bij de tweede patiënt was er een tracheobronchiale amyloïdose met hemoptoë. Amyloïdose van het res- piratoire systeem wordt slechts zelden gediagnosticeerd. Nagenoeg alle klinisch belangrijke gevallen worden veroorzaakt door lichteketenamyloïd (AL-amyloïdose). De beschreven dubbelzijdige longparen- chymafwijkingen zijn een uiting van systemische amyloïdose. De tracheobronchiale amyloïdose is daar- entegen een aandoening die meestal gelokaliseerd blijft in de luchtwegen. De systemische AL-amyloï- dose kan behandeld worden met chemotherapie en stamceltransplantatie. Het is onbekend of deze behandeling kan leiden tot een afname van longfunctiestoornissen. Tracheobronchiale amyloïdose kan behandeld worden met endobronchiale therapie, waarbij vaak herhaalde ingrepen noodzakelijk zijn. De eerste patiënt overleed 2 maanden na het stellen van de diagnose aan de gevolgen van een pneumonie. De tweede patiënt werd behandeld met endobronchiale argonplasmacoagulatie en diathermie en was bij de laatste follow-up 3 jaar klachtenvrij. Ned Tijdschr Geneeskd. 2007;151:2055-60 VU Medisch Centrum, Postbus 7057, 1007 MB Amsterdam. Afd. Longziekten: hr.W.Jacobs, hr.dr.A.Vonk Noordegraaf, hr.dr.T.G. Sutedja en hr.prof.dr.P.E.Postmus, longartsen. Afd. Pathologie: mw.dr.K.Grünberg, patholoog. Correspondentieadres: hr.W.Jacobs ([email protected]).
Transcript of Twee patiënten met een zeldzame manifestatie van ...ig.extras.bsl.nl/bestanden/file_1324.pdf · De...

2055Ned Tijdschr Geneeskd. 2007 15 september;151(37)
Nicolaas Fonteyn beschreef in 1639 een patiënt met een zo-
genaamde ‘sagomilt’.1 In retrospect betrof het waarschijn-
lijk een geval van amyloïdose. Amyloïd dankt zijn naam aan
het feit dat het proteïne blauw aankleurt door jodium. Ook
zetmeel (Latijn: amylum) kleurt met jodium blauw aan.2
Amyloïdneerslagen kunnen overal in het lichaam optreden.
De klinische verschijnselen aan de tractus respiratorius
door amyloïdose zijn divers. Ze worden echter maar zelden
gerapporteerd. Dit kan komen doordat respiratoire amy-
loïdose weinig voorkomt, maar ook doordat er niet over
wordt geschreven. Wij beschrijven hieronder 2 casussen van
patiënten met respiratoire amyloïdose.
ziektegeschiedenissen
Patiënt A, een 62-jarige man, werd opgenomen vanwege
progressieve dyspneuklachten, hoestklachten en het opge-
ven van wit, taai sputum. De voorgeschiedenis vermeldde
matig ernstig obstructief longlijden in stadium II volgens
de criteria van het Global Initiative for Chronic Obstructive
Lung Disease (GOLD; www.goldcopd.org). Longfunctie-
onderzoek 7 jaar tevoren toonde een geforceerde expiratoire
éénsecondevolume(FEV1)-waarde van 2,18 l (60% van voor-
speld). Patiënt had al 4 jaar een pacemaker wegens chrono-
trope incompetentie, dat wil zeggen dyspneuklachten bij
een te traag oplopende hartslag.3 Hij was 11 jaar tevoren ge-
stopt met roken. Lichamelijk onderzoek toonde cyanose,
forse dyspneu, bij auscultatie zachte cortonen zonder souf-
fles en over de longen zacht ademgeruis met een verlengd,
piepend exspirium.
Oriënterend bloedonderzoek toonde een verhoogde
bezinking: 56 mm/1e uur, maar geen afwijkingen van
het bloedbeeld, de elektrolyten of de nierfunctie. Arteriële
bloedgasanalyse toonde ernstige hypoxemie met een Po2
van 4,3 kPa bij ademen van kamerlucht. De thoraxfoto
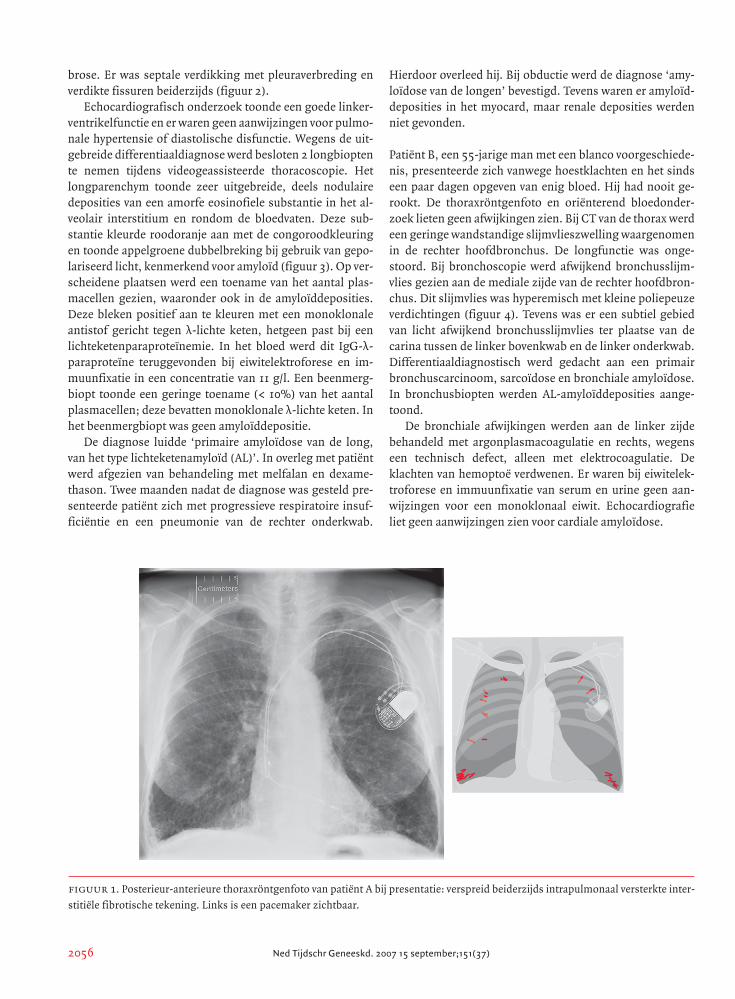
toonde een versterkte interstitiële fibrotische tekening (fi-
guur 1). Deze tekening was toegenomen in vergelijking met
die op eerdere beelden. De longfunctie was ernstig obstruc-
tief gestoord. De FEV1 was in de laatste 7 jaar sterk gedaald
en was nu 0,79 l (24% van voorspeld). De koolmonoxidedif-
fusiecapaciteit was ernstig gestoord: 2,5 mmol per kPa/min
(26% van voorspeld). De werkhypothese was dat patiënt
interstitiële longafwijkingen had bij een pre-existente COPD.
In de differentiaaldiagnose overwoog men: idiopathi-
sche pulmonale fibrose bij COPD, pulmonale fibrose bij col-
lageenziekte, lymfangitis carcinomatosa, sarcoïdose, chro-
nische extrinsieke allergische alveolitis, asbestose, systemi-
sche amyloïdose met longlokalisatie, hartfalen, en een com-
binatie van deze ziekten. Om meer informatie te krijgen
omtrent de aard van het interstitiële longbeeld werd hoge-
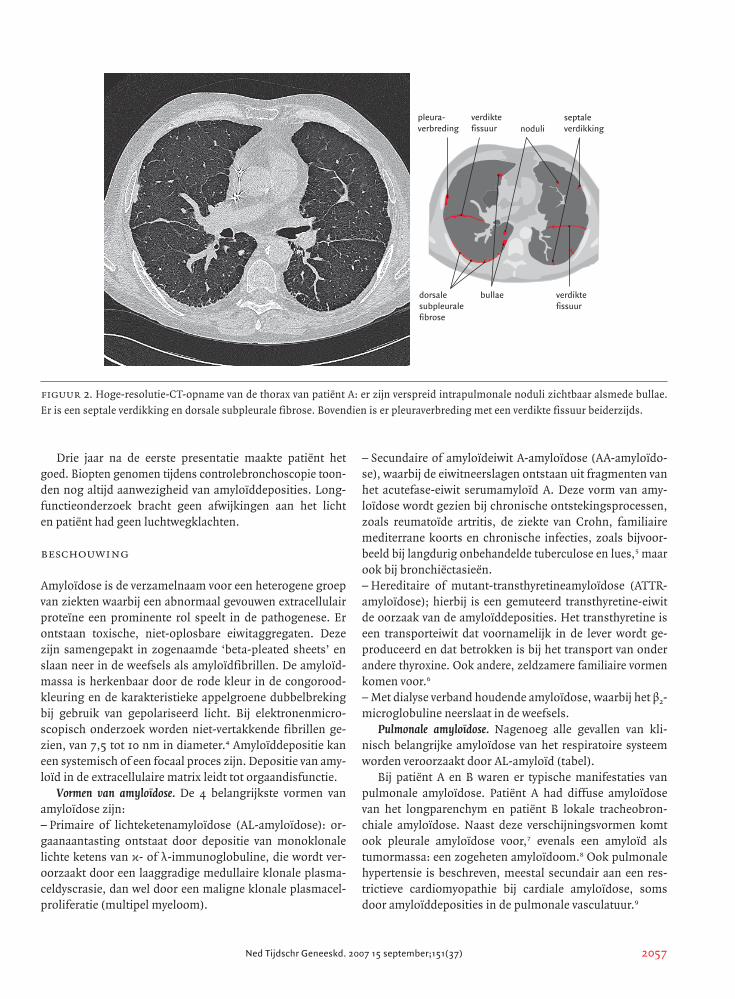
resolutie-CT van de thorax verricht. Deze liet verspreid gele-
gen, intrapulmonale noduli zien, alsook bulleus gedegene-
reerd longweefsel en met name dorsobasaal subpleurale fi-
casuïstische mededelingen
Twee patiënten met een zeldzame manifestatie van amyloïdose in het
respiratoire systeem
W.Jacobs, A.Vonk Noordegraaf, T.G.Sutedja, K.Grünberg en P.E.Postmus Zie ook de artikelen op bl. 2021 en 2032.
Bij 2 patiënten werd amyloïdose van de tractus respiratorius vastgesteld. Het betrof mannen van res-pectievelijk 62 en 55 jaar. De eerste patiënt kwam met dyspneu en dubbelzijdige longparenchymafwij-kingen. Er was een snel progressieve obstructieve longfunctiestoornis met een ernstige diffusiestoornis. Bij de tweede patiënt was er een tracheobronchiale amyloïdose met hemoptoë. Amyloïdose van het res-piratoire systeem wordt slechts zelden gediagnosticeerd. Nagenoeg alle klinisch belangrijke gevallen worden veroorzaakt door lichteketenamyloïd (AL-amyloïdose). De beschreven dubbelzijdige longparen-chymafwijkingen zijn een uiting van systemische amyloïdose. De tracheobronchiale amyloïdose is daar-entegen een aandoening die meestal gelokaliseerd blijft in de luchtwegen. De systemische AL-amyloï-dose kan behandeld worden met chemotherapie en stamceltransplantatie. Het is onbekend of deze behandeling kan leiden tot een afname van longfunctiestoornissen. Tracheobronchiale amyloïdose kan behandeld worden met endobronchiale therapie, waarbij vaak herhaalde ingrepen noodzakelijk zijn. De eerste patiënt overleed 2 maanden na het stellen van de diagnose aan de gevolgen van een pneumonie. De tweede patiënt werd behandeld met endobronchiale argonplasmacoagulatie en diathermie en was bij de laatste follow-up 3 jaar klachtenvrij.
Ned Tijdschr Geneeskd. 2007;151:2055-60
VU Medisch Centrum, Postbus 7057, 1007 MB Amsterdam.Afd. Longziekten: hr.W.Jacobs, hr.dr.A.Vonk Noordegraaf, hr.dr.T.G.Sutedja en hr.prof.dr.P.E.Postmus, longartsen.Afd. Pathologie: mw.dr.K.Grünberg, patholoog.Correspondentieadres: hr.W.Jacobs ([email protected]).
2056 Ned Tijdschr Geneeskd. 2007 15 september;151(37)
brose. Er was septale verdikking met pleuraverbreding en
verdikte fissuren beiderzijds (figuur 2).
Echocardiografisch onderzoek toonde een goede linker-
ventrikelfunctie en er waren geen aanwijzingen voor pulmo-
nale hypertensie of diastolische disfunctie. Wegens de uit-
gebreide differentiaaldiagnose werd besloten 2 longbiopten
te nemen tijdens videogeassisteerde thoracoscopie. Het
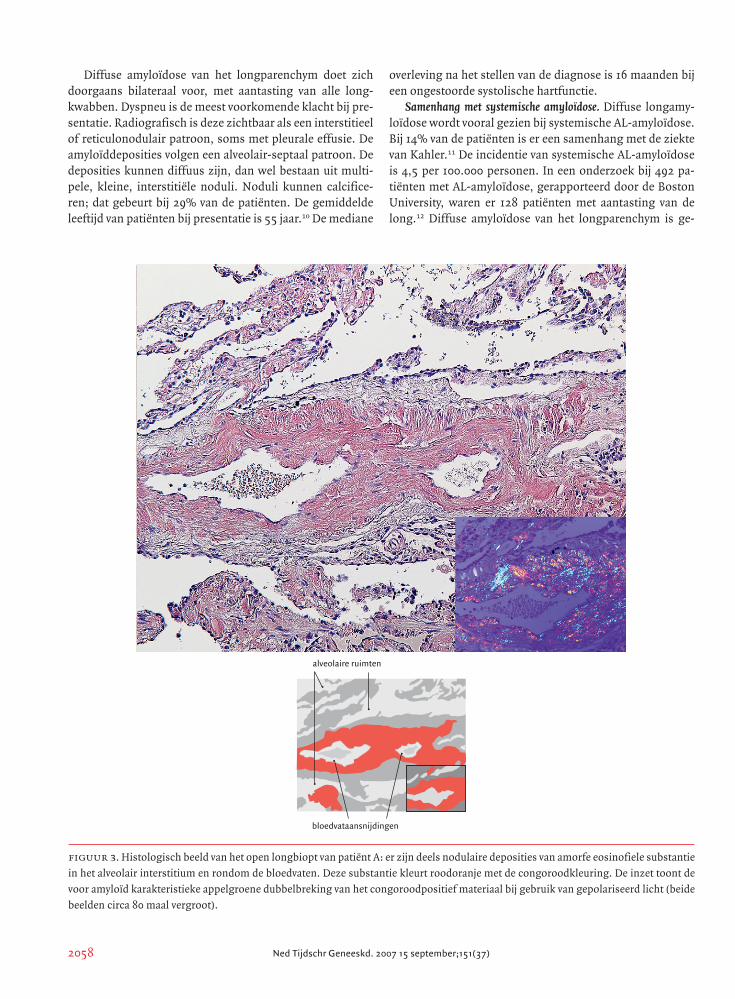
longparenchym toonde zeer uitgebreide, deels nodulaire
deposities van een amorfe eosinofiele substantie in het al-
veolair interstitium en rondom de bloedvaten. Deze sub-
stantie kleurde roodoranje aan met de congoroodkleuring
en toonde appelgroene dubbelbreking bij gebruik van gepo-
lariseerd licht, kenmerkend voor amyloïd (figuur 3). Op ver-
scheidene plaatsen werd een toename van het aantal plas-
macellen gezien, waaronder ook in de amyloïddeposities.
Deze bleken positief aan te kleuren met een monoklonale
antistof gericht tegen λ-lichte keten, hetgeen past bij een
lichteketenparaproteïnemie. In het bloed werd dit IgG-λ-
paraproteïne teruggevonden bij eiwitelektroforese en im-
muunfixatie in een concentratie van 11 g/l. Een beenmerg-
biopt toonde een geringe toename (< 10%) van het aantal
plasmacellen; deze bevatten monoklonale λ-lichte keten. In
het beenmergbiopt was geen amyloïddepositie.
De diagnose luidde ‘primaire amyloïdose van de long,
van het type lichteketenamyloïd (AL)’. In overleg met patiënt
werd afgezien van behandeling met melfalan en dexame-
thason. Twee maanden nadat de diagnose was gesteld pre-
senteerde patiënt zich met progressieve respiratoire insuf-
ficiëntie en een pneumonie van de rechter onderkwab.
Hierdoor overleed hij. Bij obductie werd de diagnose ‘amy-
loïdose van de longen’ bevestigd. Tevens waren er amyloïd-
deposities in het myocard, maar renale deposities werden
niet gevonden.
Patiënt B, een 55-jarige man met een blanco voorgeschiede-
nis, presenteerde zich vanwege hoestklachten en het sinds
een paar dagen opgeven van enig bloed. Hij had nooit ge-
rookt. De thoraxröntgenfoto en oriënterend bloedonder-
zoek lieten geen afwijkingen zien. Bij CT van de thorax werd
een geringe wandstandige slijmvlieszwelling waargenomen
in de rechter hoofdbronchus. De longfunctie was onge-
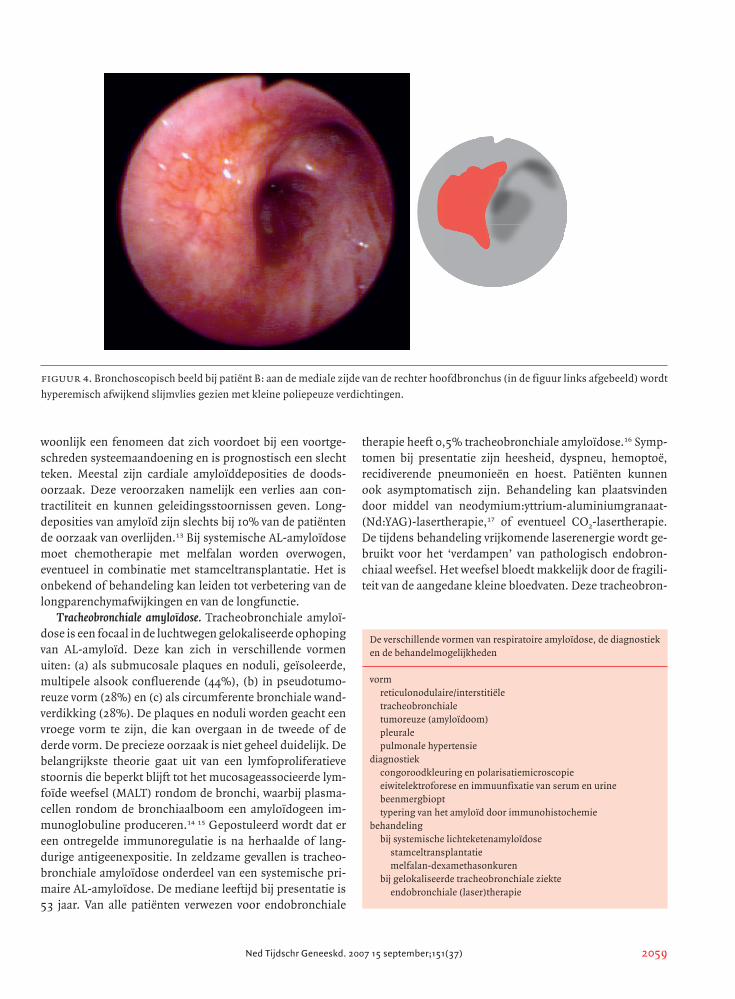
stoord. Bij bronchoscopie werd afwijkend bronchusslijm-
vlies gezien aan de mediale zijde van de rechter hoofdbron-
chus. Dit slijmvlies was hyperemisch met kleine poliepeuze
verdichtingen (figuur 4). Tevens was er een subtiel gebied
van licht afwijkend bronchusslijmvlies ter plaatse van de
carina tussen de linker bovenkwab en de linker onderkwab.
Differentiaaldiagnostisch werd gedacht aan een primair
bronchuscarcinoom, sarcoïdose en bronchiale amyloïdose.
In bronchusbiopten werden AL-amyloïddeposities aange-
toond.
De bronchiale afwijkingen werden aan de linker zijde
behandeld met argonplasmacoagulatie en rechts, wegens
een technisch defect, alleen met elektrocoagulatie. De
klachten van hemoptoë verdwenen. Er waren bij eiwitelek-
troforese en immuunfixatie van serum en urine geen aan-
wijzingen voor een monoklonaal eiwit. Echocardiografie
liet geen aanwijzingen zien voor cardiale amyloïdose.
figuur 1. Posterieur-anterieure thoraxröntgenfoto van patiënt A bij presentatie: verspreid beiderzijds intrapulmonaal versterkte inter-
stitiële fibrotische tekening. Links is een pacemaker zichtbaar.
2057Ned Tijdschr Geneeskd. 2007 15 september;151(37)
Drie jaar na de eerste presentatie maakte patiënt het
goed. Biopten genomen tijdens controlebronchoscopie toon-
den nog altijd aanwezigheid van amyloïddeposities. Long-
functieonderzoek bracht geen afwijkingen aan het licht
en patiënt had geen luchtwegklachten.
beschouwing
Amyloïdose is de verzamelnaam voor een heterogene groep
van ziekten waarbij een abnormaal gevouwen extracellulair
proteïne een prominente rol speelt in de pathogenese. Er
ontstaan toxische, niet-oplosbare eiwitaggregaten. Deze
zijn samengepakt in zogenaamde ‘beta-pleated sheets’ en
slaan neer in de weefsels als amyloïdfibrillen. De amyloïd-
massa is herkenbaar door de rode kleur in de congorood-
kleuring en de karakteristieke appelgroene dubbelbreking
bij gebruik van gepolariseerd licht. Bij elektronenmicro-
scopisch onderzoek worden niet-vertakkende fibrillen ge-
zien, van 7,5 tot 10 nm in diameter.4 Amyloïddepositie kan
een systemisch of een focaal proces zijn. Depositie van amy-
loïd in de extracellulaire matrix leidt tot orgaandisfunctie.
Vormen van amyloïdose. De 4 belangrijkste vormen van
amyloïdose zijn:
– Primaire of lichteketenamyloïdose (AL-amyloïdose): or-
gaanaantasting ontstaat door depositie van monoklonale
lichte ketens van ϰ- of λ-immunoglobuline, die wordt ver-
oorzaakt door een laaggradige medullaire klonale plasma-
celdyscrasie, dan wel door een maligne klonale plasmacel-
proliferatie (multipel myeloom).
– Secundaire of amyloïdeiwit A-amyloïdose (AA-amyloïdo-
se), waarbij de eiwitneerslagen ontstaan uit fragmenten van
het acutefase-eiwit serumamyloïd A. Deze vorm van amy-
loïdose wordt gezien bij chronische ontstekingsprocessen,
zoals reumatoïde artritis, de ziekte van Crohn, familiaire
mediterrane koorts en chronische infecties, zoals bijvoor-
beeld bij langdurig onbehandelde tuberculose en lues,5 maar
ook bij bronchiëctasieën.
– Hereditaire of mutant-transthyretineamyloïdose (ATTR-
amyloïdose); hierbij is een gemuteerd transthyretine-eiwit
de oorzaak van de amyloïddeposities. Het transthyretine is
een transporteiwit dat voornamelijk in de lever wordt ge-
produceerd en dat betrokken is bij het transport van onder
andere thyroxine. Ook andere, zeldzamere familiaire vormen
komen voor.6
– Met dialyse verband houdende amyloïdose, waarbij het β2-
microglobuline neerslaat in de weefsels.
Pulmonale amyloïdose. Nagenoeg alle gevallen van kli-
nisch belangrijke amyloïdose van het respiratoire systeem
worden veroorzaakt door AL-amyloïd (tabel).
Bij patiënt A en B waren er typische manifestaties van
pulmonale amyloïdose. Patiënt A had diffuse amyloïdose
van het longparenchym en patiënt B lokale tracheobron-
chiale amyloïdose. Naast deze verschijningsvormen komt
ook pleurale amyloïdose voor,7 evenals een amyloïd als
tumormassa: een zogeheten amyloïdoom.8 Ook pulmonale
hypertensie is beschreven, meestal secundair aan een res-
trictieve cardiomyopathie bij cardiale amyloïdose, soms
door amy loïddeposities in de pulmonale vasculatuur.9
figuur 2. Hoge-resolutie-CT-opname van de thorax van patiënt A: er zijn verspreid intrapulmonale noduli zichtbaar alsmede bullae.
Er is een septale verdikking en dorsale subpleurale fibrose. Bovendien is er pleuraverbreding met een verdikte fissuur beiderzijds.
pleura-verbreding noduli
septaleverdikking
verdiktefissuur
verdiktefissuur
bullaedorsalesubpleuralefibrose
2058 Ned Tijdschr Geneeskd. 2007 15 september;151(37)
Diffuse amyloïdose van het longparenchym doet zich
doorgaans bilateraal voor, met aantasting van alle long-
kwabben. Dyspneu is de meest voorkomende klacht bij pre-
sentatie. Radiografisch is deze zichtbaar als een interstitieel
of reticulonodulair patroon, soms met pleurale effusie. De
amyloïddeposities volgen een alveolair-septaal patroon. De
deposities kunnen diffuus zijn, dan wel bestaan uit multi-
pele, kleine, interstitiële noduli. Noduli kunnen calcifice-
ren; dat gebeurt bij 29% van de patiënten. De gemiddelde
leeftijd van patiënten bij presentatie is 55 jaar.10 De mediane
overleving na het stellen van de diagnose is 16 maanden bij
een ongestoorde systolische hartfunctie.
Samenhang met systemische amyloïdose. Diffuse longamy-
loïdose wordt vooral gezien bij systemische AL-amyloïdose.
Bij 14% van de patiënten is er een samenhang met de ziekte
van Kahler.11 De incidentie van systemische AL-amyloïdose
is 4,5 per 100.000 personen. In een onderzoek bij 492 pa-
tiënten met AL-amyloïdose, gerapporteerd door de Boston
University, waren er 128 patiënten met aantasting van de
long.12 Diffuse amyloïdose van het longparenchym is ge-
figuur 3. Histologisch beeld van het open longbiopt van patiënt A: er zijn deels nodulaire deposities van amorfe eosinofiele substantie
in het alveolair interstitium en rondom de bloedvaten. Deze substantie kleurt roodoranje met de congoroodkleuring. De inzet toont de
voor amyloïd karakteristieke appelgroene dubbelbreking van het congoroodpositief materiaal bij gebruik van gepolariseerd licht (beide
beelden circa 80 maal vergroot).
alveolaire ruimten
bloedvataansnijdingen
2059Ned Tijdschr Geneeskd. 2007 15 september;151(37)
woonlijk een fenomeen dat zich voordoet bij een voortge-
schreden systeemaandoening en is prognostisch een slecht
teken. Meestal zijn cardiale amyloïddeposities de doods-
oorzaak. Deze veroorzaken namelijk een verlies aan con-
tractiliteit en kunnen geleidingsstoornissen geven. Long-
deposities van amyloïd zijn slechts bij 10% van de patiënten
de oorzaak van overlijden.13 Bij systemische AL-amyloïdose
moet chemotherapie met melfalan worden overwogen,
eventueel in combinatie met stamceltransplantatie. Het is
onbekend of behandeling kan leiden tot verbetering van de
longparenchymafwijkingen en van de longfunctie.
Tracheobronchiale amyloïdose. Tracheobronchiale amyloï-
dose is een focaal in de luchtwegen gelokaliseerde ophoping
van AL-amyloïd. Deze kan zich in verschillende vormen
uiten: (a) als submucosale plaques en noduli, geïsoleerde,
multipele alsook confluerende (44%), (b) in pseudotumo-
reuze vorm (28%) en (c) als circumferente bronchiale wand-
verdikking (28%). De plaques en noduli worden geacht een
vroege vorm te zijn, die kan overgaan in de tweede of de
derde vorm. De precieze oorzaak is niet geheel duidelijk. De
belangrijkste theorie gaat uit van een lymfoproliferatieve
stoornis die beperkt blijft tot het mucosageassocieerde lym-
foïde weefsel (MALT) rondom de bronchi, waarbij plasma-
cellen rondom de bronchiaalboom een amyloïdogeen im-
munoglobuline produceren.14 15 Gepostuleerd wordt dat er
een ontregelde immunoregulatie is na herhaalde of lang-
durige antigeenexpositie. In zeldzame gevallen is tracheo-
bronchiale amyloïdose onderdeel van een systemische pri-
maire AL-amyloïdose. De mediane leeftijd bij presentatie is
53 jaar. Van alle patiënten verwezen voor endobronchiale
therapie heeft 0,5% tracheobronchiale amyloïdose.16 Symp-
tomen bij presentatie zijn heesheid, dyspneu, hemoptoë,
recidiverende pneumonieën en hoest. Patiënten kunnen
ook asymptomatisch zijn. Behandeling kan plaatsvinden
door middel van neodymium:yttrium-aluminiumgranaat-
(Nd:YAG)-lasertherapie,17 of eventueel CO2-lasertherapie.
De tijdens behandeling vrijkomende laserenergie wordt ge-
bruikt voor het ‘verdampen’ van pathologisch endobron-
chiaal weefsel. Het weefsel bloedt makkelijk door de fragili-
teit van de aangedane kleine bloedvaten. Deze tracheobron-
figuur 4. Bronchoscopisch beeld bij patiënt B: aan de mediale zijde van de rechter hoofdbronchus (in de figuur links afgebeeld) wordt
hyperemisch afwijkend slijmvlies gezien met kleine poliepeuze verdichtingen.
De verschillende vormen van respiratoire amyloïdose, de diagnostiek
en de behandelmogelijkheden
vorm
reticulonodulaire/interstitiële
tracheobronchiale
tumoreuze (amyloïdoom)
pleurale
pulmonale hypertensie
diagnostiek
congoroodkleuring en polarisatiemicroscopie
eiwitelektroforese en immuunfixatie van serum en urine
beenmergbiopt
typering van het amyloïd door immunohistochemie
behandeling
bij systemische lichteketenamyloïdose
stamceltransplantatie
melfalan-dexamethasonkuren
bij gelokaliseerde tracheobronchiale ziekte
endobronchiale (laser)therapie

2060 Ned Tijdschr Geneeskd. 2007 15 september;151(37)
chiale bloedingen zijn goed te behandelen met lasercoagu-
latie en lokale applicatie van epinefrine.
In de door ons beschreven casus werd de tracheobron-
chiale amyloïdosis behandeld met diathermische coagulatie
en argonplasmacoagulatie. De coagulatie bij diathermie
wordt bereikt door het toedienen van een hoogfrequente
elektrische stroom via een probe. Bij de argonplasmacoagu-
latie gebeurt dit door het toedienen van een monopolaire
elektrische stroom met argongas als geleidingsmedium.
Spontane resolutie van amyloïddeposities is gerappor-
teerd. Tracheobronchiale amyloïdose is echter meestal een
chronische aandoening. Herhaalde bronchoscopische in-
terventies kunnen noodzakelijk zijn. Uiteindelijk kunnen
de luchtwegen uitgebreid gecompromitteerd raken, met de
dood tot gevolg. Dit gebeurt bij circa een derde van de pa-
tiënten met aantasting van de proximale luchtwegen of ern-
stige aantasting van de middelgrote luchtwegen, en gemid-
deld 9 jaar na het stellen van de diagnose. De mortaliteit is
in recent beschreven patiëntengroepen laag. Bij 32 patiën-
ten gevolgd gedurende een gemiddelde periode van 84
maanden waren er 2 sterfgevallen ten gevolge van tracheo-
bronchiale amyloïdose.18 In casuïstische mededelingen zijn
goede resultaten van radiotherapie beschreven. Het is niet
bekend of endobronchiale therapie zinvol is bij asymptoma-
tische tracheobronchiale amyloïdose.18-20
conclusie
Klinisch belangrijke amyloïdose van het respiratoire sys-
teem treedt vrijwel uitsluitend op door depositie van het
AL-amyloïd. De klachten bij presentatie zijn divers. Dubbel-
zijdige longparenchymafwijkingen met dyspneuklachten
zijn de belangrijkste pulmonale uitingen van systemische
AL-amyloïdose. Tracheobronchiale amyloïdose is daaren-
tegen meestal geen systeemaandoening en ontstaat door
lokale ophopingen van AL-amyloïd.
Belangenconflict: geen gemeld. Financiële ondersteuning: geen gemeld.
Aanvaard op 23 januari 2007
Literatuur
1 Fonteyn N. Responsionum & curationum medicinalium liber unus. Amsterdam: Typis Ioannis Ianssonii; 1639.
2 Kyle RA. Amyloidosis: a convoluted story. Br J Haematol. 2001;114:529-38.
3 Gentlesk PJ, Markwood TT, Atwood JE. Chronotropic incompetence in a young adult: case report and literature review. Chest. 2004;125:297-301.
4 Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-96.
5 Lobel TC, Thije PA ten. Amyloiedafzetting in de long bij algemeene amyloïdose. Ned Tijdschr Geneeskd. 1921;65:1234-40.
6 Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898-909.
7 Berk JL, Keane J, Seldin DC, Sanchorawala V, Koyama J, Dember LM, et al. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest. 2003;124:969-77.
8 Desai RA, Mahajan VK, Benjamin S, Ordstrand HS van, Cordasco EM. Pulmonary amyloidoma and hilar adenopathy. Rare manifestations of primary amyloidosis. Chest. 1979;76:170-3.
9 Dingli D, Utz JP, Gertz MA. Pulmonary hypertension in patients with amyloidosis. Chest. 2001;120:1735-9.
10 Hui AN, Koss MN, Hochholzer L, Wehunt WD. Amyloidosis present-ing in the lower respiratory tract. Clinicopathologic, radiologic, im-munohistochemical, and histochemical studies on 48 cases. Arch Pathol Lab Med. 1986;110:212-8.
11 Utz JP, Swensen SJ, Gertz MA. Pulmonary amyloidosis. The Mayo Clinic experience from 1980 to 1993. Ann Intern Med. 1996;124:407-13.
12 Berk JL, O’Regan A, Skinner M. Pulmonary and tracheobronchial amyloidosis. Semin Respir Crit Care Med. 2002;23:155-65.
13 Cordier JF, Loire R, Brune J. Amyloidosis of the lower respiratory tract. Clinical and pathologic features in a series of 21 patients. Chest. 1986;90:827-31.
14 Thompson LD, Derringer GA, Wenig BM. Amyloidosis of the larynx: a clinicopathologic study of 11 cases. Mod Pathol. 2000;13:528-35.
15 Chen KT. Amyloidosis presenting in the respiratory tract. Pathol Annu. 1989;24:253-73.
16 Shah H, Garbe L, Nussbaum E, Dumon JF, Chiodera PL, Cavaliere S. Benign tumors of the tracheobronchial tree. Endoscopic characteris-tics and role of laser resection. Chest. 1995;107:1744-51.
17 Russchen GH, Wouters B, Meinesz AF, Janssen S, Postmus PE. Amy-loid tumour resected by laser therapy. Eur Respir J. 1990;3:932-3.
18 Piazza C, Cavaliere S, Foccoli P, Toninelli C, Bolzoni A, Peretti G. Endoscopic management of laryngo-tracheobronchial amyloidosis: a series of 32 patients. Eur Arch Otorhinolaryngol. 2003;260:349-54.
19 Capizzi SA, Betancourt E, Prakash UB. Tracheobronchial amyloido-sis. Mayo Clin Proc. 2000;75:1148-52.
20 O’Regan A, Fenlon HM, Beamis JF, Steele MP, Skinner M, Berk JL. Tracheobronchial amyloidosis: the Boston University experience from 1984 to 1999. Medicine. 2000;79:69-79.
Abstract
Two patients with a rare manifestation of amyloidosis in the respiratory system. – Amyloidosis of the respiratory tract was diagnosed in 2 pa-tients. The patients were men, 62 and 55 years of age. The first patient presented with dyspnoea and diffuse parenchymal lung abnormalities. There was a rapidly progressive obstructive lung function disorder and a severe diffusion impairment. The second patient had haemoptysis due to tracheobronchial amyloidosis. Amyloidosis of the respiratory system is rarely diagnosed. Nearly all cases of clinically relevant respiratory amyloi-dosis are due to light chain amyloid (AL amyloidosis). The described dif-fuse lung parenchymal abnormalities are a manifestation of systemic AL amyloidosis. On the other hand, tracheobronchial amyloidosis is a dis-order which usually remains localised in the airways. Systemic AL amyloi-dosis may be treated with chemotherapy or stem cell transplantation. It is unknown whether this treatment leads to a decrease of pulmonary func-tion abnormalities. Tracheobronchial amyloidosis can be treated by en-dobronchial therapy. Often this treatment is required repeatedly. The first patient died 2 months after diagnosis due to pneumonia. The second patient was treated with endobronchial argon plasma coagulation and diathermy and has been symptom-free for 3 years since.Ned Tijdschr Geneeskd. 2007;151:2055-60












