
UNIVERSITEIT GENT FACULTEIT FARMACEUTISCHE...
63
UNIVERSITEIT GENT FACULTEIT FARMACEUTISCHE WETENSCHAPPEN Vakgroep Farmaceutische Analyse Laboratorium voor Analytische Chemie Academiejaar 2009 – 2010 VALIDATIE VAN EEN IONENCHROMATOGRAFISCHE METHODE VOOR DE BEPALING VAN CALCIUM Sofie STALMANS Eerste master geneesmiddelenontwikkeling Promotor Prof. dr. L. Thienpont Commissarissen Prof. dr. T. De Beer Dr. C. Stove
Transcript of UNIVERSITEIT GENT FACULTEIT FARMACEUTISCHE...

UNIVERSITEIT GENT
FACULTEIT FARMACEUTISCHE WETENSCHAPPEN
Vakgroep Farmaceutische Analyse
Laboratorium voor Analytische Chemie
Academiejaar 2009 – 2010
VALIDATIE VAN EEN IONENCHROMATOGRAFISCHE
METHODE VOOR DE BEPALING VAN CALCIUM
Sofie STALMANS
Eerste master geneesmiddelenontwikkeling
Promotor Prof. dr. L. Thienpont
Commissarissen Prof. dr. T. De Beer
Dr. C. Stove


UNIVERSITEIT GENT
FACULTEIT FARMACEUTISCHE WETENSCHAPPEN
Vakgroep Farmaceutische Analyse
Laboratorium voor Analytische Chemie
Academiejaar 2009 – 2010
VALIDATIE VAN EEN IONENCHROMATOGRAFISCHE
METHODE VOOR DE BEPALING VAN CALCIUM
Sofie STALMANS
Eerste master geneesmiddelenontwikkeling
Promotor Prof. dr. L. Thienpont
Commissarissen Prof. dr. T. De Beer
Dr. C. Stove

AUTEURSRECHT
“De auteur en de promotor geven de toelating deze masterproef voor consultatie beschikbaar
te stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder de
beperkingen van het auteursrecht, in het bijzonder met betrekking tot de verplichting
uitdrukkelijk de bron te vermelden bij het aanhalen van de resultaten uit deze masterproef.”
27/05/2010
Promotor Auteur
Prof. dr. L. Thienpont Sofie Stalmans

DANKWOORD
Als eerste wil ik mijn promotor prof. L. Thienpont bedanken voor het aanreiken van het
onderwerp van de thesis en het ter beschikking stellen van de ionenchromatograaf. Bedankt
voor de bezorgdheid toen het toestel het even liet afweten, de goede opvolging van de
onderzoeksstage en de verbetering van thesis.
Dr. D. Stöckl verdient een bijzonder dankwoord voor z’n eindeloze inzet en enthousiasme bij
het deskundig uitleggen van de statistiek en voor de nuttige suggesties en adviezen bij het
schrijven van de thesis. Hij was steeds bereid om ons te helpen en om onze vragen te
beantwoorden en was altijd te vinden voor zowel wetenschappelijke als levensbeschouwelijke
discussies.
Verder wil ik ook een woord van dank richten aan Hedwig Stepman en Sofie Van Houcke
voor de dagelijkse begeleiding en hun geduld toen het fout liep met het toestel. Bedankt voor
het snel oplossen van het probleem. Ik apprecieer het dat jullie steeds klaarstonden voor ons.
Dr. Katleen Van Uytfanghe wil ik bedanken voor het nalezen van de thesis en de nuttige tips.
Ook wil ik Brigitte, Hilde en Tanja bedanken voor hun vriendelijkheid en de goede zorgen in
het labo bij praktische probleempjes.
Natuurlijk verdienen mijn medestudenten Renate, Hanne, Stefanie, Elise en Lode ook een
dankwoord voor de leuke sfeer in het labo, de toffe babbels en de hulp bij het verwerken van
de resultaten.
Tot slot dank ik mijn ouders, broer, familie, vrienden en vriendinnen voor hun interesse,
aanmoedigingen, steun en hulp bij het schrijven van deze thesis.

INHOUDSOPGAVE
1. INLEIDING............................................................................................................................1
1.1. STRUCTUUR EN CHEMISCHE EIGENSCHAPPEN VAN CALCIUM.....................1
1.2. VOORKOMEN ...............................................................................................................1
1.3. FUNCTIE.........................................................................................................................2
1.4. METHODEVALIDATIE ................................................................................................3
2. OBJECTIEVEN .....................................................................................................................6
3. MATERIALEN EN METHODEN ........................................................................................7
3.1. MATERIALEN ...............................................................................................................7
3.2. STANDAARDEN EN STALEN.....................................................................................7
3.2.1. Stockoplossing ..........................................................................................................7
3.2.2. Stalen gebruikt voor de verschillende validatie-experimenten .................................7
3.3. APPARATUUR.............................................................................................................10
3.3.1. Instrument ...............................................................................................................10
3.3.2. Randapparatuur .......................................................................................................11
3.4. METHODEN.................................................................................................................11
3.4.1. Systeemfunctiecontrole ...........................................................................................11
3.4.2. Systeemgeschiktheidstest ........................................................................................11
3.4.3. Analyse....................................................................................................................12
3.5. VALIDATIE-EXPERIMENTEN..................................................................................13
3.5.1. Lineariteit ................................................................................................................13
3.5.2. Opstellen kalibratiecurve ........................................................................................14
3.5.3. Imprecisie ................................................................................................................14
3.5.4. Aantoonbaarheidsgrens ...........................................................................................15
3.5.5. Juistheid...................................................................................................................16
3.5.6. Methodevergelijking ...............................................................................................17
3.6. DATAVERWERKING EN STATISTIEK ...................................................................18
3.7. SPECIFICATIES ...........................................................................................................19
3.8. LITERATUURONDERZOEK......................................................................................19
4. RESULTATEN EN DISCUSSIE.........................................................................................21
4.1. METHODEN.................................................................................................................21
4.1.1. Systeemfunctiecontrole ...........................................................................................21

4.1.2. Systeemgeschiktheidstest ........................................................................................21
4.2. VALIDATIE-EXPERIMENTEN..................................................................................22
4.2.1. Lineariteit ................................................................................................................22
4.2.2. Kalibratiecurve........................................................................................................23
4.2.3. Imprecisie ................................................................................................................25
4.2.4. Aantoonbaarheidsgrens ...........................................................................................27
4.2.5. Juistheid...................................................................................................................28
4.2.6. Methodevergelijking ...............................................................................................31
4.3. LITERATUURONDERZOEK......................................................................................33
4.3.1. Opzoeken wetenschappelijke literatuur ..................................................................33
4.3.2. Methodevalidatie in de in vitro diagnostica ............................................................34
4.3.2.1. Protocollen en statistiek....................................................................................34
4.3.2.2. Specificaties......................................................................................................38
4.3.2.3. Beslissen ...........................................................................................................40
4.3.3. Andere bepalingsmethoden voor calcium...............................................................41
4.3.4. Farmaceutisch gebruik van calcium........................................................................43
5. CONCLUSIE........................................................................................................................45
6. REFERENTIELIJST ............................................................................................................47

DEFINITIES
De definities van de volgende begrippen zijn overgenomen uit de “Vocabulaire International
de Métrologie – Concepts fondamentaux et généraux et termes associés” (VIM, 2008).
- Measurand:
Quantity intended to be measured.
- Measurement procedure:
Detailed description of a measurement according to one or more measurement
principles and to a given measurement method, based on a measurement model and
including any calculation to obtain a measurement result.
NOTE: A measurement procedure is sometimes called a standard operating procedure,
abbreviated SOP.
- Analytical selectivity:
Property of a measuring system, used with a specified measurement procedure,
whereby it provides measured quantity values for one or more measurands such that
the values of each measurand are independent of other measurands or other quantities
in the phenomenon, body, or substance being investigated.
NOTE: Selectivity as used in physics is a concept close to specificity as it is
sometimes used in chemistry.
- Detection limit:
Measured quantity value, obtained by a given measurement procedure, for which the
probability of falsely claiming the absence of a component in a material is β, given a
probability α of falsely claiming its presence.
NOTE: IUPAC recommends default values for α and β equal to 0.05.
- Bias of measurements:
Estimate of a systematic error.
NOTE: A systematic measurement error is a component of measurement error that in
replicate measurements remains constant or varies in a predictable manner.

- Trueness of a measurement:
Closeness of agreement between the average of an infinite number of replicate
measured quantity values and a reference quantity value.
NOTE 1: Measurement trueness is not a quantity and thus cannot be expressed
numerically, but measures for closeness of agreement are given in ISO 5725.
NOTE 2: Measurement trueness is inversely related to systematic measurement error,
but is not related to random measurement error.
- Accuracy of a measurement:
Closeness of agreement between a measured quantity value and a true quantity value
of a measurand.
NOTE 1: The concept ‘measurement accuracy’ is not a quantity and is not given a
numerical quantity value. A measurement is said to be more accurate when it offers a
smaller measurement error.
NOTE 2: The term “measurement accuracy” should not be used for measurement
trueness and the term measurement precision should not be used for ‘measurement
accuracy’, which, however, is related to both these concepts.
NOTE 3: ‘Measurement accuracy’ is sometimes understood as closeness of agreement
between measured quantity values that are being attributed to the measurand.
- Precision of a measurement:
Closeness of agreement between indications or measured quantity values obtained by
replicate measurements on the same or similar objects under specified conditions.
NOTE 1: Measurement precision is usually expressed numerically by measures of
imprecision, such as standard deviation, variance or coefficient of variation under the
specified conditions of measurement.
NOTE 2: The ‘specified conditions’ can be, for example, repeatability conditions of
measurement, intermediate precision conditions of measurement or reproducibility
conditions of measurement (see ISO 5725-3:1994).
NOTE 3: Measurement precision is used to define measurement repeatability,
intermediate measurement precision and measurement reproducibility.

- Repeatability
Measurement precision under a set of repeatability conditions of measurement.
NOTE: A repeatability condition of a measurement is a condition of measurement, out
of a set of conditions that includes different locations, operators, measuring systems
and replicate measurements on the same or similar objects.
- Reproducibility:
Measurement precision under reproducibility conditions of measurement.
NOTE: A reproducibility condition is a condition of measurement, out of a set of
conditions that includes different locations, operators, measuring systems and replicate
measurements on the same or similar objects
De metrologische termen worden in het Nederlands vertaald volgens het CMA-6-A-
Prestatiekenmereken (Compendium voor Monsternemening en Analyse in uitvoering van het
afvalstoffendecreet en bodemsaneringsdectreet, 2010):
- Accuracy: Accuraatheid
- Limit of detection: Aantoonbaarheidsgrens
- Limit of quantification: Bepalingsgrens
- Linearity: Lineariteit
- Precision: Precisie
- Repeatability: Herhaalbaarheid
- Reproducibility: Reproduceerbaarheid
- Ruggedness/robustness: Robuustheid
- Trueness: Juistheid
- Uncertainty of measurement: Meetonzekerheid
- Working/measuring range: Werkgebied of bereik

LIJST MET GEBRUIKTE AFKORTINGEN
°C Graden Celcius
µg Microgram
µL Microliter
µm Micrometer
µS Microsiemens
BCFI Belgisch Centrum voor Farmacotherapeutische Informatie
BI Betrouwbaarheidsinterval
Ca Calcium
CaCl2.2H2O Calciumchloridedihydraat
cal Calorie
CE Capillaire Elektroforese
CL Confidentielimiet
CLSI Clinical and Laboratory Standards Institute
conc. Concentratie
CV Variatiecoëfficiënt (coefficient of variation)
CVT Totale variatiecoëfficiënt (total coefficient of variation)
CVwr Binnen analyseserie variatiecoëfficiënt (within run coefficient of variation)
d. Dag
EP Evaluatieprotocol (Evaluation Protocol)
FAAS Vlam (Flame) Atomaire Absorptie Spectrofotometrie
g Gram
GLR Gewogen lineaire regressie
HPLC High Performance Liquid Chromatography
ICH International Conference on Harmonization
IQC Interne kwaliteitscontrole (Internal Quality Control)
ISO International Organization for Standardization
JCTLM Joint Committee for Traceability in Laboratory Medicine

L Liter
LoB Limit of the Blank (blancolimiet)
LoD Limit of Detection (aantoonbaarheidsgrens)
LoQ Limit of Quantification (bepalingsgrens)
MgSO4.7H2O Magnesiumsulfaatheptahydraat
mg Milligram
min Minuten
mL Milliliter
mm Millimeter
mmol Millimol
n Aantal
ng Nanogram
OLR Ordinary Least-square Regression
psi Pound-force per Square Inch (1 psi = 6,9 kPa)
PI Predictie-interval
P-waarde Probabiliteitswaarde
r. Replicaten
Swr Binnen analyseserie standaarddeviatie (within run standard deviation)
ST Totale standaarddeviatie
SD Standaarddeviatie
SE Systematic Error (systematische fout)
SI Système International
S/R Signaal-over-ruisverhouding
TE Total Error (totale fout)
tR Retentietijd
USP United States Pharmacopoeia
VIM Vocabulaire International de Métrologie

1
1. INLEIDING
1.1. STRUCTUUR EN CHEMISCHE EIGENSCHAPPEN VAN CALCIUM
Calcium (Ca) is een aardalkalimetaal dat voor het eerst werd geïsoleerd door Davy in
1808. Het heeft een molaire massa van 40,08 g/mol en twee valenties. De dichtheid bedraagt
1,54 g/cm3. In de natuur komen zes verschillende isotopen van calcium voor, waarvan 40Ca
het meest abundante is (97%). Calcium wordt in de natuur nooit alleen teruggevonden, maar
steeds onder de vorm van zouten. De belangrijkste natuurlijke bron van calcium is kalksteen.
Voor commercieel gebruik wordt metallisch calcium geproduceerd door elektrolyse van
calciumchloride (Merck Index, 1996).
Calcium is een glanzend zilverwit metaal en heeft een kubisch vlakgecentreerde
kristalstructuur onder 300°C. Het metaal is harder dan natrium, maar zachter dan aluminium
of magnesium. Het smeltpunt van calcium is 850°C en het kookpunt is gelegen bij 1440°C.
De elektrische resistiviteit bij 20°C is 3,4 µOhm⋅cm. Calcium is minder reactief dan de
alkalimetalen en de andere aardalkalimetalen. Metallisch calcium is brandbaar (Merck Index,
1996).
Calcium reageert met water, alcoholen en verdunde zuren met vorming van
waterstofgas. Het reageert ook met de halogenen. Metallisch calcium is oplosbaar in vloeibare
ammoniak, waarin het een blauwe oplossing vormt, maar is inert en onoplosbaar in benzeen
en kerosine. Contact tussen calcium en alkalische hydroxiden of carbonaten kan het best
vermeden worden, wegens het ontploffingsgevaar (Merck Index, 1996).
1.2. VOORKOMEN
Calcium is een wijdverspreid element op aarde. Het is met 3,64% het vijfde meest
voorkomende element in de aardkorst. Zeewater bevat ongeveer 400 g/ton calciumchloride
(Merck Index, 1996). Ook planten en bomen zijn rijk aan calcium. Daarom vormen diverse
groenten en fruit samen met melk en zuivelproducten een belangrijk calciumbron voor mens
en dier. Calcium is namelijk het meest voorkomende mineraal in het menselijk lichaam. Het
ligt voor 98% opgeslagen in het skelet en de tanden, waarvan 1% vrij uitwisselbaar is met de
extracellulaire lichaamsvloeistof. Hierin komt het voor als een divalent kation (Ca2+). In de
extracellulaire vloeistof circuleert calcium in drie verschillende fracties. De helft van het Ca2+
komt voor in de geïoniseerde vorm en is biologisch de meest belangrijke fractie. 40% van het

2
calcium is gebonden aan eiwitten, voornamelijk albumine, en 10% is gecomplexeerd met
anionen zoals citraat, bicarbonaat, sulfaat, fosfaat en lactaat (Tortora & Derrickson, 2006;
Bushinsky & Monk, 1998).
1.3. FUNCTIE
Calciummineralen in de bodem en water vormen een belangrijke voedingsbron voor
planten en bomen. Calcium behoort tot één van de essentiële nutriënten voor planten. Het
vervult zowel een structurele als een tweede boodschappersfunctie in de plantencel.
Complexen van calcium en organische verbindingen zoals de fosfaten en carboxylaten van
fosfolipiden, proteïnen, suikers en pectines verstevigen de celwand en het celmembraan van
de plantencel (Maathuis, 2009).
Doordat calcium zeer makkelijk onoplosbare zouten vormt met sulfaten en fosfaten, is
de cytoplasmatische vrije concentratie van Ca2+ zeer laag. Daarom is het divalent kation
geschikt als tweede boodschappermolecule en zullen verschillende extracellulaire stimuli
zorgen voor een verandering in de concentratie van vrij cytoplasmatisch Ca2+, met een
celrespons tot gevolg (Maathuis, 2009).
Calcium speelt een vitale rol in de cellen van alle levende wezens. In het menselijk
lichaam vervult calcium diverse functies. Als tweede boodschapper zorgt het voor de
overdracht van signalen tussen het plasmamembraan en het intracellulair milieu. In de
extracellulaire vloeistof is calcium een belangrijke cofactor voor de stollingsfactoren en de
adhesiemoleculen tijdens het bloedstollingsproces (Power et al., 1999). In de botten en tanden
vormt calcium samen met de aanwezige fosfaten een kristallijn netwerk van minerale zouten.
Op deze manier draagt calcium bij tot de stevigheid van het skelet en het gebit. Daarnaast
speelt calcium ook een belangrijke rol bij de vrijstelling van neurotransmitters t.h.v. de
synaptische spleet. Tot slot is calcium essentieel voor het behoud van de spiertonus en de
exciteerbaarheid van het zenuw- en spierweefsel (Tortora & Derrickson, 2006; Bushinsky &
Monk, 1998).
Het is belangrijk dat de serumconcentraties van calcium vrij constant gehouden
worden (normale totale calciumconcentratie: 2,10 - 2,55 mmol/L). De calciumhomeostase
wordt geregeld door calcium zelf via een calciumreceptor en verschillende hormonen,
waarvan het parathyroïdhormoon en het 1,25-dihydroxyvitamine D3 de belangrijkste zijn.
Indien dit regelmechanisme van de calcemie faalt, kan de serumconcentratie van calcium te

3
hoog (hypercalcemie) of te laag (hypocalcemie) worden. Dit wordt veroorzaakt door diverse
pathologieën (Bushinsky & Monk, 1998).
1.4. METHODEVALIDATIE
In deze masterthesis wordt een ionenchromatografische methode voor de bepaling van
calcium gevalideerd. Mehodevalidatie vormt het eerste niveau in de kwaliteitswaarborging
van een analytisch laboratorium. Analytische kwaliteitswaarborging omvat de volledige reeks
van metingen die moeten worden uitgevoerd, opdat het labo steeds kan garanderen dat de
afgeleverde data van hoge kwaliteit zijn. Analytische methodevalidatie speelt een belangrijke
rol in het onderzoeken van de kwaliteit van een analytische methode. Het laat toe om de
analytische kwaliteit numeriek uit te drukken (Taverniers et al., 2004).
De ISO 9000 definitie van methodevalidatie luidt: “De bevestiging dat de methode
voldoet aan de vereisten voor specifiek gebruik of toepassing, door het aanleveren van
objectief bewijs.” Validatie houdt vier stappen in:
1) Bepalen van het beoogd gebruik van de methode.
2) Definiëren van de gewenste specificaties van de prestatiekenmerken.
3) Leveren van objectief bewijs m.b.v. de data bekomen tijdens de validatie-
experimenten.
4) Statistische analyse van de bekomen data, opdat kan bevestigd worden dat er aan
de vooropgestelde eisen wordt voldaan (Stöckl et al., 2009).
Telkens er een nieuwe methode ontwikkeld wordt, moet ze gevalideerd worden. In
sommige omstandigheden is revalidatie noodzakelijk. Bijvoorbeeld indien er wijzigingen zijn
aangebracht aan de methode om de bepaling van een analyt te optimaliseren, indien een
methode wordt toegepast in een ander laboratorium, door andere laboranten, bij gebruik van
andere toestellen of indien er aanwijzigingen zijn dat een bepaalde methode verandert in de
tijd. Er moet dan worden nagegaan of de prestatiekenmerken nog steeds voldoen aan de
vooropgestelde specificaties. De mate waarin de (re)validatie moet uitgevoerd worden is
afhankelijk van de aard van de wijzigingen die werden aangebracht en waarvoor de methode
zal gebruikt worden (Chandran & Singh, 2007).
Een methodevalidatie verloopt steeds volgens een vooraf opgesteld stappenplan,
waarvan een voorbeeld staat in TABEL 1.1.

4
TABEL 1.1. VOORBEELD VAN EEN VALIDATIEPLANa
1. Definieer de toepassing, het doel en het bereik van de methode.
2. Definieer de prestatiekenmerken en hun aanvaardingscriteria.
3. Stel een validatieprotocol op of een uitvoeringsprocedure van de validatie.
4. Bepaal de gebruikte materialen: bv. standaarden, reagentia en stalen.
5. Voer de validatie-experimenten uit.
6. Rapporteer de validatie-experimenten en resultaten in een validatiedocument.
7. Interpreteer de data en trek conclusies uit de resultaten van de statistische testen. a(Stöckl, 2007a)
Tijdens de eerste stap van het validatieplan wordt de toepassing, het doel en het bereik
van de methode bepaald. Het analytisch systeem wordt beschreven: het doel en het type van
de methode, het verwachte concentratiebereik van de analyt(en) en de staalmatrix (Taverniers
et al., 2004). Afhankelijk van het domein waarin de analytische methode wordt toegepast, het
bedoeld gebruik, het type van de methode (bv. identificatie of kwantitatieve test), gelden er
andere specificaties (Chandran & Singh, 2006). Er kunnen voor eenzelfde methode dus
verschillende aanvaardingscriteria bestaan voor gebruik in de klinische biologie tegenover in
de voedingssector. De verschillende prestatiekarakteristieken van een analytische methode,
die tijdens een validatie kunnen geëvalueerd worden zijn: precisie, juistheid,
aantoonbaarheidsgrens (LoD, “limit of detection”), bepalingsgrens (LoQ, “limit of
quantification”), meetbereik, lineariteit, terugvinding, totale fout, selectiviteit/specificiteit en
robuustheid (Taverniers et al., 2004).
Hoe deze prestatiekenmerken moeten geëvalueerd worden, is vastgelegd in
protocollen. Internationale organisaties zoals de “International Organization for
Standardization” (ISO), “de International Conference on Harmonization” (ICH) en het
“Clinical and Laboratory Standards Institute” (CLSI) bieden dergelijke protocollen aan. De
ISO-protocollen gelden meer algemeen, terwijl de evaluatieprotocollen (EP-protocollen) van
de CLSI specifiek zijn voor het domein van de in vitro diagnostica. De ICH-protocollen zijn
bedoeld voor de industrie. In deze procollen staat beschreven welke materialen nodig zijn en
hoe de validatie-experimenten moeten uitgevoerd worden.

5
Tot slot moet geanalyseerd worden of de resultaten van de statistische analyse van de
bekomen meetresultaten voldoen aan de vooropgestelde specificaties. Deze kunnnen zowel
statistisch, analytisch als toepassingsgericht zijn. Om te besluiten of aan de specificaties voor
een bepaalde toepassing voldaan is, wordt gebruik gemaakt van betrouwbaarheidsintervallen
en/of statistische significantietesten. De statistische testen zijn bij alle drie de specificaties
hetzelfde, de interpretatie verloopt echter verschillend. Bij statistische specificaties wordt er
vergeleken met de nulhypothese, in geval van analytische specificaties met een maat voor een
stabiele prestatie en bij toepassingsgerichte specifaties wordt er vergeleken met een
validatielimiet.
Onder het hoofdstuk literatuuronderzoek (4.3.) wordt verder ingegaan op de
methodevalidatie in het domein van de in vitro diagnostica.

6
2. OBJECTIEVEN
In het praktische deel van deze masterthesis wordt een methodevalidatie uitgevoerd. Er wordt
nagegaan of de ionenchromatografische methode geschikt is voor de bepaling van calcium.
De lineariteit, precisie, juistheid en de aantoonbaarheidsgrens van de methode worden
geëvalueerd. De validatie-experimenten worden gepland, uitgevoerd, statistisch
geïnterpreteerd en gerapporteerd.
Voor het uitvoeren van de metingen wordt steeds gecontroleerd of het systeem optimaal
functioneert en of het geschikt is om de metingen uit te voeren. Er wordt ook nagegaan welk
kalibratiemodel het meest robuust en juist is om de concentratie van calcium te berekenen uit
de bekomen piekoppervlakte. In een methodevergelijkingstudie wordt de
ionenchromatografische methode beschouwd als de referentiemethode en vergeleken met een
hiërarchisch lagere routinemethode. A.d.h.v. gesimuleerde meetresultaten wordt de functie
van methodevergelijking in methodevalidatie geïllustreerd.
Het tweede deel van de thesis bestaat uit een literatuuronderzoek. Hierin wordt uitgebreid
ingegaan op methodevalidatie in het domein van de in vitro diagnostica. De verschillende
protocollen worden uiteengezet, alsook welke specificaties er gelden. Daarna volgt hoe kan
beslist worden op basis van de statistische evaluatie van de meetresultaten of een methode
gevalideerd is of niet. Verder worden andere analysemethoden voor calcium beschreven met
hun voor- en nadelen en wordt het gebruik van calcium in de farmaceutische context
geplaatst.

7
3. MATERIALEN EN METHODEN
3.1. MATERIALEN
Voor de aanmaak van de stockoplossing, de stalen en de testmix wordt CaCl2.2H2O
van Sigma – Aldrich (St. Louis, USA) gebruikt, met een minimale zuiverheid van 99%.
Testmix bevat ook MgSO4.7H2O van Sigma – Aldrich, dat 100% zuiver is. Als oplosmiddel
wordt steeds gebruik gemaakt van ultrazuiver water (VWR BDH Prolabo®, Leuven, België).
De stalen worden aangemaakt in doorzichtige plastieken potjes met draaidop of maatkolven
van 100 mL van Nalgene® (Roskilde, Denemarken).
Het eluens voor de ionenchromatograaf wordt aangemaakt in plastieken maatkolven
van 1000 mL van Brand (Wertheim, Duitsland). Eerst wordt 400 mg L-histidine (Sigma –
Aldrich, St. Louis, USA) afgewogen en overgebracht in een droge maatkolf. Om het L-
histidine op te lossen wordt ongeveer 250 mL ultrazuiver water toegevoegd en wordt de
oplossing gezwenkt. Als alle histidinekristallen opgelost zijn, wordt 800 µL 95-97% H2SO4
van Acros Organics (Geel, België) toegevoegd, waarna met ultrazuiver water wordt
aangelengd tot 1000 mL.
3.2. STANDAARDEN EN STALEN
3.2.1. Stockoplossing
Voor de stockoplossing wordt 0,1485 g CaCl2.2H2O afgewogen op een analytische
balans in een maatkolf van 100 mL. Vervolgens wordt er aangelengd tot 100 mL met
ultrazuiver water. Voor de aanmaak van de stockoplossing wordt gravimetrisch te werk
gegaan. Aan de hand de massa’s toegevoegde CaCl2.2H2O en solvent wordt de exacte
concentratie van de stockoplossing berekend (1493 µg/g).
3.2.2. Stalen gebruikt voor de verschillende validatie-experimenten
Uitgaande van de stockoplossing worden de stalen voor de hieronder beschreven
validatie-experimenten aangemaakt. Hierbij wordt ook gravimetrisch te werk gegaan. Aan de
hand van de massa’s afgewogen oplossing en oplosmiddel wordt de exacte concentratie van
de stalen berekend. Alle stalen worden bewaard in de koelkast bij 4°C.

8
3.2.2.1. Lineariteit
In het CLSI-EP6-A (2003) protocol zijn er verschillende mengprotocollen beschreven
voor de aanmaak van de lineariteitstalen. Ze staan samengevat in TABEL 3.1.
TABEL 3.1. OVERZICHT VAN DE MOGELIJKE MENGPROTOCOLLEN VOOR DE
AANMAAK VAN DE LINEARITEITSTALEN VOLGENS HET CLSI-EP6 PROTOCOL.
Mengprotocol Alternatief mengprotocol
1. laag 1. laag
2. laag + hoog (3:1) 2. laag medium: laag + medium (1:1)
3. laag + hoog (2:2) 3. medium: laag + hoog (1:1)
4. laag + hoog (1:3) 4. hoog medium: hoog + medium (1:1)
5. hoog 5. hoog
Voor deze masterthesis wordt een dynamisch bereik van 3 – 200 µg/g gekozen. Op
basis hiervan wordt bepaald welke concentratie elke lineariteitstaal moet hebben.
Lineariteitstaal 5 wordt aangemaakt door 13,0 mL stockoplossing te pipetteren in een
maatkolf van 100 mL en aan te lengen met ultrazuiver water tot 100 mL. Vervolgens wordt
lineariteitstaal 1 aangemaakt uit lineariteitstaal 5 door 1888 µL ervan in een maatkolf aan te
lengen met ultrazuiver water tot 100 mL. Daarna worden de andere lineariteitstalen
aangemaakt volgens het alternatief mengprotocol. A.d.h.v. de massa’s van de samenstellende
stalen en/of solvent, wordt de exacte concentratie van de lineariteitstalen berekend.
3.2.2.2. Kalibratiecurve
De kalibratoren, die worden geïnjecteerd om de kalibratiecurve op te stellen, worden
op dezelfde manier aangemaakt zoals onder 3.2.2.1. Lineariteit.
3.2.2.3. Imprecisie
Voor de evaluatie van de imprecisie zijn volgens het CLSI-EP5-A protocol (1999)
twee stalen met lage en hoge concentratie nodig. Voor het lage interne kwaliteitscontrolestaal
(IQC, “internal quality control”) moet er 3,0 mL van de stockoplossing aangelengd worden
tot 100 mL om een concentratie van 44,55 µg/g te verkrijgen. Het hoge IQC-staal (148,51

9
µg/g) wordt aangemaakt door 10,0 mL van de stockoplossing aan te lengen tot 100 mL in een
maatkolf.
3.2.2.4. Aantoonbaarheidsgrens
Voor het LoD-staal wordt een concentratie gekozen, die bij het meten aanleiding geeft
tot een signaal-over-ruisverhouding (S/R) van vijf à zes. Hierdoor wordt vermeden dat er
gecensureerde data’s verkregen worden. Om een dergelijk staal aan te maken, wordt van
lineariteitstaal 1 de gemiddelde S/R bepaald van 10 metingen (=148,84). Vervolgens wordt de
verdunning berekend die nodig is om een signaal-over-ruisverhouding te verkrijgen van vijf
of zes. Eerst wordt lineariteitstaal 1 13 maal verdund en wordt de S/R berekend van het
verdunde staal (=16). Vervolgens wordt het verdunde lineariteitstaal 1 nogmaals driemaal
verdund. Bij het meten van het staal is de S/R 6,1.
3.2.2.5. Juistheid
De juistheidstalen worden ter beschikking gesteld door het labo Analytische Chemie.
3.2.2.6. Systeemgeschiktheidstesten
De testmix voor het testen van de systeemgeschiktheid wordt ter beschikking gesteld
door het labo Analytische Chemie.
3.2.2.7. Overzicht
De concentraties van de verschillende stalen gebruikt bij de methodevalidatie staan
samengevat in TABEL 3.2.
TABEL 3.2. OVERZICHT VAN DE CONCENTRATIES VAN DE VERSCHILLENDE STALEN
GEBRUIKT BIJ DE METHODEVALIDATIE VOOR DE BEPALING VAN CALCIUM.
Prestatiekenmerk Staal Concentratie
Lineariteit Lin – 1 3,660 µg/g
Lin – 2 51,85 µg/g
Lin – 3 99,74 µg/g
Lin – 4 149,6 µg/g
Lin – 5 196,0 µg/g

10
TABEL 3.2. VERVOLG
Kalibratiecurve Cal – 1 3,595 µg/g
Cal – 2 51,01 µg/g
Cal – 3 98,11 µg/g
Cal – 4 145,1 µg/g
Cal – 5 192,5 µg/g
Imprecisie Lage IQC 45,07 µg/g
Hoge IQC 150,0 µg/g
Aantoonbaarheidsgrens LoD 0,09335 µg/g
Juistheid Onbekende 1 9,586 µg/g
Onbekende 2 24,75 µg/g
Onbekende 3 77,82 µg/g
Onbekende 4 122,4 µg/g
Onbekende 5 146,6 µg/g
Systeemgeschiktheidstesten Testmix:
Calcium
Magnesium
109,8 µg/g
107,9 µg/g
3.3. APPARATUUR
3.3.1. Instrument
Voor de bepaling van calcium wordt gebruik gemaakt van de DX-500
ionenchromatograaf van Dionex (Sunnyvale, USA). Het eluens wordt door de IP20
isocratische pomp over twee IonPac CG10 Guard kolommen (4 x 50 mm (interne diameter x
lengte kolom), harspartikels 8,5 µm) gepompt; beide zijn ook van Dionex. In het systeem is
ook een hulppomp aanwezig, die de kolomschakeling mogelijk maakt (= “front-cutting”). Het
ionenchromatografisch systeem is uitgerust met een CD20 conductiviteitsdetector van
Dionex. De meting van de conductiviteit van de ionen in de mobiele fase na
chromatografische scheiding gebeurt na suppressie van de conductiviteit van de

11
achtergrondelektrolyt door de CSRS 300 (4 mm)-suppressor (“Cation Self-Regenerating
Suppressor”) van Dionex. De suppressor zet het sterk zure eluens om in H2O, H2 en O2, die
afgeleid worden naar het afvalvat. Hierdoor zullen enkel de calcium- en magnesiumionen uit
het staal aanleiding geven tot een verandering in detectorsignaal.
De stalen worden manueel geïnjecteerd m.b.v. een spuit (1,0 mL) van Becton,
Dickinson and company (Madrid, Spanje) met een naald van Alltech (Lokeren, België). Er
wordt steeds 500 µL geïnjecteerd, waarvan er 40 µL op de kolom gebracht wordt (= volume
injectieloop).
De toestellen worden gestuurd door de Chromeleon®-software versie 6.8. van Dionex.
Hiermee worden ook de chromatogrammen geregistreerd en de pieken geïntegreerd.
3.3.2. Randapparatuur
Voor de aanmaak van de stalen wordt de analytische balans Mettler AT261 Delta
Range® 353 van Mettler-Toledo SA gebruikt (Greifensee, Zwitserland). De
meetnauwkeurigheid van deze balans bedraagt 10-5 g.
Voor het pipetteren worden twee pipetten gebruikt: de autoclaveerbare Calibra 832
pipet (1-10 mL) en de macropipette Calibra 832 (0,2-2 mL); beide zijn van Socorex
(Ecublens, Zwitserland).
3.4. METHODEN
3.4.1. Systeemfunctiecontrole
Voor het uitvoeren van de experimenten worden eerst een aantal systeemparameters
geëvalueerd, namelijk de druk, de achtergrondconductiviteit, de flow door de suppressor en
de suppressordruk. Door te vergelijken met de normale waarden voor deze parameters (de
zogenaamde “bench mark”) en door eventuele aanpassingen aan te brengen, kan verzekerd
worden dat het systeem optimaal functioneert. Dit is belangrijk om correcte meetwaarden
voor de stalen te bekomen.
3.4.2. Systeemgeschiktheidstest
Nadat is bevestigd dat het systeem optimaal functioneert, wordt gecontroleerd of het
chromatografische en suppressiesysteem geschikt is om de metingen uit te voeren. Hiervoor

12
wordt de testmix geïnjecteerd. Van het bekomen chromatogram worden zowel voor calcium
en magnesium verschillende chromatografische parameters geëvalueerd en vergeleken met
vooraf bepaalde specificaties. Volgende parameters worden door de Chromeleon®-software
berekend: retentietijd (min), piekoppervlakte (µS min), piekhoogte (µS), capaciteitsfactor
(k’), resolutie, asymmetriefacor en het aantal theoretische platen (N). De laatste vier
parameters worden berekend volgens de USP-formules in TABEL 3.3.
TABEL 3.3. OVERZICHT VAN DE USP-FORMULES VAN DE CHROMATOGRAFISCHE
PARAMETERS GEËVALUEERD TIJDENS DE SYSTEEMGESCHIKTHEIDSTEST.
Chromatografische parameter Formule Verklaring symbolen
Resolutie R = 2 ( tR-tP / w + wP )
tR = retentietijd
tP = retentietijd vorige piek
w = piekbreedte1
wP = breedte vorige piek1
Aantal theoretische platen N = 16 (tR / w)2 tR = retentietijd
w = piekbreedte1
Asymmetriefactor As = w0,05 / (2 a0,05)
w0,05 = piekbreedte op 5% piekhoogte
a0,05 = breedte van de linkerhelft
van de piek (van startpunt tot
loodlijn uit top) op 5%
piekhoogte
Capaciteitsfactor (k’) k’ = (tR/t0) – 1 tR = retentietijd
t0 = dode tijd
1 de piekbreedte is de tijd tussen twee punten op de basislijn, waar de twee raaklijnen aan de piek de basislijn snijden.
3.4.3. Analyse
Gedurende de analyse van de stalen wordt het debiet van de pomp ingesteld op 1,10
mL/min en bedraagt de druk ± 780 psi. Het systeem is isocratisch en er wordt gewerkt bij
kamertemperatuur.
Het injectievolume bedraagt 500 µL, waarvan er 40 µL over de kolom gebracht wordt.
Gedurende de eerste 48 seconden van de totale analysetijd (= 5 min) loopt het staal enkel over
de eerste kolom. Calcium en magnesium zullen weerhouden worden, terwijl andere
13
componenten, waarin we niet geïnteresseerd zijn, zullen elueren en afgeleid worden naar het
afvalvat. Na 48 seconden grijpt er m.b.v. een hulppomp een kolomschakeling plaats, zodat het
staal ook over de tweede kolom gebracht wordt, waar calcium en magnesium van elkaar
worden gescheiden. Vervolgens kan de concentratie van calcium berekend worden, door de
bekomen piekoppervlakte in te vullen in de vergelijking van de kalibratiecurve, die vooraf is
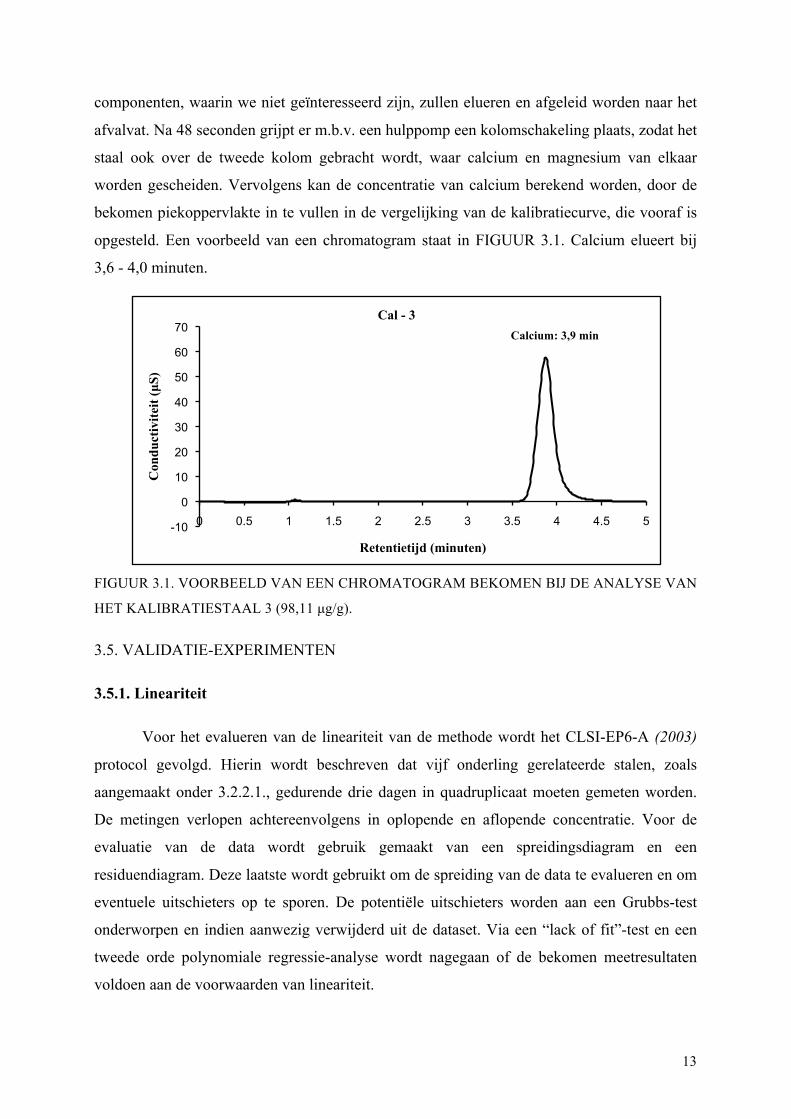
opgesteld. Een voorbeeld van een chromatogram staat in FIGUUR 3.1. Calcium elueert bij
3,6 - 4,0 minuten.
FIGUUR 3.1. VOORBEELD VAN EEN CHROMATOGRAM BEKOMEN BIJ DE ANALYSE VAN
HET KALIBRATIESTAAL 3 (98,11 µg/g).
3.5. VALIDATIE-EXPERIMENTEN
3.5.1. Lineariteit
Voor het evalueren van de lineariteit van de methode wordt het CLSI-EP6-A (2003)
protocol gevolgd. Hierin wordt beschreven dat vijf onderling gerelateerde stalen, zoals
aangemaakt onder 3.2.2.1., gedurende drie dagen in quadruplicaat moeten gemeten worden.
De metingen verlopen achtereenvolgens in oplopende en aflopende concentratie. Voor de
evaluatie van de data wordt gebruik gemaakt van een spreidingsdiagram en een
residuendiagram. Deze laatste wordt gebruikt om de spreiding van de data te evalueren en om
eventuele uitschieters op te sporen. De potentiële uitschieters worden aan een Grubbs-test
onderworpen en indien aanwezig verwijderd uit de dataset. Via een “lack of fit”-test en een
tweede orde polynomiale regressie-analyse wordt nagegaan of de bekomen meetresultaten
voldoen aan de voorwaarden van lineariteit.
-10
0
10
20
30
40
50
60
70
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5 5
Con
duct
ivite
it (µ
S)
Retentietijd (minuten)
Cal - 3 Calcium: 3,9 min

14
De “lack of fit”-test is een F-test voor lineariteit. Ze evalueert of de variantie tussen de
verschillende stalen groter is dan de variantie tussen de verschillende metingen van een staal
(= afwijking van lineariteit). De tweede orde polynomiale regressie-analyse is een t-test die
nagaat of de coëfficiënt van x2 in een tweedegraadsvergelijking significant verschillend is van
nul. Indien de berekende probabiliteitswaarde (P-waarde) groter is dan 0,05, dan is de
coëfficiënt niet significant verschillend van nul en hebben we een lineaire vergelijking van de
vorm: y = ax + b.
3.5.2. Opstellen kalibratiecurve
Onder paragraaf 3.5.1. wordt bepaald of de methode voldoet aan de specificaties voor
lineariteit. Afhankelijk van de conclusie van deze evaluatie, zal een lineaire of tweedegraads
kalibratiecurve gebruikt worden.
Vervolgens wordt bepaald welk regressiemodel de meest robuuste en juiste is om
a.d.h.v. de piekoppervlakte de concentratie van het analyt in het staal te berekenen. Indien de
methode voldoet aan de voorwaarden voor lineariteit, wordt de regressievergelijking van de
kalibratiecurve berekend via een “ordinary least square regression” (OLR)-analyse, een OLR-
regressie-analyse waarbij de trendlijn wordt geforceerd door nul en via een gewogen lineaire
regressie-analyse (GLR). Gedurende vijf meetdagen wordt de vergelijking van de
kalibratiecurve op deze drie manieren berekend en worden ze gebruikt om de concentratie van
het LoD-staal en de imprecisiestalen te berekenen. Na vijf dagen wordt de gemiddelde
concentratie berekend voor de drie regressiemodellen. De methode met de laagste
variatiecoëfficiënt (CV, “coefficient of variation”) is de meest robuuste. De meest juiste
methode is deze waarvan de terugvinding van de gravimetrisch bepaalde concentratie van de
stalen (= gemiddelde experimenteel berekende concentratie – werkelijke concentratie) het
dichtst 100% nadert.
3.5.3. Imprecisie
Volgens het CLSI-EP5-A (1999) protocol moeten twee IQC-stalen gemeten worden
om de precisie van de meetprocedure te evalueren. Gedurende 20 dagen wordt één staal met
lage concentratie en één staal met hoge concentratie in tweevoud gemeten.
Voor de evaluatie van de binnen analyseserie imprecisie worden de verschillen tussen
de duplicaten uitgezet in een puntendiagram voor zowel het lage als het hoge IQC-staal. In het

15
puntendiagram kan de spreiding van de meetresultaten visueel geëvaleerd worden. Potentiële
uitschieters worden onderworpen aan een Grubbs-test. Indien een uitschieter aanwezig is,
wordt het meetresultaat dat het meest afwijkt verwijderd uit de dataset. Het daggemiddelde
wordt vervangen door het weerhouden meetresultaat.
Vervolgens worden in een puntendiagram de daggemiddelde concentraties uitgezet
van de meetresultaten van zowel het lage als het hoge IQC-staal. In dit puntendiagram kan
aan de hand van de spreiding van de punten de tussendag imprecisie geëvalueerd worden.
Opnieuw wordt de grafiek visueel gecontroleerd op de aanwezigheid van uitschieters, die
bevestigd worden door een Grubbs-test. Eventueel aanwezige uitschietende daggemiddelden
worden verwijderd uit de dataset.
De specificaties voor de imprecisie zijn 2% voor de binnen analyseserie CV (CVwr,
within run CV) en 5% voor de totale CV (CVT). Er is aan de specificaties voldaan, indien de
experimentele CV’s < gespecifieerde CV’s. Als de experimentele CV’s groter zijn dan de
doelwaarden voor een stabiel proces moet een Chi2-test uitgevoerd worden. Dit is een één
steekproef-F-test, die nagaat of het 95%-betrouwbaarheidsinterval van de variantie van een
dataset de doelwaarde van de variantie voor een stabiel proces insluit. M.a.w. er wordt getest
of de onderlimiet van het 95%-betrouwbaarheidsinterval onder de doelwaarde gelegen is. Er
is aan de specificaties voldaan, indien de experimentele Chi2–waarde ≤ de kritische Chi2-
waarde.
3.5.4. Aantoonbaarheidsgrens
Voor de evaluatie van de aantoonbaarheidsgrens van de methode wordt een generisch
protocol gevolgd. Er worden 20 metingen in singlicaat uitgevoerd op een staal dat aanleiding
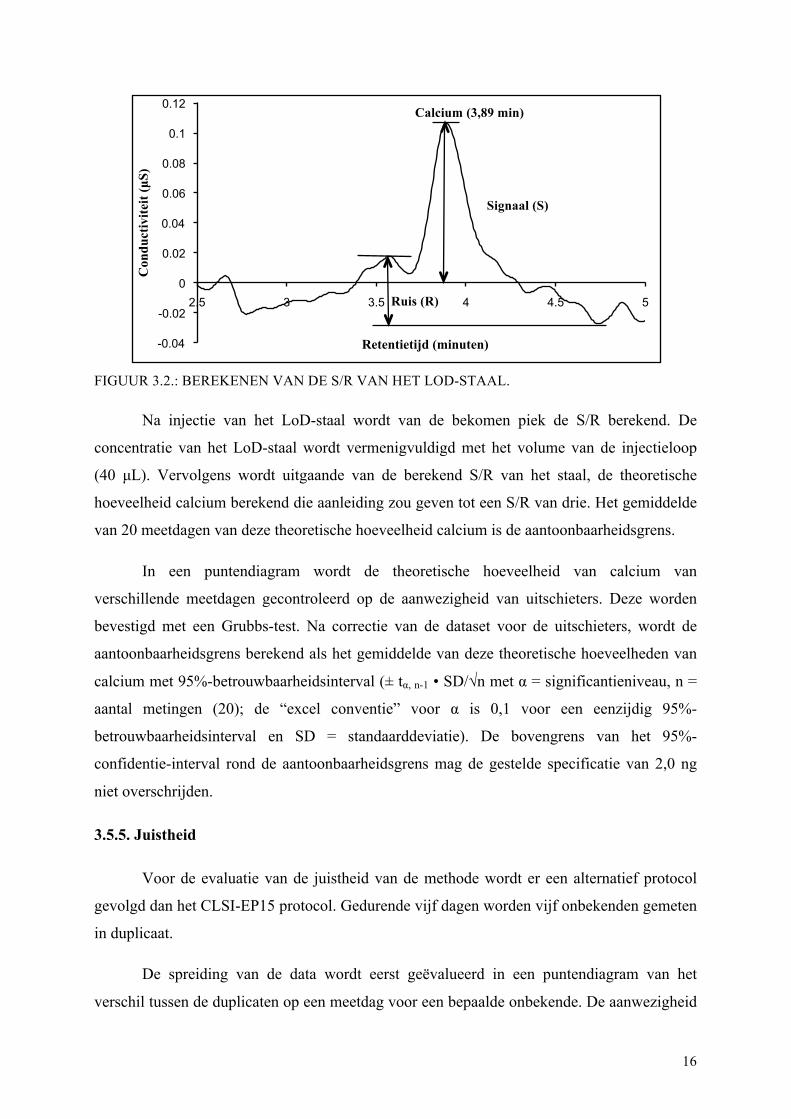
geeft tot een S/R van vijf à zes. In FIGUUR 3.2. staat een voorbeeld van hoe de S/R manueel
berekend wordt. In het voorbeeld is S = 4,2 cm en R = 1,8 cm; de S/R bedraagt dus 2,3. Bij de
evaluatie van de aantoonbaarheidsgrens wordt de S/R berekend door de Chromeleon®
software. Deze berekent voor dit LoD-staal een S/R van 3,9. Noteer dat de ruis (R) hier gelijk
is aan de ruis en de onstabiliteit op de basislijn.
16
FIGUUR 3.2.: BEREKENEN VAN DE S/R VAN HET LOD-STAAL.
Na injectie van het LoD-staal wordt van de bekomen piek de S/R berekend. De
concentratie van het LoD-staal wordt vermenigvuldigd met het volume van de injectieloop
(40 µL). Vervolgens wordt uitgaande van de berekend S/R van het staal, de theoretische
hoeveelheid calcium berekend die aanleiding zou geven tot een S/R van drie. Het gemiddelde
van 20 meetdagen van deze theoretische hoeveelheid calcium is de aantoonbaarheidsgrens.
In een puntendiagram wordt de theoretische hoeveelheid van calcium van
verschillende meetdagen gecontroleerd op de aanwezigheid van uitschieters. Deze worden
bevestigd met een Grubbs-test. Na correctie van de dataset voor de uitschieters, wordt de
aantoonbaarheidsgrens berekend als het gemiddelde van deze theoretische hoeveelheden van
calcium met 95%-betrouwbaarheidsinterval (± tα, n-1 • SD/√n met α = significantieniveau, n =
aantal metingen (20); de “excel conventie” voor α is 0,1 voor een eenzijdig 95%-
betrouwbaarheidsinterval en SD = standaarddeviatie). De bovengrens van het 95%-
confidentie-interval rond de aantoonbaarheidsgrens mag de gestelde specificatie van 2,0 ng
niet overschrijden.
3.5.5. Juistheid
Voor de evaluatie van de juistheid van de methode wordt er een alternatief protocol
gevolgd dan het CLSI-EP15 protocol. Gedurende vijf dagen worden vijf onbekenden gemeten
in duplicaat.
De spreiding van de data wordt eerst geëvalueerd in een puntendiagram van het
verschil tussen de duplicaten op een meetdag voor een bepaalde onbekende. De aanwezigheid
-0.04
-0.02
0
0.02
0.04
0.06
0.08
0.1
0.12
2.5 3 3.5 4 4.5 5
Con
duct
ivite
it (µ
S)
Retentietijd (minuten)
Signaal (S)
Ruis (R)
Calcium (3,89 min)

17
van de op visuele basis vermoede uitschieters wordt bevestigd door een Grubbs-test. Na
correctie van de dataset voor de uitschieters, wordt in een puntendiagram de verdeling van de
daggemiddelden van elke onbekende onderzocht. Indien potentiële uitschieters worden
opgemerkt, worden de daggemiddelden onderworpen aan een Grubbs-test en de uitschieters
verwijderd. Vervolgens wordt het verschildiagram van de aangepaste dataset beschouwd. Het
verschil tussen de daggemiddelde concentratie en de gemiddelde concentratie van de
onbekende wordt hierin uitgezet in functie van de gemiddelde concentratie. Er zouden geen
uitschieters meer mogen opgemerkt worden.
Om de juistheid van de methode te evalueren, wordt gebruik gemaakt van het
procentueel verhoudingsdiagram, waarin de terugvinding (zie formule 3.1) van de
doelconcentratie in de experimentele concentratie wordt uitgezet voor elke onbekende.
Terugvinding (%) = 100 (gemeten waarde / doelwaarde) ± 95 % BI (3.1)
Met BI = betrouwbaarheidsinterval
De terugvinding met zijn 95%-betrouwbaarheidsinterval mag de limiet van 95 %-105 %
terugvinding niet overschrijden opdat voldaan zou zijn aan de specificaties. Dit kan ook getest
worden met een eenzijdige één steekproef-t-test. Indien niet aan de specificaties voldaan is,
kan met een “power”-analyse berekend worden hoeveel metingen moeten uitgevoerd worden
opdat de terugvinding met zijn 95%-betrouwbaarheidsinterval wel binnen de limieten ligt.
3.5.6. Methodevergelijking
Bij deze methodevergelijking worden de analyseresultaten van een routinemethode
vergeleken met die van een referentiemethode, uitgevoerd op hetzelfde staal. Volgens het
CLSI-EP9-A (1995) protocol moeten minstens 40 stalen willekeurig in duplicaat gemeten
worden verspreid over vijf dagen. In totaal worden 80 meetresultaten gesimuleerd via de
excelfile “DataGeneration”, ter beschikking gesteld door het labo Analytische Chemie. Bij
ionenchromatografie, de referentiemethode, wordt het bereik van de meetwaarden begrensd
door de serumwaarden van calcium bij hypo-en hypercalcemie ( 1 – 4 mmol/L; Bushinsky &
Monk, 1998). Als routinemethode wordt een fotometrische kleurtest gebruikt voor de
kwantitatieve bepaling van calcium in serum (calcium oCPC-test) met een Beckman Coulter
analyser (Beckman Coulter®, 2009). De methode heeft een totale CV van 1,5%.

18
Om te evalueren of aan de specificaties voldaan is, wordt gebruik gemaakt van het
Bland – Altmandiagram en de lineaire regressie-analyse. De Bland – Altman benadering voor
de interpretatie van de methodevergelijkingstudie, maakt gebruik van een verschildiagram.
Hierin wordt het procentueel verschil tussen de meetresultaten van de routine- en de
referentiemethode uitgezet in functie van de meetresultaten van de referentiemethode. De
berekeningen die moeten gemaakt worden zijn het gemiddeld verschil tussen de methoden en
het 1,96 CV van de individuele verschillen met hun respectievelijke 95%-
betrouwbaarheidsintervallen. Bij de interpretatie van het Bland – Altmandiagram kan visueel
onderzocht worden of het gemiddeld verschil met zijn 95%-betrouwbaarheidsinterval ≤ de
specificatie voor de systematische fout en of het 1,96 CV met zijn 95%-
betrouwbaarheidsinterval ≤ de limieten van de totale fout.
De uitkomst van de Bland – Altman-test wordt ook getoetst met de lineaire regressie-
analyse. Indien het intercept van de lineaire regressievergelijking verschillend is van nul en de
richtingscoëfficiënt van één, dan wijst dit respectievelijk op een constante en proportionele
systematische fout. Indien deze aanwezig zijn, moet er nagegaan worden of deze fouten
kleiner zijn dan de limieten voor de systematische en totale fout. Daarvoor wordt van de
laagste (min (x)) en de hoogste (max (x)) concentraties, bekomen met de referentiemethode,
de y-waarde voor de routinemethode voorspeld a.d.h.v. de lineaire regressievergelijking.
Vervolgens wordt nagegaan of die voorspelde y-waarde met zijn 95%-
betrouwbaarheidsinterval binnen de specificatie van de systematische fout gelegen is. Het
confidentie-interval drukt de onzekerheid uit op de trendlijn van de meetwaarden. Het 95%-
predictie-interval rond dit punt wordt ook berekend en moet binnen de limieten voor de totale
fout gelegen zijn. Het predictie-interval omvat met 95% zekerheid de volgende
meetresultaten.
Op deze manier kan onderscheid gemaakt worden tussen de prestatie van de
routinemethode bij hoge en lage calciumconcentraties in serum.
3.6. DATAVERWERKING EN STATISTIEK
Het opstellen van de kalibratiecurve en het berekenen van de concentratie die
overeenstemt met een bepaalde piekoppervlakte, gebeurt m.b.v. Microsoft Office Excel versie
2003 (Microsoft Corporation, USA).

19
Voor de statistische analyse van de meetresultaten wordt gebruik gemaakt van het
Excelbestand “MethVal” ter beschikking gesteld door dr. D. Stöckl (STT Consulting). Er
wordt eveneens gebruik gemaakt van “Method validation with confidence” (Stöckl, 2007a) en
“Laboratory statistics & graphics with EXCEL” (Stöckl, 2007b). Deze statistiekboeken
worden ter beschikking gesteld door Dr. D. Stöckl.
De “lack of fit”-test en de tweede orde polynomiale regressie-analyse, die gebruikt
worden bij de evaluatie van de lineariteit van de methode en GLR-analyse gebeuren m.b.v.
CBStat5 versie 5.1.0 (2005, Kristian Linnet, Risskov, Denemarken).
De power-analyse, die bij de evaluatie van de juistheid wordt gebruikt om het aantal
metingen te bepalen opdat de terugvinding binnen de specificaties ligt, gebeurt m.b.v.
G*Power versie 3.1.2. (F. Faul et al., Universiteit van Düsseldorf, Duitsland).
3.7. SPECIFICATIES
In TABEL 3.4. staan de specificaties van de verschillende prestatiekenmerken, die
tijdens deze methodevalidatie worden geëvalueerd.
TABEL 3.4.: OVERZICHT VAN DE SPECIFICATIES VOOR DE VERSCHILLENDE
PRESTATIEKENMERKEN DIE WORDEN GEËVALUEERD TIJDENS DE
METHODEVALIDATIE.
Prestatiekenmerk Specificatie
Precisie – binnen-analyseserie CV 2 %$
Precisie – totale CV 5 %$
Lineariteit 5 %*
Juistheid 5%*
Aantoonbaarheidsgrens 2,0 ng*
Methodevergelijking – Systematische fout 5%*
Methodevergelijking – Totale fout 15 %*
$Doelwaarde voor stabiel proces; *Limiet
3.8. LITERATUURONDERZOEK
In TABEL 3.5. wordt een overzicht gegeven van de geraadpleegde informatiebronnen tijdens
het literatuuronderzoek.

20
TABEL 3.5.: OVERZICHT VAN DE GERAADPLEEGDE INFORMATIEBRONNEN.
Algemene zoekopdrachten - Google
Wetenschappelijk zoeken - PubMed
- Web of Science
- Google Scholar
- Belgisch Centrum voor Farmacotherapeutische Informatie (BCFI)
- Farmacotherapeutisch Kompas
- Food and Drug Administration (FDA)
- Westgard QC
Fabrikanten - Dionex
- Sigma – Aldrich
- Beckman Coulter
- Roche
Kennis gebaseerd zoeken - Merck Index
- United States Pharmacopoeia (USP)

21
4. RESULTATEN EN DISCUSSIE
4.1. METHODEN
4.1.1. Systeemfunctiecontrole
Gedurende alle meetdagen is de gemiddelde druk op de kolom en de standaarddeviatie
781 ± 27,1 psi. De druk schommelt tussen 749 psi en een maximale druk van 843 psi. De
achtergrondconductiviteit bedraagt gemiddeld 3,4 ± 0,6 µS, wat de normale waarde van 3,5
µS benadert. De druk op de suppressor is gemiddeld 14,6 ± 1,8 psi, wat ruimschoots onder de
maximale druk van 25 psi gelegen is. Er is een stijgende trend in de druk op de suppressor te
zien: initeel was een druk van 13 psi voldoende om een goede flow door de suppressor te
genereren. Uiteindelijk moest de druk opgedreven worden tot 18 psi. De flow bedraagt
gemiddeld 3,8 ± 0,5 mL/min, wat dichtbij de normale waarde van 4 mL/min ligt.
De functie van het systeem is op alle meetdagen geschikt voor het uitvoeren van de
metingen. Alle systeemparameters schommelen rond de gespecifieerde waarden.
4.1.2. Systeemgeschiktheidstest
De resultaten van de verschillende chromatografische parameters berekend bij het
injecteren van de testmix, staan samengevat in TABEL 4.1.
TABEL 4.1. GEMIDDELDE RESULTATEN ± STANDAARDDEVIATIE VAN DE
VERSCHILLENDE CHROMATOGRAFISCHE PARAMETERS VAN DE TESTMIX.
Parameter Calcium Limieten Magnesium Limieten
tR (min) 3,82 ± 0,09 3,8 ± 0,2 2,30 ± 0,06 2,3 ± 0,2
Piekoppervlakte (µS min) 20,22 ± 1,21 > 17 11,74 ± 0,75 > 9
Piekhoogte (µS) 85,92 ± 4,98 > 70 65,49 ± 3,97 > 50
k’ 7,32 ± 0,19 > 6,5 4,01 ± 0,12 > 3,5
Resolutie 4,60 ± 0,11 > 4 4,60 ± 0,11 > 4
Asymmetriefactor 1,08 ± 0,04 < 1,1 0,92 ± 0,03 < 1,15
Aantal theoretische platen 1718 ± 73 > 1550 1000 ± 53 > 900
22
Uit TABEL 4.1. kan afgeleid worden dat zowel voor calcium als magnesium de
retentietijd steeds binnen het gestelde interval ligt. De piekoppervlakte, de piekhoogte, de
capaciteitsfactor, de resolutie en het aantal theoretische platen liggen voor beide ionen op elke
meetdag boven de gespecifieerde minima. De asymmetriefactor van de calcium- en
magnesiumpiek is op elke meetdag kleiner dan het gestelde maximum. Het systeem wordt
steeds geschikt bevonden om de metingen uit te voeren.
4.2. VALIDATIE-EXPERIMENTEN
4.2.1. Lineariteit
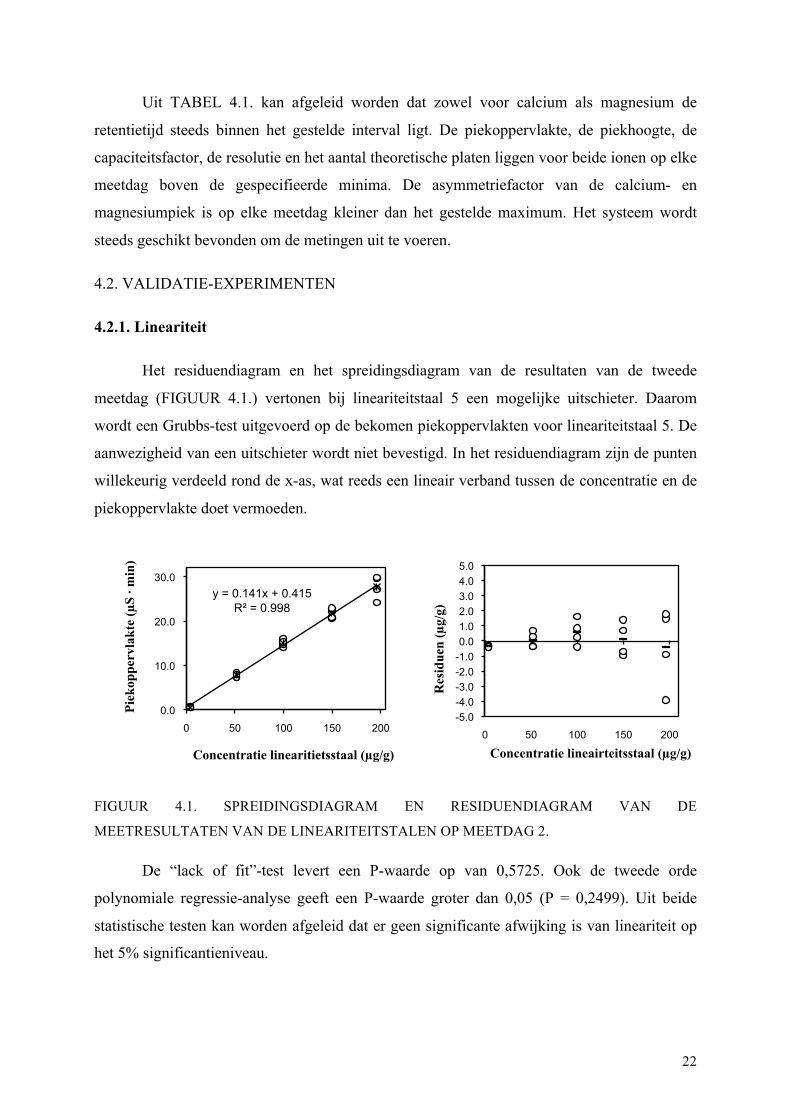
Het residuendiagram en het spreidingsdiagram van de resultaten van de tweede
meetdag (FIGUUR 4.1.) vertonen bij lineariteitstaal 5 een mogelijke uitschieter. Daarom
wordt een Grubbs-test uitgevoerd op de bekomen piekoppervlakten voor lineariteitstaal 5. De
aanwezigheid van een uitschieter wordt niet bevestigd. In het residuendiagram zijn de punten
willekeurig verdeeld rond de x-as, wat reeds een lineair verband tussen de concentratie en de
piekoppervlakte doet vermoeden.
FIGUUR 4.1. SPREIDINGSDIAGRAM EN RESIDUENDIAGRAM VAN DE
MEETRESULTATEN VAN DE LINEARITEITSTALEN OP MEETDAG 2.
De “lack of fit”-test levert een P-waarde op van 0,5725. Ook de tweede orde
polynomiale regressie-analyse geeft een P-waarde groter dan 0,05 (P = 0,2499). Uit beide
statistische testen kan worden afgeleid dat er geen significante afwijking is van lineariteit op
het 5% significantieniveau.
y = 0.141x + 0.415 R² = 0.998
0.0
10.0
20.0
30.0
0 50 100 150 200
Piek
oppe
rvla
kte
(µS
· min
)
Concentratie linearitietsstaal (µg/g)
-5.0 -4.0 -3.0 -2.0 -1.0 0.0 1.0 2.0 3.0 4.0 5.0
0 50 100 150 200
Res
idue
n (µ
g/g)
Concentratie lineairteitsstaal (µg/g)

23
Uit de statistische analyse van de meetresultaten van dag 1 en 3 kunnen dezelfde
conclusies getrokken worden. Er kan dus geconcludeerd worden dat de methode voldoet aan
de specificaties voor de lineariteit op het 5%-significantieniveau. Er mag een lineaire
kalibratiecurve gebruikt worden.
4.2.2. Kalibratiecurve
Uit de evaluatie van de lineariteit van de methode (4.2.1.) volgt dat een lineaire
kalibratiecurve mag gebruikt worden. Nu moet er nagegaan worden welk lineair
regressiemodel de meest robuuste en juiste is: het OLR – regressiemodel, het OLR – model
waarbij de trendlijn wordt geforceerd door nul of het GLR – regressiemodel.
De concentraties van het LoD-staal, het lage en hoge IQC-staal worden berekend
a.d.h.v. deze drie regressievergelijkingen gedurende vijf meetdagen. Vervolgens worden de
variatiecoëfficiënten van de concentraties en de terugvinding van de gravimetrisch bepaalde
concentratie van de stalen berekend. De resultaten staan samengevat in TABEL 4.2.
TABEL 4.2.: VARIATIECOËFFICIËNTEN VAN DE CONCENTRATIES BEREKEND VIA DE
VERSCHILLENDE REGRESSIEVERGELIJKINGEN EN DE TERUGVINDING VAN DE
WERKELIJKE CONCENTRATIE VAN HET LOD-STAAL EN DE IQC- STALEN.
OLR OLR geforceerd door 0 GLR
LoD
CVa (%) 142,5 13,83 60,01
Terugvinding (%) 366 210 -630*
Lage IQC
CVa (%) 1,4 0,8 2,0
Terugvinding (%) 97,4 102,8 97,1
Hoge IQC
CVa (%) 1,6 1,6 1,9
Terugvinding (%) 99,3 99,3 100,2
a variatiecoëfficiënt; *een negatieve terugvinding omdat via het GLR regressiemodel een negatieve concentratie wordt berekend.

24
Uit TABEL 4.2. kan afgeleid worden dat de variatiecoëfficiënt van zowel de LoD- als
de lage IQC-concentraties berekend via het OLR-regressiemodel met trendlijn geforceerd
door nul het kleinst is (respectievelijk 13,8% en 0,8%). De concentraties van beide stalen,
berekend op de verschillende meetdagen, variëren minder bij dit regressiemodel dan bij de
andere. De variatiecoëfficiënten van de concentraties van het hoge IQC-staal berekend via de
drie kalibratiemodellen verschillen nauwelijks van elkaar (CVOLR en OLR geforceerd door nul = 1,6%
en CVGLR =1,9%). Er kan dus geconcludeerd worden dat voor het berekenen van lage
concentraties het OLR-regressiemodel met trendlijn geforceerd door nul het meest robuust is.
Bij stalen met hoge calciumconcentraties is er nauwelijks verschil in robuustheid tussen de
verschillende kalibratiemodellen.
Uit TABEL 4.2. kan ook afgeleid worden dat de terugvinding van de werkelijke LoD-
concentratie bij het OLR-regressiemodel met trendlijn geforceerd door nul het minst afwijkt
van 100% (210%). Voor beide IQC-stalen is de terugvinding van de werkelijke concentratie
voor de drie kalibratiemodellen ongeveer gelijk aan elkaar (een afwijking van ±3% bij het
lage IQC-staal en ±0,5% bij het hoge IQC-staal). Bij het berekenen van zeer lage
concentraties is het OLR-regressiemodel met trendlijn geforceerd door nul het meest juiste.
Bij hogere concentraties is er geen onderscheid tussen de verschillende kalibratiemodellen.
Voor het opstellen van de kalibratiecurve wordt het OLR-regressiemodel met trendlijn
geforceerd door nul gebruikt. Het is het meest robuuste en juiste kalibratiemodel over een
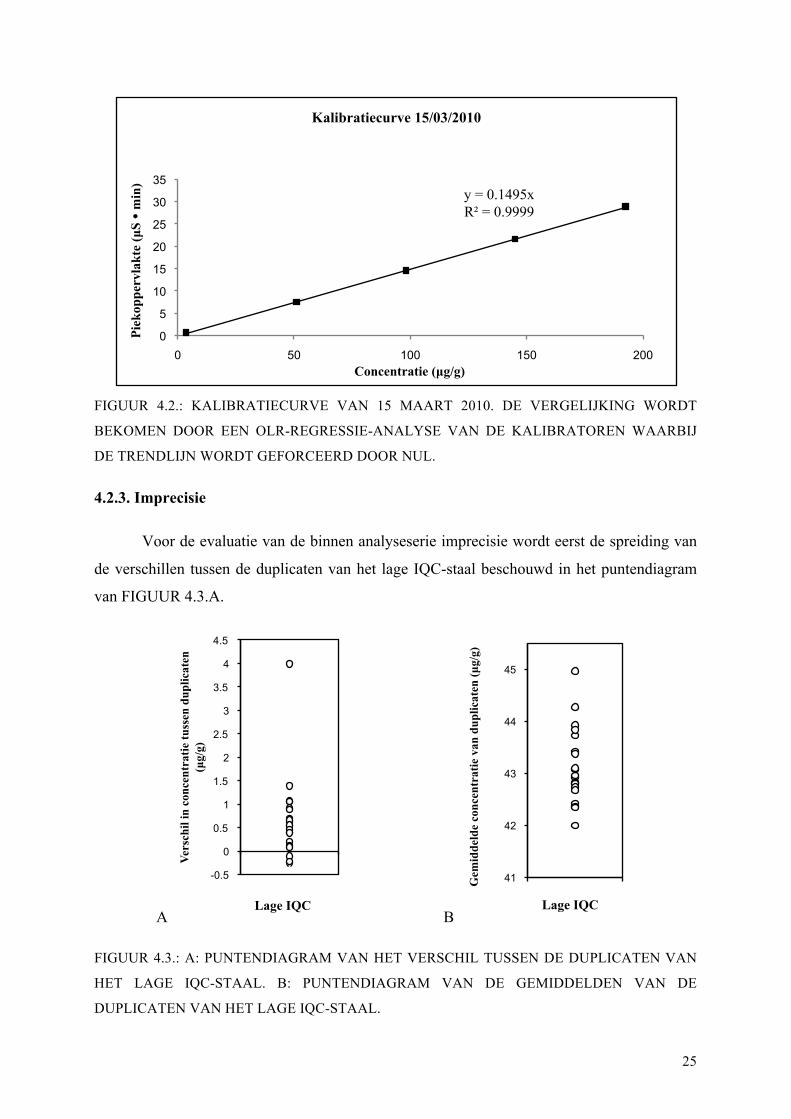
groot concentratiebereik. In FIGUUR 4.2. staat een voorbeeld van een kalibratiecurve, die
gebruikt wordt om de concentratie uit de piekoppervlakte te berekenen.
25
FIGUUR 4.2.: KALIBRATIECURVE VAN 15 MAART 2010. DE VERGELIJKING WORDT
BEKOMEN DOOR EEN OLR-REGRESSIE-ANALYSE VAN DE KALIBRATOREN WAARBIJ
DE TRENDLIJN WORDT GEFORCEERD DOOR NUL.
4.2.3. Imprecisie
Voor de evaluatie van de binnen analyseserie imprecisie wordt eerst de spreiding van
de verschillen tussen de duplicaten van het lage IQC-staal beschouwd in het puntendiagram
van FIGUUR 4.3.A.
A B
FIGUUR 4.3.: A: PUNTENDIAGRAM VAN HET VERSCHIL TUSSEN DE DUPLICATEN VAN
HET LAGE IQC-STAAL. B: PUNTENDIAGRAM VAN DE GEMIDDELDEN VAN DE
DUPLICATEN VAN HET LAGE IQC-STAAL.
-0.5
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
1
Vers
chil
in c
once
ntra
tie tu
ssen
dup
licat
en
(µg/
g)
Lage IQC
41
42
43
44
45
Gem
idde
lde
conc
entr
atie
van
dup
licat
en (µ
g/g)
Lage IQC
y = 0.1495x R² = 0.9999
0
5
10
15
20
25
30
35
0 50 100 150 200
Piek
oppe
rvla
kte
(µS
min
)
Concentratie (µg/g)
Kalibratiecurve 15/03/2010

26
In FIGUUR 4.3.A. is bovenaan duidelijk een uitschieter te zien, die ook door de
Grubbs-test wordt bevestigd. Het meetresultaat dat het meest afwijkt van de andere resultaten,
wordt verwijderd uit de dataset en het daggemiddelde wordt vervangen door het weerhouden
meetresultaat. Na correctie van de dataset vertonen de punten een regelmatige spreiding. In
FIGUUR 4.3.B. staat het puntendiagram van de daggemiddelden voor de evaluatie van de
tussendag imprecisie. De punten vertonen geen onregelmatigheden.
De meetresultaten van het hoge IQC-staal worden op dezelfde manier geëvalueerd. De
verschillen tussen de de duplicaten zijn regelmatig verdeeld. In de dataset van de
daggemiddelde concentraties van het hoge IQC-staal is er mogelijk een uitschieter aanwezig.
De Grubbs-test is positief en het overeenkomstig daggemiddelde wordt uit de dataset
verwijderd.
In TABEL 4.3. staan de berekende gemiddelden, de binnen analyseserie en totale
standaarddeviaties (Swr/ST) en variatiecoëfficiënten (CVwr/CVT) van het lage en hoge IQC-
staal samengevat.
TABEL 4.3.: RESULTATEN VAN DE BEREKENINGEN BIJ DE EVALUATIE VAN DE
MEETRESULTATEN VAN HET LAGE EN HOGE IQC-STAAL. DE SPECICATIE VOOR CVWR =
2% EN CVT = 5%.
Gemiddelde (µg/g) Swr (µg/g)a ST (µg/g)b CVwr (%) CVT (%)
Lage IQC 43,22 0,78 0,89 1,80 2,06
Hoge IQC 148,91 1,03 2,63 0,69 1,76 a binnen analyseserie standaarddeviatie; b totale standaarddeviatie.
Uit TABEL 4.3. kan afgeleid worden dat de binnen analyseserie CV zowel voor het
lage (1,80%) als het hoge IQC-staal (0,69%) lager is dan de doelwaarde van 2% voor een
stabiel proces. De totale CV van het lage (2,06%) en hoge IQC-staal (1,76%) ligt ook onder
de doelwaarde van 5%.
Er kan geconcludeerd worden dat de binnen analyseserie imprecisie niet significant
verschillend is van de doelwaarde voor een stabiel proces (CVwr = 2%) voor zowel het lage
als het hoge IQC-staal op het 5% significantieniveau. Ook de totale imprecisie van de
meetresultaten van het lage en het hoge IQC-staal verschilt niet significant van de doelwaarde
27
voor een stabiel proces van 5% op het 5%-significantieniveau. De methode voldoet aan de
specificaties voor de imprecisie.
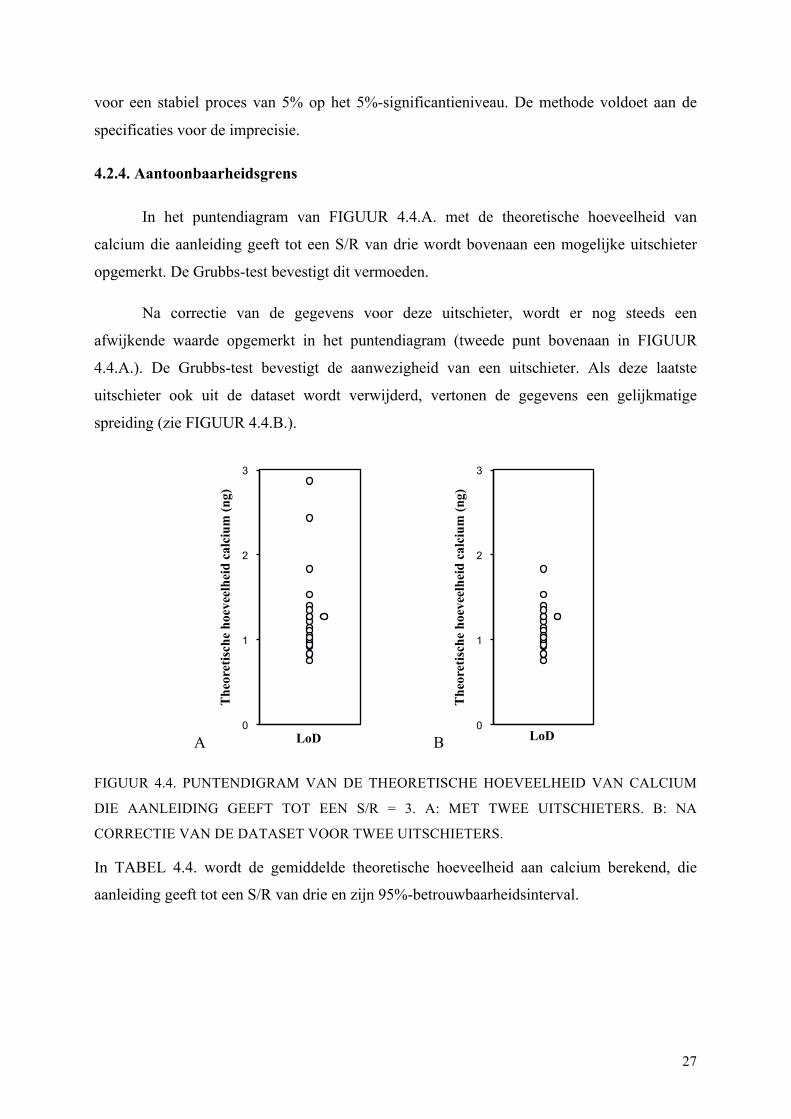
4.2.4. Aantoonbaarheidsgrens
In het puntendiagram van FIGUUR 4.4.A. met de theoretische hoeveelheid van
calcium die aanleiding geeft tot een S/R van drie wordt bovenaan een mogelijke uitschieter
opgemerkt. De Grubbs-test bevestigt dit vermoeden.
Na correctie van de gegevens voor deze uitschieter, wordt er nog steeds een
afwijkende waarde opgemerkt in het puntendiagram (tweede punt bovenaan in FIGUUR
4.4.A.). De Grubbs-test bevestigt de aanwezigheid van een uitschieter. Als deze laatste
uitschieter ook uit de dataset wordt verwijderd, vertonen de gegevens een gelijkmatige
spreiding (zie FIGUUR 4.4.B.).
A B
FIGUUR 4.4. PUNTENDIGRAM VAN DE THEORETISCHE HOEVEELHEID VAN CALCIUM
DIE AANLEIDING GEEFT TOT EEN S/R = 3. A: MET TWEE UITSCHIETERS. B: NA
CORRECTIE VAN DE DATASET VOOR TWEE UITSCHIETERS.
In TABEL 4.4. wordt de gemiddelde theoretische hoeveelheid aan calcium berekend, die
aanleiding geeft tot een S/R van drie en zijn 95%-betrouwbaarheidsinterval.
0
1
2
3
The
oret
isch
e ho
evee
lhei
d ca
lciu
m (n
g)
LoD 0
1
2
3 T
heor
etis
che
hoev
eelh
eid
calc
ium
(ng)
LoD

28
TABEL 4.4.: BEREKENEN VAN DE GEMIDDELDE ABSOLUTE HOEVEELHEID CALCIUM
EN ZIJN 95%-BETROUWBAARHEIDSINTERVAL.
Gemiddelde theoretische hoeveelheid calcium - 95% BIa + 95% BIa
1,14 ng 1,0 ng 1,2 ng a BI = betrouwbaarheidsinterval.
Uit TABEL 4.4. kan afgeleid worden dat de aantoonbaarheidsgrens gelijk is aan 1,14
ng met 95%-betrouwbaarheidsinterval [1,0 ng; 1,2 ng]. De bovengrens van het 95%-
confidentie-interval overschrijdt de specificatie van 2,0 ng niet. Er kan geconcludeerd worden
dat de methode voldoet aan de specificatie voor de aantoonbaarheidsgrens.
4.2.5. Juistheid
In het puntendiagram van het verschil tussen de duplicaten van een meetdag wordt
enkel bij onbekende 2 een uitschieter waargenomen (FIGUUR 4.5.A.). De aanwezigheid
ervan wordt bevestigd door de Grubbs-test. De waarde die het meest afwijkt van de
doelwaarde wordt beschouwd als uitschieter en verwijderd uit de dataset. Vervolgens wordt
de spreiding van de daggemiddelden van de onbekenden geëvalueerd in een ander
puntendiagram. In FIGUUR 4.5.B. is duidelijk te zien dat bij onbekende 5 één uitschieter
aanwezig is, die ook door de Grubbs-test wordt bevestigd.
A B
FIGUUR 4.5. A: PUNTENDIAGRAM VAN HET VERSCHIL IN CONCENTRATIE TUSSEN DE
DUPLICATEN OP DE VIJF MEETDAGEN VAN ONBEKENDE 2. B: PUNTENDIAGRAM VAN
DE GEMIDDELDE CONCENTRATIE VAN ONBEKENDE 5 OP DE VERSCHILLENDE
MEETDAGEN.
0
0.5
1
1.5
2
1
Vers
chil
tuss
en d
uplic
aten
op
1 m
eetd
ag
(µg/
g)
Onbekende 2 148
149
150
151
152
153
154
Dag
gem
idde
lde
conc
entr
atie
(µg/
g)
Onbekende 5
29
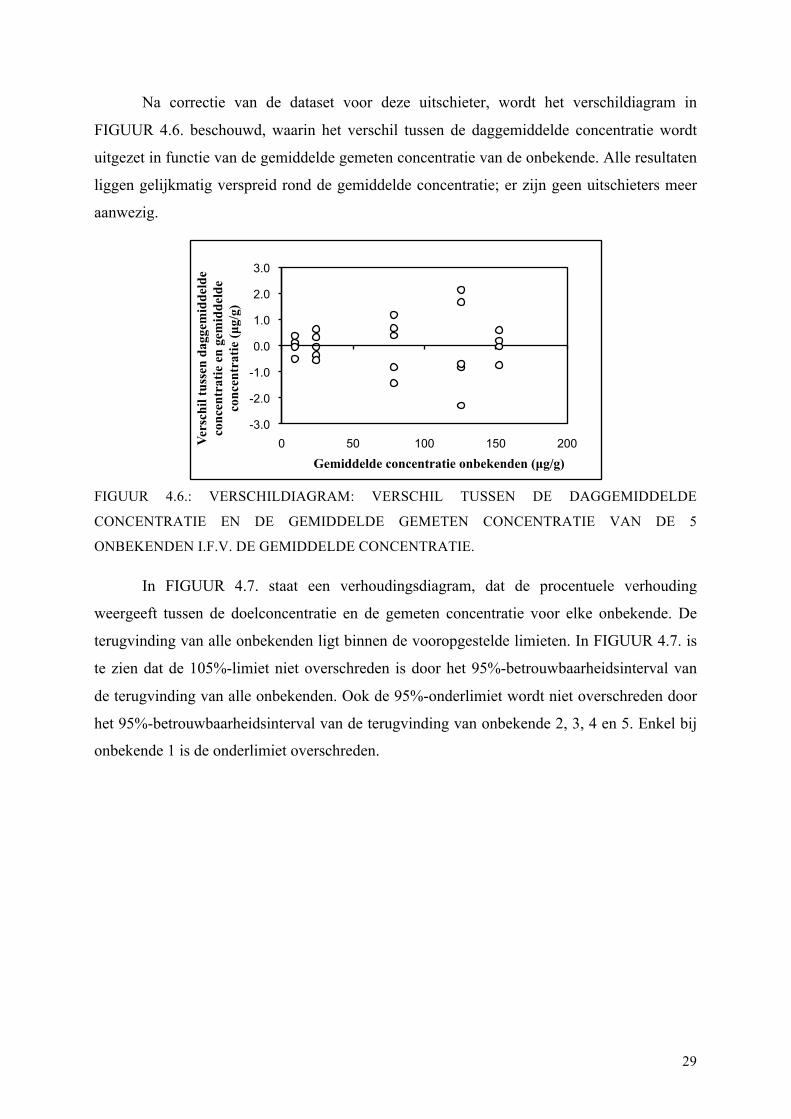
Na correctie van de dataset voor deze uitschieter, wordt het verschildiagram in
FIGUUR 4.6. beschouwd, waarin het verschil tussen de daggemiddelde concentratie wordt
uitgezet in functie van de gemiddelde gemeten concentratie van de onbekende. Alle resultaten
liggen gelijkmatig verspreid rond de gemiddelde concentratie; er zijn geen uitschieters meer
aanwezig.
FIGUUR 4.6.: VERSCHILDIAGRAM: VERSCHIL TUSSEN DE DAGGEMIDDELDE
CONCENTRATIE EN DE GEMIDDELDE GEMETEN CONCENTRATIE VAN DE 5
ONBEKENDEN I.F.V. DE GEMIDDELDE CONCENTRATIE.
In FIGUUR 4.7. staat een verhoudingsdiagram, dat de procentuele verhouding
weergeeft tussen de doelconcentratie en de gemeten concentratie voor elke onbekende. De
terugvinding van alle onbekenden ligt binnen de vooropgestelde limieten. In FIGUUR 4.7. is
te zien dat de 105%-limiet niet overschreden is door het 95%-betrouwbaarheidsinterval van
de terugvinding van alle onbekenden. Ook de 95%-onderlimiet wordt niet overschreden door
het 95%-betrouwbaarheidsinterval van de terugvinding van onbekende 2, 3, 4 en 5. Enkel bij
onbekende 1 is de onderlimiet overschreden.
-3.0
-2.0
-1.0
0.0
1.0
2.0
3.0
0 50 100 150 200 Vers
chil
tuss
en d
agge
mid
deld
e co
ncen
trat
ie e
n ge
mid
deld
e co
ncen
trat
ie (µ
g/g)
Gemiddelde concentratie onbekenden (µg/g)

30
FIGUUR 4.7.: PROCENTUEEL VERHOUDINGSDIAGRAM: TERUGVINDING VAN DE
DOELWAARDE VOOR ELKE ONBEKENDE. DE LIMIET IS 5 %.
Dat de grens van 95%-105% terugvinding overschreden is, kan ook aangetoond
worden met een eenzijdige één steekproef-t-test. Wanneer de uiterste waarden van het 95%-
betrouwbaarheidsinterval van de terugvinding de limieten niet overschrijden, wordt bij deze
test een P-waarde < 0,05 bekomen. Worden ze wel overschreden, dan is P > 0,05. Toegepast
op onze validatieresultaten wordt bij het toetsen t.o.v. de 105%-limiet voor alle onbekenden
een P-waarde < 0,05 gevonden. Bij het toetsen t.o.v. 95%-limiet wordt enkel voor onbekende
1 een P-waarde > 0,05 berekend (P = 0,2083). De 95%-limiet is dus overschreden door het
95%-betrouwbaarheidsinterval rond de terugvinding van onbekende 1. De conclusies
genomen uit FIGUUR 4.7. worden bevestigd.
Uit FIGUUR 4.7. kan ook afgeleid worden dat voor onbekende 1 het 95%-
betrouwbaarheidsinterval rond de gemiddelde concentratie opmerkelijk breder is dan bij de
andere onbekenden. Indien meer metingen zouden uitgevoerd worden, zal het interval
versmallen, waardoor het wel binnen de specificaties kan komen liggen. Via een power-
analyse wordt berekend dat daarvoor 54 metingen nodig zouden zijn.
Er kan dus geconcludeerd worden dat de methode niet voldoet aan de specificaties
voor de juistheid, omdat bij onbekende 1 (9,586 µg/g) de 5%-limiet voor de terugvinding
overschreden is. Het 95%-betrouwbaarheidsinterval van de terugvinding van onbekende 1
overlapt de 95%-limiet. De terugvinding met zijn 95%-betrouwbaarheidsinterval van de
andere onbekenden ligt wel binnen de 95%-105%-limieten. Mogelijke oorzaken van de
onjuistheid van de methode kunnen onder andere de onstabiliteit van het systeem, fouten bij
1 2
3 4
5
90
95
100
105
110
0 1 2 3 4 5
Teru
gvin
ding
(%) v
an d
e do
elw
aard
e
Onbekende
Terugvinding (%)
Specificatielimiet
31
het aanmaken van de stalen en/of kalibratoren en fouten bij het opstellen van de
kalibratiecurve zijn. Volgens het resultaat van de power-analyse zouden er 54 metingen van
onbekende 1 nodig zijn, opdat de methode wel zou voldoen aan de specificaties van de
juistheid.
4.2.6. Methodevergelijking
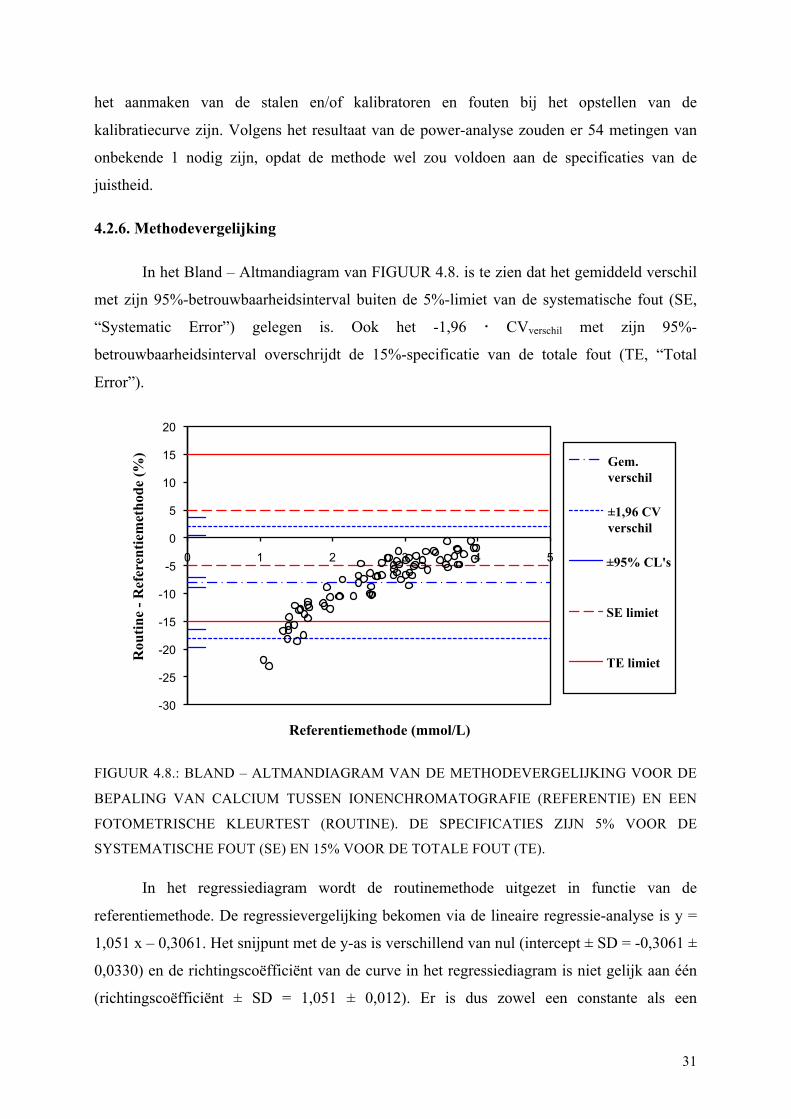
In het Bland – Altmandiagram van FIGUUR 4.8. is te zien dat het gemiddeld verschil
met zijn 95%-betrouwbaarheidsinterval buiten de 5%-limiet van de systematische fout (SE,
“Systematic Error”) gelegen is. Ook het -1,96 CVverschil met zijn 95%-
betrouwbaarheidsinterval overschrijdt de 15%-specificatie van de totale fout (TE, “Total
Error”).
FIGUUR 4.8.: BLAND – ALTMANDIAGRAM VAN DE METHODEVERGELIJKING VOOR DE
BEPALING VAN CALCIUM TUSSEN IONENCHROMATOGRAFIE (REFERENTIE) EN EEN
FOTOMETRISCHE KLEURTEST (ROUTINE). DE SPECIFICATIES ZIJN 5% VOOR DE
SYSTEMATISCHE FOUT (SE) EN 15% VOOR DE TOTALE FOUT (TE).
In het regressiediagram wordt de routinemethode uitgezet in functie van de
referentiemethode. De regressievergelijking bekomen via de lineaire regressie-analyse is y =
1,051 x – 0,3061. Het snijpunt met de y-as is verschillend van nul (intercept ± SD = -0,3061 ±
0,0330) en de richtingscoëfficiënt van de curve in het regressiediagram is niet gelijk aan één
(richtingscoëfficiënt ± SD = 1,051 ± 0,012). Er is dus zowel een constante als een
-30
-25
-20
-15
-10
-5
0
5
10
15
20
0 1 2 3 4 5
Rou
tine
- Ref
eren
tiem
etho
de (%
)
Referentiemethode (mmol/L)
Gem. verschil
±1,96 CV verschil
±95% CL's
SE limiet
TE limiet
32
proportionele systematische fout aanwezig op de meetresultaten bekomen via de
routinemethode. In TABEL 4.5. wordt geëvalueerd of die fouten nog binnen de
vooropgestelde limieten voor de systematische en totale fout gelegen zijn.
TABEL 4.5.: RESULTATEN VAN DE LINEAIRE REGRESSIE-ANALYSE BIJ DE
METHODEVERGELIJKING VAN EEN REFERENTIEMETHODE MET EEN
ROUTINEMETHODE VOOR DE BEPALING VAN CALCIUM. DE SPECIFICATIE VOOR
SYSTEMATISCHE FOUT IS 5% EN VOOR DE TOTALE FOUT 15%.
Minimale x-waardea
(1,05 mmol/L)
Maximale x-waardea
(3,98 mmol/L)
Gemiddelde systematische fout (%) -24,0 -2,6
- BIb (%) -25,8 -3,0
+ BIb (%) -22,3 -2,2
Gemiddelde totale fout (%) -24,0 -2,6
- PIc (%) -31,3 -4,5
+ PIc (%) -16,8 -0,7 a voor de minimale en maximale x-waarde, bekomen via de referentiemethode, wordt a.d.h.v. de lineaire
regressievergelijking de y-waarde voor de routinemethode voorspeld en wordt de systematische en totale fout
erop bepaald. b BI = betrouwbaarheidsinterval; c PI = predictie-interval.
Uit TABEL 4.5. kan worden afgeleid dat bij lage concentraties van calcium de 5%-
limiet voor de systematische fout overschreden is (gemiddelde SE = -24,0% met
betrouwbaarheidsinterval (BI): [-25,8%; -22,3%]). De systematische fout op de voorspelde y-
waarde voor de routinemethode blijft binnen de vooropgestelde specificatie bij hoge
calciumconcentraties (gemiddelde SE = -2,6% met BI: [-2,2%; -3,0%]). Voor de totale fout is
bij lage concentraties van calcium de 15%-limiet overschreden (gemiddelde TE = -24,0% met
predictie-interval (PI): [-31,3%; -16,8%]). Er is wel aan de specificatie van de totale fout
voldaan bij de meetresultaten van een staal met hoge calciumconcentratie (gemiddelde TE = -
2,6% met PI: [-4,5%; -0,7%]).
Uit het Bland – Altmandiagram zou kunnen geconcludeerd worden dat de
routinemethode niet voldoet aan de specificaties op het 5%-significantieniveau. Zowel de
limiet van de systematische (5%) als van de totale fout (15%) zijn overschreden. Dit besluit
kan echter genuanceerd worden m.b.v. de lineaire regressie-analyse, waaruit blijkt dat enkel

33
bij lage calciumconcentraties niet voldaan is aan de specificaties voor de systematische en de
totale fout. De algemene conclusie van de methodevergelijking luidt dus dat bij hoge
calciumconcentraties de routinemethode niet significant verschillend is van de
referentiemethode op het 5% significantieniveau. Bij lage concentraties van calcium in het
staal is de bias op de meetresultaten bekomen via de routinemethode te hoog en de
nauwkeurigheid van de metingen te laag.
4.3. LITERATUURONDERZOEK
4.3.1. Opzoeken wetenschappelijke literatuur
De CLSI protocollen zijn niet beschikbaar via het internet en worden ter beschikking gesteld
door dr. Stöckl. Informatie over het farmaceutisch gebruik van calcium wordt opgezocht via
het BCFI, het Farmacotherapeutisch Kompas en de Merck Index. In TABEL 4.6. staan de
bronnen, zoekwoorden en de bekomen resultaten samengevat voor het opzoeken van
literatuur.
TABEL 4.6. SAMENVATTING VAN DE VERSCHILLENDE ZOEKOPDRACHTEN.
Bron Zoekwoorden Aantal resultaten Opmerkingen
Methodevalidatie in de in vitro diagnostica
Google Scholar “method validation” +
CLSI 115
1 zeer geschikt artikel: via
referenties ervan verder
gezocht
Web of Science “method validation” 2440 Niet specifiek genoeg
Web of Science “method validation” +
“in vitro diagnostic” 1 Geschikt artikel
Westgard QC “specifications” 51 Publicaties met gewenste
specificaties
Andere bepalingsmethoden van calcium
Google “Determination
calcium” 7480000 Eerste 2 pagina’s bekeken
Web of Science “Determination
calcium” 2200 Veel goede publicaties

34
TABEL 4.6.: VERVOLG.
Functie calcium
Google Scholar Calcium + health 2080000 Eerst resultaat was zeer geschikt, via de referentielijst verder gezocht
PubMed Calcium + plants 9493
Eerst resultaat was een goede review, maar niet beschikbaar. Via de lijst van gerelateerde artikels verder gezocht.
Farmaceutische context van calcium
Calcium +
pharmaceutical
excipient
314000 Eerste pagina’s bekeken,
geschikte informatie
Google Ringer solution +
baxter 107000 Eerste pagina bekeken.
4.3.2. Methodevalidatie in de in vitro diagnostica
4.3.2.1. Protocollen en statistiek
De belangrijkste internationale organisatie die protocollen aanbiedt voor de
methodevalidatie in het domein van de in vitro diagnostica is het Clinical and Laboratory
Standards Institute (CLSI). De Société Française de Biologie Clinique biedt ook
validatieprotocollen aan, maar nationale aanbevelingen zijn natuurlijk ondergeschikt aan de
internationale zoals deze van het CLSI. Het voordeel van het beschikbaar zijn van vastgelegde
protocollen is dat zij toelaten verschillende evaluaties met elkaar te vergelijken, omdat de
validaties op dezelfde manier werden uitgevoerd (Vassault et al., 1986).
De evaluatieprotocollen (EP-protocollen) van de CLSI bevatten zowel de validatie-
experimenten die moeten uitgevoerd worden (bv. type staal, aantal stalen, aantal metingen,...)
als de bijhorende statistiek. De CLSI biedt protocollen aan voor de evaluatie van de
aantoonbaarheidsgrens (LoD, EP17-A), de bepalingsgrens (LoQ, EP17-A), de lineariteit
(EP6-A), de interferentie (EP7-P), de juistheid (EP9-A en EP15-A2), de precisie (EP5-A en
EP15-A2) en de totale analytische fout van de methode (EP-21-A). In TABEL 4.7. staan deze
protocollen samengevat voor de verschillende prestatiekenmerken. De verschillende validatie-
protocollen beschrijven hoe de ontwikkelaar van een methode (meestal de fabrikant) de

35
verschillende prestatiekenmerken kan bepalen. Daarnaast wordt ook beschreven hoe de
gebruiker van de methode (de laboratoria) de gestelde waarde door de fabrikant kan
verifiëren.
TABEL 4.7.: SAMENVATTING VAN DE CLSI-PROTCOLLEN VOOR DE
METHODEVALIDATIE.
Prestatiekenmerk Experimenteel protocol Testen CLSI-protocol
Aantoonbaarheidsgrens (LoD)
Blanco’s en stalen met zeer lage analytconc. Validatie: n = 60 (beide stalen) Verificatie: n = 20 (beide stalen)
Validatie: n.v.t. Verificatie: ≤ 5% metingen < LoB (LoD) 3 metingen > gestelde LoB (LoB)
EP17-A (2004)
Bepalingsgrens (LoQ)
Validatie: 3-5 stalen met zeer lage analytconc./ referentiemat. met conc. > LoD n = 40 Verificatie: Stalen met conc. = gestelde LoQ n = 25
Validatie: 1-steekproef t-test Verificatie: Aantal meetwaarden dat doelwaarde totale fout overschrijdt ≤ toegelaten (zie tabellen)
EP17-A (2004)
Lineariteit 5-7 conc., equidistaal in gesteld bereik 2r. × 1d.a
- Polynomiale regressie-analyse: regressie + t-test - 1-steekproef t-test
EP6-A (2003)
Precisie
Validatie: Lage en hoge conc. 2r. × 20d. a(minstens) Verificatie: Lage en hoge conc. 3r. × 5d.a
Verificatie: Chi2-test
EP5-A (1999) EP15-A2 (2006)
Juistheid (methodevergelijking)
Validatie: ≤ 40 patiëntstalen 2r. × 5d.a (willekeurig) zowel test- als ref. methode Verificatie: 1e procedure 20 patiëntstalen 2r. × 3-4d.a (5-7 stalen/d.) 2e procedure 1 lage + hoge conc. 2r.a × 3-5 analyseseries
Validatie: Regressie-analyse Biasdiagram (Bland – Altman) Verificatie: 1-steekproef t-test
EP9-A (1995) EP15-A2 (2006)
Totale fout n = ≤ 120 patiëntstalen
Biasdiagram (Bland – Altman) “Mountain plot” Tolerantie-interval
EP21-A (2003)

36
TABEL 4.7.: VERVOLG.
Interferentie
Gepaarde-verschil methode: Teststalen met potentiële interfererende stof + controlestalen (= patiëntstalen) afwisselend in 1 serie (n = afh verschillende factoren) Dosis-respons methode: Serie stalen met verschillende conc. van de interfererende stof. 1 analyseserie (n, idem boven) Analyse bias methode: Testserie met interfererende stof + controleserie (= patiëntstalen) n =10, 2r. × 1d.a
Gemiddelde + SD Lineaire regressie- analyse Biasdiagram
EP7-P (1986)
a r. = replicaten en d. = dagen.
Met het EP21-A protocol biedt de CLSI ook een protocol aan voor de evaluatie van de
totale analytische fout van een methode. Het domein van de in vitro diagnostica is daardoor
een uitzondering t.o.v. de andere analytische domeinen, die meestal geen totale fout evalueren
tijdens een methodevalidatie (Stöckl et al., 2009).
De totale analytische fout is een maat voor de nauwkeurigheid van een methode en
drukt het verschil uit tussen een meetresultaat en de ware waarde van dit meetresultaat (Stöckl
et al., 2009). De totale analytische fout omvat zowel de toevallige fouten (imprecisie en
interferentie) als de systematische fouten. In het EP21-A protocol wordt de totale fout van een
methode bepaald door ze te vergelijken met een andere methode, bij voorkeur een
referentiemethode (CLSI-EP21-A, 2003).
De reden waarom er specifiek in dit domein wel protocollen beschikbaar zijn voor de
totale fout, is dat vanuit het standpunt van de artsen de totale fout de meest frequent gebruikte
prestatiekenmerk is van een methode. Een onjuist meetresultaat kan namelijk aanleiding tot
geven tot foutieve medische beslissingen. Bovendien kan door het bepalen van de totale fout
eenvoudig en kosten-effectief de geschiktheid van de methode beoordeeld worden.
Daarentegen zijn de fabrikanten meer geïnteresseerd in de bronnen van de totale analytische
fout. Indien deze gekend zijn, kan de methode geoptimaliseerd worden zodat de totale fout
vermindert en de kwaliteit van de meetresultaten verbetert (Krouwer, 2002). Indien de

37
verschillende foutenbronnen gekend zijn, kan naast een methodevergelijkingstudie de totale
analytische fout ook bepaald worden via een simulatiemodel (bij een bepaalde verdeling van
die foutenbronnen), waarbij de verschillende foutenbronnen gecombineerd worden om de
totale fout te berekenen (Aronsson et al., 1974). Deze methode is echter zeer arbeidsintensief.
Een zeer belangrijk aspect in het domein van de in vitro diagnostica is de
traceerbaarheid van de meetresultaten. Dankzij de traceerbaarheid in de
laboratoriumgeneeskunde worden accurate meetresultaten verkregen die vergelijkbaar zijn
onafhankelijk van de tijd, de plaats en het meetsysteem. Door het verwezenlijken van de
metrologische traceerbaarheid van de meetresultaten wordt voldaan aan de klinische
basisbehoeften en wordt de patiëntenzorg en de controle en preventie van ziekten aanzienlijk
verbeterd (Vesper & Thienpont, 2009).
De VIM-definitie (“Vocabulaire International de Métrologie”, 2008) van de
metrologische traceerbaarheid luidt: “Eigenschap van een meetresultaat, waarbij het
resultaat gerelateerd is met een bepaalde referentie door een ononderbroken reeks van
kalibraties, die elk bijdragen tot een meetonzekerheid.” Traceerbaarheid wordt dus verkregen
door een meetresultaat in verband te brengen met een opgegeven referentie d.m.v. een
ononderbroken aaneenschakeling van kalibraties. Deze opgegeven referentie kan gaan van
een bedrijfsstandaard tot gecertificeerd primair referentiemateriaal, dat een bepaalde SI-
eenheid voorstelt. Het “Joint Committee for Traceability in Laboratory Medicine” (JCTLM)
erkent dergelijk referentiemateriaal en referentiemeetprocedures en houdt een databank bij
met de materialen en methoden, gebruikt om een traceerbaarheidsketen tot stand te brengen
(Vesper & Thienpont, 2009).
De ononderbroken aaneenschakeling van kalibraties is een alternerende procedure om
waarden toe te kennen aan materiaal dat wordt gebruikt om een hiërarchisch lagere methode
te kalibreren. Aan het begin van de reeks meetprocedures bevindt zich de referentiemethode,
die gebruikt wordt om met de laagste meetonzekerheid het referentiemateriaal te meten. De
reeks meetprocedures wordt afgesloten door de routinemethode, die met de hoogste
meetonzekerheid de routinestalen meet. Op deze manier is het meetresultaat van een
routinestaal herleidbaar tot de SI-eenheid, die gebaseerd is op de zuivere analytstandaard. In
de praktijk helpen referentielaboratoria met het linken van meetresultaten, afkomstig van
routinelaboratoria of fabrikanten, aan hiërarchisch hogere standaarden (Vesper & Thienpont,

38
2009). In FIGUUR 4.9. staat een voorbeeld van de traceerbaarheidsketen ontwikkeld voor de
bepaling van cortisol in serum (Vesper & Thienpont, 2009).
FIGUUR 4.9.: VOORBEELD VAN EEN TRACEERBAARHEIDSKETEN VOOR DE BEPALING
VAN CORTISOL IN SERUM. DE PIJLEN NAAR LINKS VERWIJZEN NAAR HET
TOEKENNEN VAN EEN WAARDE GEBRUIK MAKEND VAN DE MEETPROCEDURE. DE
PIJLEN NAAR RECHTS DUIDEN HET GEBRUIK VAN HET MATERIAAL VOOR
KALIBRATIE AAN. (VESPER & THIENPONT, 2009).
4.3.2.2. Specificaties
Het vastleggen van de specificaties voor de verschillende prestatiekenmerken heeft
een bepalende rol in de methodevalidatie. Het hangt namelijk van die aanvaardingscriteria af
of uiteindelijk besloten wordt of de methode gevalideerd is of niet (Taverniers et al., 2004).
Het doel van deze specificaties is dat kan nagegaan worden of een methode voldoende
betrouwbare resultaten kan opleveren, zodat er geen fouten kunnnen gemaakt worden bij de
interpretatie ervan in de context van het stellen van een diagnose. Elk meetresultaat kent een
zekere variabiliteit die zowel pre-analytisch (bv. bij staalname), analytisch (door imprecisie
en bias) als biologisch-fysiologisch (intra- en interindividueel) van aard kan zijn.
Zoals de CLSI protocollen aanbiedt, zijn er geen vastgelegde specificaties
beschikbaar. In de literatuur zijn talrijke publicaties en reviews terug te vinden met richtlijnen

39
voor de gewenste analytische specificaties voor methoden in het domein van de in vitro
diagnostica (vb. Ricos et al., 1999; Fraser et al., 1992; en Vassault et al., 1999). Doelwaarden
voor specificaties zijn voornamelijk beschikbaar voor de totale fout, de imprecisie (SD) en de
bias. Gedurende vele jaren en nu nog steeds is er veel gediscussieerd over o.b.v. welke
modellen die specificaties moeten vastgelegd worden (Krouwer, 2002).
In 1999 werd op een conferentie in Stockholm hierover een overeenkomst gesloten.
De hiërarchie van de modellen, waarop de analytische specificaties moeten gebaseerd zijn,
werd vastgelegd. Het beste model is deze gebaseerd op de klinische “outcome” (resultaat).
Hierbij wordt de invloed van de analytische kwaliteit op een patiënt in een bepaalde klinische
situatie geëvalueerd (betere diagnose, verbetering van de ziekte,...). Dit effect is echter
moeilijk te bewijzen, waardoor het model weinig bruikbaar is in de praktijk. Op de tweede
plaats in de hiërarchie staat het model waarbij het effect van de analytische prestatatie op
klinische beslissingen in het algemeen wordt geëvalueerd. Deze specificaties zijn gebaseerd
op de biologische variatie van de analytconcentratie en op de analyse van de opinie van
artsen. Specificaties gebaseerd op expertopinies staan op de derde plaats, gevolgd door deze
gebaseerd op doelwaarden voor de specificaties vastgelegd door de regulatoire organen
(Kenny et al., 1999).
Als laatste in de hiërarchie staan de specificaties gebaseerd op de “state of the art”
(Kenny et al., 1999). Deze specificaties weerspiegelen de analytische prestatie die op een
bepaald moment verkregen is in een bepaald aantal laboratoria. Verschillende publicaties
raden het af om op basis hiervan specificaties op te stellen omdat de “state of the art” niet
stabiel is. Afhankelijk van de methoden die in gebruik zijn kan de analytische prestatie
verbeteren of verslechteren (Vassault et al., 1999; Stöckl et al., 1995).
Het meest geschikte model voor het vastleggen van de specificaties is, zoals eerder
vermeld, weinig bruikbaar in de praktijk. De meeste auteurs raden aan om de specificaties te
baseren op de biologische variabiliteit: de intra- en interindividuele variabiliteit van de
analytconcentratie. In verschillende publicaties wordt gesteld dat de toegelaten analytische
imprecisie gelijk is aan de helft van de intra-individuele variabiliteit (Vassault et al., 1999;
Stöckl et al.,1995). De fractie 1/2de is arbitrair gekozen en kan soms tot aanvaardingscriteria
leiden die niet haalbaar zijn met de huidige methoden. Bovendien kan het ook zijn dat door de
grote intra-individuele variabiliteit zeer grote limieten gelden. Als de specificaties niet stroken
met de realiteit, kunnen de specificaties toch best gebaseerd worden op de “state of the art”

40
(Vassault et al., 1999). Volgens Stöckl et al. (1995) zou de bias < 0,25 ⋅ (CV2intra-individueel +
CV2 inter-individueel)1/2 moeten zijn.
De reden waarom de specificaties het best kunnen gebaseerd zijn op de biologische
variatie van de analytconcentratie, is dat het makkelijk te begrijpen en toe te passen is, er veel
data beschikbaar zijn en de biologische variatie geografisch constant is. Bovendien voldoet de
analytische kwaliteit aan de meeste klinische noden indien aan deze criteria voldaan is (Stöckl
et al., 1995).
In TABEL 4.8. staan verschillende voorbeelden uit de literatuur van gewenste
specificaties, bepaald o.b.v. de biologische variabiliteit, voor de bepaling van calcium in
serum.
TABEL 4.8.: VERSCHILLENDE GEWENSTE SPECIFICATIES VOOR DE BEPALING VAN
CALCIUM IN SERUM.
Artikel Imprecisie (%) Bias (%) Totale fout (%)
1a 1,0 0,8 2,4
2b 0,9 0,7 2,2
3c 1,2 1,7 2,3 a(Ricos et al., 1999); b(Fraser et al., 1992; Petersen et al., 1996); c(Vassault et al., 1999)
Uit TABEL 4.8. kan afgeleid worden dat de verschillende specficaties min of meer
met elkaar overeenstemmen. In de literatuur worden geen specificaties teruggevonden voor de
aantoonbaarheidsgrens, lineariteit, totale en binnen analyseserie imprecisie en de juistheid,
zoals bij de methodevalidatie voor de bepaling van calcium in deze masterthesis. Deze
specificaties worden bepaald via een voorstudie of zijn gebaseerd op specificaties van analoge
methoden, die beschikbaar zijn in de literatuur. In het domein van de in vitro diagnostica
moeten dus o.b.v. de gewenste specificaties voor de imprecisie, totale fout en bias die
beschikbaar zijn in de literatuur, de limieten en de doelwaarden voor de verschillende
prestatiekenmerken vastgelegd worden.
4.3.2.3. Beslissen
Na het uitvoeren van de statistische testen waarbij de verschillende
prestatiekenmerken van de methode worden vergeleken met hun limiet of de doelwaarde voor

41
een stabiel proces, kan besloten worden of de methode gevalideerd is of niet. Indien alle
prestatiekenmerken voldoen aan de gewenste specificaties, kan de methode aangewend
worden in het laboratorium. Als er een statistische significante afwijking is van de gewenste
specificaties, moet de oorzaak opgespoord worden en de methode aangepast, opdat er toch
aan voldaan is.
De statistische significantietesten die vandaag meestal worden uitgevoerd gedurende
een methodevalidatie zijn de zogenaamde nulhypothese-testen. De huidige kritiek hierop is
dat hierbij enkel rekening wordt gehouden met de type I-fout (of α-fout), namelijk de kans op
het foutief verwerpen van de nulhypothese (vandaar ook producent-risico genoemd). Hierdoor
zal een methode die meer imprecies is makkelijker gevalideerd worden dan een meer precieze
methode (Feinberg, 2007). In de toekomst zullen powerberekeningen de nieuwe trend zijn in
methodevalidatie in de in vitro diagnostica. Zij laten toe om de uitkomst van een test te
voorspellen en houden ook rekening met de type II- of β-fout, die de kans uitdrukt om
verkeerdelijk de nulhypothese te weerhouden (patiënt-risico).
Een andere trend in de in vitro diagnostica is de methodevalidatie, gebaseerd op
accuraatheidsprofielen. Hierbij wordt een interval berekend, dat een bepaald percentage van
toekomstige meetresultaten bevat. Dat interval wordt dan vergeleken met een
aanvaardingsinterval, dat werd vastgelegd door de gebruiker van de methode. Op deze manier
kan eenvoudig beslist worden of de methode gevalideerd is of niet (Feinberg, 2007). Deze
benadering van methodevalidatie is nog steeds in ontwikkeling (Stöckl et al., 2009).
4.3.3. Andere bepalingsmethoden voor calcium
In TABEL 4.9. staan enkele voorbeelden van andere bepalingsmethoden voor calcium
samengevat met een voorbeeld uit de literatuur en hun voor- en nadelen.
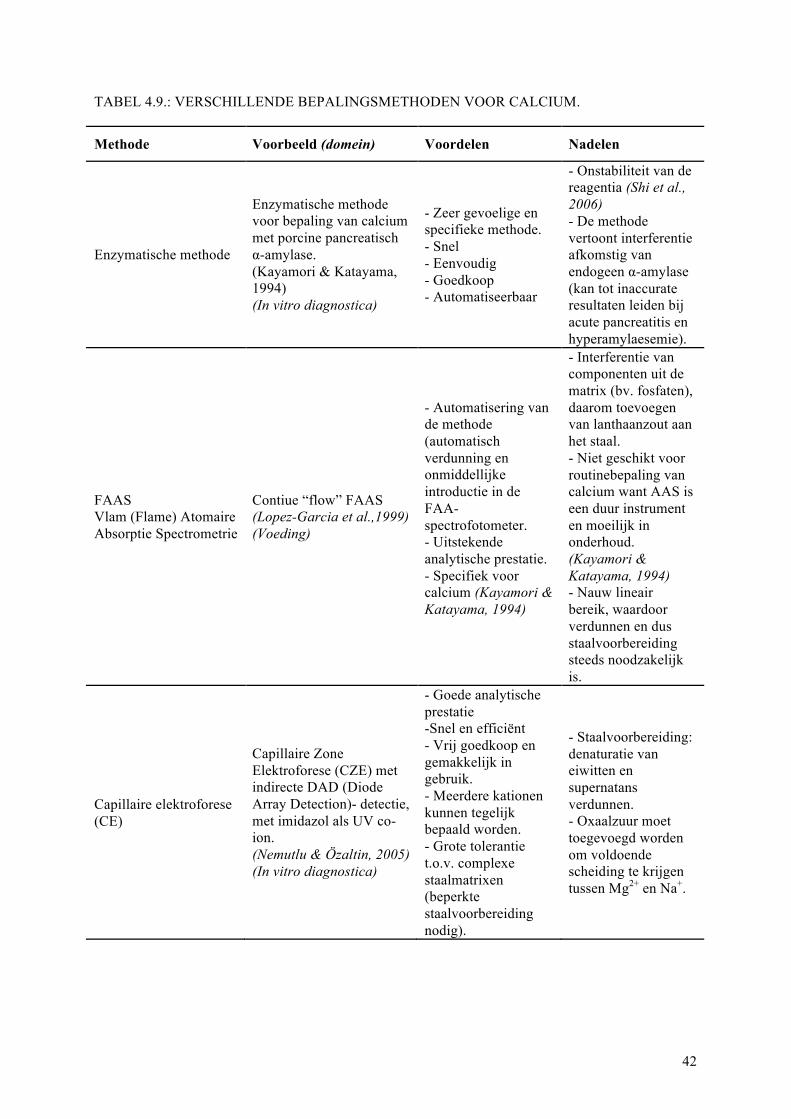
42
TABEL 4.9.: VERSCHILLENDE BEPALINGSMETHODEN VOOR CALCIUM.
Methode Voorbeeld (domein) Voordelen Nadelen
Enzymatische methode
Enzymatische methode voor bepaling van calcium met porcine pancreatisch α-amylase. (Kayamori & Katayama, 1994) (In vitro diagnostica)
- Zeer gevoelige en specifieke methode. - Snel - Eenvoudig - Goedkoop - Automatiseerbaar
- Onstabiliteit van de reagentia (Shi et al., 2006) - De methode vertoont interferentie afkomstig van endogeen α-amylase (kan tot inaccurate resultaten leiden bij acute pancreatitis en hyperamylaesemie).
FAAS Vlam (Flame) Atomaire Absorptie Spectrometrie
Contiue “flow” FAAS (Lopez-Garcia et al.,1999) (Voeding)
- Automatisering van de methode (automatisch verdunning en onmiddellijke introductie in de FAA-spectrofotometer. - Uitstekende analytische prestatie. - Specifiek voor calcium (Kayamori & Katayama, 1994)
- Interferentie van componenten uit de matrix (bv. fosfaten), daarom toevoegen van lanthaanzout aan het staal. - Niet geschikt voor routinebepaling van calcium want AAS is een duur instrument en moeilijk in onderhoud. (Kayamori & Katayama, 1994) - Nauw lineair bereik, waardoor verdunnen en dus staalvoorbereiding steeds noodzakelijk is.
Capillaire elektroforese (CE)
Capillaire Zone Elektroforese (CZE) met indirecte DAD (Diode Array Detection)- detectie, met imidazol als UV co-ion. (Nemutlu & Özaltin, 2005) (In vitro diagnostica)
- Goede analytische prestatie -Snel en efficiënt - Vrij goedkoop en gemakkelijk in gebruik. - Meerdere kationen kunnen tegelijk bepaald worden. - Grote tolerantie t.o.v. complexe staalmatrixen (beperkte staalvoorbereiding nodig).
- Staalvoorbereiding: denaturatie van eiwitten en supernatans verdunnen. - Oxaalzuur moet toegevoegd worden om voldoende scheiding te krijgen tussen Mg2+ en Na+.

43
TABEL 4.9.: VERVOLG.
HPLC/Colorimetrische methode
HPLC met koolstof stationaire fase en een mobiele fase met o-cresolftaleïne (OCPC) complexvormer en spectrofotometrische detectie. (Paull et al.,1997) (milieu)
- OCPC is zeer selectief voor divalente kationen - Gekleurd complex OCPC-Ca2+ kan spectrofotometrisch gedetecteerd worden. - Eenvoudig - Relatief vrij van interferentie. - Goede reproduceerbaarheid.
- Scheiding van calcium en magnesium duurt vrij lang (6-10 min). - Kortdurende stabiliteit van OCPC in het systeem (Shi et al., 2006).
Potentiometrische bepaling
Indirecte potentiometrische bepaling van Ca2+ en Mg2+ m.b.v. metallische cobaltelektrode in fosfaatbuffer carrier gebruik makend van “flow injection analysis”. (Chen & Adams, 1998) (milieu)
- Goedkoop - Snelle respons. - Eenvoudige opstelling. - Hoge gevoeligheid - - Selectieve bepaling van Ca2+ en Mg2+ (indien maskerende agentia toegevoegd).
- Andere ionen vertonen interferentie. - Maskerende agentia moeten toegevoegd worden om Ca2+ en Mg2+ van elkaar te onderscheiden.
4.3.4. Farmaceutisch gebruik van calcium
Het gebruik van calcium in de farmaceutische context is vrij beperkt. Verschillende
calciumzouten worden teruggevonden in preparaten ter preventie van fracturen ten gevolge
van osteoporose. Ze worden ook gebruikt voor de behandeling van osteoporose in associatie
met andere geneesmiddelen zoals vitamine D en de bisfosfonaten. In TABEL 4.10. staan
voorbeelden van deze preparaten en de samenstellende calciumzouten (BCFI, 2010).
TABEL 4.10.: VOORBEELDEN VAN PREPARATEN TER PREVENTIE VAN FRACTUREN
T.G.V. OSTEOPOROSE EN VOOR DE BEHANDELING VAN OSTEOPOROSE.
Naam specialiteit (fabrikant) Samenstellende calciumzouten
Cacit (Procter & Gamble) Calciumcarbonaat
Sandoz Calcium (Sandoz) Calciumglubionaat (ampullen), calciumcarbonaat
+ calciumgluconolactaat (bruistabletten)
(BCFI, 2010)
Een tweede groep van geneesmiddelen die calciumzouten bevat, zijn de antacida.
Deze preparaten verminderen de pijnklachten t.g.v. dyspepsie, gastritis, pyrosis en lichte

44
refluxsymptomen. Naast calciumcarbonaat bevatten de antacida nog aluminium-, magnesium-
en/of natriumzouten.
Daarnaast zijn er nog een aantal preparaten die worden aangewend ter behandeling
van stoornissen van de water- en elektrolythuishouding. Orale multivitaminepreparaten, die
geïndiceerd zijn bij patiënten met malabsorptie of intraveneus samen met totale parenterale
voeding, bevatten onder ander calciumcarbonaat (BCFI, 2010). Fosfaatchelatoren zijn
preparaten o.b.v. calciumcarbonaat, - acetaat of -lactogluconaat, die worden toegediend aan
volwassenen met chronische nierinsufficiëntie onder nierdialyse, om hyperfosfatemie tegen te
gaan. (BCFI, 2010; Farmacotherapeutisch Kompas, 2010). Ter preventie of behandeling van
calciumdeficiëntie kunnen preparaten o.b.v. calciumcarbonaat, - lactaat, -citraat, -glubionaat
en gluconaat oraal ingenomen worden. De laatste twee calciumzouten kunnen samen met
calciumchloride ook parenteraal toegediend worden (Farmacotherapeutisch Kompas, 2010;
Merck Index, 1996).
Hydrogelen o.b.v. calciumgluconaat en chloorhexidine worden lokaal aangewend als
antidoot bij een fluorwaterstofzuurintoxicatie (Farmacotherapeutisch Kompas, 2010).
Verschillende calciumzouten worden gebruikt als hulpstof in de farmaceutische
industrie. De belangrijkste zijn de calciumsulfaten, -carbonaten en -fosfaten, die als vulstof
gebruikt worden tijdens de productie van tabletten (Merck Index, 1996). Calciumcarbonaat
wordt o.w.v. zijn capaciteit om zuur te neutraliseren gebruikt als excipiënt in tabletten,
waarvan het actief bestanddeel door zijn zuur karakter de maagwand kan aantasten.
Calciumcarbonaat helpt dus de zure pH te bufferen (Specialty Minerals Inc., 2010).
Calciumfosfaten worden ook in vaccins gebruikt als hulpstof. Het zout adsorbeert de
oplosbare antigenen, waardoor het als partikel kan worden gepresenteerd aan het
immuunsysteem (Relyveld et al., 1985).
Ca2+-ionen (CaCl2) zijn ook aanwezig in isotone oplossingen die intraveneus worden
toegediend. Een voorbeeld zijn de Ringer-oplossingen die o.a. gebruikt worden voor het
onderhouden en het herstel van de vocht- en elektrolytbalans, voor de toediening van
geneesmiddelen, chemotherapie en parenterale voeding en voor de transfusie van
bloedprodukten (Baxter, 2010). Volgens de USP (2009) mag de concentratie van calcium in
deze Ringer-oplossingen schommelen tussen 2,05 – 2,45 mmol/L.
Tot slot is calcium het tegenion van diverse actieve bestanddelen in geneesmiddelen
(Merck Index, 1996; BCFI, 2010).

45
5. CONCLUSIE
Gedurende de validatie-experimenten wordt het systeem steeds optimaal bevonden en
is het geschikt voor het uitvoeren van de metingen. Uit de evaluatie van de lineariteit blijkt
dat de methode voldoet aan de specificatie. Voor het opstellen van de lineaire kalibratiecurve
wordt het OLR-regressiemodel met trendlijn geforceerd door nul gebruikt. Het is het meest
robuuste en juiste kalibratiemodel voor het berekenen van de concentratie uit de
piekoppervlakte over een breed concentratiebereik. Tijdens de evaluatie van de precisie van
de methode is de experimentele CV steeds kleiner dan de doelwaarde voor een stabiel proces
voor beide IQC-stalen. De methode voldoet aan de specificaties voor de binnen analyseserie
en de totale precisie.
De aantoonbaarheidsgrens van de methode is 1,14 ng. De bovengrens van het 95%-
betrouwbaarheidsinterval overschrijdt de limiet van 2,0 ng niet. De aantoonbaarheidsgrens is
dus conform met de specificatie. De methode voldoet echter niet aan de eisen voor de
juistheid. Onbekende 1 overschrijdt de 5%-limiet voor de terugvinding. Indien er 54 metingen
zouden uitgevoerd worden op onbekende 1, zou de methode volgens de power-analyse wel
voldoen aan de specificaties voor de juistheid. De methode voldoet dus niet aan alle
specificaties.
Een methodevergelijking laat toe om na te gaan of de systematische en totale fout van
een methode voldoet aan de specificaties door ze te vergelijken met een hiërarchisch hogere
methode. Uit de simulatie blijkt dat dat de routinemethode bij lage calciumconcentraties niet
voldoet aan de specificaties voor de systematische en totale fout. Bij hoge concentraties zijn
de methoden niet significant verschillend op het 5% significantieniveau. Het is belangrijk om
tijdens de methodevergelijking zowel het Bland – Altmandiagram als de lineaire regressie-
analyse te beschouwen. Wanneer er inderdaad constante of proportionele verschillen zijn
tussen de twee methoden, laat de lineaire regressie-analyse toe om de prestatie van de
methode te evalueren bij verschillende concentratieniveaus. Op deze manier kan de conclusie
genomen uit het Bland – Altmandiagram genuanceerd worden.
In het domein van de in vitro diagnostica beschrijven de CLSI-protocollen de
validatie-experimenten en de bijhorende statistiek voor de evaluatie van de verschillende
prestatiekenmerken tijdens een methodevalidatie. In tegenstelling tot de andere analytische
domeinen, wordt in dit domein ook de totale analytische fout geëvalueerd tijdens een
methodevalidatie.

46
Voor het vastleggen van de specificaties bestaat er nog veel discussie binnen het
domein, maar er is reeds een bepaalde hiërarchie van modellen overeengekomen, waarop de
specficaties moeten gebaseerd zijn. De meeste auteurs raden aan om de specificaties van de
verschillende prestatiekenmerken te baseren op de biologische variabiliteit van de
analytconcentratie. Het vastleggen van specificaties vormt een kritisch onderdeel in de
methodevalidatie. Ze bepalen of een methode uiteindelijk slaagt of niet tijdens de statistische
evaluatie van de resultaten van de validatie-experimenten.
In de literatuur worden er nog andere bepalingsmethoden voor calcium
teruggevonden. De belangrijkste zijn FAAS, capillaire elektroforese, de potentiometrische, de
colorimetrische en de enzymatische methoden. In de farmaceutische context wordt calcium
steeds onder de zoutvorm teruggevonden in o.a. de antacida, preparaten ter behandeling van
osteoporose, fosfaatchelatoren en Ringer-oplossingen.

47
6. REFERENTIELIJST
Aronsson, T.; de Verdier, C. H.; Groth, T. (1974). Factors influencing the quality of analytical
methods – a systems analysis, with use of computer simulation. Clin. Chem., 20, 738-748.
Bushinsky, D. A.; Monk, R. D. (1998). Calcium. Lancet, 352, 305-311.
Calcium oCPC-test package insert (2009). Beckman Coulter®, Brea, USA.
Chandran, S.; Singh, R. S. P. (2007). Comparison of various international guidelines for
analytical method validation. Pharmazie, 62, 4-14.
Chen, Z. L.; Adams, M. A. (1998). A metallic cobalt electrode for the indirect potentiometric
determination of calcium and magnesium in natural waters using flow injection analysis.
Talanta, 47, 779-786.
EP5-A – Evaluation of Precision Performance of Clinical Chemistry Devices (1999). Clinical
and Laboratory Standards Institute (CLSI), Wayne (PA), USA.
EP6-A – Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical
Approach (2003). Clinical and Laboratory Standards Institute (CLSI), Wayne (PA), USA.
EP7-P – Interference Testing in Clinical Chemistry (1986). Clinical and Laboratory
Standards Institute (CLSI), Wayne (PA), USA.
EP9-A – Method Comparison and Bias Estimation Using Patient Samples (1995). Clinical
and Laboratory Standards Institute (CLSI), Wayne (PA), USA.
EP15-A2 – User Verification of Performance for Precision and Trueness (2006). Clinical and
Laboratory Standards Institute (CLSI), Wayne (PA), USA.
EP17-A – Protocols for Determination of Limits of Detection and Limits of Quantitation
(2004). Clinical and Laboratory Standards Institute (CLSI), Wayne (PA), USA.
EP21-A – Estimation of Total Analytical Error for Clinical Laboratory Methods (2003).
Clinical and Laboratory Standards Institute (CLSI), Wayne (PA), USA.
Feinberg, M. (2007). Validation of analytical methods based on accuracy profiles. J.
Chromatogr. A, 1158, 174-183.

48
Fraser, C. G.; Petersen, P. H.; Ricós, C.; Haeckel, R. (1992). Proposed quality specifications
for imprecision and inaccuracy of analytical systems for clinical chemistry. Eur. J. Clin.
Chem. Clin. Biochem., 30, 311-317.
ISO 9000: Quality management systems – Fundamentals and vocabulary, 3rd edition (2005).
International Organization for Standardization (ISO), Genève, Zwitserland.
Kayamori, Y.; Katayama, Y. (1994). Enzymatic Method for Assaying Calcium in Serum and
Urine with Porcine Pancreatic α-Amylase. Clin. Chem., 40, 781-784.
Kenny, D.; Fraser, C. G.; Petersen, P. H.; Kallner, A. (1999). Consensus agreement. Scand. J.
Clin. Lab. Invest., 59, 585.
Krouwer, J. S. (2002). Setting Performance Goals and Evaluating Total Analytical Error for
Diagnostic Assays. Clin. Chem., 48, 919-927.
López-García, I.; Viñas, P.; Blanco, C.; Hernández-Córdoba, M. (1999). Fast determination
of calcium, magnesium and zinc in honey using continuous flow flame atomic absorption
spectrometry. Talanta, 49, 597-602.
Maathuis, F. J. M. (2009). Physiological functions of mineral macronutrients. Curr. Opin.
Plant Biol., 12, 250-258.
Nemutlu, E.; Özaltin, N. (2005). Determination of magnesium, calcium, sodium and
potassium in blood plasma samples by capillary zone electrophoresis. Anal. Bioanal. Chem.,
383, 833-838.
Paull, B.; Macka, M.; Haddad, P. R. (1997). Determination of calcium and magnesium in
water samples by high-performance liquid chromatography on a graphitic stationary phase
with a mobile phase containing o-cresolphthalein complexone. J. Chromatogr. A, 789, 329-
337.
Petersen, P. H.; Ricós, C.; Stöckl, D.; Libeer, J. C.; Baadenhuijsen, H.; Fraser, C. G.;
Thienpont, L. M. (1996). Proposed guidelines for the internal quality control of analytical
results in the medical laboratory. Eur. J. Clin. Chem. Clin. Biochem., 34, 983-999.
Power, M. L.; Heaney, R. P.; Kalkwarf, H. J.; Pitkin, R. M.; Repke, J. T.; Tsang, R. C.;
Schulkin J. (1999). The role of calcium in health and disease. Am. J. Obstet. Genecol., 181,
1560-1569.

49
Relyveld, E. H.; Ickovic, M. R.; Hénocq, E.; Garcelon, M. (1985). Calcium – phosphate
adjuvanted allergens. Ann. Allergy, 54, 521-529.
Ricós, C.; Alvarez, V.; Cava, F.; García-Lario, J. V.; Hernández, A.; Jiménez, C. V.;
Minchinela, J.; Perich, C.; Simon, M. (1999). Current databases on biological variation: pros,
cons and progress. Scand. J. Clin. Lab. Invest., 59, 491-500.
Shi, L.; Liu, X.; Li, H.; Xu, G. (2006). Determination of Total Calcium in Plasma by Flow
Injection Analysis with Tris(2,2’-bipyridyl)ruthenium(II) Electrochemiluminescent Detection.
Electroanal., 18, 1584-1589.
Stöckl, D. (2007a). Method validation with confidence. STT Consulting, Horebeke, België.
Stöckl, D. (2007b). Laboratory Statistics & Graphics with EXCEL®. STT Consulting,
Horebeke, België.
Stöckl, D.; Baadenhuijsen, H.; Fraser, C. G.; Libeer, J. C.; Petersen, P. H.; Ricos, C. (1995).
Desirable Routine Analytical Goals for Quantities Assayed in Serum – Discussion paper from
members of the External Quality Assessment (EQA, Working Group A) on analytical goals of
laboratory medicine. Eur. J. Clin. Chem. Clin. Biochem., 33, 157-169.
Stöckl, D.; D’Hondt, H.; Thienpont, L. M. (2009). Method validation across the disciplines –
Critical investigation of major validation criteria and associated experimental protocols. J.
Chromatogr. B, 877, 2180-2190.
Taverniers, I.; De Loose, M.; Van Bockstaele, E. (2004). Trends in quality in the analytical
laboratory. II. Analytical method validation and quality assurance. Trac-Trends Anal. Chem.,
23, 535-552.
The Merck Index twelfth edition on CD-rom 12:1 (1996). Merck & Co., Inc., Whitehouse
Station, New Jersey, USA.
Tortora, G. J.; Derrickson, B. (2006). Principles of Anatomy and Physiology, 11th edition.
John Wiley & Sons, Inc., Hoboken, USA, Chapter 27.
United States Pharmacopoeia (USP). Volume 32, NF 27 (2009).

50
Vassault, A.; Grafmeyer, D.; de Graeve, J.; Cohen, R.; Beaudonnet, A.; Bienvenu, J. (1999).
Analyses de biologie médicale: spécifications et normes d’acceptabilité à l’usage de la
validation de techniques. Ann. Biol. Clin., 57, 685-695.
Vassault, A.; Grafmeyer, D.; Naudin, C.; Dumont, G.; Bailly, M.; Henny, J.; Gerhardt, M. F.;
Georges, P. (1986). Commission “For validation of methods” of the Société Française de
Biologie Clinique – Protocol for the validation of methods (document B, stage 3). Ann. Biol.
Clin., 44, 686-745.
Vesper, H. W.; Thienpont, L. M. (2009). Traceability in Laboratory Medicine. Clin. Chem.,
55, 1067-1075.
Vocabulaire International de Métrologie – Concepts fondamentaux et généraux et termes
associés, 3e édition (2008). Joint Committee for Guides in Metrology (JCGM), Genève,
Zwitserland.
http://www.baxter.be/nl/therapies/solution_therapy/sub/iv_therapy.html (03/05/2010).
http://www.bcfi.be/ (01/05/2010).
http://www.emis.vito.be/sites/default/files/pagina/referentielabo_bodem_CMA_6-A.pdf
(08/05/2010).
http://www.fk.cvz.nl/ (01/05/2010).
http://www.bipm.org/jctlm/ (09/05/2010).
http://www.specialtyminerals.com/specialty-applications/specialty-markets-for-
minerals/pharmaceuticals/ (30/04/2010).



















