
tics Project (1)...
25
BIOINFORMATICS PROJECT BY: MIT KOTECHA
-
Upload
mit-kotecha -
Category
Documents
-
view
218 -
download
0
Transcript of tics Project (1)...

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 1/25
BIOINFORMATICSPROJECT
BY: MIT KOTECHA

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 2/25
CONTENTS….
Aim Introduction
Softwares used for modeling
Sequence alignment & homologymodeling
Validation of model
Docking Docking Results
Conclusions

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 3/25
Structure prediction of chitinase(enzymethat degrades chitin layers) isolated fromthe pathogen Aphanocladium Album ,using
web based bioinformatics tools ,molecularmodeling softwares and servers.
To find effective inhibitor for this enzyme by
docking.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 4/25
INTRODUCTION Degrades chitin layer by breaking down the glycosidic
bonds.
Mostly used for digestion purposes, this enzyme isfound in a wide category of organisms includingorganisms as primitive as bacteria & fungi and asadvanced as human beings.
Most often the chitinase in pathogens falls underfamily 18.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 5/25
Clinical references associate chitinase with variousallergies (both in animals and plants )including
asthma for which exposure to high levels of chitinase isobserved to be a cause (though the reason for infectionis not clear yet.
In microorganisms, chitinase is found as to be a
primary metabolite , hence inhibiting chitinase willinhibit the growth of the microorganisms. Therefore
we can form antibiotic drugs via inhibiting chitinase.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 6/25
Aphanocladium Album ,though a weak pathogen, is
associated with the brown spots growing on the caps
of mushrooms.And so this particular fungus is found to be
pathogen of another fungus.
Pathogen Activity of the
Given Fungus

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 7/25
SOFTWARES
Modeling involves sequence alignment (single or multiple
depending on the requirements) followed by homology modelling orthreading(2’ structure prediction).
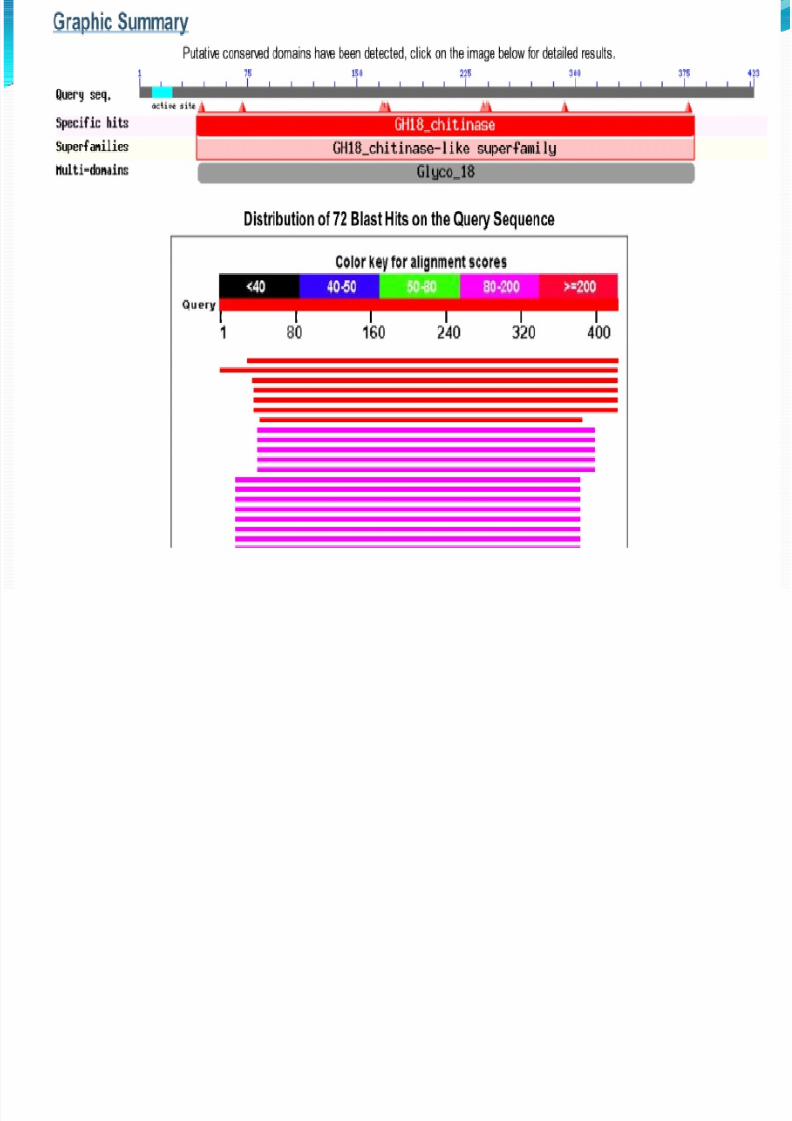
From sequence alignment (BLAST) we can find out whether or
not the target sequence has a crystal structure assigned in PDB.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 8/25
Homology Modeling (automated)can be done by SWISS MODEL,
PHYRE, GENO3D .
The acquired models are to be verified for validation.
This can be done either by VERIFY-3D or by Ramachandran plot
(wincoot).

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 9/25
Sequence of the query protein obtained from protein
database of NCBI( www.ncbi.nlm.nih.gov) .
This sequence is put into BLAST programme ofNCBI and the closest homologue is determined to be“crystal structure of a chitinase CrChi1 from thenematophagous fungus Clonostachys rosea”( pdb
code 3G6L).
Sequence alignment and
homology modelling

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 10/25
8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 11/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 12/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 13/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 14/25
BLAST indicates 2 things.
1.The target sequence doesn’t have a
known structure.2.For the homology modelling ,if to be
done manually , the protein with max
sequence identity(70%). After BLAST we need to do the homology
modelling.
As mentioned earlier the automatedhomology modeling was performed by usingthree programs: SWISS MODEL, PHYRE,and GENO3D.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 15/25
The validation for the structure models obtained fromvarious software tools (SWISS MODEL, PHYRE,GENO3D), was done by analyzing the psi-phiRamachandran plot generated by wincoot.
The compatibility of the predicted structures with thegiven sequence was also verified using VERIFY-3D.
The entire process of verification pointed towards thestructure predicted by swiss-model.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 16/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 17/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 18/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 19/25

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 20/25
Molecular docking is a widely-used
computational tool for the study of molecular
recognition, which aims to predict the bindingmode and binding affinity of a complex formedby two or more constituent molecules withknown structures.
•An important type of molecular docking isprotein-ligand docking because of its therapeuticapplications in modern structure-based drugdesign.
DOCKING

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 21/25
Inhibitor is a substance that inhibits the action of anenzyme.
By finding inhibitors to the given protein and making aprotein- inhibitor complex helps a lot in antibiotic drugpreparation.
To calculate the exact position where the inhibitorshould bind ,having minimum energy ineractions , we
use computational docking.
For the protein of interest , the suitable ligands arefound to be methyl xanthine derivatives (Caffeine,Threophylline) & allosamidin.

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 22/25
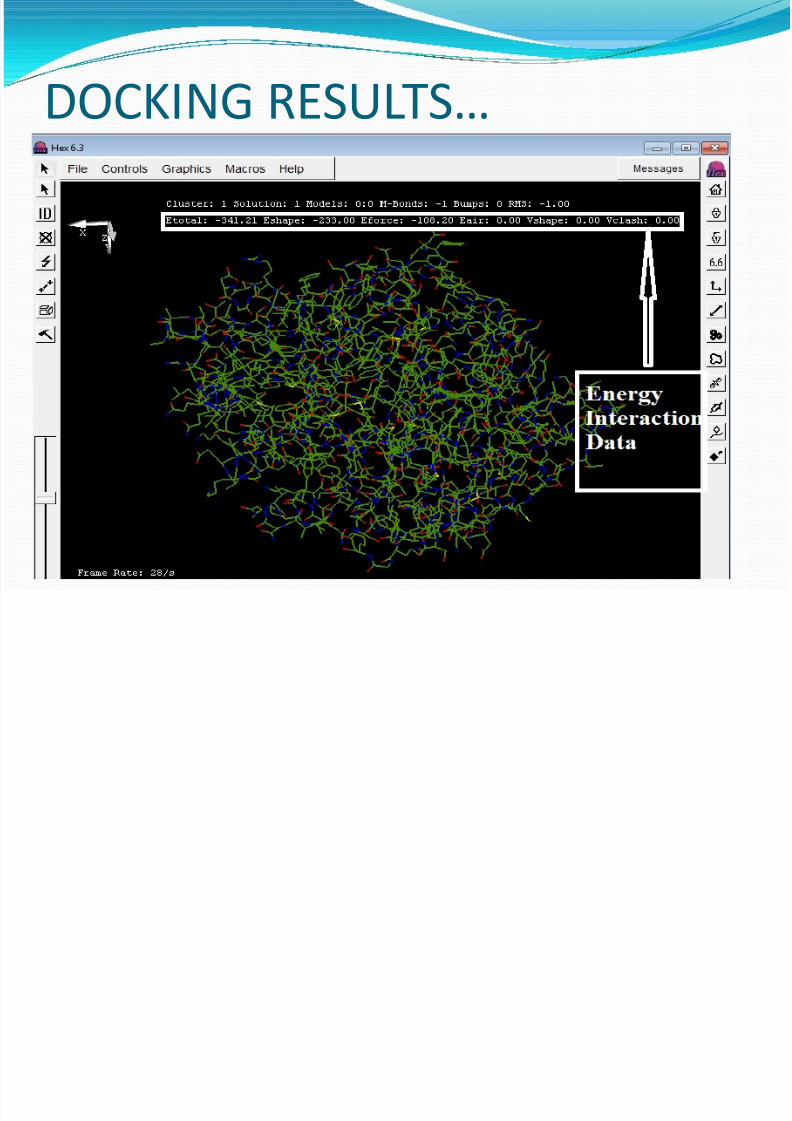
We docked three different ligands with theobtained 3D structure of the given proteinusing Hex.
This was done to find the energy interactionsbetween the ligands and the given protein.
The functions performed by Hex in order tofind the best possible protein-ligandinteraction are: SPF Transform, FFT streric scan,FFT Final Search , MM Refinement, TotalDockings.
8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 23/25
DOCKING RESULTS…

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 24/25
DOCKING RESULTS (contd…)
Energy Interactions of Each Complex

8/3/2019 tics Project (1)...
http://slidepdf.com/reader/full/tics-project-1 25/25
DOCKING CONCLUSIONS The comparision of the total energies of interaction shows
that the best inhibitor(ligand) for the given proteinmolecule is Allosamidin.
Allosamidin has the minimum amount of interactionenergy ,i.e. the interaction between the given protien andallosamidin is most feasible as compared to other ligandsdocked.
Hence ,out of the given 3 molecules, Allosamidin turns out
to be the most potent inhibitor. APPLICATION :This process is used to make Anti-fungal
drugs these days by inhibiting the chitinase function infungi.







![Lg Project Ppt[1]](https://static.fdocuments.nl/doc/165x107/577d23a81a28ab4e1e9a6a6b/lg-project-ppt1.jpg)




![Project 1 Adviesrapport Finisol[1]](https://static.fdocuments.nl/doc/165x107/55721171497959fc0b8efb4a/project-1-adviesrapport-finisol1.jpg)





