
onderzoek naar aard en omvang van integriteitsschendingen ...

Grondslagen van de chemie
Deel A: de wereld van atomen en moleculen
A1: de atoomstructuurDe eigenschappen van licht zijn de start voor het onderzoek naar de atoomstructuur. Licht is slechts een kleine fractie van het totale elektromagnetische spectrum. Deze straling is plant zich transversaal voor, dit betekent dat de uitwijking van de amplitude van het veld loodrecht gebeurt op de voortplantingsrichting van de golf. Het omgekeerde hiervan noemt men een longitudinale golf en hun uitwijking van de amplitude is volgens de voortplantingsrichting.Het wisselende elektromagnetische veld ondergaat cycli, het aantal cycli per tijdseenheid noemt men de frequentie van de golf en wordt uitgedrukt in Hertz. Het wordt ook gekenmerkt door de golflengte λ, dit is de lengt tussen 2 maxima of minima. Naast de golfeigenschappen heeft licht ook een deeltjeskarakter, het wordt dan een stroom van energiepakketjes genoemd; fotonen. Hiermee kan men het foto-elektrische effect verklaren: er kan namelijk alleen een elektron losgeslagen worden uit een metaal als de minimale energie om dit te onttrekken wordt toegediend. Hieruit hebben Compton en later ook Broglie 2 verbanden gevonden die deze 2 beschrijvingen van licht met elkaar in verband brengen, de resulterende toepassingen zijn de elektronenmicroscoop en elektronendiffractie. Met zichtbaar licht kunnen we enkel deeltjes onderscheiden met een minimale grootte ongeveer gelijk aan de golflengte van dit licht, dit noemt men de diffractie limiet. Wilt men dan kleinere deeltjes bekijken dan heeft men licht met een kleinere golflengte nodig.
Het onzekerheidsbeginsel van Heisenberg zegt dat we nooit met zekerheid tegelijkertijd de positie en de impuls van het waargenomen deelt kunnen bepalen. Hierdoor kan het atoommodel van Bohr dus niet opgaan omdat met dit model een exacte waarde voor de energieniveaus en de straal van het elektron rond de kern kan berekenen. Omdat het elektron golfeigenschappen heeft, kunnen we werken met de golfvergelijking van Schrödinger. Deze vergelijking gaat niet in tegen het onzekerheidsbeginsel omdat de oplossing ervan de waarschijnlijkheid aangeeft om een elektron op de bepaalde plaats x te vinden. De oplossing van de vergelijking kan men vinden door het invoeren van de kwantumgetallen:
1. Hoofdkwantumgetal n n= 1,2,3,4,5,…. Het geeft de hoofdschil aan waarin het elektron zich bevindt (K,L,M,N,O,P,Q,… schil. Het bepaalt de energie van het elektron: K<L<M<N<O<P<Q. Het bepaalt de uitgestrektheid van de elektronenwolk of orbitaal. Het bepaalt het aantal nodale vlakken*: n-1.
(*)= een nodaal vlak is een vlak waarin de waarschijnlijkheid om elektronen te vinden nul is2. Nevenkwantumgetal l
l<n het bepaalt de vorm van het orbitaal: l=0,1,2,3 komt overeen met s-, p-, d- en f-
orbitaal. Voor één-elektronsystemen geldt dat alle orbitalen dezelfde energie bezitten en dus
op hetzelfde energieniveau zitten. Men noemt dit “ontaarde” orbitalen. Voor meer-elektronsystemen is er een opheffing van de ontaarding: de s-, p-, d- en
f-orbitalen hebben niet meer hetzelfde energieniveau.

3. Magnetisch kwantumgetal ml -l< ml < +l Bepaalt de richting in de ruimte Alle orbitalen met hetzelfde magnetisch kwantumgetal hebben dezelfde energie,
buiten een magneetveld4. Spinkwantumgetal ms
Dit kwantumgetal volgt niet uit de oplossing van de Schrödingervergelijking. De 2 mogelijke
waarden, ± 12 , worden in de energiediagramma’s weergegeven door ↑ en ↓.
In een meer-elektronsysteem is er een repulsieterm, ontstaan uit de onderlinge afhankelijkheid van de elektronen, die er voor zorgt dat de Schrödingervergelijking niet meer exact oplosbaar is. Voor de beschrijving van de atoomstructuur mag er echter hetzelfde systeem worden gebruikt als in één-elektronsystemen, er moeten echter een aantal regels worden gevolgd:
1. De regel van Pauli: “Elk elektron moet een uniek set van 4 kwantumgetallen hebben”. Dit betekend dat er maar 2 elektronen in 1 orbitaal kunnen. Dit zijn gepaarde elektronen.
2. Het Aufbau-principe: doordat de ontaarding van de orbitalen wegvalt krijgen de orbitalen een bepaald energieniveau: s<p<d<f. hierbij komt nog eens dat als er een magneetveld is, de nevenschillen (vb. 2px, 2py en 2pz) ontaard zijn. Dit kan er toe leiden dat de volgorde van energie bepaalt door het hoofdkwantumgetal niet meer wordt gerespecteerd. Het niveau van een 4s-orbitaal is namelijk lager dan dat van een 3d-orbitaal. De elektronen worden dus ingevuld via het volgend schema:
3. De regel van Hund: de verschillende orbitalen moeten eerst worden ingevuld met elektronen die dezelfde spin bezitten. Dit komt omdat de elektronen elkaar afstoten en de laagste energietoestand wordt verkregen als de elektronen zover mogelijk apart zijn, vb. 1 in 2px, 1 elektron in 2py en 1 in 2pz.
Een zijn wel een paar uitzonderingen op deze regels, dit komt omdat volledig bezette of halfbezette schillen energetisch gunstiger zijn door hun symmetrische verdeling van elektronen rond de kern. Deze uitzonderingen komen alleen voor in de D- en f-blokken van het PSE.
A2: periodiciteit van atomaire eigenschappenDe atomen in de groepen en periodes van het PSE delen gemeenschappelijke eigenschappen.
1. De atoomstraal

De atoomstraal wordt voor metalen en atomen in vaste toestand omschreven als de helft van de afstand tussen de kernen. Voor niet-metalen is het de helft van de afstand tussen de kernen in een covalente binding.
De atoomstraal neemt geleidelijk af van links naar rechts in een periode De atoomstraal neemt geleidelijk toe van boven naar onder in een groep
2. De ionstraalBij het afgeven van een elektron zal het atoom de vorige edelgasconfiguratie aannemen, hierdoor is de kationstraal dus kleiner als de atoomstraal. Bij het opnemen van een elektron wordt de volgende edelgasconfiguratie aangenomen, de anionstraal is dus groter dan de atoomstraal.
Binnen een periode neemt de ionstraal af van links naar rechts, met een sprongsgewijze toename bij overgang van kation naar anion.
Binnen een groep is er een geleidelijke toename van de ionstraal van boven naar onder omdat het aantal schillen toeneemt.
3. De ionisatie-energie of ionisatiepotentiaalDit is de energie die moet worden toegevoegd om een elektron aan een neutraal atoom te onttrekken in de gasfase.
De ionisatiepotentiaal neemt geleidelijk toe van links naar rechts in een periode,De atoomstraal neemt af van links naar recht in een periode terwijl de effectieve lading toeneemt. De attractieve Coulomb-interactie wordt dus groter van links naar rechts in een periode, zodat de energie nodig om het elektron te onttrekken ook toeneemt
De ionisatiepotentiaal neemt geleidelijk af van boven naar onder in een groep, De atoomstraal stijgt ook van boven naar onder in een groep zodat het makkelijker wordt om het elektron te onttrekken; de attractieve Coulomb-interactie wordt steeds geringer.
4. De elektronenaffiniteitDit is de energie die vrijkomt wanneer een elektron wordt toegevoegd aan het atoom in de gasfase oftewel de energie vereist om een elektron te onttrekken aan een eenwaardig negatief ion in de gasfase.
De elektronenaffiniteit neemt geleidelijk toe van links naar rechts in een periode De elektronenaffiniteit neemt heel geleidelijk af van boven naar onder in een groep
De redenen hiervoor zijn dezelfde als bij de ionisatiepotentiaal.5. De elektronegativiteit
Dit geeft de relatieve neiging weer van een ion om elektronen aan te trekken. Een goede basis voor deze waarde is ionisatiepotentiaal : hoe groter de IP hoe moeilijker een elektron kan worden onttrokken. Men kan ook met de elektronenaffiniteit werken: hoe groter deze is, hoe groter de neiging om een elektron naar zich toe te trekken.
De elektronegativiteit neemt toe van links naar rechts in een periode De elektronegavititeit neemt af van boven naar onder in een groep
Al deze eigenschappen bepalen de fysische en chemische kwaliteiten van de metalen en niet-metalen. Deze zijn hier weergeven:


A3: De chemische bindingEen atoom zal de octetstructuur willen bereiken, het kan dit doen op 2 manieren: de ionbinding en de covalente binding.Bij de ionbinding zullen beide atomen de octetstructuur bereiken door de uitwisseling van valentie-elektronen. Één atoom zal elektronen afstaan en de andere zal deze opnemen. Deze binding kan alleen plaatsvinden tussen atomen met een groot verschil in ∆EN of maw tussen een metaal en een niet-metaal. Het metaal zal door het afstaan van elektronen de vorige edelgasconfiguratie aannemen terwijl het niet-metaal de volgende edelgasconfiguratie aanneemt. Naast de edelgasconfiguraties zijn er voor de ionen ook nog andere stabiele configuraties:
Anionen: Anionen zullen altijd de volgende edelgasconfiguratie aannemen door het opnemen
van elektronen Kationen:
Kationen kunnen een verschillend aantal elektronen hebben op de buitenste schil en toch stabiel zijn.
0 of 2 elektronen: de configuratie van H+ of He 8 elektronen: de configuratie van de edelgassen 18 elektronen: dit noemt de pseudo-edelgasconfiguratie, ns²np6nd10 met n= 3,4 of 5 (18+2) elektronen: het inerte-elektronenpaar-effect, ns²np6nd10(n+1)s² met n=3,4,5 13 elektronen: ns²np6nd5 met n=3,4,5
Kwalitatief kan de ionbinding verklaard worden als de uitwisseling van elektronen waardoor de betrokken atomen een stabielere elektronenstructuur verkrijgen. Deze verlaging in energie kan men kwantitatief berekenen met een formule:
∆H 3D= −14 πε
∙ e2
R 0∙Navo ∙ Madelungc te
Met ε= 8,854*10-12 F/mE= 1,602*10-19 CR0= som van de 2 ionstralenNavo= 6,022*1023 1/molMadelungsconstante: specifiek voor elke kristalstructuur.
De sterkte van een ionbinding wordt vooral bepaald door de lading van de stabiele ionen.Ook is er een verband tussen de ionenstralen r+/r- , het coördinatiegetal en de coördinatiestructuur voor kristallen en ionbindingen:
De eigenschappen van de ionbinding zijn een gevolg van de Coulombkrachten tussen de kationen en de anionen. Dit zijn de eigenschappen:
1. Hoog smeltpunt en dus vast bij kamertemperatuur2. Hoog kookpunt3. Geringe vluchtigheid4. Slechte elektrische geleider bij kristallijne en amorfe toestand maar goede geleider indien
opgelost5. Goed oplosbaar in H2O

Als de atomen niet instaat zijn om elektronen uit te wisselen (∆EN is niet groot genoeg), kunnen ze de octetstructuur toch bekomen door elektronen in gemeenschap te stellen. Dit noemt men de covalente binding. Voor de beschrijving van de covalente binding heeft men de Lewisstructuur nodig en de regels ervan kennen:
1. Het skelet van het molecule opstellen2. Alle atomen verbinden met een bindend elektronenpaar3. Alle atomen de octetstructuur geven4. Voor elk atoom het aantal elektronen op de buitenste schil beschouwen5. Voor elke atoom het aantal elektronen tellen (het bindend elektronenpaar wordt gesplitst)6. Indien de elektronen in punt 5 hoger zijn dan in punt 4 worden aan het betreffende atoom
dubbele bindingen gegeven.De nadelen van de Lewisstructuur zijn:
Men moet het skelet van het molecule kennen Radicalen hebben een ongepaard elektron Er is geen info over de ruimtelijke structuur Er is geen info over de relatieve reactiviteit van de binding (enkelvoudig, dubbel of
drievoudig Bij geconjugeerde systemen moet men het begrip resonantie gebruiken, dit betekent dat een
molecule verschillende Lewisstructuren heeft en de realiteit een samensmelting is van deze structuren
De valentiebindingstheorie kan tot een totaal verkeerde elektronenconfiguratie leidenMaar vanuit de Lewisstructuur kan men wel de bindingspartners en het aantal vrije elektronen vinden en zo een structuur verkrijgen die voldoet aan de regels maar niet noodzakelijk overeen komt met de realiteit.
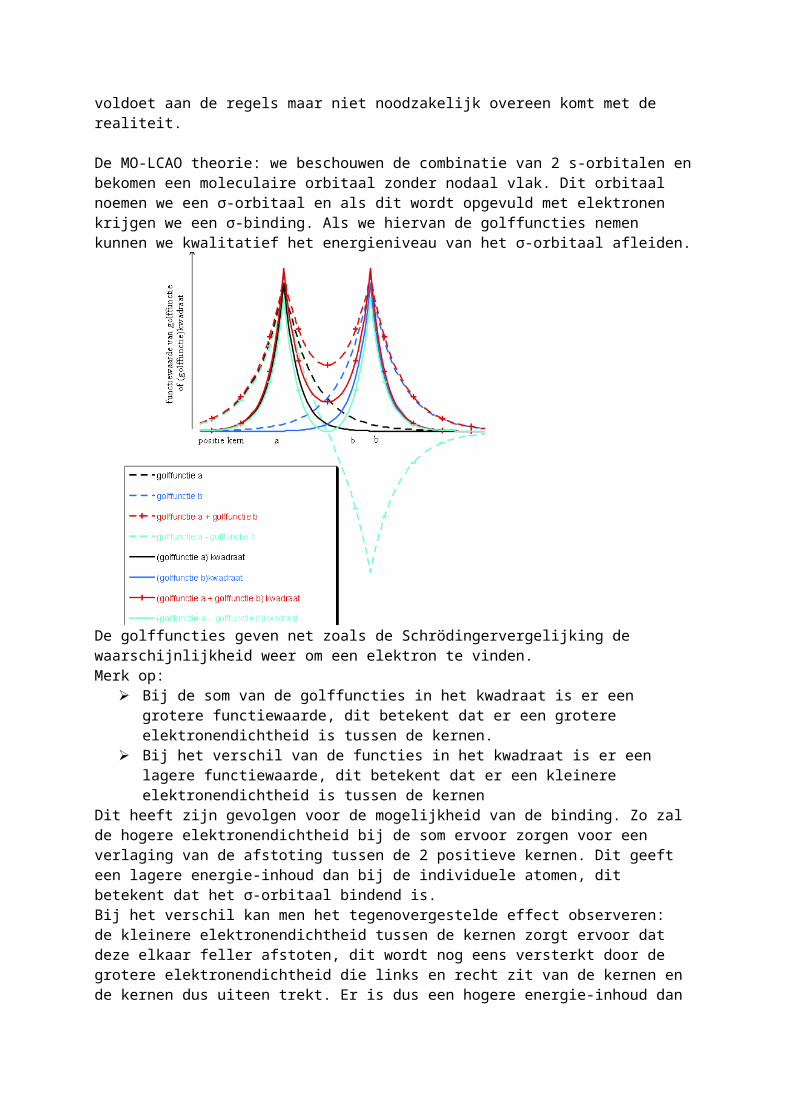
De MO-LCAO theorie: we beschouwen de combinatie van 2 s-orbitalen en bekomen een moleculaire orbitaal zonder nodaal vlak. Dit orbitaal noemen we een σ-orbitaal en als dit wordt opgevuld met elektronen krijgen we een σ-binding. Als we hiervan de golffuncties nemen kunnen we kwalitatief het energieniveau van het σ-orbitaal afleiden.
De golffuncties geven net zoals de Schrödingervergelijking de waarschijnlijkheid weer om een elektron te vinden.

Merk op: Bij de som van de golffuncties in het kwadraat is er een grotere functiewaarde, dit betekent
dat er een grotere elektronendichtheid is tussen de kernen. Bij het verschil van de functies in het kwadraat is er een lagere functiewaarde, dit betekent
dat er een kleinere elektronendichtheid is tussen de kernenDit heeft zijn gevolgen voor de mogelijkheid van de binding. Zo zal de hogere elektronendichtheid bij de som ervoor zorgen voor een verlaging van de afstoting tussen de 2 positieve kernen. Dit geeft een lagere energie-inhoud dan bij de individuele atomen, dit betekent dat het σ-orbitaal bindend is.Bij het verschil kan men het tegenovergestelde effect observeren: de kleinere elektronendichtheid tussen de kernen zorgt ervoor dat deze elkaar feller afstoten, dit wordt nog eens versterkt door de grotere elektronendichtheid die links en recht zit van de kernen en de kernen dus uiteen trekt. Er is dus een hogere energie-inhoud dan bij de individuele atomen en men krijgt een antibindend orbitaal wat men voorstelt met σ*. Dit anti-bindend orbitaal heeft wel een nodaal vlak.We hebben nu 2 moleculaire orbitalen waarin we net zoals bij de atomaire orbitalen elektronen kunnen instoppen. Deze theorie is normaal alleen voor één-elektron systemen maar voor meer-elektron systemen volgen we dezelfde regels als bij atomaire orbitalen. Als we dit nu beschouwen voor een één-elektron systeem namelijk de binding van 2 H2
+ ionen, zien we het volgende:
Als we 1 elektron in het orbitaal zetten zal H2+ verlagen in energie van 1s naar σ1s, het ion
wordt dus gevormd. Bij 2 elektronen is er een dubbele verlaging in energie en de vorming van het molecule vindt
dus zeker plaats. Bij 3 elektronen zal er echter 1 elektron in het antibindende orbitaal terechtkomen, dit heeft
als gevolg dat de gemaakte energiewinst voor de helft verloren gaat. De binding gaat echter nog door aangezien er netto nog winst is.
Bij 4 elektronen zal er geen binding plaatsvinden omdat er 2 elektronen in het antibindend orbitaal terechtkomen. Hierdoor gaat de energiewinst volledig verloren en is er geen fysische reden voor de binding aan te gaan.
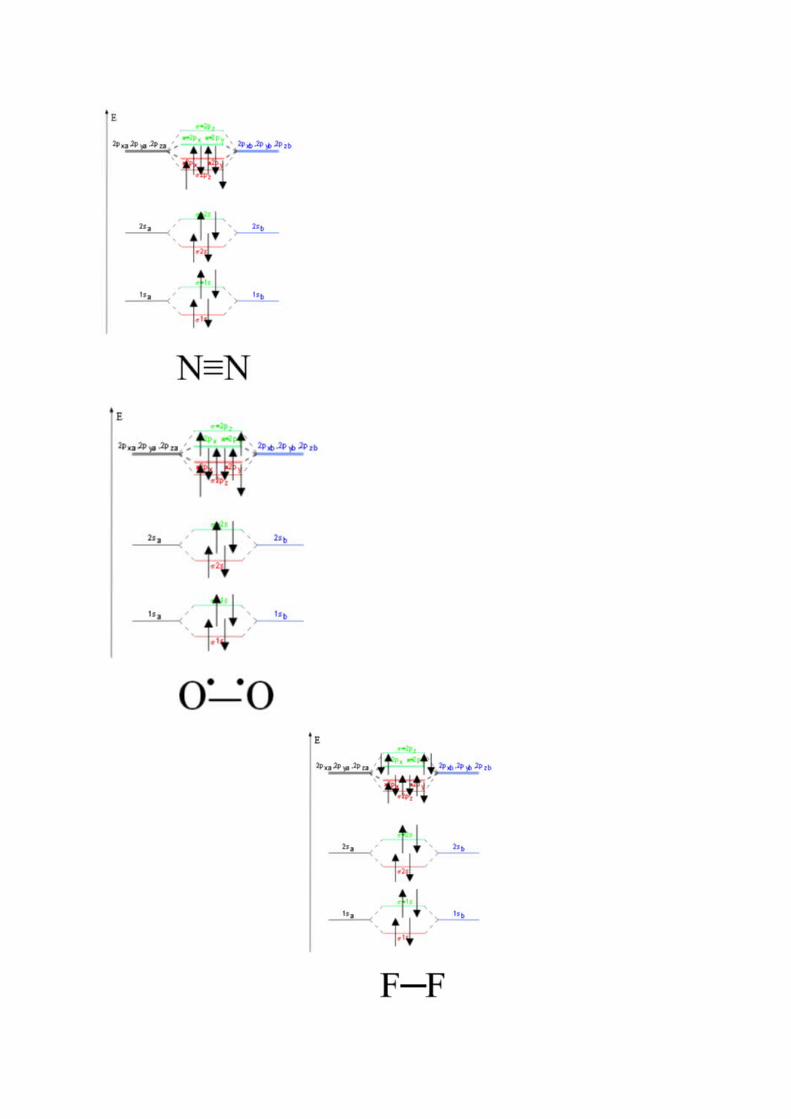
Naarmate de orbitalen stijgen zijn er meer opties, het 2s orbitaal heeft hetzelfde schema als een 1s maar een p-orbitaal heeft meer mogelijkheden. Er zijn namelijk 3 p-orbitalen per atoom, 1 orbitaal anders reageren namelijk volgens de moleculaire as. Deze orbitalen hebben geen nodaal vlak. De 2 andere orbitalen hebben een andere richting van de as, dit heeft als gevolg dat er nog wel interactie gebeurt tussen de atomen maar deze is geringer. De extra bindingsterkte is dus niet dezelfde kracht als een σ-orbitaal maar is lager en men noemt dit een π-orbitaal. De 3 p-orbitalen (px, py, pz) hebben allemaal een bindend en antibinden orbitaal dat kan worden opgevuld.

Nu blijkt dat sommige atomen in staat zijn hun orbitalen samen te voegen: men noemt dit hybridisatie. Een aantal voorbeelden:
1) Methaan: Centraal koolstofatoom heeft 1s²2s²2px
12py1 als elektronenstructuur. Dit betekent
dat het C-atoom maar 2 bindingen aankan. Koolstof kan 4 bindingen aangaan doordat de 2s en 2p orbitalen hybridiseren tot 4
sp3-orbitalen. 2) Stikstof
De elektronenstructuur van stikstof levert wel 3 bindingen op maar niet de juiste waargenomen geometrische vorm (trigonale piramide). Deze piramide is niet zoals bij koolstof een tetraëder omdat het niet-bindende orbitaal meer plaats inneemt als de bindende orbitalen.
Dit gebeurt ook bij zuurstof, alleen heeft dit atoom 2 niet-bindende orbitalen waardoor de hoek in de ruimte nog groter wordt
3) Boortrifluoride Volgens de elektronenstructuur kan boor maar 1 binding aangaan, experimenteel
werd er echter de mogelijkheid tot 3 bindingen ondervonden. Boor heeft maar 3 elektronen op de laatste schil en dit leidt dus tot de hybridisatie
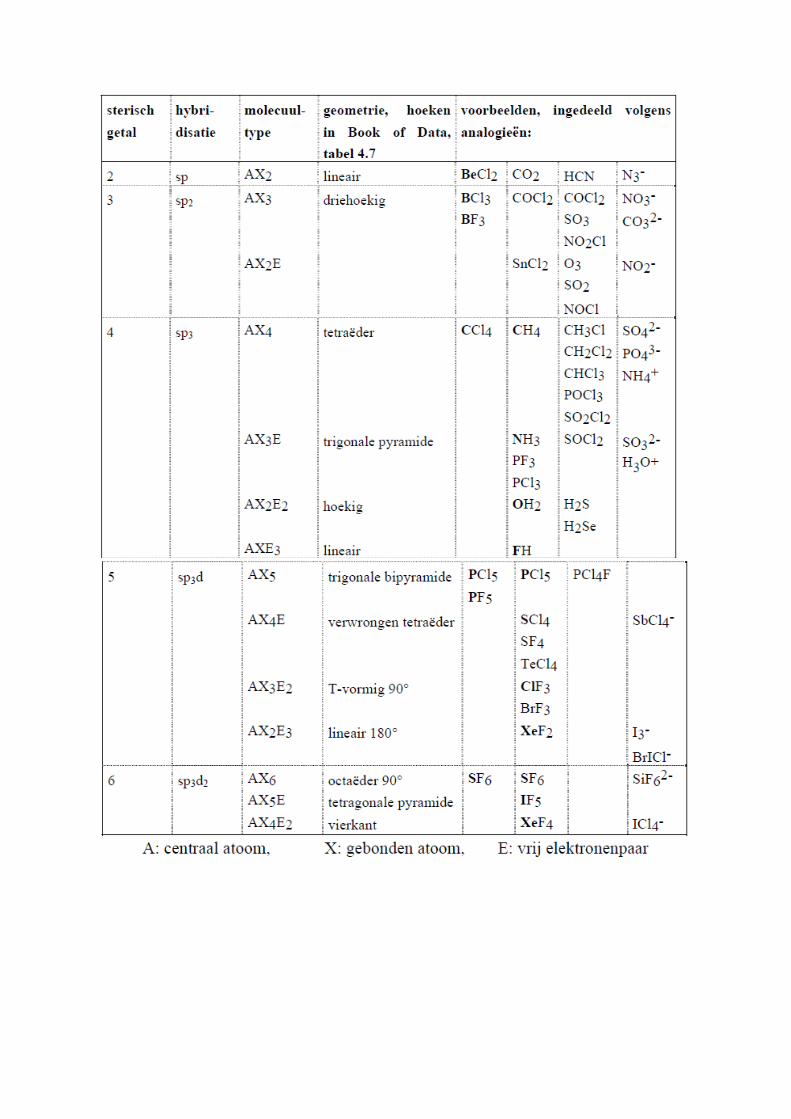
van 3 sp2-orbitalen.Belangrijk om te weten is dat er een verband is tussen het sterisch getal(= het aantal andere atomen dat rechtstreeks op het centrale atoom gebonden staat plus het aantal vrije elektronenparen op dit centrale atoom) , de hybridisatie en geometrie ervan:

De sterkte van de covalente binding worden bepaald door ∆EN van de atomen in de binding (1) en de aard van binding en van de atomen (2).
(1) Het verschil in de elektronegativiteit van de atomen geeft de zogenaamde polariteit van de binding. Bij een verschil zal er een dipoolmoment ontstaan voor de binding. Bij de di-atomaire homo-atomaire moleculen is er geen verschil in EN lading. Deze bindingen zullen een apolaire verbinding vertonen terwijl de di-atomaire hetero-atomaire moleculen een polaire binding geven. Om bij moleculen met meerdere partners de polariteit van de binding te weten, stelt men de dipoolmomenten voor met vectoren en telt men deze op. Als de som van de vectoren elkaar opheffen dan is het molecule apolair.
(2) Hiermee wordt bedoeld of de binding enkel, dubbel, σ of π is. Ook de grootte van de atomen is van belang, zo zal een groter atoom resulteren in een zwakkere covalente binding. Dit komt omdat de kern verder van gemeenschappelijke atomen ligt en deze er dus minder mee zijn verbonden. De aard van binding helpt ook mee, zo heeft een driedubbele binding meer energie nodig om doorbroken te worden maar door de 2 π-orbitalen zal deze reactiever en dus onstabieler zijn dan een enkelvoudige binding.
Atomen die een covalente binding aangaan, zullen niet zoals bij de ionbinding verbonden zijn met de andere atomen in het rooster. Ze worden alleen bij elkaar gehouden door de zwakke cohesiekracht. Dit heeft zijn gevolgen voor de kenmerken van de covalente binding:
1. Een minder harde rooster2. Lagere kook/ smelt temp dus vluchtig3. Goede oplosbaar in apolaire solventen4. Slechte geleider
In de chemie worden er verschillende formules gebruikt om stoffen te benoemen. Dit leid tot isomerie, verschillende chemische stoffen met verschillende structuurformule hebben dezelfde brutoformule.
1. Structurele isomerie Functie-isomerie: Wanneer voor eenzelfde brutoformule 2 verschillende chemische
functies kunnen geschreven worden. Bv: C2H6O, dit kan zowel een alcohol zijn (ethanol) als een ether (dimethylehter).
Ketenisomeren: Wanneer er voor eenzelfde brutoformule verschillende ketenstructuren kunnen worden geschreven. Bv: pentaan heeft 3 vormen: n-pentaan, isopentaan en neopentaan
Plaatsisomeren: Wanneer er een andere substituent dan H op koolstof voorkomt, kan men deze op verschillende plaatsen in de keten zetten. Dit kan ook mogelijk zijn met dubbele bindingen. Bv: 1-penteen en 2-penteen.
2. Ruimetelijke isomerie of stereo-isomerie Geometrische of cis-tran-isomerie:Door de dubbele binding is er ook een andere
soort isomerie mogelijk. De dubbele zorgt er namelijk voor dat er geen vrije rotatie mogelijk is tussen de 2 koolstofatomen. Wanneer 2 groepen aan dezelfde kant van de dubbele binding staan noemt men dit het cis-isomeer. Het tegenovergestelde noemt men het trans-isomeer. Deze 2 isomeren hebben elk hun eigen chemische kenmerken
Optische isomerie: deze structuren verschillen in de ruimte van elkaar maar ze moeten ook elkaars spiegelbeeld zijn. Deze isomeren hebben een specifieke fysische eigenschap: ze zorgen voor een draaiing van het polarisatievlak van gepolariseerd licht wanneer dit licht in contact komt met een oplossing of vloeistof van het molecule. De optische isomerie komt doordat er op een koolstofatoom 4 verschillende substituenten zitten. Dit koolstofatoom noemt men chiraal en zorgt voor 2 niet-indentieke mogelijkheden, de enantiomeren. Een mengsel van 2 enantiomeren noemt men een racemisch mengsel.

Deel B: gassen, vloeistoffen, vaste stoffen en mengsels
B1: inter-moleculaire krachtenEr zijn ook krachten die werken tussen de moleculen zelf ipv tussen de atomen van een molecule, men noemt dit de intermoleculaire krachten. De aard en de grootte van deze krachten word bepaald door de aard van het molecule. Er zijn 4 verschillende intermoleculaire krachten:
1. Londonkrachten of dispersiekrachtenDit zijn de kleinste inter-moleculaire krachten. Niet elk atoom heeft een permanent dipoolmoment maar elk atoom heeft door fluctuaties in de elektronenverdeling een fluctuerend dipoolmoment. Dit zorgt dan voor een kleine positieve of negatieve lading die dan fluctuerende dipoolmomenten van andere atomen gaan aantrekken.
2. Dipool-dipool interacties of KeesomkrachtenDeze inter-moleculaire kracht komt alleen voor bij polaire moleculen omdat deze een permanente dipool hebben. De waarde van deze kracht is groter en zorgt ook voor aantrekkingskracht tussen de atomen. Hier komt nog eens bij dat deze kracht moet worden opgeteld bij de Londonkrachten die er ook aanwezig zijn.
3. DebijekrachtenEen molecule met een permanent dipoolmoment kan in een andere molecule ook een dipool induceren, deze tijdelijk dipool kan dan reageren met het permanente dipool.
4. De waterstofbrugDe waterstofbrug is specifiek en komt alleen voor tussen een H-atoom gebonden aan een elektronegatief atoom en een elektronegatief atoom met een vrij elektronenpaar. Het zal naast de binding met het elektronegatief zal het H-atoom een elektrostatische brug maken met het atoom dat een vrij elektronenpaar heeft. De bruggen hebben een grootte invloed op de fysische eigenschappen van een stof.
B2: gassen en mengselsDe gassen en gasmengsels nemen de vorm en volume aan van hun behouder. De gassen komen ,op uitzondering van H, allemaal rechts voor in het PSE. Ze worden gekenmerkt door een apolaire binding. Het zijn niet-metalen en hebben dus een kleine ∆EN, dit heeft als gevolg dat alleen de zwakke Londonkracht inwerkt op de moleculen. Deze intermoleculaire kracht is te zwak om de gassen te laten condenseren.In de chemie is de ideale gaswet van groot belang, om deze te kunnen toepassen moet men eerste een ideaal gas kunnen beschrijven:
1. Het gas bestaat uit een groot aantal deeltjes2. De gasdeeltjes zijn puntmassa’s zonder eigen volume3. De gasdeeltjes bewegen snel, rechtlijnig en botsen met elkaar en met de wanden van de
behouder.4. De gasdeeltjes vertonen elastische botsingen, er is geen verlies van kinetische energie5. De energie van temperatuursverhoging wordt omgezet in kinetische energie
Als de druk laag genoeg is en de temperatuur hoog dan voldoen reële gassen aan dit model. In de chemie is de druk van belang, er wordt standaard gewerkt met 1 atmosfeer of 1013,25 hPa.De ideale gaswet is een gevolg van de fysische en chemische gaswetten die doorheen de tijd zijn ontdekt:
1. De fysische gaswetten De wet van Boyle en Mariotte: P1V1= P2V2 bij een constante temperatuur (=isotherm) De wet van Charles: V1/T1= V2/T2 bij een constante druk (isobaar) Wet van Guy-lussac: P1/T1= P2/T2 bij een constant volume (isochoor)


2. De chemische gaswetten De wet van Guy-lussac:
“Tussen de volumes van met elkaar reagerende gassen of de gevormde gasvormige producten bestaan eenvoudige volumeverhoudingen.”
De wet van Dalton en Guy-lussac:“Bij eenzelfde temperatuur en druk bevat eenzelfde volume gas eenzelfde aantal atomen.” Deze interpretatie is echter in strijd met de atoomtheorie van Dalton zelf, want dit zou betekenen dat 2 waterstofatomen reageren met 1 zuurstofatoom om 2 moleculen water te vormen. Een molecule water zou dan een halve atoom zuurstof hebben.
De wet van Avogadro en Ampère:“Bij eenzelfde temperatuur en druk bevat eenzelfde volume gas eenzelfde aantal moleculen.” Deze kleine verandering lost het conflict van Dalton op terwijl aan zijn atoomtheorie niets moest veranderd worden.
Wet van Dalton:“de totale gasdruk is de som van de partiële drukken van alle componenten van het gasmengsel.”
3. De gevolgenUit de combinatie van de verbanden tussen druk, temperatuur, volume en aantal mol van
een gas is de ideale gaswet geboren: PV=nRT met de gasconstante R= 8,315 J
K ∙mol of
0,08206L ∙atmK ∙mol . Uit deze gaswet valt te berekenen hoeveel volume 1mol gas inneemt (22,4
L bij 0°C) en hoeveel mol er gevormd moet worden om een bepaald volume op te vullen.We moeten echter rekening mee houden dat de druk en temperatuur niet op het juiste niveau zijn om de gassen als ideaal te kunnen benoemen:
1. De gasmoleculen zijn geen puntmassa’s maar nemen ook een volume in2. De gasmoleculen oefenen cohesiekrachten op elkaar uit.3. Het volume van het gas moet verminderd worden met het co-volume van de gasmoleculen
zelf4. De druk moet vermeerderd worden met de cohesiedruk die de onderlinge
aantrekkingskracht tussen de gasmoleculen in rekening brengtDit resulteerde in de van der Waalsvergelijking waarbij deze 2 factoren zijn bij ingebracht.
(P+a n2
V 2 ) ∙ (V−nb )=nRT
Dit is met b het co-volume van de gasmoleculen en a n2
V 2 de cohesiedruk.
VANAF HIER ALLEEN LEZEN, NIET KENNEN!!!B3: vloeistoffenVloeistoffen hebben een constant volume en net zoals gassen nemen ze de vorm aan van hun behouder. In de vloeistof zijn de inter-moleculaire krachten groter als bij gassen, dit heeft als gevolg dat de individuele moleculen in contact met elkaar staan. De vloeistoffen worden gekenmerkt door 2 specifieke eigenschappen:
1. Viscositeit:“de weerstand die een vloeistof ondervindt tegen het vloeien, deze wordt veroorzaakt door de inter-moleculaire krachten.”
2. Oppervlaktespanning en capillariteit:

Deze 2 kenmerken worden ook veroorzaakt door de inter-moleculaire krachten in combinatie met de aanwezigheid van andere stoffen. Doordat vloeistoffen een constant volume hebben is er boven dit volume lucht aanwezig, de inter-moleculaire krachten tussen de moleculen in de vloeistof is groter als die met de lucht. Hierdoor is de netto kracht gericht naar het binnenste van de vloeistof (=oppervlaktespanning). Capillariteit ligt aan de behouder: als de adhesiekracht groter is als de cohesiekracht dan heeft men een holle meniscus en andersom verkrijgt men een bolle meniscus.
B4: vaste stoffenVaste stoffen hebben een constante vorm en volume en zijn onafhankelijk van hun behouder. Ze worden ingedeeld volgens de oorzaak van hun vast fase of in amorf/ kristallijn.
1. MetalenDeze stoffen zijn gebonden via de metaalbinding en de elektronen kunnen zich vrij bewegen in het rooster. Zijn vervormbaar, niet oplosbaar in water, goede geleiders.
2. Ionische vaste stoffenDeze stoffen ontstaan door de sterke aantrekking van anion en kation in het ionenrooster. Zie eerder
3. Netwerk vaste stoffenDe atomen zijn covalent gebonden over het ganse netwerk van de stof en dus niet alleen met de atomen binnen het moleculen. Deze stoffen zijn hard, onvervormbaar, broos en onoplosbaar in water. Van een atoom kan meer als 1 vorm voorkomen, bv koolstof; de configuratie ervan in grafiet en diamant is verschillend wat ook verschillende eigenschappen geeft.
4. Moleculaire vaste stoffenDeze stoffen bestaan uit individuele atomen bij elkaar gehouden door de sterke cohesiekrachten. Ze hebben een laag tk en ts, voorbeeld ijs.
Kristallijne stoffen zijn stoffen met een uiterst regelmatige kristalstructuur. Een amorfe stof is het tegenovergestelde hiervan en heeft dus geen regelmatige rooster.
B5: mengsels Onderscheid mengsels en verbinding
Mengsels kunnen dmv fysische eigenschappen gescheiden worden; hun samenstelling kan variëren; hun eigenschappen zijn bepaald door de componenten
Verbindingen kunnen niet meer dmv fysische eigenschappen opgedeeld worden maar enkel door scheidkundige technieken; hun samenstelling ligt vast; hun eigenschappen zijn verschillend van deze van hun bestanddelen.
Onderscheid heterogeen en homogeen In heterogene mengsels zijn niet alle eigenschappen overal identiek, er zijn
grensvlakken of interfases tussen de domeinen met verschillende eigenschappen:

In homogene mengsels zijn alle eigenschappen overal identiek; de verdeling van de bestanddelen is doorgevoerd tot op moleculaire schaal; de individuele bestanddelen kunnen niet meer onderscheiden worden.
Onderscheid fase en component Een fase is een homogeen deel van een systeem met dezelfde samenstelling en
dezelfde eigenschappen binnen dat deel en met bepaalde fysische grenzen Een component is een zuivere stof waaruit een fase is samengesteld.
Voorbeeld. ijs en water in gesloten systeem: 1 component; het water en 2 fasen; vast en vloeibaar.

Deel C: chemische kinematica
C1: concentratie en snelheidReacties worden bepaald door de reactiesnelheid, waar 2 definities voor bestaan:
1) De snelheid van verbruik van een reagens wordt gegeven door de vermindering van het aantal moleculen reagens (Nreagens) per eenheid van tijd en bij een gegeven temperatuur.
2) De snelheid van vorming van een product wordt gegeven door de toename van het aantal moleculen product per eenheid van tijd en bij een gegeven temperatuur.
Om de snelheid van een reactie weer te geven gebruikt men stochiometrische coëfficiënten die de relatieve vorming of gebruik van het reagens of product weer geven. De gemiddelde snelheid wordt
dan: ¿ v>¿− 1Nr
∙ ∆ reagens∆t
= 1Np
∙ ∆ product∆ t .
Om de ogenblikkelijke snelheid te weten gebruikt men de limiet naar t=0 en krijgt men dus de helling van de raaklijn. Experimenteel stelde men vast dat de snelheid evenredig is met de concentratie zelf:v=k ∙ [ A ]n [B ]m met A en B de reagentia. Om de snelheidconstante en de macht voor een reagens te bepalen, houdt men alle parameters buiten de concentratie van het reagens constant en meet men voor verschillende concentraties de snelheid. Dit noemt men initiële kinetica. Een andere techniek, de lopende kinetica, meet men de snelheid van de reactie terwijl deze loopt. Dit is een pseudo-techniek omdat men de parameters niet helemaal constant kan houden.Voor n= 0,1,2 spreekt men van nulde-orde, eerste-orde en tweede-orde reactie. De bijbehorende snelheidsconstante wordt vaak aangeduid met k0, k1, k2. De som van de exponenten van de reagentia wordt de totale orde genoemd. De som van de coëfficiënten noemt men de moleculariteit van de reactie. Als er tussen deze 2 een verschil is, is het mechanisme van de reactie anders als de reactievergelijking weergeeft.
C2: de botsingstheorie en factoren die de reactiesnelheid beïnvloeden Experimenteel is er vastgesteld dat :
1. Er meer gasmoleculen zijn met een grotere snelheid bij hogere temperatuur.2. Bij hogere temperatuur zijn er meer gasmoleculen met een grotere kinetische energie.
Hieruit heeft men de botsingstheorie gehaald die geld voor gassen en solventen als men de kinetische energie van de moleculen in het solvent meetelt,
Een chemische reactie kan slechts gebeuren als Er een botsing gebeurt tussen de reagensmoleculen. Deze reagensmoleculen de minimale energie hebben bereikt. De botsing van de reagensmoleculen de juiste orientatie heeft.
Deze theorie geeft ook de factoren die de reactiesnelheid bepalen en deze kunnen we dan beïnvloeden:
i. Via het aantal botsingen tussen de reagensmoleculen De concentratie van het reagens: hoe groter de concentratie, hoe groter de kans op
de botsingen. De totale druk of de partieeldruk van de een reagens (enkel gasfase): als de druk
stijgt, daalt het volume en stijgt de concentratie De verdelingstoestand van het reagens (enkel heterogene reacties): botsingen zijn
enkel mogelijk aan de interfase( gas-vloeistof, gas-vast, vloeistof-vast). Als dit de beperkende factor is heeft de reactie een nulde-orde.
ii. Via de minimale energie van de botsingA. Voor de vorming van het product moeten de bindingen tussen de reagens moleculen
verbroken worden. hiervoor is de activeringsenergie nodig. De toestand waarbij sommige bindingen van de reagensmoleculen gedeeltelijk zijn verbroken noemt men

de transitietoestand en heeft een grote energie-inhoud. Deze energie wordt bekomen uit de kinetische energie van de moleculen. Dit betekent dat alleen moleculen met een groteren kinetische als activeringsenergie deze toestand kunnen bereiken. Een katalysator kan de activeringsenergie verlagen en zo de reactiesnelheid verhogen.
iii. Via de onderlinge oriëntatie van de reagensmoleculenB. Niet alle botsingen van het reagens produceert productmoleculen. Soms moeten de
reagensmoleculen onder een bepaalde hoek botsen. De fractie moleculen met de juiste oriëntatie kan men berekenen uit de Arrheniusvergelijking, de fractie wordt voorgesteld met de factor A.
k=Ae xp−EactRT met k de snelheidsconstant
De werking van een katalysator berust op het verlagen van de activeringsenergie. Van de katalysator heeft men 2 soorten :
C. De homogene katalyseHierbij bevinden de reagentia en de katalysator zich in dezelfde fase, een voorbeeld hiervan is het loden-kamerprocéde. Dit procedé toont aan hoe verbranding van zwavelhoudende stoffen leid tot de vorming van gasvormig waterstofsulfaat. Dit is ook de oorzaak van zure regen.
D. De heterogene katalyseHierbij bevinden de reagentia en de katalysator zich in verschillende fase. Dit is vaak gas en een vaste stof met een groot specifiek oppervlak.
C3: reactiemechanismesEen globaal reactiemechanisme is een opeenvolging van 3 elementaire reactiestappen:
1. De monomoleculaire reactie met een eerste-orde kineticaBv.: O3 → O2 + O
2. De bimoleculaire reactie met een tweede-orde kineticaBv.: O3 + O → 2O2
3. De termoleculaire reactie met een derde-orde kineticaBv.: O2+ O2+ O2 → 2O3
Deze stap komt niet vaak voor omdat de kans op een gelijktijdige botsing van 3 moleculen gering.
De reactiesnelheid in een globaal mechanisme wordt bepaald door de traagste elementaire stap, deze noemt de snelheidsbepalende stap en heeft de hoogste activeringsenergie.Volgens het stationariteitsprincipe zullen de elementaire stappen voor de snelheidsbepalende stap in evenwicht zijn maw de vormingssnelheid is gelijk aan de snelheid van de terugreactie plus de 2de reactie:

Deel D: chemische thermodynamica
D1: het dynamische evenwichtAls een reactie zijn chemische evenwicht heeft bereikt lijken de concentraties van de reagentia macroscopisch niet meer te veranderen. Op microscopische niveau zal zowel de voorwaartse als de terugwaartse reactie met even grote snelheid opgaan. Dit noemt men het dynamische evenwicht.
D2: de evenwichtsconstanteDe algemene uitdrukking voor de evenwichtsconstante van een algemene reactie is
K ev ,C=[C ]ev
c ∙ [ D ]evd
[ A ]eva ∙ [B ]ev
b met a,b, c en d de stochiometrische coëfficiënten. Dit noemt men de wet van
Guldberg en Waage en kan voor gassen ook uitgedrukt worden in hun partiële druk. Constante concentraties zoals van zuivere vloeistoffen worden in de evenwichtsconstante zelf verrekend zodat die niet in de uitdrukking van de evenwichtsconstante zelf voorkomt, dit geldt ook voor zuivere vaste
stoffen. Het verband tussen de KC en de Kp wordt afgeleid van de ideale gaswet: P= nV∙ R ∙T=cRT
of algemeen K p=KCRT ∆ngas. ∆ngas wordt gedefinieerd als het einde- het begin van de som van de stochiometrische coëfficiënten van de gasvormige reagentia of producten. De gasconstante wordt uigedrukt in liter.atm/mol.K. De waarde van de evenwichtsconstante geeft aan hoeveel het evenwicht naar links of naar rechts ligt en of de reactie dus weinig reagerend is of aflopend. Een evenwichtsconstante rond 1 duidt erop dat er bij evenwicht evenveel reagentia is als product.
Naast de evenwichtsconstante heeft men ook het reactiequotiënt. Deze verschillen doordat het
reactiequotiënt altijd geldig tijdens de reactie en niet alleen tijdens het evenwicht: Q=[C ]c ∙ [ D ]d
[A ]a ∙ [B ]b.
Een reactie kan normaal niet voorbij zijn evenwicht reageren maar men kan dit wel theoretisch beschouwen. Dit is dan een omgekeerde reactie waarbij de reagentia de producten worden en de producten de reagentia; teller wordt noemer en noemer teller; en Q wordt terug kleiner als Kev.Er zijn 3 omzettingsgraden die bij evenwicht ifv beginconcentratie gelden:
1. Dissociatiereactie: AB ↔A+B Hierbij is de dissociatiegraad αev de verhouding van het aantal gedissocieerde
moleculen tot het oorspronkelijk aantal moleculen. αC0 is de concentratie gedissocieerde moleculen (1-α) is de fractie niet-gedissocieerde moleculen (1-α)C0 is de concentratie niet-gedissocieerde moleculen

Hieruit volgt dat αC0 +(1-α)C0= C0 de totale beginconcentratie is en als α klein is geldt:
. Dit betekend dat bij een dissociatiereactie waarbij een groter aantal moleculen wordt gevormd, neemt bij toenemende beginconcentratie de dissociatiegraad α af

2. Associatiereactie: A+B↔AB Hierbij is de associatiegraad α de verhouding van het aantal geassocieerde moleculen
A of B tot het oorspronkelijk aantal moleculen A of B αC0 de concentratie geassocieerde moleculen (1-α) de fractie niet-geassocieerde moleculen (1-α)C0 de concentratie niet associeerde moleculen
Als α klein is geldt: 3. Omzettingsreactie: A+B↔C+D
Hierbij is de omzettingsgraad α de verhouding van de omgezette moleculen C of D tot het oorspronkelijke aantal moleculen A of b.
αC0 de concentratie gevormde moleculen (1-α) de fractie gevormde moleculen (1-α)C0 de concentratie niet gevormde moleculen
Als α klein is geld:K c ≈α 2
1 of α = √K c
D3: energetische aspecten van het chemisch evenwicht.Een toestandsfunctie wordt enkel bepaald door de huidige toestand van het systeem. Toestandfuncties zijn de energie E, de kinetische energie Ekin, inwendige energie U en enthalpie H. Ze worden weergegeven dmv hoofdletters. De potentiële energie van de reagentia en producten wordt binnen de chemie de inwendige energie U genoemd, er geldt nagenoeg Epot= U. Om U te berekenen kijkt men naar de 1ste wet van de thermodynamica: “de totale energie van een geïsoleerd systeem is constant”. Dit betekent dat energie niet vernietigd of gecreëerd kan worden maar enkel omgezet kan worden in andere vormen. Een verandering van inwendige energie-inhoud gaat dus gepaard met een uitwisseling van warmte q en/of arbeid w:
U=q+wDe vorm van arbeid bij chemische processen is de arbeid die geleverd wordt door het systeem bij een volume-expansie tegen een buitendruk, w is dan gelijk aan –Pext*∆V. Wanneer we bij een constant volume kunnen werken (bv een calometrische bom) dan zal de arbeid nul worden. Bij een constant volume is de uitgewisselde warmte q dus gelijk aan de verandering in inwendige energie U:
(∆U ) v=qQ kan men berekenen met q=Cv ∙∆T . ∆T moet men experimenteel vaststellen door de temperatuur te meten van het reagerende systeem. De warmtecapaciteit berekend men uit de specifieke warmtecapaciteit c van zuivere stoffen maal de massa van deze stoffen in het systeem. De eenheid van de warmtecapaciteit is cal en 1 cal komt overeen met 4,184 joule.

Het blijkt echter makkelijker te zijn om de druk constant te houden dan het volume, met als gevolg dat de uitgewisselde warmte niet overeenkomt met de verandering in inwendige energie-inhoud U. Als men werkt bij een constante druk definieert men daarom een nieuwe toestandsfunctie: de enthalpie H. De enthalpie H wordt gegeven door:H=U+P ∙V . Hieruit volgt dat als men het verschilt pakt en voor de inwendige energie q+(-Pext*V) invult:∆ H=∆ (U+PV )∆H=∆U+∆PV +P∆V∆H=q+ (−P ∆V )+∆ PV+P ∆V∆H=q+P∆V
Als men nu bij een constante druk werkt is de uitgewisselde warmte gelijk aan de verandering in enthalpie.Het verband tussen de enthalpie en inwendige warmte wordt dus gegeven door ∆ H=∆U+P∆V . Dit betekent voor stoffen, vloeistoffen en oplossingen dat ∆H≈∆U. Voor gassen geldt dat ∆V= ∆ngas*(RT/P) en dit zorgt ervoor dat ∆H= ∆U+ ∆ngas*RT.Bij een exotherme reactie is de enthalpie-inhoud van de producten lager dan die van de reagentia. Bij een endotherme reactie is de energie-inhoud van de producten hoger dan die van de reagentia. ∆H r is gedefinieerd als de enthalpie-inhoud van de producten min de enthalpie-inhoud van de reagentia. Bij een exotherme reactie is ∆Hr dus negatief en bij endotherme reactie dus positief.

D4: entropische aspecten van het chemische evenwichtEen spontaan proces is irreversibel, terwijl een reversibel proces alleen kan plaatsvinden door een uitwendige drijfveer. Experimenteel is er vastgesteld dat de qrev> qirr. Dit betekent dat de reversibel uitgewisselde warmte een toestandsfunctie is die niet afhangt van de manier waarop die toestand
werd bereikt. We gebruiken dit om de nieuwe toestandsfunctie, entropie S, te definiëren:
∆ S=QrevT
Deze definitie legt geen verband met de moleculaire aspecten van het chemische evenwicht. Daarvoor kan men de entropie beter definiëren als de hoeveelheid wanorde van een systeem. Deze hoeveelheid kan gekwantificeerd worden op basis van de waarschijnlijkheid van voorkomen van een hoeveelheid wanorde. Er kan ook een verband aangetoond worden tussen de entropie S en de waarschijnlijkheid W van een toestand met dergelijke hoeveelheid wanorde:S=Kb∙ lnW . Voor de entropie zijn de 2de en 3de wet van de thermodynamica van toepassing, nl:
1. Een spontaan proces gaat gepaard met een entropietoename.2. Bij het absolute nulpunt (O K) is de entropie nul in perfect geordende kristallen.
De waarden voor standaardentropie van een mol zuivere stof is op te zoeken (BOD tabel: 5.2, 5.3, 5.5)
D5: vrije energie GAls we een spontaan proces veronderstellen dan is ∆H= q= qirr, hieruit volgt dat:
Er is dus een nieuwe toestandsfunctie G, de vrije energie. En deze is dus gedefinieerd als G= H-TS. Uit het spontaan proces blijkt dat ∆G altijd kleiner is als nul en als we bij constante temperatuur werken geldt: ∆G= ∆H- T∆S.
Verband tussen de standaard-vrije-energieverandering en de evenwischtsconstante:G=H−TSG=U+PV −TS∆G=∆U+V ∆ P+P∆V−S ∆T−T ∆ S
Bij isotherm omstandigheden geldt (∆T=O)∆G=∆U+V ∆ P+P∆V−T ∆ S
We nemen nu reversibele omstandigheden zodat T∆S= qrev en uit de eerste wet geldt q= ∆U+P∆V : ∆G=V∆P. als we hiervan de integraal berekenen (dus dG=VdP) krijgt men G= G°+RT
ln p1atm . De “°” verwijst naar de ondergrens van 1 atm, de temperatuur is dus nog gekozen.
Dit leidt dan tot het volgende:

En bij evenwicht kunnen we dan Qp vervangen door Kp, waarmee men een verband krijgt tussen G en Kp:
Spontane reacties zullen optreden wanneer een verlaging van vrije energie optreedt ùaw wanneer de verandering van vrije energie negatief is.

Deel E: evenwichtsreacties
E1: zuur-base-reacties als protontransferreactiesVolgens de huidige opvattingen kan men een zuur HA definiëren als een protondonor, het zal na deprotonatie de geconjugeerde base A- worden. Zo kan men ook een base A- definiëren als een protonacceptor die na protonatie een geconjugeerd zuur HA wordt. Zo krijgt men dus koppels van zuren en hun geconjugeerde basen en omgekeerd. De zuur-base-reactie wordt dan een protontransferreactie door het koppelen van een proton-donor met een proton-acceptor. Dit is de Brönsted-Lowry theorie. Voor een zuur op zich kan men dan altijd de halfreactie van de protonafgifte schrijven: HAH+ + A- en voor een base kan men de omgekeerde halfreactie schrijven zodat men ook terug protonafgifte heeft. Uiteraard zal de neiging om een proton af te geven veel sterker zijn voor een gewoon zuur dan voor het geconjugeerde zuur van een base.
In deze tabel staan de zuur-base-reactie’s geordend. Ze kan worden geïnterpreteerd als een reeks van halfreacties gerangschikt volgens afnemend vermogen om het proton af te splitsen(linkse kolom). Omgekeerd is het uiteraard ook een reeks van halfreacties van protonopname, gerangschikt volgens toenemend vermogen van protonopname. Nu kan men 2 van deze zuur-base-reactie’s koppelen, hierbij zal het sterkste zuur blijven staan als protonafgifte en de reactie van het zwakkere zuur zal een protonopname worden.In de tabel komt water 2 keer voor, eenmaal als de zwakke geconjugeerde base van het zuur H3O+ en één keer als het zwakke geconjugeerde zuur van de base OH-. Dit betekent dat water dus zowel een proton kan opnemen als afgeven, dit wordt amfiprotisch genoemd. Een gevolg hiervan is dat alle zuren sterker als H3O+ hun proton afgeven aan water met andere woorden, het sterkste zuur dat in water kan voorkomen is H3O+. Omgekeerd geldt ook dat alle basen sterken als OH- in water een proton zullen opnemen en de sterkste base dus OH- is in water. Deze 2 gevolgen noemt men het nivellerend effect van water. Naast water zijn ook NH3 en CH2COOH2
+ amfiprotisch, deze zullen dus ook een nivellerend effect vertonen als ze worden gebruikt als solvent.In puur water zal er autoprotolyse optreden, dit kan men beschrijven mbv de Kw. 2H2O↔H3O
+¿+OH−¿¿¿; Kw= ¿= 10^-14

Dit is een evenwichtsvergelijking en is dus altijd geldig. Aangezien het product constant is kan men afleiden dat er tussen de concentratie H3O+ en OH- een omgekeerde evenredigheid bestaat.
De definitie van pH van een oplossing is: pH= -log[H3O+]. Voor zuiver water is de enige bron van H3O+ de autoprotolyse en moet dus de concentratie H3O+ gelijk zijn aan OH-. De pH van zuiver water is dus –log[H3O+]= -logKw
1/2=-log(10-14)1/2= 7. Voor oplossingen zuurder als water ligt de pH hier dus onder, oplossingen meer basisch als water ligt dit tussen de 7 en 14. Aangezien Kw afhankelijk is van temperatuur kan men denken dat water bij andere temperaturen zuurder of meer basisch is, dit klopt echter niet: er blijft altijd evenveel H3O+ als OH- in water. Alleen zal het evenwicht naar rechts verschuiven bij een stijgende temperatuur.Uit de autoprotolyse van water kan men afleiden dat pH+pOH= Kw bij 25°C. Zoals gezegd zullen sterke zuren/basen een proton afgeven/ opnemen in water. Dit geldt echter niet voor de zuren/basen die tussen H3O+ en OH- staan. Deze stoffen hebben een evenwicht in water omdat niet alle moleculen hun proton zullen overdragen aan het solventmolecule. Men kan dan de volgende evenwichten schrijven in water:
1. Ka= ¿¿2. Kb= ¿¿
Nu kan men zien dat er tussen de Kb en de Ka een verband is in water namelijk: Ka*Kb=Kw.
Zuren in waterDe sterkte van een zuur of hoe gemakkelijk een proton kan worden afgesplitst, wordt bepaald door de polariteit en de sterkte van de binding.
1. Binaire zuren Binnen een periode: hoe meer elektronegatief X, hoe sterker zuur HX is. Binnen een groep: hoe zwakker de HX binding, hoe sterker het zuur. De sterke zuren
gaan volledig dissociëren in water, de zwakke niet. De enige sterke zijn dus HI, HBr en HCl.
2. Oxozuren: HxX(OH)yOZBv H2SO4= S(OH)2O2
Deze zuren kunnen enkel het proton afsplitsen dat gebonden is op het zuurstofatoom.
Hoe groter de elektronegativiteit van X, hoe sterker het oxozuur. Hoe meer zuurstofatomen het zuur heeft, hoe groter het effecten van de
elektronegativiteit van zuurstof en hoe sterker het zuur. Bij de afsplitsing van meerdere protonen geldt dat de 2de dissociatie altijd zwakker is
dan de eerste en de derde zwakker dan de 2de. Dit komt omdat er een proton moet worden afgesplitst van een zuurrest die reeds negatief geladen is, en dus moeilijker om te ontrekken.
Verbindingen met zure of basische eigenschappen in waterEr zijn verschillende verbindingen die eens opgelost in water, zure of basische eigenschappen ontwikkelen.
A. Oxides en hydroxides Alkalimetaaloxides (groep 1 in PSE), deze oxiden zijn goed oplosbaar in H2O. Ze zullen
genivelleerd worden tot OH- en basisch reageren in water. Aardalkalimetaaloxides (groep 2), deze oxiden reageren analoog in water. Ze zijn
echter minder goed oplosbaar door de ladingsdichtheid op hun metaal. De metalen zelf (OT=0) zullen in water ook basisch reageren, dit komt omdat het reductantia zijn die door het afgeven van elektronen(Brönsted-Lowry theorie), hun voorliggende edelgasconfiguratie bereiken.
Transitie-elementen met lagere OT zoals chroom()oxide en mangaan()oxide reageren ook basisch in water.

Transitie-elementen met hogere OT(bv chroom()oxide) en koolstof reageren zwak zuur in water.
Niet-metalen in hun hoogste oxidatietrap zullen sterk zuur reageren in water. Ze worden zuuranhydrides genoemd. Wanneer er bij de reactie met water niet alleen een protontransfer gebeurt maar ook een redoxreactie, spreekt men van een meng-anhydride.
Dan zijn er ook nog oxiden die amfoteer kunnen reageren, dit betekent dat ze zowel zuur als basisch kunnen reageren afhankelijk van het toegevoegde reagens: bij een zuur reagens reageren ze basische en omgekeerd. Deze elementen bevinden zich tussen de aardalkalimetalen en de niet-metalen(diagonaal relaties).
Voor de eigenschappen van oxiden en hydroxiden (zuur of basisch) is er een heel duidelijk verband met
1. De plaats van het element in de tabel van Mendeljev: metaal, niet-metaal, transitie-element, diagonaalrelatie.
2. En ook met de oxidatietrap van het transitie-element of (niet-)metaal:Lage OT=basisch oxide, hoge OT= zuur oxide, intermediar OT= amfoteer. En dus algemeen kan men zeggen hoe hoger de oxidatietrap, hoe sterker zuur.
B. ZuurhalogenidenDe zuurhalogeniden zijn sterk reactieve stoffen. Opgelost vertonen ze zure eigenschappen:
Met water hydrolyseren ze tot 2 verschillende zuren:Bv: SO2Cl2 + H2O 2HCl + H2SO4
Met een hydroxidebase worden ze geneutraliseerd tot 2 verschillende zouten:Bv: POCl3 + 6NaOH 3NaCl + Na3PO4 + 3H2O
Omdat het zo een reactieve stoffen zijn, worden ze altijd in situ gemaakt voor captive use. Dit betekent dat men ze ter plaatste maakt en onmiddellijk verder gebruikt de effectieve bereiding van een zuurchloriden is gebaseerd op het reactieve chloorgas.
C. KationenAlle kationen die geconjugeerd zuur zijn van een neutrale base zijn zuur. Metaalkationen met een hoge ladingsdichtheid kunnen in water ook zuur reageren, bv Al3+. Door de hoge ladingsdichtheid kunnen deze kationen niet vrij bestaan, maar moeten ze worden omgeven door watermoleculen. Dit vermindert de ladingsdichtheid omwille van het toegenomen volume. Van deze watermoleculen kan er echter een proton afgesplitst worden om de ladingsdichtheid te verlagen door het verlagen van de lading zelf.
Bv: D. Anionen
Analoog aan de kationen zijn de anionen die geconjugeerd base zijn van een zwak zuur basisch. Echter wel op te merken is dat de anionen die geconjugeerde base zijn van een sterk zuur neutraal reageren. Dit komt omdat deze ionen een lage ladingsdichtheid hebben en houdt ook verband met de edelgasconfiguratie.
pH-berekenen in waterige oplossingenwe onderscheiden 2 gevallen voor de berekening van de pH: enerzijds een zwak zuur of base en anderzijds de oplossing van een polyprotisch zuur. Voor een sterk zuur/base is de pH direct af te leiden uit de beginconcentratie zelf.
A. pH van een zwak zuur/baseVoor de berekening van de pH van een zwak zuur bij evenwicht in water en gekende beginconcentratie en evenwichtsconstante kan men 4 vergelijkingen opstellen:
1. De evenwichtsvergelijking: Ka=¿¿2. Het ionenproduct van water:Kw=¿ 3. De ladingsbalans: ¿4. De massabalans: [ HA ]0=¿
!!Al deze vergelijkingen zijn bij evenwicht van het zuur!!Dit stelsel kan men oplossen (4 vergelijkingen en 4 onbekenden), het omvormbaar tot een 3de graads vergelijking waarbij de [H3O+] de onbekende is. Deze vergelijking heeft maar 1 oplossing die chemisch zinvol is. Met deze derde graadsvergelijking kan men dan oplossingen berekenen voor alle combinaties van [HA]0 en Ka. het oplossen van de vergelijkingen kan men ook doen door bepaalde aannames te maken:
Aanname 1: sterk zuur (grote Ka:pKa0) en de autoprotolyse van water wordt teruggedrongenDe gevolgen van deze aanname zijn dat het zuur volledig dissocieert ([HA]ev=0) en dat de autoprotolyse van water wordt terug gedrongen. Dit betekent dat er maar 2 onbekenden over zijn ([A-]ev en [H3O+]ev). Hierdoor worden vergelijking 1 en 2 zinloos waardoor men vindt dat: [H3O+]ev =[HA]0.Bij een te kleine [HA]0 mogen we echter de autoprotolyse niet verwaarlozen omdat we dan een basische pH krijgen.
Aanname 2: sterk zuur (grote Ka:pKa0) en de autoprotolyse van water wordt niet teruggedrongenNu zal het zuur volledig dissociëren zonder dat de autoprotolyse wordt terug gedrongen. Dit betekent dat er drie onbekenden zijn: [H3O+]ev, [A-]ev en [OH-]. Vergelijking 1 wordt zinloos en uit de andere drie vinden we een 2de graadsvergelijking (0= [H3O+]ev
2 - [HA]0*[H3O+]ev- Kw) waaruit de enige zinvolle
oplossing [H3O+]ev=[HA ]0+√[ HA ]0
2+4K w
2 is. Bij een grote beginconcentratie
wordt dit [H3O+]ev =[HA]0. Aanname 3: zwak zuur (kleine Ka:pKa0) en de autoprotolyse van water
wordt verwaarloosdWeinig zuur zal dissociëren, de [A-]ev is klein maar nog verschillend van nul. De autoprotolyse wordt terug gedrongen, [OH-]=0. Hieruit volgt dat vergelijking 2 zinloos wordt en de drie andere kunnen we combineren tot ¿¿(of pH= ½(pKa+p[HA]0). Ook hier komt weer het probleem terug dat bij een te kleine [HA]0 een basische pH wordt verkregen.
Aanname 4: zwak zuur (kleine Ka:pKa0) en de autoprotolyse van water wordt niet verwaarloosdWeinig zuur zal dissociëren zoals daarjuist maar omdat de autoprotolyse niet wordt terug gedrongen blijven er 4 onbekenden en krijgen we ¿¿.
Aanname 5: enkel de autoprotolyse van water wordt terug gedrongen.Deze aanname wordt gemaakt zodat het gebied van de berekenbare pH groter wordt. Doordat er geen autoprotolyse is, is er ook geen [OH-]. De vergelijking 2 wordt dus zinloos en met de andere vindt men een 2de graadsvergelijking waarvan de enige zinvolle oplossing ¿¿.

Voor de berekingen met zwakke basen moet men de [OH-] bepalen, men krijgt dan de pOH waarmee men dan met 14-pOH=pH vindt.
B. pH van een polyprotisch zuurzie slide 27 tot 33 van deel E1
HydrolyseWe weten dat de ionen van zouten basisch of zuur kunnen reageren in oplossing. Voor berekingen ivm kunnen we dus hetzelfde doen als bij zwakke zuren/basen. We spreken van de hydrolyse en de Kh. Als het zout opgelost is dienen we alleen rekening te houden met de ionen die een geconjugeerd
zuur of base zijn. De waarde van de hydrolyseconstante Kh is te berekenen als Kw
Ka. Om hier nu de pH
uit te berekenen maken we 2 aannames:1. We verwaarlozen de autoprotolyse: [OH-]ev= [geconjugeerde base]ev
2. Er is geringe hyrdolyse: [geconjugeerde base]ev= beginconcentratie zoutNa doorvoering van deze aannames en enige afleiding(p169 in cursus): ¿¿. Hier kunnen we ook de hydrolysegraad γ invoeren, analoog aan de dissociatiegraad; de hydrolysegraad γ is gedefinieerd als de fractie van de oorspronkelijke aanwezige geconjugeerde base die gehydrolyseerd is:
γ= [ geconjugeerdebase ]C zout
=√ Kb
C zout
.
Voor een hydrolysereactie waarbij alle ionen zure/basische eigenschappen hebben, kunnen we schrijven: ¿¿.We kunnen ook besluiten dat
Buffers en pH-indicatorenEen buffer wordt in de chemie gedefinieerd als een waterige oplossing waarvoor de pH realtief constant blijft bij verdunning en bij toevoeging van zuur of base.Een buffer bestaat uit een zwak zuur/base+ het zout van dit zuur/base. Een vaak gebruikt buffersyteem is dat van is de mengoplossing van het zwakke azijnzuur (CH3COOH) en natriumacetaat(CH3COONa). De aanwezigheid van het zuur zal er voor zorgen dat de hydrolyse terg wordt gedrongen ([CH3COO-]ev= CCH3COONa) en het zout dient om de zuurdissociatie terug te dringen ([CH3COOH]ev= CCH3COOH). Om de pH van een buffer te weten kunnen we de Henderson-Hasselbalch of
bufferformule gebruiken: pH=p Ka−log(C zuur
C zout)=p Ka+log (
C zout
C zuur) of
pH=14−p Kb−log (C zout
Cbase).
De eigenschappen van een buffer:1. Een verdunning heeft geen effect op de pH2. Het ± constant blijven van de pH bij toevoegen van een zuur of base.
Bewijs constant blijven pH: zie p 173-174
Regels voor het maken van een buffer:

1. Kies een zwak zuur met zijn pKa zo dicht mogelijk bij de pH van de te maken buffer. De buffer werkt dan effectief in een gebied met een pH-bereik van pKa+1.
2. Neem een zo groot mogelijk concentratie van zuur en zout, rekeninghouden met de oplosbaarheid (zorg ervoor dat alles opgelost is).
Bloed als buffer:Bloed is bufferen tegen melkzuur dat wordt aangemaakt in de spieren, het bevat een een overmaat base (HCO3
-). Het kan dus alleen bufferen voor zuur en niet base. Bloed werkt als buffer in een pH tussen 7,35 en 7,45. Bij spierarbeid wordt er dus melkzuur aangemaakt wat sterker zuur is dan H2CO3. Het zal afgevoerd worden langs conversie in de buffer waarna het als CO2 zal wordt afgevoerd.
Bij een overmatige melkzuurproductie spreekt men van metabolische acidosis, wanneer hierdoor de ademhaling wordt bemoeilijkt(buiten adem geraken) spreekt men ook van ademhalingsacidosis. Een natuurlijke tegenreactie hiervoor is hyperventilatie waarmee het lichaam dan het teveel aan koolzuuranhydride doet vrijzetten, men noemt dit ademhalings-alkalosis.
Een makkelijke manier om de pH van een oplossing te bepalen is via een pH-indicator. Deze pH-indicatoren zijn zwakke zuren of basen waarvan de geprotoneerde en gedeprotoneerde vorm een sterk verschillende kleur hebben. Men voegt de indicator in geringe concentratie toe aan de oplossing waarvan men de pH wilt bepalen of wilt titreren. Uit log ¿¿¿kunnen we afleiden dat de zuurconcentratie niet wordt bepaald door de concentratie indicator. Wel wordt de verhouding [In-]/[HIn](kleur van de oplossing) bepaald door de zuurconcentratie en de zuurdissociatieconstante van de indicator.Men kan de precisie opdrijven door te werken met meerdere indicatoren die achtereenvolgens in een kleiner pH-gebied actief zijn. Of men kan een universele indicator gebruken, deze maakt gebruik van verschillende dissociatieconstante. Het is een papierstrip die afhankelijk van de pH een andere kleur vertoont.
Titraties De afleidingen van dit deel zijn niet te kennen, de grafieken wel. Bij een titratie gaat men een oplossing toevoegen tot dat men visueel een kleuromslag krijgt van de indicator, theoretisch is dan de titratie-index gelijk aan 1. Bij een zuur-base titratie van eenzelfde molariteit betekent dat Nb*Vb= Na*Va, dit punt noemt men het equivalentiepunt. Wanneer het EP is bereikt is er een grote verandering in pH. Op de grafiek gaat men dan een sterke stijging of daling van de pH, men noemt dit ook het omslaggebied. Bij meer verdunde zuuroplossingen begint de pH-curve bij hogere waarden maar bij gebruik van geconcentreerde base als titrans eindigt de curve bij een hoge pH. Daarom kiest men bij deze titraties een basische indicator, waarvan het actieve gebied hoog ligt, dit geldt ook andersom.Bij een zwak zuur men een sterke base of omgekeerd hebben 4 gevallen tijdens de titratie:
1. Dit is voordat men begint met titreren: α=0, er is alleen zwak zuur aanwezig en de pH= 1/2pKa- ½ logCa.
2. Dit is tijdens de titratie: 0α1, er is al gedeeltelijk zwak zuur geneutraliseerd en er is dus hydrolyse (pH= 1/2pKa+ ½ pKw+ ½ log Cz. Er is ook een gedeeltelijk buffer-effect.
3. α= 1, al het zuur is geneutraliseerd in zout. Er is volledige hydrolyse en ook word het EP bereikt bij een pH verschillende van het neutralisatiepunt (pH=7).
4. α 1, er is enkel zout en base in de oplossing, het verdere pH-verloop gebeurt zoals bij sterk zuur/sterke base

E2: redoxreacties en elektrontransferreacties
Oxidantia en reductantiaEen oxidans is een elektronacceptor en dus in staat 1 of meer elektronen op te nemen, na de elektronopname wordt het een geconjugeerd reductans: Oxm++n*e- → Red(m-n)+.Een reductans is een elektrondonor en in staat om 1 of meer elektronen af te geven, na de elektronafgave wordt het een geconjugeerd oxidans: Red(m-n)+ → Oxm++n*e-.Het samenbrengen van een oxidantia en hun geconjugeerde reductantia en viceversa noemt men de oxidoreductiereactie. Dit is een elektrontransferreactie door het koppelen van een elektronacceptor met een elektrondonor. Voor een oxidans kan men altijd de reactie schrijven via een elektronopname, een reductie, terwijl men voor het reductans ook de omgekeerde reactie van elektronopname kan schrijven. Uiteraard zal hier nu de neiging om elektronen op te nemen veel kleiner zijn voor dit geconjugeerde oxidans dan voor het echte oxidans. Ook hier kunnen we net zoals bij protonreacties een tabel opstellen van oxidans/reductanskoppels geordend naar afnemende neiging van elektronopname:
De derde kolom geeft de standaardpotentiaal (E0) aan, een maat voor de neiging van het oxidans om elektronen op te nemen. De volledige reductiehalfreacties staan gerangschikt in volgorde van afnemende oxidatiesterkte of toenemende reductiesterkte. Dit toont ook aan dat het geconjugeerde reductans van een sterk oxidans zwak is terwijl het geconjugeerde reductans van een zwak oxidans sterk is.Regels voor het schrijven van een reductiehalfreactie:
1. bepaal de oxidatietrap van het oxiderend/reducerend element. Dit wordt weergeven door romeinse cijfers in superscript.
2. Bepaal de juiste verschijningsvorm van het element: gebeurt de reactie in zuur of basisch milieu?
3. Breng de ladingsbalans in orde: het aantal elektronen neutraliseren met H+(zuur milieu en linkse kant van de reactie) of OH- (basisch milieu en rechtse kant van de reactie).Bv:F2+ 2e-+2H+ → 2HF O2+2e- → HO2
-+ OH-
4. Breng de massabalans in orde: voeg H2O toe zodat het aantal waterstof- en zuurstofatomen aan elke kant van de reactie klopt (in zuur milieu aan de rechterkant H2O, in basisch milieu aan de linkerkant

In een reductiehalfreactie zal er geen opname van elektronen gebeuren zonder dat de reductans de elektronen levert. Bij het schrijven van een redoxreactie moet 1 van de 2 halfreacties omgedraaid worden: de reductiehalfreactie met het grootste oxiderend vermogen of met andere woorden met de grootste E0-waarde blijft als reductie staan, de reductiehalfreactie met het kleinste oxiderend vermogen wordt omgedraaid. Dat noemt men dan de essentiële ionische redoxvergelijking, in deze vergelijking moet men zorgen dat het aantal uitgewisselde elektronen gelijk zijn in beide halfreacties.
Galvanische cellenWanneer we 2 elektrontransferreacties met een verschillende standaardreductiepotentiaal laten gebeuren in een aparte beker maar met een elektrisch contact voor de geleiding van elektronen dan verkrijgt men een galvanische cel. Tengevolge het verschil in reductiepotentiaal ontstaat er een spanningsverschil tussen de bekers. Bij standaardomstandigheden(1atm, 298K, 1M) is het spanningsverschil gelijk aan het verschil in standaardreductiepotentiaal. Een gekende cel is het Daniell element, deze cel gebruikt koper en zink als spanningsopwekkers. De kring tussen de 2 elementen wordt gesloten door een geleider, vaak een zoutbrug of semipermeabele scheiding. Deze geleiders laten wel uitwisseling van ionen toe zonder dat de oplossing gemengd worden.De benaming van de elektrodes in een galvanische cel volgt een simpele regel:
Aan de anode gebeurt de oxidatie(AO)Aan de kathode gebeurt de reductie(KR)
Galvanische cellen worden schematisch weergegeven door middel van de essentieel deelnemende deeltjes, de scheiding wordt weergegeven door ||, men geeft ook de concentratie mee als deze gekend is:
In het Daniell element spelen de koper en zinkstaafjes tegelijk de chemische rom van de reductantia van de 2 halfreacties en de fysische rol van elektrode. Dit is echter niet altijd het geval, meestal kiest men een inert metaal, bv platina, dit zal dan geen chemische rol spelen in de elektrontransferreactie. Door het verschil in neiging van het reductans ontstaat een potentiaalverschil. Deze kan berekend worden aan de hand van de standaardredoxpotentialen van beide halfreacties:
∆ E0=Ekathode0 −Eanode
0
Men neemt altijd de meest positieve min de minst positieve zodat men steeds een positief potentiaalverschil verkrijgt. De celpotentiaal ∆E0 is enkel te meten als een potentiaalverschil voor een totale redoxreactie en niet voor redoxhalfreacties. We kunnen deze echter wel vinden aan de hand van een referentiepotentiaal. Deze referentie is de standaard-waterstof-elektrode waaraan een standaardreductiepotentiaal van 0,00V is toegekend. Als we nu een galvanische cel maken met de standaard-waterstof-elektrode en een andere halfreactie onder standaardomstandigheden krijgen we de ongekende celpotentiaal van de halfreactie.Wet van Nernst:“Bij het aflopen van 2 spontane reacties in aparte halfcellen verlaagt de concentratie aan de kathode en verhoogt de concentratie aan de anode. Dit resulteert in een verlaagde celpotentiaal. Er zal echter zolang er een potentiaalverschil is, stroom blijven vloeien, en al er reductie aan de kathode en oxidatie aan de anode blijven optreden tot de concentraties aan de anode en kathode zo zijn dat er geen potentiaal verschil meer bestaat en de reacties zijn afgelopen.

Zie afleiding p195/196 voor formule:K ev=10nF∆ E0
2,303RT=10n∆E 0
0,059 .
De Nernst-vergelijkingVoor een redoxhalfreactie is het mogelijk een Nernstvergelijking te schrijven als de 2 halfreacties van
de algemene vorm zijn: E=E0− 0,059n
∗log ( [red ]r
[ox ]O). Voor de afleiding zie p197.
Zo kan men een effectieve reductiepotentiaal berekenen voor een halfreactie, onder gelijk welke omstandigheden van concentratie van deelnemende deeltjes. Dit is het grote belang van de Nernstvergelijking.Samenvattend kunnen we zeggen dat de reductiepotentiaal wordt bepaald door
1. De aard van het redoxstelsel;2. De temperatuur T;3. Het aantal uitgewisselde elektronen n;4. De concentratie van de betrokken ionen in oplossing of de partieeldruk van de betrokken
gassen.Met de Nernstvergelijking kan men ook een alternatieve afleiding geven voor het verband tussen de evenwichtsconstante en de standaardreductiepotentiaal. We kunnen dit doen met het Daniell element:
We zien dus dat bij het verbinden van de 2 halfcellen er een spanningsverschil zal zijn van 1,10V. dit zal echter afnemen volgens de wet van Nernst: Cu2+ wordt gereduceerd en Zn geoxideerd tot het potentiaalverschil nul is. Dan is het evenwicht bereikt en wordt ∆E0 waardoor men een oneindige evenwichtsconstante krijgt:
In het algemeen geldt K ev=10
n∆ E0
0,059 met als gevolg dat hoe meer positief ∆E0 is, hoe meer de reactie
aflopend naar rechts is en omgekeerd. Wanneer we aannemen dat een reactie in de praktijk als aflopend kan worden beschouwd voor een evenwichtsconstante van minstens 103 kunnen we afleiden dat ∆E0≈0,2/n V. bij een verschil van 200mV in standaard reductiepotententiaal mogen we een redoxreactie als aflopend beschouwen.
ConcentratiecellenEen concentratiecel bestaat uit 2 compartiment die identiek zijn op de concentratie van de reagentia na. Via wet van Nernst is er echter wel een potentiaalverschil ∆E als gevolg van het verschil in concentratie. Echter enkel zolang er in verschil is in concentratie, zal ∆E verschillend van 0V zijn en er een reactie optreden.
A. Primaire cellen

Een primaire cel is een cel die niet oplaadbaar is en waarvan de elektrontransferreactie enkel spontaan kan verlopen tot de bronspanning op nul gevallen is. De reactieproducten van de spontane redoxreactie vermengen zich in de elektrolytpasta. De spontane redoxreactie kan niet worden omgekeerd door een elektrolyse.
1. Leclanché celRedoxreactie:
Zn¿2¿2MnO2+2N H 4+Zn+2N H3→2MnO (OH )+Zn¿
De anode van deze cel bestaat uit Zn dat geoxideerd wordt tot Zn2+, gecomplexeerd in Zn(NH3)4
2+ in de elektroytpasta. De kathode is een grafietstaaf in contact met MnO2, dat gereduceerd wordt tot MnO(OH). Het elektrolyt is een vaste pasta die geleiding verzekert en bestaat uit NH4Cl en ZnCl2. De beginspanning bedraagt 1,5V.
2. Alkaline batterijRedoxreactie:
Zn¿2¿2MnO2+Zn+4OH−¿→2MnOO−¿+Zn ¿¿¿
De anode bestaat uit Zn dat geoxideerd wordt tot Zn2+, dat als Zn(OH)42-
voorkomt. De kathode is identiek aan die van de Leclanché cel. Het elektrolyt is KOH, een sterke base die wat ervoor zorgt dat MnOO- wordt gevormd. De beginspanning is 1,5V.
3. Lithium batterijRedoxreactie:
L i+¿+e−¿ →Li ¿¿I 2+2e−¿→2 I−¿¿ ¿2 Li+ I 2→2L i+¿+2 I−¿ ¿¿
De kathode bestaat uit een I2 complex, dat gereduceerd wordt tot I-. de anode bestaat uit Li, dat geoxideerd wordt tot Li+. Er is geen pasta maat LiI-kristallen scheiden de anode en de kathode, terwijl diffusie van Li+ door de kristallen toch de geleiding verzekert. Hierdoor is de inwendige weerstand heel groot, waardoor geen grote stroom kan vloeien, wat een lange levensduur tot gevolg heeft. De celpotentiaal en vermogen van de cel zijn zeer groot
Het verloop van redoxreacties kan in omgekeerde zin gestuurd worden door het aanleggen van een tegenspanning die groter is dan de celpotentiaal. De grootte van de tegenspanning moet minstens de celpotentiaal bedragen naast 3 andere factoren:
1) Het elektrische circuit bevat ook een weerstand. Er is ook een spanningsval over deze weerstand, dus moet de tegenspanning vermeerderd worden met deze spanningsval.
2) De elektrolysereactie zelf zal als gevolg hebben dat de concentraties van de deelnemende deeltjes in die zin evolueren dat de celpotentiaal, volgens de Nernst-vergelijking zal toenemen.
3) Het mechanisme van de elektrontransferreactie aan de elektrode-oppervlakken is zodanig dat een hogere spanning een versnelling van de reactie als gevolg heeft. Bij een spanning infinitesimaal hoger dan de celspanning zal de elektrolyse oneindig traag verlopen. Vooral bij gasvorming op inerte elektroden is een zogenaamde overspanning nodig om merkbaar reactie te hebben.

Het uitgewisselde elektron in de redoxreactie staat in verband met een hoeveelheid materie die gereduceerd of geoxideerd wordt. Er is dus een verband tussen de elektrische stroom en de massa omgezet.
a) Eerste wet: de massa van een product gevormd in een elektrolyse is evenredig met de totale lading Q die verbruikt werd: m= K1Q= K1It
b) Tweede wet: de massa van een product gevormd in een elektrolyse is rechtstreeks evenredig met de relatieve molecuulmassa M en omgekeerd evenredig met het aantal uitgewisselde elektronen: m= K2* (M/n)Samen geeft dit: m= (I*t*M)/(96500*n).
B. Secundaire cellenEen secundaire cel is wel oplaadbaar, de redoxreactie wordt omgedraaid mbv een tegenspanning.
1. Ni-Cd batterijRedoxreactie:
¿¿Cd ¿De reactieproducten van de spontane reaxreactie worden als vaste stof op de elektrodes afgezet en vermengen dus niet. Dit laat toe om de richting van de reactie om te keren: het heropladen van batterijen. Het gebruiken van deze batterijen resulteert in problemen van fysische aard: de elektrodes worden bedekt met reactieproduct of gedeeltelijk ontdaan van product. Het regelmatig geheel ontladen en opnieuw opladen heeft een positief effect omwille van fysische redenen.
2. Ni-MH batterijRedoxreactie:
¿¿H20+M+e−¿→MH+O H−¿ ¿¿
De vervanging van het cadmium door een metaalhydride laat toe om meer energie te stockeren. Hier schrijven we formeel de reductie van +1H tot 0H. het metaal M speelt dan enkel de rol van matrix om de H op te slaan en speelt dus niet mee in de redoxreactie. Daarom is M ook vaak speciaal vervaardigd om een heel groot specifiek oppervlak te hebben.Merk op dat de werking van de batterij dus gebaseerd is op waterstof als energiedrager en het metaal de fysische drager is. Als standaardreductiepotentiaal kunnen we deze van waterstof in basisch milieu nemen wat de batterij een bronspanning geeft van 1,26V. Ook CH4 is een energiedrager die geoptimaliseerd is om waterstof onder de vorm van een hydride vast te leggen. Hierbij kan koolstof dan ook nog eens omgezet worden tot CO2, die ook energie levert. Doordat de drager verdwijnt is deze batterij niet heroplaadbaar.
3. LoodaccuDe celspanning van deze batterij bedraagt ongeveer 2V. door de cellen in serie te plaatsen kan men 6; 12 of 24 volt verkrijgen. Het heropladen gebeurt door een tegenspanning die verkregen wordt van de alternator en dan op gepaste wijze gestabiliseerd wordt.

E3: verband tussen proton- en elektrontransferreactiesHet formele verband tussen de protontransferreacties, beschreven door de pH van de oplossing, en de elektrontransferreacties, gekenmerkt door hun reductiepotentiaal E, wordt gegeven door de Nernstvergelijking. Wanneer er protonen voorkomen, kan uit de Nernstvergelijking de pH-afhankelijkheid berekend worden. Als men de reductiepotentiaal in functie van de pH plot, krijgt men een verschillende helling naargelang het aantal protonen dat in de halfreactie voorkomt. Dit betekent dat de rol van oxidans en reductans kan omkeren bij een bepaalde pH.Het meer conceptuele verband is dat er in beide reactie een elementair deeltje wordt uitgewisseld: het proton, positief, en het elektron, negatief. Wanneer we met deze tegengestelde lading rekening houden en de reacties schrijven in analoge richting dan zien we dat zuur en oxidans links staan en dat bas en reductans rechts vallen. Een zuur wil namelijk altijd een positieve lading afsplitsen of een negatieve lading opnemen. Zowel een protontransferreactie als een elektrontransferreactie wordt dus gedreven door de neiging tot het bereiken van de edelgasconfiguratie.
Bv: C l2+2e−¿→2C l−¿¿¿HCL→H+¿+C l−¿ ¿¿
Er wordt dus een volledig bezette schil rond Cl gevormd. Voor waterstof leidt dit tot het vormen van H+ dat niet op zichzelf kan voorkomen, het proton komt dan ook gesolvateerd voor: het wordt omgeven door 4 solvent moleculen water die binden met een vrij elektronen paar (H9O4
+) zodat ook het proton de octetstructuur bekomt. Dit is een afwijkende octetstructuur omdat er geen eerste schil voorkomt met 1 elektronenpaar. Dit is een gevolg van de lading die leidt tot een contractie van de schillen. Bij het ontbreken van de lading is er wel een eerste schil. Men kan dus zeggen dat OH- zowel reductans als base is en H+ zuur en oxidans. De richting van de halfreactie blijft wel afhangen van de sterkte van het andere reagens. Water kan dus zowel base, reductans, zuur of oxidans zijn wanneer er respectievelijk een sterker zuur, oxidans, base of reductans aanwezig is. Voor water zijn de belangrijkste reductiepotentialen -0,41V voor de reductie van H+ naar H2 en O,82V voor de oxidatie van H2O tot O2. Dit heeft als gevolg dat wanneer water de rol van solvent speel voor bv een reductans met een reductiepotentiaal kleiner dan -0,41V zonder sterker oxidans, water gereduceerd wordt waterstofgas.
Wanneer de verschijningsvorm van het atoom met veranderende oxidatietrap zelf een functie is van het milieu kan men niet zomaar de standaardreductiepotentiaal herberekenen, men heeft dan ook een zuur-base evenwicht tussen de geprotoneerd en gedeprotoneerde vorm. Uit de opgave kunnen we na afleiding (p216-218) besluiten dat van de 2 standaardreductiepotentialen van dezelfde reductiehalfreacties maar de ene keer in zuur milieu en de andere keer in basisch milieu de zuurdissociatieconstante van het evenwicht tussen de geprotoneerde en gedeprotoneerde vorm valt te berekenen. Dit kan men analoog toepassen op de Kb.Afhankelijk van de omstandigheden kan de richting van een redoxreactie omgekeerd worden. Dit is in het algemeen het geval wanneer de reductiepotentiaal E0 van de 2 redoxhalfreacties een verschillende pH-afhankelijkheid vertonen. Waar de E(pH) rechten elkaar kruisen is ∆E0= 0 en treedt er geen reactie op.Wanneer er verschillende oxidatietrappen mogelijk zijn van een bepaald element zijn er reductiehalfreacties te schrijven voor elk van de oxidatietrappen. De verschijningsvorm met de hoogste oxidatietrap zal het sterkste zuur en oxidans zijn, die met het laagste oxidatietrap zal het de sterkste base/ reductans zijn.

Berekenen van de standaardreductiepotentiaal van een gecombineerde halfreactie:1. We bekijken de verandering in vrije energie met een tussenliggende stap
Afleiding:
Zie slide 39-41 voor de verdere uitwerking van een voorbeeld.2. Om complicaties met moleculen die meer als 1 atoom hebben dat van OT verandert, kan
men voor het berekenen van de E0-waarde doen alsof men werkt met halve moleculen. Zo
kan men n gelijkstellen aan ∆OT en krijgt men E3=∆OT 1E1+∆OT 2E2
∆OT3.
Bv:
Men met deze formules in meerdere stappen werken om zo een oplossing te verkrijgen van een gecombineerde reductiehalfreactie die meerdere OT bevat. Soms kan het ook zijn dat bepaalde vormen van een atoom met een oxidatietrap niet stabiel zijn. Deze vorm zal dan disproportioneren naar een hoger en een lagere oxidatietrap. Met andere woorden, als een molecule disproportioneert zal het een spontane redoxreactie ondergaan waarbij het molecule zowel als oxidans en reductans optreedt. Dit gebeurt als de vrije energie negatief is, bij 1 elektron reacties kan men meer specifiek zeggen als E1 E2.
Katalyse bij redoxreactiesWe kunnen nu thermodynamische berekeningen uitvoeren op redoxreacties, dit zegt ons echter nog niets over de effectieve snelheid van de reactie. (zie voorbeeld p 231-232 van H2O2)Een katalysator zal de redoxreactie versnellen. Een thermodynamische voorwaarde die we kunnen stellen is dat alle reductiehalfreacties als tussenreactie mogelijk zijn als hun standaardreductiepotentiaal ligt tussen de waarden van het redoxstelsel dat men wilt katalyseren.Als men dan de bv de elektrolyse van water wilt katalyseren moet men wel opletten dat het zout dat men toevoegt als elektroliet geen ionen heeft die makkelijker oxideren dan zuurstof of makkelijker reduceren dan waterstof. De elektrolyse een industrieel belangrijke bereidingstechniek van elementen in reactieve oxidatietrappen. Zo zorgt de elektrolyse van NaCl of MgCl2 voor de vorming van het reactieve Cl2. Men gebruikt hiervoor een Downscel, hierin moeten de zouten wel gesmolten zijn en niet opgelost. Een ander voorbeeld van industriële elektrolyse is het bereiden van een zuiver

metaal, dit gebeurt door reductie van het metaalkation met positieve oxidatietrap naar het metaal met oxidatietrap 0. Zo elektrolyseert men zinksulfaat om tot metallische zink te bereiden. Men reduceert hierbij Zn2+ tot Zn en tegelijkertijd gebeurt een oxidatie, of naar zuurstof of met overspanning bij geconcentreerd sulfaat naar peroxodisulfaat (S2O8
2-). De benodigde tegenspanning wordt bepaald door de effectieve concentratie van de ionen, de pH, de temperatuur, het elektrodemateriaal en de celweerstand. Deze elektrolyse is typisch voor de bereiding van non-ferro metalen.
Redoxindicatoren Deze indicatoren worden gebruik om de celpotentiaal (van een halfcel) benaderend te bepalen en het EP te bepalen indien dit niet potentiometrisch gebeurt of het titrans zelf geen kleuromslag vertoont. Ze zijn goed oplosbare zwakke oxidantia of reductantia waarvan de de geoxideerde of gereduceerde vorm een sterk verschillende kleur hebben. De indicator wordt toegevoegd in geringe concentratie aan de oplossing. De concentratie van de indicator is voldoende laag zodat deze niet mee doet in het redoxstelsel en dus niet de celpotentiaal bepaalt. Het is zelfs omgekeerd de celpotentiaal bepaalt de concentraties van de geoxideerde en gereduceerde vorm van de indicator. De verhouding van de concentratie van de gereduceerde vorm tov de concentratie van de geoxideerde vorm wordt bepaald in door de heersende celpotentiaal E en de standaardreductiepotentiaal E0 voor de redoxindicator.
log[ℜd ind ][Ox ind ]
= n0,059
(E oxred
0 −E)
RedoxtitratiesOok hier gelden de algemene voorwaarden voor titraties:
I. Alle reagentia moeten oplosbaar zijn in hetzelfde solvent(fysisch)II. De gebruikte reactie moet sterk aflopend zijn(thermodynamisch)
III. De gebruikte reactie moet voldoende snel zijn (kinetisch)Aan de hand van voorwaarde kunnen we stellen dat we dus altijd een sterk titrans (oxidans of reductans) moeten nemen want net zoals bij een pH-titratie (sterk+ zwak) zal zwak zuur+zwak base niet lukken. Volgens de wet van Nernst zal een oplossing met een bepaalde concentratie aan reductans een bepaalde potentiaal vertonen. Wanneer alleen het reductans/oxidans aanwezig is zou de potentiaal in principe - of + zijn. Door onzuiverheden wordt waarde niet ± maar een relatief kleine of grote waarde. De analyse van de halfcelpotentiaal gedurende het verloop van de titratie wordt gemaakt in functie van de fractie f van weggereageerd reductans (met f= v/V waarin v het toegevoegd volume is en V het beginvolume). De fractie wordt gelijk aan 1 wanneer (bij oplossingen met gelijke beginconcentraties) evenveel volume oxidans v toegevoegd is als er oorspronkelijk reductans V was.
Tijdens de titratie maar voor het EP geldt: Ered /ox=E red/ox0 −0,059
nlog 1−f
f .
Voorbij het EP moet men rekenen met het titrans dat men toevoegd als red/ox. Bij het EP is f=1 en moet men een aparte berekening uitvoeren omdat men de log(0) krijgt, er geldt:
[Red1]=[Red2][O1]=[Ox2]
Kev=[Red1]2/[Ox1]2= 10(n∆E0)/(0,059)
Ered/ox= Ered/ox0 +(0,059)/2* Log (Kev)1/2
Dit is de theorie bij een titratie waarbij de beide redoxstelsel hetzelfde aantal elektronen uitwisseld. Als er niet dezelfde elektronen worden uitgewisseld zal de 2de helft van de titratiecurve veranderen. Zo zal bij het EP de potentiaalsprong groter worden naarmate we in zuurder milieu werken, het

eerste deel van de curve is pH-onafhankelijk, het 2de niet. De potentiaal bij het EP wordt gegeven
door de vergelijking E=
n∗Ered 1 /ox 10 +n∗E red 2/ox2
0
ntot−0,059
n totlog
[O x1 ][ ℜd1 ]
∗[O x2 ]
[ ℜd2 ]∗[H ].
E4: neerslag reactiesHet oplossen van een vaste stof in een solvent geeft een homogeen mengsel. De graad van vermenging tussen vaste stof en solvent wordt tijdens het oplossen doorgedreven tot op het moleculaire niveau. Elk molecule van de vaste stof wordt uiteindelijk omgeven door solventmoleculen. Het oplossen kan enkel gebeuren mits er voldoende intermoleculaire interactie is tussen het solvent en de op te lossen stof. Bij water zullen vooral elektrostatishe interacties en waterstofbruggen van belang zijn. Als er echter teveel op te lossen stof toegevoegd wordt ten opzichte van het solvent zal er een deel blijven bestaan als vaste stof. Men noemt de oplossing dan verzadigd. Een aantal moleculen combineren een alifatische staart met een polaire geladen kop. De staart is hydrofoob, terwijl de kop hydrofiel is. Deze moleculen noemt men amfifiel of amfipatisch en zijn in staat om vet te dispergeren in water: de alifatische staart interageert met het vet en maakt het oppervlak ervan hydrofiel zodanig dat het vet “oplost” in water. Vanwege zijn polaire aarde is water een goed solvent voor ionbindingen. Het zijn de aparte anionen en kationen in het solvent voorkomen en gestabiliseerd worden. Een zoutmolecule zal in water dissociëren in zijn ionen, als de ladingsdichtheid van hiervan niet te groot is, kunnen ze apart voorkomen en worden ze gestabiliseerd. Als de ladingsdichtheid te groot is ka het zout niet dissociëren en lost het niet op, een voorbeeld hiervan is Ca3(PO4). De oplosbaarheid van een stof is de maximale hoeveelheid van de stof die kan opgelost worden in een bepaald solvent. De maximale hoeveelheid is de heoveelheid die in een verzadigde oplossing voorkomt. Voor moeilijk oplosbare zouten wordt de molariteit genomen. De molariteit van de opgeloste stof in een verzadigde oplossing is dan de oplosbaarheid S van dit zout. Het oplosbaarheidsproduct is gelijk aan het product van de concentraties van de opgeloste ionen verheven tot de macht gelijk aan hun stoichiometrische coëfficiënt: K sp=¿¿. De oplosbaarheid in zuiver water wordt berekend als de concentratie van de eenwaardige ionen, bv SCaF2= [Ca2+]=1/2[F-]. Uit de gegeven oplosbaarheid kan men het oplosbaarheidsproduct berekenen en omgekeerd. De oplosbaarheid wordt beïnvloed door de aanwezigheid van één van de ionen die door de oplossingsreactie gevormd wordt. De waard van het oplosbaarheidsproduct is een constante die bij een bepaalde temp altijd dezelfde waarde heeft. De oplosbaarheid van bv. AgCl in 0,1 mol NaCl is lager dan in zuiver water. Een dergelijk effect bij oplosbaarheden van zouten wordt het “gemeenschappelijk-ion effect” genoemd.
Het vormen van een selectieve neerslag of argimetrie:Wordt uitgelegd adhv Ag2CrO4 en AgCl, waarbij AgCl eerst wordt neergeslagen.
1. Tot welke Ag+-concentratie komt er geen AgCl-neerslag voor?¿
Bij concentratie kleiner dan X zal er geen enkele neerslag zijn, groter dan deze concentratie heeft men zilverchloride neerslag.
2. Vanaf welke Ag+-concentratie slaat zilverchromaat neer?¿
Bij deze concentratie heeft men zilverchromaat dat neerslaat3. Hoeveel chloride ionen zijn dan nog in oplossing?
¿

Als deze concentratie te verwaarlozen is tov de beginconcentratie, heeft men “alle” zilverchloride laten neerslaan. Men kan dus de rode neerslag gebruiken als indicator voor een neerslagtitratie met Cl- in een zout.
Neerslagreacties worden ook beïnvloed door het toevoegen van een reagens dat een minder oplosbare verbinding doet ontstaan. Dit gebeurt bv als men bij lood(2)chloride voldoende joodionen toevoegt, dan krijgt men een omzetting naar lood(2)jodide.
Verband tussen pH en oplosbaarheidBij een neerslagreactie waarin als anionen de geconjugeerde base van zwakke zuren betrokken zijn verschuift de ligging van het evenwicht in functie van de pH. Een neerslag kan uiteraard rechtstreeks opgelost worden door een inwerking van een sterk zuur dat wegreageert met een sterke geconjugeerde base ter vorming van een zwakker zout of de algemene regel van een sterk zuur verdrijft een zwak zuur uit zijn zout.
Metalen met eenzelfde concentratie neerslaan bij verschillende pH-waarden: zie voorbeeld p258-259
Men kan kationen scheiden door het selectief neerslaan van metaalionen met de H2S-methode bij een bepaalde pH. Hiervoor gebruikt men de methode van argimetrie waarbij men dan nog een buffer moet aanmaken om de pH constant te houden. Zie p260-261
De oplosbaarheid van Mg(OH)2 in zuiver water is klein. Men kan deze verhogen door het toevoegen van een reagens dat met 1 der opgeloste ionen wegreageert. Voor Mg(OH)2 kunnen ammoniumionen met de hydroxide ionen ammoniak en water vormen dat uit de oplossing kan ontsnappen. Hiervoor berekend men eerst het maximum aantal OH-ionen adhv Ksp, dan houdt men rekening met de Kb van het ammoniumevenwicht waaruit men dan met de concentratie ammonium haalt. Wanneer we een algemene halfreactie, als reductie geschreven, van een metaal M beschouwen samen met de neerslagvorming van het zout MA dan zal door neerslagvorming de concentratie oxidans(metaalkation) afnemen en ook het oxiderend vermogen terwijl het reducerend vermogen toeneemt.Bv:
De concentratie van het oxidans is in het 2de geval hoger en dus ook het oxiderend vermogen. Men kan ook zien dat uit het meten van een celpotentiaal de waarde van heel kleine oplosbaarheidsproducten kan bepalen.

De calomelelektrode (SCE) is een praktische uitvoering van een standaardelektrode met een welbepaalde potentiaal, de standaard waterstof elektrode met potentiaal 0V is moeilijk te realiseren. De SCE berust op :
De reductiepotentiaal voor deze halfcel kan men dus constant houden als met Cl-concentratie constant houdt. Dit kan gebeuren in een oplossing die verzadigd is aan goed oplosbare KCL. Bij verzadiging mag men een effectieve concentratie van 1,87M Cl-ionen nemen, dit geeft een halfcelpotentiaal van 0,25V.Aan deze referentie elektrode kunnen we ook een concentratiegevoelige halfcel verbinden om een potentiaalverschil te meten dat in functie is van de concentratie van een oxidans of reductans. De celpotentiaal wordt gegeven door ∆E= ESCE-EH+/H2=0,25+ 0,059pH

E5: complexreactiesComplexreacties worden gedefinieerd als evenwichtsreacties tussen een complex ion en zijn bestanddelen. Deze bestanddelen zijn een positief centraal ion(kation van een tranisitiemetaalelement) en een aantal omringende anionen of neutrale moleculen met een vrij elektronenpaar. Dit laatste noemt men de liganden van het complex. Men kan de evenwichtsreactie schrijven als een associatie die wordt gedefinieerd door een stabiliteitsconstant Kst of men kan ze schrijven als een dissociatie met de dissociatieconstant KD. Deze 2 zijn elkaars inverse. Aan een complex moet men meestal nog een anion toevoegen om een neutraal moleculen te bekomen.De associatie/dissociatie gebeurt meestal stapsgewijs maar men kan altijd een totale associate/dissociatie reactie schrijven waarbij Kst/KD= Kst/D 1+Kst/ D 2+…. De indeling van complexen gebeurt meestal via het aantal bindingsplaatsen per ligand. Een nevenindeling gebeurt daarna op basis van de lading van het ligand:
I. Ééntandig ligand/ monodentaat: 1 bindingsplaats Neutrale moleculen: bv NH3, CO, NO, H2O Eenwaardig negatief geladen: OH-, F-, Cl-, Br-,… Tweewardig negatief geladen: O2-, S2-,…
II. Tweetandig ligand/ bidentaat: 2 bindingsplaatsen Neutraal molecule: H2N-CH2-CH2-NH2
Tweewaardig negatief: -OOC-COO-, -OOC-CH2-CH2-COO-,…III. Drietandig ligand/ tridentaat: 3 bindingsplaatsen
Bv: H2N-CH2-CH2-NH-CH2-CH2-NH2
IV. Zestandig ligand/ hexadentaat: 6 bindingsplaatsenBv: EDTA4-: ethyleendiaminetetraacetaat
In de benaming wordt het aantal liganden weergegeven door mono, di, tri, tetra, penta of hexa. De liganden worden alfabetisch benoemd maar de telwoorden zijn niet bepalend. Daarna komt het centrale metaalkation waarvan de OT in haakjes wordt weergegeven. Negatieve complexen krijgen een de uitgang –aat.Het centrale metaalkation is vaak een transitiemetaalkation. Nochtans zullen alle kationen in meer of mindere mate complexreacties ondergaan. Een speciale klasse van complexe ionen zijn de tetrahydroxocomplexen van amfotere elementen, deze kationen zullen bij het toevoegen van een base eerste een onoplosbare hyrdoxideneerslag vormen maar bij verder toeveogen van de base een oplosbaar tetrahydroxocomplex. Het coördinatiegetal is het aantal atomen (van liganden) dat rechtstraak op het centrale metaalkation is gebonden. Het is dus gelijk aan het aantal ééntandige liganden maar het is verschillend van het aantal liganden wanneer er meer dan 1 bindingsplaats is.Een van de drijfveren om het vormen van een complex is het spreiden van de lading over een groter volume. De transitiemetaalkationen vertonen in het algemeen een grotere neiging tot het vormen van complexen, dit komt omdat deze kationen een gedeeltelijk gevulde d-orbitalen hebben waarin het vrije elektronenpaar terecht kan, hier is de drijfveer dus het bereiken van de edelgasconfiguratie (voobeelden zie p 278). Wanneer een complex ion gevormd zou worden dat een elektron teveel heeft voor een edelgasconfiguratie dan zal dit complex ion gemakkelijker geoxideerd worden dan het niet-gecomplexeerde kation.
Factoren die Kst beïnvloeden1. Factoren vanuit het centrale metaalkation
Hoe groter de lading op het niet-gecomplexeerde centrale metaalkation, hoe stabieler het gevormde complex
Hoe kleiner het volume van het centraal kation alleen, hoe groter de stabiliteitsconstante
Samenvattend: hoe groter de ladingsdichtheid van het niet gecomplexeerde kation, hoe groter de stabiliteitsconstante voor de vorming van een complex ion.

2. Factoren vanuit het ligand Een negatief geladen ligand resulteert in een stabieler complex ion want met een
negatief geladen ligand kan tegelijkertijd Coulombinteractie met het positief geladen centrale kation en neutralisatie van de lading gebeuren. Met een neutraal ligand is dit afwezig.
Hoe sterker de geconjugeerde base, hoe stabieler het complexe ion. De stabiliteit van een complex ion is groter voor een groter aantal bindingsplaatsen.
Er moeten voor de dissociatie namelijk 2 of meer bindingen gebroken worden, wat meer energie vraagt en dus thermodynamisch moeilijker is. Samenvattend kunnen we zeggen dat tweetandige liganden stabielere complexen vormen dan ééntandige liganden.
Als we aan een complex andere ionen toevoegen kan er een liganduitwisselingsreactie gebeuren waarbij we een andere complex verkrijgen. De liganduitwisselingsreactie is des te sterker naarmate het verschil in stabiliteitsconstante groter is. In realiteit zijn alle kationen in oplossing min of meer gesolvateerd, dus ook een gewone complexreactie is een liganduitwisselingsreactie waarbij het zwakke ligand water wordt verdreven.Zie p284 voor loodvergiftingAls gevolg van de mogelijkheid voor een positief geladen centraal metaalkation tot het uitwisselen van neutrale solventmoleculen voor negatief geladen ligandmoleculen kan een neutraal complex ion als tussenproduct ontstaan. Dit product is een goed oplosbaar zout maar het dissocieert niet wat het een slecht elektroliet maakt. Vooral transitiemetaalkationen bv Hg2+ vormen dergelijke stabiele complexen met anionen als liganden bv Cl-. Een metaalkation dat kan neerslaan in een moeilijk oplosbare verbinding kan in oplossing gebracht worden door het toevoegen van liganden. Soms kan hetzelfde anion aanleiding geven tot een neerslag en een complex ion. Meestal is dit het geval voor de kationen die amfotere hydroxiden vormen met OH- als ligand.De vorming van complexen met het ligand OH- zal uiteraard beïnvloed worden door de pH. Anderzijds zal de pH veranderen als een base ook als ligand kan optreden omdat er een kation aanwezig is in de oplossing. Heel in het algemeen zijn echter de centrale kationen die goede complexe ionen vormen zure kationen in water. De lading wordt uitgesmeerd over het complexe ion met water als ligand. Door het afgeven van een proton kan de lading echter verlaagd worden. omgekeerd zijn de liganden basen. Als ligand hebben zij een vrij elektronenpaar dat ofwel een proton ofwel een metaalkation met hoge ladingsdichtheid kan binden. Het feit dat het proton in waterig milieu niet als dusdanig kan voorkomen maar opgenomen wordt door water als base en voorkomt als H3O+ kan ook gezien worden als een gevolg van complexatie met water als ligand. Dan kunnen we zelfs stellen dat het proton in waterig milieu niet als H3O+ kan voorkomen maar eerder als H9O4
+.
Het verband tussen complexreacties en protontransferreacties heeft ook weer gevolgen bij de instelling van mulitpele evenwichten. Een oplossing van een goed oplosbaar zout met zuurrest is zwak zuur tgv de zure hyrdolyse. Het toevoegen van een goed oplosbar zout van een transitiemetaalkation zal de pH verder verlagen. Maar eerst ondergaat het 2de zout ook de zure hydrolyse. Het toevoegen van het transitiemetaalkation aan de oplossing van het zout met zuurrest zal ook een complexatie toelaten of beter een ligandenuitwisselingreactie. Deze reactie gebruikt de niet-zuurrest van het 1ste waardoor het eerste evenwicht naar rechts verschuift met als gevolg dat er nog een toename is van de pH.
Wanneer een centraal metaalkation, dat tevens oxidans is, gecomplexeerd wordt zal zijn effectieve concentratie verlagen. Dit zal (Nernstvergelijking) de redoxpotentiaal doen afnemen. Dit is het verband tussen complexatiereacties en elektrontransfereacties.







![bibliotecaweb20 [licensed for non-commercial use only] / Homebibliotecaweb20.pbworks.com/w/file/fetch/86022718... · 2020-06-20 · Luísa Ducla Soares ilustrações Susana Oliveira](https://static.fdocuments.nl/doc/165x107/5faac9ac0a465769352b33a2/bibliotecaweb20-licensed-for-non-commercial-use-only-2020-06-20-lusa-ducla.jpg)












