TECHNISCHE UNIVERSITÄ T MÜ NCHENmediatum.ub.tum.de/doc/1207075/1207075.pdf · Ü berexprimierung...
103
TECHNISCHE UNIVERSITÄ T MÜ NCHEN Chirurgie des Klinikums rechts der Isar Relevanz des Kinesin Motor Proteins Kif20a im Pankreaskarzinom Daniela Stangel Vollständiger Abdruck der von der Fakult ät fü r Medizin der Technischen Universit ät Mü nchen zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. Radu Roland Rad Prü fer der Dissertation: Priv.-Doz. Dr. Murat Mert Erkan (schriftliche Beurteilung) Univ.-Prof. Dr. Jö rg Kleeff (mü ndliche Prü fung) Univ.-Prof. Dr. Bernhard Kü ster Die Dissertation wurde am 17.04.2014 bei der Technischen Universit ät Mü nchen eingereicht und durch die Fakultät fü r Medizin am 17.09.2014 angenommen.
Transcript of TECHNISCHE UNIVERSITÄ T MÜ NCHENmediatum.ub.tum.de/doc/1207075/1207075.pdf · Ü berexprimierung...

TECHNISCHE UNIVERSITÄ T MÜ NCHEN
Chirurgie des Klinikums rechts der Isar
Relevanz des Kinesin Motor Proteins Kif20a im Pankreaskarzinom
Daniela Stangel
Vollständiger Abdruck der von der Fakultät fü r Medizin der Technischen Universität Mü nchen zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften
genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. Radu Roland Rad Prü fer der Dissertation: Priv.-Doz. Dr. Murat Mert Erkan (schriftliche Beurteilung)
Univ.-Prof. Dr. Jö rg Kleeff (mü ndliche Prü fung)
Univ.-Prof. Dr. Bernhard Kü ster
Die Dissertation wurde am 17.04.2014 bei der Technischen Universität Mü nchen eingereicht und durch die Fakultät fü r Medizin am 17.09.2014 angenommen.


Widmung
Fü r meine Familie


Zusammenfassung
I
Zusammenfassung
Das Pankreaskarzinom ist eine der fü nfhäufigsten Todesursachen in westlichen Ländern.
Aufgrund der relativen 5- Jahres Ü berlebensrate von annähernd 5% ist es wichtig, neue
therapeutische Strategien zu entwickeln um neue Kandidatengene mit funktioneller
Signifikanz in der Karzinogenese des Pankreas zu entwickeln. Im Rahmen des Nationalen
Genomforschungsnetzwerk Projekts “PaCa-Net” wurden Metaanalysen von „high-
throughput“ Expressionsprofilen durchgefü hrt. Fü r die vorliegende Arbeit wurde das Kinesin
Motor Protein Kif20a (Kif20a) ausgewählt, das in Vorversuchen eine 18- fache
Ü berexprimierung im Tumorgewebe gegenü ber Normalgewebe des Pankreas zeigte. Die
Tatsache, dass Kif20a eine wichtige Rolle beim Organelltransport, bei der Zellmotilität und
diversen Prozessen des Zellzyklusses spielt, sowie die stark erhö hte Expression im
Tumorgewebe macht Kif20a zu einem interessanten Zielgen fü r die Krebsforschung. Mit
dieser Arbeit sollte das Kinesin Motor Protein Kif20a sowohl hinsichtlich seiner Expression in
verschiedenen Pankreasgeweben als auch bezü glich seiner Funktion genauer untersucht
werden. Dafü r wurden im Einzelnen histologische Analysen von Normalgewebe, chronischer
Pankreatitis sowie Tumorgewebe durchgefü hrt. Dabei konnte sowohl eine stärkere
zytoplasmatische als auch nukleäre Kif20a Färbung der Krebszellen gegenü ber dem
Normalgewebe festgestellt werden. Die Analyse der Kif20a Expressionsprofile von duktalen
Adenokarzinomzelllinien (PDAC) sowie neuroendokrinen (NET) Zelllinien ergab ein sehr
heterogenes Muster, sowohl auf Transkriptions- als auch auf Proteinebene.
Interessanterweise fü hrte sowohl die Ü berregulierung mittels Expressionsvektor als auch die
Suppression des Kif20a Gens mittels siRNA-Knockdown-Strategie zu einer verminderten
Proliferations- sowie Migrationsfähigkeit von PDAC Zelllinien. Zudem konnte in Kif20a
supprimierten Tumorzellen eine verminderte Invasivität sowie Motilität festgestellt werden.
Je nach Stärke der Kif20a Runterregulierung waren die Zellen in der S-Phase des Zellzyklusses
arretiert, oder gingen in Apoptose. Tumorzellen, die mit dem Expressionsvektor transfiziert
wurden zeigten einen bi- bzw. multinukleärer Phänotyp. Erste in vitro Versuche mit dem Ziel
einer kombinierten Chemotherapie (Taxol, Gemcitabine) zeigten, dass ein zusätzlicher Effekt
auf die Proliferations- sowie Migrationsfähigkeit der Tumorzellen, bei gleichzeitiger
Suppression von Kif20a, ermittelt werden konnte. Des Weiteren wurde der Effekt der Kif20a
Expression auf das Ü berleben von Patienten mit duktalem Adenokarzinom analysiert (n=89).
Die Ergebnisse zeigten, dass es keinen Unterschied bzgl. der Kif20a Expression und dem

Zusammenfassung
II
Patientenü berleben gab. Vergleichsfärbungen von Kif20a und PCNA („Proliferating cell
nuclear antigen“) in den Patientenproben zeigte keine Korrelation zwischen Proliferation
und Kif20a Expression. In der vorliegenden Arbeit konnte gezeigt werden, dass Kif20a einen
deutlichen Einfluss auf die Pankreaskarzinogenese hat. Sowohl die Proliferation, Migration
als auch Motilität in Tumorzellen konnte nach Runterregulierung von Kif20a signifikant
reduziert werden. Jedoch konnte auch gezeigt werden, dass eine Ü berexpression des Kif20a
Gens nicht mit einer schlechten Prognose fü r Patienten mit duktalem Adenokoarzinom
korrelierte. Nichtsdestotrotz konnte in anderen Studien gezeigt werden, dass Kif20a
aufgrund seiner signifikanten Ü berexprimierung in Tumoren ein gutes Kandidatengen fü r die
Immuntherapie darstellt.

Summary
III
Summary
Pancreatic cancer is the fifth most common cancer to cause death in western societies.
Because of an overall 5- year survival rate of approximately 5%, it is almost important to
develop novel therapeutic strategies. In order to identify new candidate genes with
functional significance in pancreatic cancer biology, a meta-analysis of publically available
high-throughput gene expression datasets was performed within the framework of the
German National Genome Research Network project “PaCa-Net” . Here, we focused on the
kinesin family member 20A (Kif20A), which was consistently reported to be 18- fold over-
expressed in pancreatic cancer compared to normal pancreas. Since Kif20a is necessary for
organelle transport, cell cycle and motility, to generate mechanical force within the cell and
is strongly upregulated in pancreatic cancer, it is a good candidate as a “drugable target” in
pancreatic cancer. Within this dissertation the kinesin motor protein Kif20a was
characterized concerning its expression in different pancreatic tissue as well as regarding its
function in carcinogenesis. When analyzing sections of normal pancreas, chronic pancreatitis
and pancreatic cancer, a predominant staining of Kif20a in the cytoplasm as well as in nuclei
of cancer cells was observed. The RNA and protein expression of Kif20a in cell lines isolated
from pancreatic ductal adenocarcinoma (PDAC) and neuroendocrine tumor (NET) achieved a
very heterogeneous profile. Interestingly both over- expression and knock down of Kif20a in
PDAC cells caused a diminished proliferation and migration ability. Additional Kif20a knock
down in tumor cells leads to a reduced invasiveness and motility. Transfected cells where
either arrested in the S-phase of the cell cycle or went into apoptosis. Over- expression of
Kif20a led to a bi- and accordingly multi-nuclear phenotype of tumor cells. In evaluating the
benefit of Kif20a expression as a part of a combination chemotherapeutic therapy (taxol,
gemcitabine), it was shown, that combination of chemotherapeutics with Kif20a over-
expression or knock down, led to an additional inhibitory effect of proliferation and
migration of PDAC cells. Next to in vitro studies the effect of Kif20a expression and median
survival of patients with PDAC (n=89) was analyzed. Patients with higher Kif20a expression
had the better median survival compared to patients with lower Kif20a expression.
Additional PCNA (Proliferating cell nuclear antigen) stainings were arranged, but showed no
correlation with expression status of Kif20a. In conclusion, our in vitro analysis
demonstrated, that proliferation, motility and migration could be significantly reduced,
when Kif20a was silenced in pancreatic cancer cells. However, in a clinical cohort of PDAC

Summary
IV
patients, overexpression did not correlate with worse survival. Nonetheless, due to its
significant overexpression in cancer, Kif20a might be a good candidate for immunotherapy,
as has been shown for other tumor entities.

Abkü rzungen
V
Abkü rzungen
aa Amino acid Abb. Abbildung ATP Adenosintriphosphat bp Basenpaare bzw. beziehungsweise Cdc cell division cycle protein homolog CT Computertomographie Da Dalton DAPI 4´,6-Diamidino-2-phenylindol DNA Desoxyribonukleinsäure D.S. Daniela Stangel E.coli Escherichia coli ER Endoplasmatisches Retikulum FACS Fluorescence activated cell sorting FCS Fetal calf serum GAPDH Glycerinaldehyd-3-phosphat-Dehydrogenase h Stunde HCl Wasserstoffchlorid (Salzsäure) HPDE Human pancreatic ductal epithelial HPRT Hypoxanthin-Phosphoribosyl-Transferase IF Immunfluoreszenz IHC Immunhistochemie kbp Kilobasenpaare kDa Kilodalton M.E. Mert Erkan min Minute MRT Magnetresonanztomographie NaCl Natriumchlorid NaOH Natriumhydroxid NET Neuroendocrine Tumor NGS Normal goat serum nm Nanometer PCR Polymerase chain reaction PCNA Proliferating cell nuclear antigen PDAC Pancreatic ductal adenocarcinoma Pen Penicillin PFA Paraformaldehyd PI Probidiumiodid PLK Polo like Kinase 1 RNA Ribonukleinsäure RNAseA Ribunuklease A Strep Streptomycin Tab. Tabelle TM Schmelztemperatur vgl. vergleiche z.B. zum Beispiel z.T. zum Teil α -SMA α -smooth muscle actin

Inhaltsverzeichnis
VI
Inhaltsverzeichnis
Zusammenfassung ................................ ................................ ................................ ............. I
Summary ................................ ................................ ................................ .......................... III
Abkü rzungen ................................ ................................ ................................ .................... V
Inhaltsverzeichnis ................................ ................................ ................................ ............ VI
1 Einleitung ................................ ................................ ................................ ..................... 8
1.1 Pankreas ................................ ................................ ................................ ............................ 8 1.1.1 Aufbau und Funktion ............................................................................................................................ 8 1.1.2 Pankreaskarzinom ................................................................................................................................ 9 1.1.3 Epidemiologie und Risikofaktoren ...................................................................................................... 11 1.1.4 Therapieverfahren .............................................................................................................................. 12 1.1.5 Gemcitabine........................................................................................................................................ 13 1.1.6 Paclitaxel ............................................................................................................................................. 14
1.2 Motorproteine ................................ ................................ ................................ .................. 16 1.2.1 Kinesine............................................................................................................................................... 16 1.2.2 Kinesin Motorprotein Kif20a .............................................................................................................. 20
2 Material und Methoden ................................ ................................ .............................. 23
2.1 Material ................................ ................................ ................................ ........................... 23 2.1.1 Chemikalien ........................................................................................................................................ 23 2.1.2 Geräte und Verbrauchsmaterialien .................................................................................................... 24 2.1.3 Puffer, Medien und Lö sungen ............................................................................................................ 25 2.1.4 Organismen......................................................................................................................................... 26 2.1.5 Primer ................................................................................................................................................. 27 2.1.6 siRNA und Vektoren ............................................................................................................................ 27 2.1.7 Antikö rper ........................................................................................................................................... 28 2.1.8 Kits und Marker .................................................................................................................................. 28 2.1.9 Software.............................................................................................................................................. 29
2.2 Methoden ................................ ................................ ................................ ........................ 30 2.2.1 Mikrobiologische Methoden .............................................................................................................. 30 2.2.2 Arbeiten mit eukaryotischen Zellen .................................................................................................... 30 2.2.3 Immunologische Methoden ............................................................................................................... 36 2.2.4 Proteinanalytik .................................................................................................................................... 38 2.2.5 In vitro Analysen ................................................................................................................................. 41 2.2.6 Biologische Analysen .......................................................................................................................... 43
3 Ergebnisse ................................ ................................ ................................ .................. 47
3.1 Analyse der Kif20a Expression in humanem Pankreasgewebe und Karzinomzelllinien......... 47 3.1.1 Kif20a Expression in humanem Pankreasgewebe .............................................................................. 47 3.1.2 Kif20a Expression in humanen Karzinomzelllinien des Pankreas ....................................................... 49
3.2 Lokalisation des Kif20a Proteins in PDAC-Zelllinien ................................ ............................ 51
3.3 Etablierung einer temporären Kif20a Suppression in PDAC- Zelllinien................................ . 52 3.3.1 Funktionelle Analysen von Tumorzellen des Pankreas bei gleichzeitiger Kif20a Suppression ........... 54
3.4 Etablierung einer temporären Kif20a Hochregulierung in PDAC-Zelllinien ........................... 59 3.4.1 Funktionelle Analysen von Tumorzellen des Pankreas bei gleichzeitiger Kif20a Hochregulierung .... 61
3.5 Funktionelle Analysen von PDAC- Zelllinien nach Taxolbehandlung bei gleichzeitiger Kif20a Suppression ................................ ................................ ................................ ........................... 63
3.5.1 Einfluss der Doppelbehandlung auf die Proliferation der Tumorzellen ............................................. 64 3.5.2 Effekt der Doppelbehandlung auf die Migration der Tumorzellen..................................................... 66

VII
3.6 Funktionelle Analysen von PDAC- Zelllinien nach Gemcitabinebehandlung bei gleichzeitiger Kif20a Suppression ................................ ................................ ................................ ................. 67
3.6.1 Effekte der Doppelbehandlung auf die Proliferation der Tumorzellen .............................................. 67 3.6.2 Wirkung der Doppelbehandlung auf die Migration der Tumorzellen ................................................ 68
3.7 Funktionelle Analysen von PDAC- Zelllinien nach Behandlung mit Paprotrain ..................... 69 3.7.1 Auswirkung der Paprotrain Behandlung auf die Proliferation der Karzinomzellen ............................ 69
3.8 Korrelation der Kif20a Expression mit dem Überleben von Patienten mit PDAC .................. 70
3.9 Korrelation der Kif20a Expression mit der Proliferation im Gewebe von Patienten mit duktalem Adenokarzinom ................................ ................................ ................................ ...... 72
4 Diskussion ................................ ................................ ................................ ................... 74
4.1 Kif20a Expression im Pankreasgewebe und Tumorzellen des Pankreas ............................... 75
4.2 Funktionelle Analysen von PDAC Zelllinien nach Kif20a Suppression ................................ .. 77
4.3 Funktionelle Analysen von PDAC Zelllinien nach Kif20a Hochregulierung............................ 80
4.4 Funktionelle Analysen von PDAC Zelllinien nach Kombinationsbehandlung von Chemotherapeutika bei gleichzeitiger Kif20a Suppression ................................ ....................... 81
4.5 Wirkung von Paprotrain auf die Proliferation von PDAC Zelllinien ................................ ...... 84
4.6 Korrelation der Kif20a Expression mit dem Überleben von Patienten mit duktalem Adenokarzinom ................................ ................................ ................................ ...................... 85
4.7 Ausblick ................................ ................................ ................................ ............................ 86
5 Literaturverzeichnis ................................ ................................ ................................ ..... 87
6 Tabellenverzeichnis ................................ ................................ ................................ ..... 96
7 Abbildungsverzeichnis ................................ ................................ ................................ . 97
8 Danksagung ................................ ................................ ................................ ................ 99
1 Einleitung
8
1 Einleitung
1.1 Pankreas
1.1.1 Aufbau und Funktion
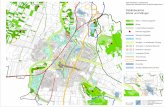
Die Bauchspeicheldrü se, auch Pankreas (von gr. Pá n: ganz und kréas: Fleisch) genannt ist ein
retroperitoneales Organ, dass circa 13- 18 cm lang, 2- 3 cm dick, 5 cm breit und in einem
erwachsenen Menschen circa 80 g schwer ist. Das Organ verläuft quer vor der Wirbelsäule
auf Hö he des 1.-2. Lendenwirbels. Man unterteilt das Pankreas in drei Abschnitte: den
Pankreaskopf (Caput pancreatis), den Kö rper (Corpus pancreatis) sowie die Schwanzregion
(Cauda pancreatis). Dabei windet sich der zweite und dritte Abschnitt des Zwö lffingerdarms
(Duodenum) um die Kopfregion des Pankreas wohingegen die Milz sich in unmittelbarer
Nähe zur Schwanzregion des Pankreas befindet (Abb. 1 A). Hinter der Kopfregion bildet sich
die Pfortader (Vena portae hepatis). Von der Aorta, die hinter dem Pankreas verläuft,
zweigen zwei Arterien ab, die zur Versorgung von Oberbauchorganen, wie der Leber, Milz,
Magen und grö ßeren Darmabschnitten dienen. Durch die gesamte Länge des Pankreas
verläuft der Pankreasgang (Ductus wirsungianus, Ductus pankreaticus major), der durch
kleinere Zuflü sse das Sekret des Pankreas aufnimmt und sich auf Hö he der Kopfregion mit
dem Gallengang (Ductus choledochus) vereint. Beide Gänge mü nden dann an der Papilla
vateri (Papilla duodeni major) ins Duodenum. Der Zufluss von Gallen- bzw. Pankreassaft wird
ü ber einen Schließmuskel, dem Sphincter oddi (Musculus sphincter ampullae
hepaticopancreaticae) reguliert. Dieser ö ffnet sich nahrungsabhängig und entlässt somit
Galle und Pankreassaft ins Duodenum [1].
Abb. 1: Schematische Darstellung des Pankreas. A, Anatomie des Pankreas. B, der exokrine Teil des Pankreas (aus: „Pancreatic cancer biology and genetics“(Nature Review, 2001)[2]).

1 Einleitung
9
Die Mehrheit des Pankreasgewebes (ca. 80%) besteht aus einem exokrinen Teil. Dabei
handelt es sich um ein verzweigtes Netzwerk von azinären und duktalen Zellen, die
Verdauungssäfte produzieren und in den gastrointestinalen Trakt einleiten. Dabei sind die
Azinuszellen in funktionelle Einheiten entlang der duktalen Zellen angeordnet. Zwischen
duktalen und azinären Zellen befinden sich zudem die zentroazinären Zellen (Abb. 1 B).
Durch Stimulation des Magens bzw. des Duodenums produzieren die Azinuszellen ein
Zymogen, dass aus zahlreichen inaktiven Vorstufen von Enzymen (z.B. Trypsinogen,
Chymotrypsinogen, α -Amylase, Ribo- und Desoxyribonukleasen, Lipasen) besteht [1]. Durch
das Einleiten des Sekrets in das duktale Lumen werden die Enzyme anschließend aktiviert.
Neben der Funktion als exokrine Drü se hat die Bauchspeicheldrü se auch endokrine
Funktionen. Der endokrine Anteil besteht aus Inselzellen (Langerhans´sche Zellen), die ü ber
das ganze Organ verteilt sind und die je nach sezerniertem Hormon in die folgenden
Kategorien eingeteilt werden kö nnen: α -Zellen (Glucagon), β-Zellen (Insulin), δ-Zellen
(Somatostatin), ε-Zellen (Ghrelin) sowie die PP-Zellen (pankreatisches Polypeptid). Dabei
regulieren Glucagon und Insulin als Antagonisten den Blutzuckerspiegel. Somatostatin
hemmt die Freisetzung dieser beiden Hormone sowie die exokrine Funktion des Pankreas.
Neben dem appetitanregenden Ghrelin hemmt das pankreatische Polypeptid die Enzym-
und Hydrogencarbonat-Produktion der Bauchspeicheldrü se sowie die Motilität des Darms
und des Gallenflusses [3-5].
1.1.2 Pankreaskarzinom
Das Pankreaskarzinom ist eine Neoplasie der Bauchspeicheldrü se, dessen Art anhand der
Zelldifferenzierung klassifiziert wird. Dabei unterscheidet man zwischen Tumoren, die auf
eine duktale, eine azinäre oder endokrine Differenzierung zurü ckzufü hren sind. Tumore,
duktalen Ursprungs stellen den grö ßten Prozentsatz dar. Neben dem duktalen
Adenokarzinom und seinen Varianten gehö ren zudem das intraduktale papillär-muzinö se
Karzinom, muzinö s-zystischer Tumor, serö ses Zystadenom sowie seltene Tumore wie das
medullär oder gemischte duktale-endokrine Karzinom. Die häufigsten Pankreastumore mit
einer azinären Differenzierung sind das Azinuszellkarzinom [6], sowie das Pankreatoblastom
[7]. Beide Tumore kö nnen dabei neoplastische neuroendokrine Zellen beinhalten, die bei
einer Anzahl von mehr als 30% der Tumorzellpopulation als gemischte Azinus-Endokrine
Karzinome bezeichnet werden [8]. Zu den Tumoren mit endokriner Differenzierung, die circa
1- 2% der bö sartigen Tumore des Pankreas ausmachen, gehö ren das Insulinom (45%), das

1 Einleitung
10
Gastrinom (20%), das Glukagonom (13%), das VIPoma (auch als Verner Morrison Syndrom
bezeichnet) sowie das Somatostatinom (5%) [9]. Das duktale Adenokarzinom, das die
häufigste Krebsart des Pankreas ausmacht ist eine genetische Erkrankung. Die Anhäufung
von Mutationen in Schlü ssel-Onkogenen sowie Tumor-Suppressorgenen lassen diese
Krebsart zu einer sehr komplexen heterogenen sowie genetisch instabilen Krankheit werden
[10-12]. Klinische und histologische Studien gehen davon aus, dass es drei mö gliche
Progressionswege geben kö nnte, die zu einem duktalen Adenokarzinom f ü hren kö nnen [13-
15]. Dabei unterscheidet man bislang zwischen drei gut charakterisierten Vorläuferläsionen:
• Pankreatische Interepitheliale Neoplasien (PanIN)
• Intraduktale Papilläre Muzinö se Neoplasien (IPMN)
• Muzinö sen Zystischen Neoplasien (MCN)
Die bislang am besten untersuchte Vorläuferläsion ist die der PanIN (Abb. 2) [16, 17]. Diese
kö nnen wiederum in drei Grade unterteilt werden: die flache duktale hyperplastische Läsion
PanIN-1A, die papilläre duktale Hyperplasie PanIN-1B, die flache und papilläre Hyperplasie
mit Zellpolarisationsverlust PanIN-2 sowie die schwere duktale Hyperplasie [18].
Abb. 2: Tumorprogressionsmodell des duktalen Adenokarzinoms sowie damit verbundene genetische Aberrationen in zeitlicher Abfolge; modifiziert nach Bardeesy et al. und Chang et al. [2, 19].
1 Einleitung
11
In den frü hen Läsionen wird dabei anfänglich das Onkogen KRAS aktiviert, dessen
Mutationshäufigkeit während des zeitlichen Verlaufs der PanIN- Progression zunimmt. Die
meisten Mutationen manifestieren sich jedoch vor allem in PanIN-3 Läsionen wie zum
Beispiel die Inaktivierung der Tumor-Suppressorgene CDKN2A/INK4A, p16, p53,
DPC4/SMAD4 oder BRCA2 [20]. Dabei weisen ca. 90% der Pankreastumore eine KRAS
Mutation auf, bei 95% kann eine Inaktivierung des CDKN2A Gens mit einem daraus
folgenden p16 Verlust festgestellt werden und bei 50- 75% der Tumore ist das p53 Gen
mutiert. Ein Verlust des SMAD4/MADH4 Gens wird in ~50% der Fälle detektiert [21].
1.1.3 Epidemiologie und Risikofaktoren
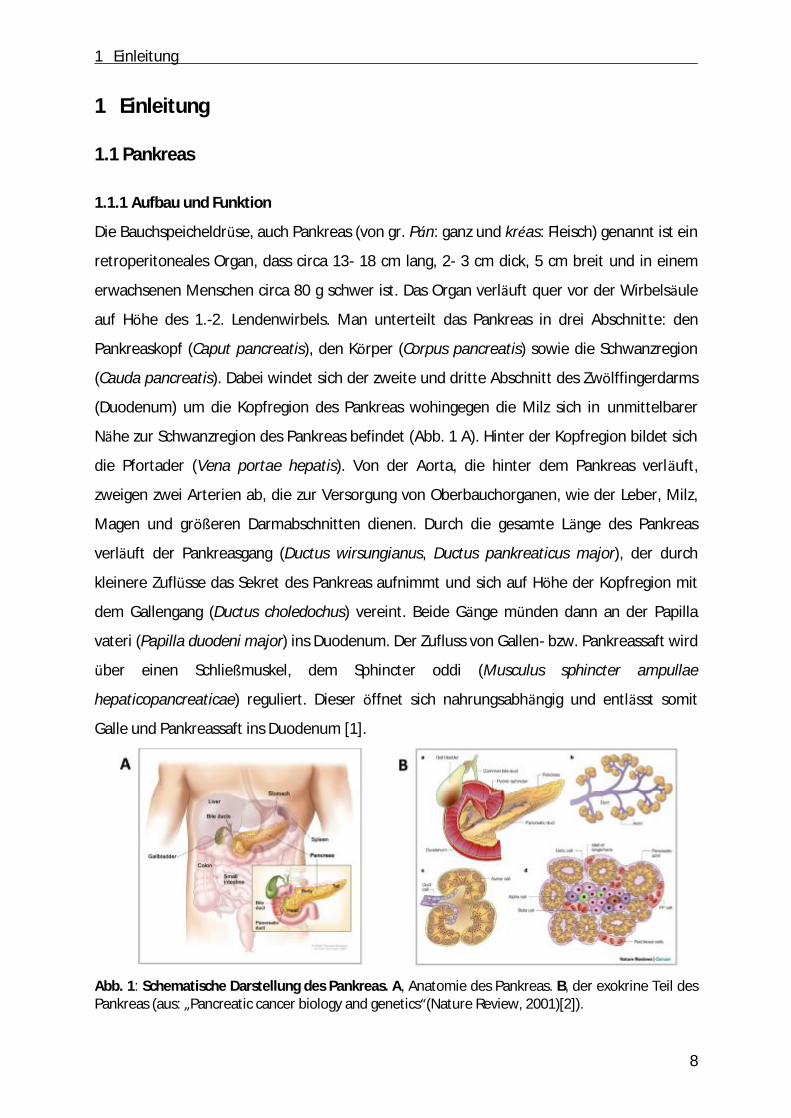
Mit circa 15.000 Neuerkrankungen jährlich stellt das Pankreaskarzinom einer der zehn
häufigsten Tumorarten in Deutschland dar (Abb. 3).
Abb. 3: Prozentualer Anteil der häufigsten Tumorlokalisationen aller Krebsneuerkrankungen in Deutschland 2010 [22].
Aufgrund der schlechten Prognose, die auf die späte Erkennung des Tumors sowie das
teilweise schlechte Ansprechen auf Chemotherapien zurü ckzufü hren ist, ist das
Pankreaskarzinom mit jährlich 7000 Todesfällen in Deutschland die vierthäufigste
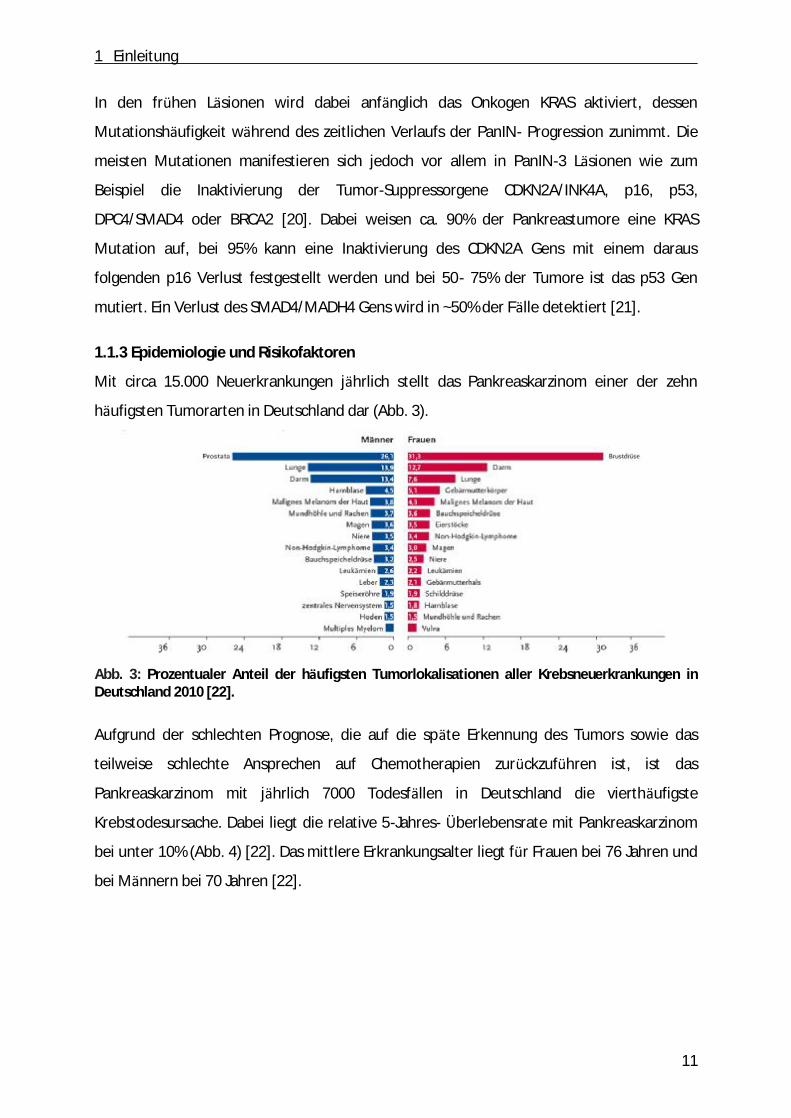
Krebstodesursache. Dabei liegt die relative 5-Jahres- Ü berlebensrate mit Pankreaskarzinom
bei unter 10% (Abb. 4) [22]. Das mittlere Erkrankungsalter liegt fü r Frauen bei 76 Jahren und
bei Männern bei 70 Jahren [22].
1 Einleitung
12
Abb. 4: Vergleich der relativen 5-Jahres-Überlebensraten, nach Lokalisation und Geschlecht, Deutschland 2009 - 2010 (Periodenanalyse) [22].
Zu den gesicherten Risikofaktoren fü r Bauchspeicheldrü senkrebs zählen ein erhö hter
Tabakkonsum oder passiv rauchen sowie starkes Ü bergewicht [23-27]. Alkohol steht nur
indirekt im Verdacht, da die Wahrscheinlichkeit an einer chronischen Pankreatitis zu
erkranken stark erhö ht ist [28, 29]. Patienten mit langjähriger chronischer Pankreatitis oder
Diabetes mellitus Typ II haben eine erhö hte Wahrscheinlichkeit an einem Tumor zu
erkranken [30, 31]. Welche Rolle Umweltfaktoren, beruflich bedingte Schadstoffbelastungen
oder Lebensmittel haben, ist bislang noch nicht geklärt. Studien ü ber den Zusammenhang
von Tee, Kaffee oder der Konsum von Milchprodukten als mö gliche Risikofaktoren konnten
bislang nicht bestätigt werden [32-35]. Zahlreiche Studien konnten zudem genetische
Prädispositionen als Risikofaktoren entschlü sseln. Aktivierung des Onkogens Kras bei
gleichzeitiger Inaktivierung von Tumorsuppressorgenen wie p53, DPC4, p16 oder BRCA2
stehen in direktem Zusammenhang mit der Entstehung von Pankreastumoren [36-40]. Auch
Patienten mit einem Mamma- oder Ovarialkarzinom, zystischer Fibrose oder hereditärer
Pankreatitis haben ein erhö htes Risiko ein Pankreaskarzinom zu entwickeln [41].
1.1.4 Therapieverfahren
Die Frü herkennung von kleinen Pankreastumoren, die anschließend resiziert werden
kö nnen, erhö ht die 5- Jahres Ü berlebensrate der Patienten von 5% auf ca. 20- 30% [42, 43].

1 Einleitung
13
Allerdings sind nur etwa 15- 20% der Fälle resizierbar aufgrund der späten Detektion des
Tumors [44]. Die häufigsten Diagnosearten sind dabei bildgebende Verfahren wie
Computertomographie (CT), endoskopischer Ultraschall oder Magnetresonanztomographie
(MRT). Als Therapie fü r fortgeschrittenen Bauchspeicheldrü senkrebs gelten bislang
Bestrahlung sowie Chemotherapie, wobei seit 1997 Gemcitabine als Gold-Standard
verwendet wird [45]. Kombinationsstudien von Gemcitabine mit anderen
Chemotherapeutika erzielten bislang keine befriedigenden Ergebnisse [46-48]. Lediglich die
Kombination von Gemcitabine mit Erlotinib erbrachte einen zusätzlichen Nutzen. Das
mediane Ü berleben stieg hier bei der Kombinationstherapie auf 6,24 Monate an, im
Gegensatz zur Therapie mit Gemcitabine plus einem Placebo (medianes Ü berleben: 5,91
Monate)[49]. Erfolgsversprechend scheint auch das Kombinationspräparat FOLFIRINOX
(Oxaliplatin, Fluoruracil und Leucovorin) zu sein, dass eine Steigerung des Gesamt ü berlebens
auf ü ber 11 Monate im Vergleich zu Gemcitabine erbrachte [50]. Zusätzliche palliative
Therapieverfahren zur Stabilisierung des Krankheitsbildes sowie der Erhö hung der
Lebensqualität sind somit unabdingbar.
1.1.5 Gemcitabine
Seit 1997 wird Gemcitabine (Gemzar© ) als Standardtherapie beim metastasierendem
duktalen Adenokarzinom eingesetzt. Aufgrund von besseren Ü berlebensraten der
behandelten Patienten (5,65 Monate vs. 4,41 Monate) sowie gesteigertem klinischen
Ansprechen, lö ste Gemcitabine das bis dahin als Standardtherapie verwendete 5-fluorouracil
(5-FU) ab [45, 51]. Gemcitabine (2`, 2`-difluoro 2`-deoxycytidin, dFdC) ist ein Pyrimidin
Nukleosid Analogon (Abb. 5), das von den Zellen vermehrt ü ber spezifische
Nukleosidtransporter aufgenommen wird.
Abb. 5: Chemische Struktur von Gemcitabine; aus Mini et al. [52].

1 Einleitung
14
Anschließend wird die Monophosphatform durch Deoxycytidinkinasen (dCKs) in die aktiven
Di- und Triphosphate phosphoryliert. Diese hoch aktiven Formen beeinflussen die DNA
Synthese innerhalb der Zelle z.B. durch Inhibierung der Ribonukleotid Reduktase RRM1 oder
durch Inhibierung der DNA Polymerase [52, 53]. Aufgrund der Resistenzen gegenü ber
Gemcitabine erhö hte sich die Lebensrate der behandelten Patienten lediglich um ca. 6
Monate [53, 54]. Deshalb wurde in verschiedenen Studien die kombinierte Gabe von
Gemcitabine mit zytotoxischen (Fluorpyrimidin- oder Topoisomerase-Inhibitoren [55-60]
oder biologischen Substanzen wie Marimastat [60], Tipifarnip [61] oder Cetuximab [47]
untersucht. Allerdings konnten auch bei der zusätzlichen Gabe von zytotoxischen Substanzen
die Ü berlebensraten der Patienten mit fortgeschrittenem Pankreaskarzinom nicht signifikant
erhö ht werden [49, 62-64]. Lediglich Patienten mit einer guten Konstitution profitieren
teilweise von den Kombinationstherapien. Heinemann et al. konnten eine Steigerung der
Ü berlebensrate von Patienten feststellen, die eine Kombinationstherap ie aus Gemcitabine
und einem Platinderivat oder Fluorpyrimidin erhielten [65]. Auch die Metaanalyse von
Sultana et al. konnte einen Vorteil der Kombinationstherapie von Gemcitabine und einer
Platinverbindung verzeichnen [66]. Aufgrund der steigenden Resistenz von Tumorzellen
gegenü ber Chemotherapeutika ist es notwendig neue Therapieansätze zu erforschen und zu
entwickeln [67, 68].
1.1.6 Paclitaxel
Taxane sind Diterpene, die von Pflanzen der Gattung Taxus produziert werden [69]. Dabei
handelt es sich um natü rliche Zytostatika, die heutzutage chemisch synthetisiert werden. Die
zwei häufigsten Taxane, die zur Behandlung von Patienten mit unterschiedlichen soliden
Tumoren eingesetzt werden sind Paclitaxel sowie Docetaxel. Paclitaxel wurde erstmals in
den frü hen 60-iger Jahren aus der Borker, der Pazifischen Eibe (Taxus brevifolia, Familie:
Taxaceae) isoliert und die chemische Struktur 1971 genauer beschrieben [70] (Abb. 6).

1 Einleitung
15
Abb. 6: Chemische Struktur von Paclitaxel (5β ,20-Epoxy-1,2α ,4,7β ,13α -hexahydroxytax-11-en-9-one4, 10-diacetate2-benzoate13-ester mit (2R,3S)-N-benzoyl-3-phenyllisoserine); aus Singla et al. [71].
Dabei handelt es sich um ein diterpenes Pseudoalkaloid mit der Summenformel C47H51NO14
und einer molekularen Masse von 853 Da. In der Krebstherapie wird Paclitaxel vom National
Cancer Institute (NCI) als der grö ßte signifikante Fortschritt der letzten 20 Jahren gehandelt.
Dabei war die Entdeckung nicht zufällig, sondern das Ergebnis einer Untersuchung von mehr
als 12000 natü rlichen Komponenten, die fü r die Krebstherapie eingesetzt werden kö nnten
[72]. Von der US Food and Drug Association (FDA) wurde Paclitaxel als Medikament fü r die
Sekundärtherapie fü r metastasierenden Eierstockkrebs [73-77], als adjuvante Behandlung
von Brustkrebs mit positive Lymphknoten sowie als Sekundärtherapie fü r metastasierenden
Brustkrebs zugelassen [78]. Als Primärtherapie in Kombination mit Cisplatin wird Paclitaxel
zudem bei metastasierendem nicht kleinzelligem Lungenkrebs [79] sowie bei Eierstockkrebs
[80, 81] eingesetzt. Zusätzlich wird Paclitaxel als Sekundärtherapie bei Patienten mit
fortgeschrittenem Pankreaskarzinom und einer Gemcitabinresistenz eingesetzt [82-84].
Paclitaxel ist ein Zytostatika, dessen Wirkungsweise erstmals 1979 beschrieben wurde [85].
Dabei handelt es sich um ein Mikrotubuli stabilisierenden Wirkstoff, der an die Untereinheit
des Tubulin bindet und somit die Polymerisierung der Mikrotubuli bewirkt [86-90]. Infolge
dessen kann die normale Formation der Mitosespindel, die entscheidend f ü r die Mitose ist,
nicht mehr stattfinden [91, 92]. Dadurch kommt es zu Chromosomenbrü chen, sowie zur
Inhibierung der Zellreplikation sowie Zellmigration. Paclitaxel inhibiert die Zellreplikation
durch das Blockieren des Zellzyklus in der späten G2- und/oder M- Phase. Ein weiterer
Wirkmechanismus von Paclitaxel ist die Induktion der Apoptose von Zellen durch Inhibierung
des Apoptose-Inhibitor-Protein Bcl2 [85].

1 Einleitung
16
1.2 Motorproteine
Fü r den Transport von Organellen oder Vesikeln in eukaryotischen Zellen wurden bislang
drei große Familien von molekularen Motorproteinen identifiziert, die mittels ATP-Hydrolyse
chemische Energie in mechanische Energie umwandeln (Abb. 7). Neben den Myosinen, die
ihre molekulare Fracht entlang der Aktinfilamente transportieren, benö tigen Dyneine und
Kinesine Mikrotubuli [93-96].
Abb. 7: Schematische Darstellung der drei „Prototypen“ von Motorproteinen. Kinesin und Dynein, die mit Mikrotubuli assoziiert sind, sowie Dynein, das Aktinfilamente bindet. Die Motordomäne ist dabei in gelb dargestellt, assoziierte Polypeptide in rosa sowie die Stielregion in blau; aus Woehlke et al. [97].
Neben dem Transport von Organellen und Vesikeln spielen die Kinesine (KIF Proteine) zudem
eine entscheidende Rolle im Zellzyklus, da sie an der Bildung der Spindelfasern, der
Chromosomen Segregation sowie der Zytokinese beteiligt sind [98]. Im folgenden Abschnitt
sollen die Kinesin-Proteine genauer betrachtet werden.
1.2.1 Kinesine
Nach der erstmaligen Entdeckung der Kinesine 1985 [99] sind bis dato 45 murine und
humane Kinesin-Proteine entdeckt worden, die in 14 Superfamilien (Kinesin-1 bis Kinesin-14)
unterteilt werden [100, 101]. Untersuchungen der Proteinstruktur von Kinesin-1 haben
gezeigt, dass Kinesine neben der Motordomäne drei weitere Strukturen aufweisen (Abb. 8).
Neben einer Halsregion (~40 aa), die meist Subtypen spezifisch ist und die Motordomäne
(Kopfregion) mit einer „coiled-coil“-Stielregion verbindet, weisen Kinesine zudem eine
Schwanzregion auf. Die Kopfregion, die bis zu 360 Aminosäuren lang sein kann, enthält
neben der ATP- eine Mikrotubuli-Bindedomäne. Diese Motor-Domäne ist entweder am C-

1 Einleitung
17
oder N-terminalen Ende oder zentral in der Mitte des Proteins lokalisiert (C-Kinesine, N-
Kinesine, M-Kinesine [102-107].
Abb. 8: Übersicht der Kinesin Superfamilien und deren Struktur; alle Mitglieder der Kinesin-Superfamilie besitzen eine konservierte Motordomäne (hier: hellblau); die Position der Motordomäne entspricht dabei der direkten Motilität des Proteins. Kinesine mit einer N-terminalen Motordomäne wandern dabei zum Plus-Ende der Mikrotubuli, wohingegen Kinesine mit einer C-terminalen Motordomäne zum Minus-Ende der Mikrotubuli wandern. Kinesine mit einer zentralen Motordomäne destabilisieren die Mikrotubuli sowohl am Minus- als auch am Plus-Ende; Manche Kinesine enthalten zudem eine „coiled-coil“-Region fü r die Oligomerisierung (hier: schwarz); KHC= „Kinesin heavy chain“; KLC=“Kinesin light chain“; aus Verhey et al. [108].
Ihre Aufgabe besteht in der ATP-Hydrolyse, um mechanische Energie fü r die Bewegung
entlang von Mikrotubuli zu generieren. Kinesine mit einer N-terminalen Motordomäne
transportieren Molekü le entlang des Plus-Endes der Mikrotubuli, wohingegen Kinesine mit
einer C-terminalen Motordomäne in die entgegengesetzte Richtung wandern. KIF-Proteine,
die eine zentrale Motordomäne besitzen, destabilisieren Mikrotubuli in einem weitaus
grö ßeren Teil als sich an ihnen fortzubewegen [109-113]. Neueste Untersuchungen haben
jedoch gezeigt, dass Kinesin-8 und Kinesin-14 Motoren die Fähigkeit besitzen an Mikrotubuli
entlang zu wandern, als auch diese zu depolymerisieren [112, 114, 115]. Fü r die Bewegung
an Mikrotubuli wird dabei die Konformationsveränderung der Kopfregion an die Halsregion

1 Einleitung
18
ü bersendet. Dabei wird je nach Position der Subtypen spezifischen Halsregion die
Bewegungsrichtung festgelegt [103, 105].
Abb. 9: Mechanismus der Kinesin Bewegung entlang von Mikrotubuli. Mittels dieses „hand-over-hand“ Mechanismus bewegen sich Kinesine pro „Schritt“ 16,6 nm fort. In grau dargestellt sind die Untereinheiten der Mikrotubuli, grü n bzw. blau sind die beiden Kopfdomänen; aus Yildiz et al [96].
Dabei binden die beiden Kopfregionen eines Homodimers an die Untereinheiten der
Mikrotubuli und wandern in einem „hand-over-hand“ Mechanismus an diesen entlang.
Dabei verbrauchen Kinesine pro „Schritt“ zwei ATPs und legen eine Strecke von 16,6 nm
zurü ck (Abb. 9)[96]. Die Dimerbildung mancher Kinesine erfolgt ü ber die Stielregion, mittels
derer Untereinheiten des Holoenzyms interagieren kö nnen. Gegenü ber der Kopfregion
befindet sich die Schwanzregion, welche Transportmolekü le wie Proteine, Lipide oder
Nukleinsäuren bindet [116-118]. Aufgrund der unterschiedlichen Strukturen besitzen die
Kinesin-Familien spezifische Aufgaben innerhalb der Zelle. Während Kinesin-1, -2, -3, -6 und -
14 Funktionen im Organelltransport haben, besteht die Aufgabe von Kinesin-4, -5, -6, -7, -8,
-10, -12, -13 und -14 vor allem in der Mitose (Tab. 1). Hier werden sie fü r die
Spindelformation, fü r chromosomale und nukleäre Bewegungen sowie fü r die Zytokinese
benö tigt. Studien haben ergeben, dass Kinesin-11 fü r die Signaltransduktion unabdingbar ist
[119]. Die genaue Funktion von Kinesin-9 ist bislang noch nicht geklärt [120].
1 Einleitung
19
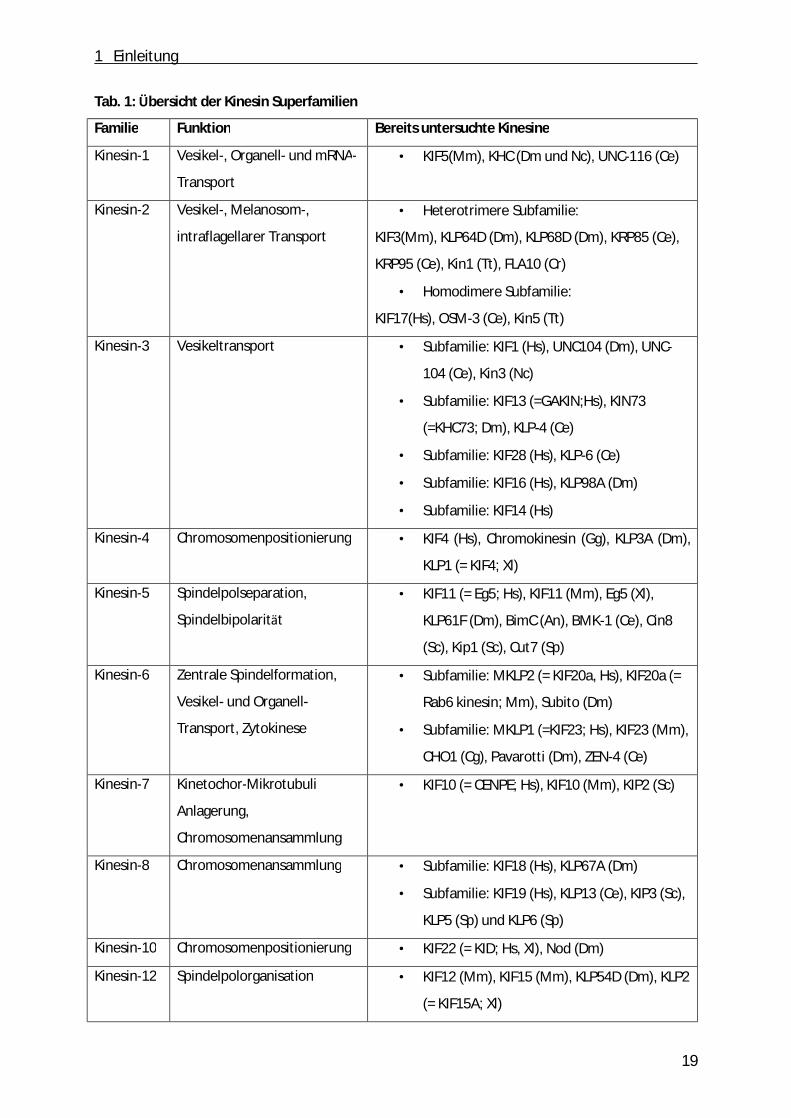
Tab. 1: Übersicht der Kinesin Superfamilien
Familie Funktion Bereits untersuchte Kinesine
Kinesin-1 Vesikel-, Organell- und mRNA-
Transport
• KIF5(Mm), KHC (Dm und Nc), UNC-116 (Ce)
Kinesin-2 Vesikel-, Melanosom-,
intraflagellarer Transport
• Heterotrimere Subfamilie:
KIF3(Mm), KLP64D (Dm), KLP68D (Dm), KRP85 (Ce),
KRP95 (Ce), Kin1 (Tt), FLA10 (Cr)
• Homodimere Subfamilie:
KIF17(Hs), OSM-3 (Ce), Kin5 (Tt)
Kinesin-3 Vesikeltransport • Subfamilie: KIF1 (Hs), UNC104 (Dm), UNC-
104 (Ce), Kin3 (Nc)
• Subfamilie: KIF13 (=GAKIN;Hs), KIN73
(=KHC73; Dm), KLP-4 (Ce)
• Subfamilie: KIF28 (Hs), KLP-6 (Ce)
• Subfamilie: KIF16 (Hs), KLP98A (Dm)
• Subfamilie: KIF14 (Hs)
Kinesin-4 Chromosomenpositionierung • KIF4 (Hs), Chromokinesin (Gg), KLP3A (Dm),
KLP1 (= KIF4; Xl)
Kinesin-5 Spindelpolseparation,
Spindelbipolarität
• KIF11 (= Eg5; Hs), KIF11 (Mm), Eg5 (Xl),
KLP61F (Dm), BimC (An), BMK-1 (Ce), Cin8
(Sc), Kip1 (Sc), Cut7 (Sp)
Kinesin-6 Zentrale Spindelformation,
Vesikel- und Organell-
Transport, Zytokinese
• Subfamilie: MKLP2 (= KIF20a, Hs), KIF20a (=
Rab6 kinesin; Mm), Subito (Dm)
• Subfamilie: MKLP1 (=KIF23; Hs), KIF23 (Mm),
CHO1 (Cg), Pavarotti (Dm), ZEN-4 (Ce)
Kinesin-7 Kinetochor-Mikrotubuli
Anlagerung,
Chromosomenansammlung
• KIF10 (= CENPE; Hs), KIF10 (Mm), KIP2 (Sc)
Kinesin-8 Chromosomenansammlung • Subfamilie: KIF18 (Hs), KLP67A (Dm)
• Subfamilie: KIF19 (Hs), KLP13 (Ce), KIP3 (Sc),
KLP5 (Sp) und KLP6 (Sp)
Kinesin-10 Chromosomenpositionierung • KIF22 (= KID; Hs, Xl), Nod (Dm)
Kinesin-12 Spindelpolorganisation • KIF12 (Mm), KIF15 (Mm), KLP54D (Dm), KLP2
(= KIF15A; Xl)

1 Einleitung
20
Kinesin-13 Korrektur von falsch
organisierten Kinetochor-
Mikrotubuli,
Chromosomensegregation
• Subfamilie: KIF2A (Hs), KIF2B (Hs), MCAK (=
KIF2C; Hs), MCAK (Cg), KCM1 (= KIF2C, Xl),
KLP10A (Dm), KLP59C (Dm), KLP59D (Dm),
KLP7 (Ce)
• Subfamilie: KIF24 (Hs), Kin1 (pf)
Kinesin-14 Spindelpolorganisation,
Transport
• Subfamilie: KIFC1 (= HSET; Hs), CHO2 (Cg),
NCD (Dm), CTK2 (Xl), Kar3 (Sc)
• Subfamilie: KIFC2 (Hs), KIFC3 (Hs), KLP3 (Ce),
und eine große Anzahl an Pflanzen
spezifischen Isoformen
An= Aspergillus nidulans; Ce= Caenorhabditis elegans; Cg= Cricetulus griseus; CHO= „Chinese hamster ovary“; Cin8= „Chromosome instability protein“; Cr= Chlamydomonas reinhardtii; CTK2= „carboxy-terminal kinesin 2“, Cut7= „cell untimely torn protein“; Dm= Drosophila melanogaster, Gg= Gallus gallus; Hs= Homo sapiens; KCM1, „kinesin central motor 1“; KHC= „kinesin heavy chain“; KLP= „kinesin-like protein“; MCAK= „mitotic centromere-associated kinesin“; MKLP= „mitotic kinesin-like protein“; Mm= Mus musculus; Nc= Neurospora crassa; NCD= „non-claret dicunjtional“; OSM-3= „osmotic avoidance abnormal protein 3“, Pf= Plasmodium faiciparum; Sc= Saccharomyces cervisiae, Sp= Schizosaccharomyces pombe; Tt= Tetrahymena thermophila; Xl= Xenopus laevis; ZEN4= „ zygotic enclosure defective protein 4“; modifiziert nach Verhey et al. [108].
1.2.2 Kinesin Motorprotein Kif20a
Kif20a, auch Rabkinesin6, MKLP2 oder RAB6KIFL genannt, ist ein Kinesin Motorprotein das in
die Kinesin Superfamilie-6 eingeordnet wird. Im Vergleich zu den anderen Kinesin
Superfamilien unterscheiden sich die Kinesin-6 Mitglieder durch zwei wesentliche Merkmale.
Zum einen ist die Halsregion fü nfmal länger als bei den anderen Kinesin Superfamilien und
zum anderen befindet sich in der ATP-Bindedomäne ein „ Insert“ [120-122]. Aufgrund der
Lokalisation der konservierten Motordomäne (350aa) am N-terminalen Ende gehö rt Kif20a
zu den N-Kinesinen. Flankiert wird dieser Bereich von der ATP-Bindesequenz sowie einer
„coiled-coil“ –Region [109, 123](Abb. 10). Das Kif20a Protein ist ein Homodimer und hat eine
Länge von 890 aa, eine molekulare Grö ße von 100,15 kDa und wird posttranslational am
Serin 528 durch die „polo-like kinase 1“ (PLK1) phosphoryliert [124, 125](Wolfram Alpha,
computational knowledge engine). Das humane Kif20a ist zu 86% identisch wie das murinen
Kif20a auf dem Chromosom 5q31 lokalisiert und hat eine Grö ße von 8987 kbp [125, 126].
Viele unmittelbar angrenzende Gene sind ebenfalls an Prozessen des Zellzyklusses beteiligt,
wie das „cell division cycle protein 23 homolog“ (cdc23), sowie das „cell division cycle
protein 25 homolog“ (cdc25) [127].

1 Einleitung
21
Abb. 10: Schematische Darstellung des Kinesin Motor Proteins Kif20a (modifiziert nach Tcherniuk et al. [128]).
Aufgrund seiner Eigenschaft als Motorprotein fungiert Kif20a sowohl in zytoplasmatischen
Prozessen als auch im Nukleus während der Zellteilung. Zu den zytoplasmatischen Aufgaben
gehö rt der retrograde Transport von Golgi-Membranen zum endoplasmatischen Retikulum
(ER), sowie der Transport von Vesikeln und Organellen entlang der Mikrotubuli [129, 130].
Dabei bindet Kif20a mit dem C-terminalen Ende an die Guanosin-Triphosphat (GTP)
gebundene Form von Proteinen der Rab-Familie (hier: RAB6A und RAB6B). RAB6A bzw.
RAB6B sind GTPasen die mit dem Golgi Apparat verbunden sind und die Assoziation bzw.
Dissoziation von Kif20a an die Mikrotubuli regulieren [123, 131]. Nach der Bindung von
Kif20a an RAB6A oder RAB6B fungiert dieses dann als Motor f ü r den Transport. Zusätzliche
Funktionen hat Kif20a zudem im Zellzyklus. Hier ist Kif20a maßgeblich an der Formation der
mitotischen Spindelfasern, der Zentrosomen Separierung, der Chromosomen Aufteilung
sowie an der Zytokinese beteiligt [109, 127, 130]. Die Verteilung des Kif20a Proteins
während des Zellzyklusses steht in Korrelation mit der Expression von Cyclin B, und erlangt
ein Maximum in der M-Phase [127]. Bei genauerer Betrachtung der einzelnen Mitosestadien
konnte mittels Immunfluoreszenz festgestellt werden, dass Kif20a während der Interphase
vor allem im Zytoplasma lokalisiert ist [132]. Aufgrund der Assoziation mit RAB6A und RAB6B
konnte mittels Doppelfärbung eine deutliche Kolokalisation mit dem Golgi Apparat
detektiert werden [123, 133]. Zu Beginn der Prophase kann eine eindeutige Kernfärbung
festgestellt werden. In der Metaphase ist Kif20a im Bereich der mitotischen Spindeln
lokalisiert und wandert während der Anaphase in die Ä quatorialebene. Während der
Telophase und der Zytokinese befindet sich das Kinesin Motor Protein in der Teilungsfurche
[127, 130, 132]. Alle Prozesse des Kinesin Motor Proteins basieren auf der Fähigkeit
chemische Energie in Form von Adenosintriphosphat (ATP) zu hydrolysieren und somit in
mechanische Energie umzuwandeln [134, 135]. Analysen haben ergeben, dass Kif20a im
normalen Gewebe vor allem in der Leber, den Hoden, im adulten Knochenmark sowie dem
adulten Thymus stark exprimiert wird, wohingegen in der Plazenta und dem Herzen nur

1 Einleitung
22
geringe Mengen detektiert werden konnten [125]. Schwach bzw. gar nicht exprimiert wird
Kif20a in der Milz, Lymphknoten, Pankreas, Lunge, Gehirn, Nieren sowie der
Skelettmuskulatur [125]. Bei der Untersuchung von Tumorgewebe steigt die Expression des
Kinesin Motorproteins allerdings enorm an. Sowohl in kleinzelligem Lungenkarzinom,
Blutkrebs, Brustkrebs und Bauchspeicheldrü senkrebs konnten eine erhö hte Menge an Kif20a
detektiert werden [132, 136]. Die Tatsache, dass Kif20a eine wichtige Rolle beim
Organelltransport, bei der Zellmotilität und diversen Prozessen des Zellzyklusses spielt,
sowie die stark erhö hte Expression im Tumorgewebe macht Kif20a zu einem interessanten
Zielgen fü r die Krebsforschung.
2 Material und Methoden
23
2 Material und Methoden
2.1 Material
2.1.1 Chemikalien
Tab. 2: Chemikalien und Reagenzien
Chemikalien Firma 2-Propanol Roth 50% Ethanol Hauseigene Apotheke 6x DNA Loading Dye Thermo Scientific 70% Ethanol Hauseigene Apotheke Aceton Merck Agar Agar, Kobe I Roth Agarose Roth Amphotericin PAA Ampicillin Roth BSA Albumin Roth DAB+Chromogen (50x) DAKO Dimethylsulfoxid (DMSO) Roth DreamTaqTM Green PCR Master Mix Fermentas Dulbecco´s PBS (1x) PAA EDTA Roth Ethanol 100% Merck Ethidiumbromid Sigma Fetal bovine Serum Sigma Fluoreszenz mounting medium DAKO Gemcitabine hydrochlorid TOCRIS bioscience Glycerin Roth Glycin Roth Hämatoxylin Merck HiPerFect Transfection Reagent Qiagen HRP Substrat Buffer DAKO Isopropanol (vergällt) Roth Kristallviolett Merck LB- Medium Roth Leupeptin Sigma Methanol Roth MOPS Roth Mounting Medium DAKO Mycoplasmen-OFF Minerva Biolabs Na3VO4 Sigma Na-deoxicholat Sigma NaF Sigma Natriumchlorid Roth Natriumhydroxid Roth Natronlauge Hauseigene Apotheke NP-40 Sigma NuPAGE LDS Sample Buffer (4x) Invitrogen

2 Material und Methoden
24
NuPAGE Sample Reducing Agent (10x) Invitrogen Paprotrain AdipoGen Paraformaldehyd Roth Paraformaledhyd (8%) Hauseigene Apotheke Penicillin-Streptomycin Solution Sigma Pepstatin A Sigma PMSF Sigma Protease Inhibitor Cocktail Tablet Roche RNase AWAY Molecular Biolabs RNase freies Wasser Qiagen Roticlear Roth Salzsäure Hauseigene Apotheke SDS ultrapure Roth Taxol (Paclitaxel) Sigma Thiazolyl Blue Tetrazolium Bromid Sigma Thymidin Sigma Triton X100 Roth Trizma Base Sigma Trypan Blue Stain 0,4% GIBCO Trypsin EDTA solution (1x) Sigma Tween 20 Roth Zitronensäure Roth TEMED (N,N,N´,N´Tetraethylmethylendiamin) Serva
2.1.2 Geräte und Verbrauchsmaterialien
Tab. 3: Geräte
Name Firma Comfort No-Frost Kü hl-und Gefrierschrank Kombi Liebherr Compact XS/S und Compact M Gelelektrophorese Kammern Biometra Cop 30 Kü hlplatte Medite Medizintechnik Electrophoresis power supply ST606 GIBCO Feinwaage Sartorius analytic Guava easyCyteTM Flow Cytometer Milipore Hera cell 150 Inkubator Thermo Scientific Hera freeze Thermo Scientific Hera safe Cellhood Thermo Scientific Heraeus Multifuge 3SR+ Thermo Scientific IKA R4 basic KT/C safety control IKA Innova 43 Incubator Shaker series New Brunswick Scientific Leica RM2255 Microtom Leica LightCycler480 Roche Mikroskop Axiovert 40CFL ZEISS Mikrowelle Sharp Milli-Q Advantage Millipore-Anlage Millipore Multipipette Plus Eppendorf Mini Protean Tetra System (Gelelektrophorese-Kammer und Blotting-Kammer)
BioRad
Mini-Rocker MR-1 Peqlab Multiskan EX Thermo Scientific
2 Material und Methoden
25
Nanodrop 2000 Spectrophotometer Thermo Scientific PCR Maschine VWR pH-Meter WTW Series Inolab Pipetboy Akku IBS IBS Integra Bioscience Scanner CanonScan 4400F Canon Standard Power Pack P25 Biometra Thermomixer comfort Eppendorf Titramax 100 Heidolph Vortexer IKA MS3 basic NEOLAB Wasserbad Lauda Zentrifuge 5424 Eppendorf
Tab. 4: Verbrauchsmaterialien
Name Firma 10 µl, 20 µl, 100 µl, 200 µl, 1000 µl Filter Tips, Tip One Star Lab 24-Multiwell Insert System BD 5 ml, 10 ml, 25 ml, 50 ml cellstar serological pipettes Greiner bio-one 6-well cell culture plate Greiner bio-one 8er Deckelkette Sarstedt 96-well cell culture plate Greiner bio-one BD 24-Multiwell Insert System BD Biosciences BD Micro Fine+ Insulin syringes with sterile interior 1ml Becton Dickinson C-Chip Disposable Hemocytometer Peqlab Cell culture dishes 100x20 mm Greiner bio-one Cell culture flasks 25 cm2 Greiner bio-one Cell culture flasks 75 cm2 Greiner bio-one Cell Scraper 16 cm Sarstedt Cell Star tubes 15 ml, 50 ml Greiner bio-oneC Combitips plus 0,5 ml, 2,5 ml, 5 ml Eppendorf Cryo Tube Vials Thermo Scientific Cryo-Safe Cooler -1°C Freeze Control Bel-Art Product DAKO Pen DAKO Deckgläser 24x50 mm Menzel Gläser High Performance Chemiluminescence Film GE Healthcare Multiply-Strip 0,2 ml Kette Sarstedt Nitrocellulose Transfer Membran Whatman Parafilm Roth Pipetten P1000N, P200N, P20N, P10N, P2N Gilson Reagiergefäß 1,5 ml, 2 ml Sarstedt Schottflasche 50 ml, 500 ml, 1000 ml Schott Superfrost Ultra Plus Objektträger Thermo Scientific Tissue culture plate 24-well Becton Dickinson Wägeschale und Wägepapier Machery-Nagel
2.1.3 Puffer, Medien und Lösungen
Tab. 5: Puffer, Lösungen und Medien
Puffer Zusammensetzung 10x TBS 12,1 g TrisBaze, 85 g NaCl, ad 1000 ml H2O, ph 7,4 1x TBE Puffer (Agarosegel) 0,1 M Tris/HCl, 89 mM Borsäure, 1 mM EDTA, pH 8,5

2 Material und Methoden
26
1x TBST 100 ml 10x TBS, 0,5 ml Tween20, ad 1000 ml H2O 4x Lower Tris 1,5 M Tris/HCl, 0,4% w/v SDS, pH 8,8 4x Upper Tris 0,5 M Tris/HCl, 0,4% w/v SDS, pH 6,8 Citratpuffer 42 g Zitronensäure, ad 1000 ml H2O, pH 6,0 Laufpuffer (Western Blot) 25 nM Tris, 192 mM Glycin, 1% w/v SDS Transferpuffer (Western Blot) 29,1 g Tris, 14,7 g Glycin, 1000 ml Methanol, 0,1875 g SDS LB Medium 10 g NaCl, 10 g Bacto-Trypton, 5 g Hefeextrakt, ad 1000 ml H2O
Tab. 6: Zellkulturmedien und Zusätze
Medium Firma F12 PAA DMEM (high glucose) PAA Keratinocyten Medium GIBCO OPTI-MEM I GIBCO RPMI1640 PAA Supplements for Keratinocyte serum free medium GIBCO
Die Medien von der Firma PAA wurden fü r die weitere Verwendung in der Zellkultur mit 1%
Penicillin-Streptomycin-Lö sung sowie 10% fö talem Kälberserum versetzt.
2.1.4 Organismen
2.1.4.1 Bakterien
Tab. 7: Bakterien
Bakterienstamm Firma Escherichia coli DH5α Invitrogen
2.1.4.2 Eukaryontische Zellen
Tab. 8: Zelllinien
Zellen Herkunft Bezugsquelle CM Neuroendokrine Tumorzelllinie des Pankres Prof. Aldo Scarpa HPDE Humane duktale Epithelzellen des Pankreas Dr. Ming-Sound Tsao Panc-1 Primärer Tumor, PDAC ATCC USA QCP Neuroendokrine Tumorzelllinie des Pankres Prof. Aldo Scarpa SU86.86 Lebermetastase, PDAC ATCC USA T3M4 Lymphknotenmetastase, PDAC ATCC USA
Bei den Zelllinien CM und QCP handelt es sich um Zelllinien aus neuroendokrinen Tumoren.
HPDE Zellen sind immortalisierte duktale Epithelzellen des Pankreas. Die Zelllinien Panc-1,
SU86.86 sowie T3M4 wurden alle aus duktalen Adenokarzinomen extrahiert.

2 Material und Methoden
27
2.1.4.3 Patientengewebe
Gewebeproben von Patienten mit duktalem Adenokarzinom wurden während Resektionen
entnommen, die im Zeitraum zwischen Oktober 2007 und Januar 2011 in der Abteilung
Chirurgie, am Klinikum rechts der Isar (TU Mü nchen) durchgefü hrt wurden. Gesunde
Gewebeproben des Pankreas, die während eines Organ Donor Programms entnommen
wurden, wurden von der Universität Heidelberg zur Verfü gung gestellt. Die
Gewebesammlung wurde von der Ethikkommission der Technischen Universität Mü nchen
sowie der Universität Heidelberg geprü ft. Von allen Patienten wurden
Einverständinserklärungen eingeholt.
2.1.5 Primer
Die verwendeten Primer wurden von der Firma Invitrogen oder Metabion bezogen und in
einem gereinigten Synthesemaßstab von 0,02 µmol mit Aquadest gelö st.
Tab. 9: Eingesetzte Oligonukleotide
Oligonukleotid Sequenz Schmelz- Temperatur
Firma
HPRT „ forward“ 5`-GCAGCCCTGGCGTCGTGATTAG -3` 60°C Invitrogen HPRT „ reverse“ 5`-TCGAGCAAGACGTTCAGTCCTGT-3` 60°C Invitrogen Kif20a10th “reverse” 5`-GCGCTGTTGGCTAGGCGGT-3` 74°C Invitrogen Kif20a1st “forward” 5`-ATTCTCCCCCGGTCCCTGGC-3` 76°C Invitrogen Kif20a 3rd “reverse” 5`-AAAGGGGAACGCCGTGAGCG-3` 74°C Invitrogen Kif20a “forward” 5`-CAAGGGCCTAACCCTCAA-3` 65°C Invitrogen Kif20a “reverse” 5`-TCTGTCGTCTCTACCTCCCTAGA-3` 65°C Invitrogen β-Aktin “forward” 5`-CTTCCTGGGCATGGAGTCCT-3` 56°C Invitrogen β- Aktin “reverse” 5`-CCGCCGATCCACACAGAGTA-3` 56°C Invitrogen Myco 280 „ forward“ 5`-GGGAGCAAACAGGATTAGATACCC T-3` 55°C Metabion Myco 280 „ reverse“ 5`-TGCACCATCTGTCACTCTGTTAACCTC-3` 55°C Metabion Myco 500“ forward“ 5`-GGCGAATGGGTGAGTAACACG-3` 55°C Metabion Myco 500 “reverse” 5`-CGGATAACGCTTGCGACCTAT-3` 55°C Metabion
2.1.6 siRNA und Vektoren
Tab. 10: Vektoren und siRNA
Name Sequenz Firma Hs_KIF20A_5 Ziel Sequenz:
AAGGCCAGGTTTCTGCCAAAA Sense Strang: GGCCAGGUUUCUGCCAAAATT Antisense Strang: UUUUGGCAGAAACCUGGCCTT
Qiagen
2 Material und Methoden
28
Human cDNA clone (ORF with C-terminal GFP tag)
Origene
Precision Shuttle mammalian vector with non-tagged expression
Origene
2.1.7 Antikörper
Tab. 11: Primäre Antikörper
Antikö rper Firma Verdü nnung Cleaved Caspase 3 anti-human (Kaninchen)
Cell Signaling 1:400 (IF)
Cleaved PARP anti-human (Maus) Cell Signaling 1:1000(WB) GAPDH anti-human (Maus) Santa Cruz 1:500 (WB) Hoechst 33258 Invitrogen 1:2000 (IF) Kif20a anti-human (Kaninchen) Bethyl 1:150 (IHC), 1:1000 (WB) PCNA anti-human (Maus) Cell Signaling 1:3000 (IHC) α -SMA anti-human (Maus) DAKO 1:1000 (IHC) α -SMA anti-human (Kaninchen) abcam 1:50 (IF) α -Tubulin anti-human (Maus) abcam 1:100 (IF)
Tab. 12: Sekundäre Antikörper und Farbstoffe
Antikö rper Firma Verdü nnung Alexa Fluor 350 donkey anti-rabbit IgG Invitrogen 1:400 (IF) Alexa Fluor 594 goat anti-rabbit IgG Invitrogen 1:300 (IF) Alexa Fluor488 goat anti-mouse IgG Invitrogen 1:400 (IF) ECL Anti-mouse IgG, Horseradish Peroxidase linked F(ab`)2 Fragment
GE Healthcare 1:2000 (WB)
ECL Anti-rabbit IgG, Horseradish Peroxidase linked F(ab`)2 Fragment
GE Healthcare 1:2000 (WB)
EnVision + System-HRP labeled Polymer anti-mouse
DAKO laut Herstellerangaben
EnVision + System-HRP labeled Polymer anti-rabbit
DAKO laut Herstellerangaben
Propidiumiodid Invitrogen 2 µg/ml
2.1.8 Kits und Marker
Tab. 13: Kits
Kits Firma ECL Kit Invitrogen LightCycler 480 SYBR Green I Master Roche Pierce BCA Protein Assay Kit Thermo Scientific Plasmid Mini Kit Qiagen Revert Aid H Minus 1st Strand cDNA Synthesis Kit Fermentas RNeasy Plus Mini Kit Qiagen KAPA2G Fast ReadyMix with Dye PeqLab FITC Annexin V Apoptosis Detection Kit I BD Biosciences
2 Material und Methoden
29
Tab. 14: DNA und Protein Leiter
Name Firma Gene Ruler 100 kb DNA Ladder Fermentas Gene Ruler 1 kB DNA Ladder Fermentas Page Ruler Prestained Protein Ladder Thermo Scientific
2.1.9 Software
Tab. 15: Software
Name Firma Ascent Software Version 2.6 Thermo Scientific GraphPad Prism 5 GraphPad LightCycler480 Software 1.5 Roche SPSS Software SPSS Manual tracking program NIH Chemotaxis tool Ibidi

2 Material und Methoden
30
2.2 Methoden
2.2.1 Mikrobiologische Methoden
2.2.1.1 Kultivierung von Bakterien
In der vorliegenden Arbeit wurden ausschließlich Bakterien des Stamms Escherichia coli
DH5α (E.coli DH5α ) verwendet. Flü ssigbakterienkulturen wurden bei 37°C und 200 rpm in
LB-Medium ü ber Nacht inkubiert, Bakterienkolonien wurden bei 37°C ü ber Nacht auf
Agarplatten kultiviert.
2.2.1.2 Transformation von kompetenten E.coli Bakterien durch Hitzeschock
Der hitzekompetente Bakterienstamm Escherichia coli DH5α wurde mit dem
Expressionsvektor „Human cDNA clone (ORF with C-terminal GFP tag)” nach folgendem
Protokoll tansformiert: Zu 50 µl der Bakterien wurde 1 µl Vektor-DNA gegeben und
anschließend fü r 15 min auf Eis gestellt. Nach einem 90 sec Hitzeschock bei 42°C wurde der
Ansatz fü r 5min auf Eis inkubiert, anschließend in 250 µl SOC-Medium aufgenommen und
der Ansatz 45 min bei 37°C im Inkubator geschü ttelt (300 U/min). Die Bakterien wurden
dann zu 10% und 90% auf Agarplatten mit einem sterilen Spatel ausgestrichen und ü ber
Nacht bei 37°C im Brutschrank inkubiert. Zur Selektion der Plasmid-tragenden Bakterien
wurden die Agarplatten mit 100 µg/ml Ampicillin versetzt.
2.2.1.3 Präparation von Plasmid-DNA aus Bakterien
Zur Vervielfältigung klonierter DNA wurden die Plasmid tragenden Bakterien in einer
Ü bernachkultur vermehrt. Dafü r wurde eine Bakterienkolonie in 3 ml LB-Medium mit 10
mg/ml Antibiotikum angeimpft und ü ber Nacht bei 37°C im Inkubator bei 200 U/min
geschü ttelt. Fü r die Plasmidpräparation aus dem Bakterienstamm Escherichia coli DH5α
(Invitrogen) wurde das „Plasmid Mini Kit“ verwendet und nach Herstellerangaben verfahren.
2.2.2 Arbeiten mit eukaryotischen Zellen
2.2.2.1 Kultivierung von eukaryotischen Zellen
Humane Zellen wurden bei 37°C und einer angefeuchteten Atmosphäre von 5% CO2
kultiviert. Dreimal pro Woche wurden die adhärenten Zellen mit PBS gewaschen, mit 1x
Trypsin/EDTA von der Kulturflasche gelö st und in einem Verhältnis von 1:2 passagiert. Eine
2 Material und Methoden
31
Ü bersicht der Zelllinien und der zugehö rigen Kulturmedien ist in Tabelle 16 aufgefü hrt. Alle
Medien enthielten 1% w/v Penicillin/Streptomycin und 10% w/v fö tales Kälberserum.
Tab. 16: Übersicht der verwendeten Zelllinien
Zelllinie Herkunft Medium
CM neuroendokriner Tumor RPMI1640
HPDE immortalisierte duktale Epithelzellen Keratinozytenmedium
Panc-1 humanes duktales Adenokarzinom RPMI1640
QCP neuroendokriner Tumor RPMI1640
SU86.86 humanes duktales Adenokarzinom RPMI1640
T3M4 humanes duktales Adenokarzinom RPMI1640
2.2.2.2 Einfrieren und Auftauen von Zellen
Fü r die Kryokonservierung von Zellen wurden diese nach dem Splitten bei 800 rpm
zentrifugiert und anschließend in 1 ml Einfriermedium (= 90% FCS, 10% DMSO)
resuspendiert und in 2 ml Kryogefäße ü berfü hrt. Einfrierbehälter („Nalgene Container“) bei -
80°C gelagert. Am nächsten Tag wurden die Zellen in einen Tank mit Flü ssigstickstoff
ü berfü hrt und bis zur weiteren Verwendung bei -196°C gelagert. Zum Auftauen von Zellen
werden diese aus dem Flü ssigstickstoff genommen und im Wasserbad bei 37°C aufgetaut.
Anschließend wird durch ein Zentrifugationsschritt bei 800 rpm fü r 4 min das DMSO entfernt
und das Zellpellet in frischem Medium aufgenommen. Die Zellen werden dann in eine
Kulturflasche ü berfü hrt.
2.2.2.3 Mykoplasmentest
Um die mö gliche Kontamination von eukaryotischen Zellen mit Mykoplasmen zu
untersuchen, wurden alle 4 Wochen Mykoplasmentest durchgefü hrt. Mittels PCR Analyse
konnten dadurch befallene Zelllinien ausgesondert werden. Daf ü r wurden Ü berstände der
kultivierten Zellen genommen und nach folgendem Pipettierschema fü r die PCR Analyse
vorbereitet:
Zellkulturü berstand 2 µl
KAPA 2G Dye 12,5 µl
Primer (500 For) 0,5 µl

2 Material und Methoden
32
Primer (500 Rev) 0,5 µl
Primer (280 For) 0,5 µl
Primer (280 Rev) 0,5 µl
H2Odest 8,5 µl
Total 25 µl
Fü r die PCR wurden zwei verschiedene Primerpaare verwendet, da somit ein großes
Spektrum an Mykoplasmenarten detektiert werden konnte. Die Synthesereaktion erfolgte in
einer PCR Maschine mittels folgenden Syntheseprogramms:
Erste Denaturierung 2 min 94°C
Amplifikation 35 Zyklen 30 sec 94°C
30 sec 55°C
30 sec 72°C
Finale Elongation 2 min 72°C
2.2.2.4 RNA Isolierung aus humanen Zellen
Humane Zellen wurden in einer T75 Zellkulturflasche bis zu einer Konfluenz von 80%
kultiviert. Anschließend wurde mittels des kommerziell verfü gbaren „RNeasy Plus Mini Kit”
(Qiagen) die RNA laut Herstellerangaben extrahiert. Im finalen Schritt wurde die RNA in 30 µl
Wasser eluiert und bis zur weiteren Verwendung bei -80°C weggefroren.
2.2.2.5 Agarosegelelektrophorese von RNA
Um die Qualität der RNA zu untersuchen kann diese auf ein Agarosegel aufgetragen und
aufgetrennt werden. Das Prinzip beruht dabei auf der Wandereigenschaft der negativ
geladenen RNA-Fragmente in einem elektrischen Feld. Kurze RNA-Fragmente wandern dabei
schneller durch das Agarosegel als grö ßere RNA-Fragmente. Bei hochwertig aufgereinigter
RNA sind auf dem Agarosegel zwei Banden deutlich sichtbar. Es handelt sich hierbei um die
28S- und die 18S- ribosomale RNA (rRNA). mRNA kann ausschließlich mit sehr sensitiven
Methoden detektiert werden, da in einer gesamt-RNA Probe lediglich 1- 3% mRNA
vorhanden sind. Die Mehrheit mit >80% stellt die rRNA dar. Das Intensitätsverhältnis der

2 Material und Methoden
33
beiden Banden sollte dabei 2:1 betragen, was fü r die Reinheit der RNA spricht. Fü r ein 1%-
iges w/v Agarosegel wurden dabei 1 g Agarose, 10 ml 1x MOPS-Puffer, 5 ml Formaldehyd
und 85 ml DEPC-Wasser gemischt. Die RNA Proben (2- 10 µg) in einem Volumen von 10 µl
wurden mit 10 µl RNA-Auftragspuffer bei 75°C fü r 4 min im Heizblock erhitzt. Anschließend
wurde dem Ansatz 1 µl Ethidiumbromid hinzugefü gt und die Proben auf das Agarosegel
aufgetragen. In einer horizontalen RNA-Gelelektrophoresekammer mit 1x MOPS als
Laufpuffer wurden die Proben bei einer Spannung von 50 V fü r 2 h aufgetrennt.
Anschließend wurde das Gel unter einem UV-Transilluminator analysiert und zur
Dokumentation fotografiert.
2.2.2.6 Reverse Transkription
Um die Transkription eines Gens untersuchen zu kö nnen, muss zunächst die RNA in cDNA
umgeschrieben werden. Mit Hilfe der reversen Transkriptase PCR, die RNA mittels RNA-
abhängiger DNA-Polymerase in cDNA ü bersetzt, entsteht somit ein Ausgansprodukt dessen
spezifische Sequenzen in einer PCR weiter untersucht werden kö nnen. Die Herstellung der
cDNA (1 µg) aus RNA erfolgte mit dem „Revert Aid H Minus 1st Strand cDNA Synthesis Kit” .
Hierfü r wurden 1 µl Oligo(dT)18Primer mit 1 µg RNA gemischt und auf ein Volumen von 12 µl
mit H2Odest aufgefü llt. Der Ansatz wurde gemischt, bei 65°C fü r 5 min erhitzt und
anschließend zentrifugiert und auf Eis gestellt. In der Zwischenzeit wurde der Master Mix
nach folgendem Protokoll pipettiert:
5x Reaction Buffer 4 µl
RiboLock RNase Inhibitor (20 U/µl) 1 µl
10 mM dNTP mix 2 µl
RevertAid H Minus M-MulV Reverse Transkriptase 1 µl
Volumen 8 µl
Der Master Mix wurde zu dem Ansatz gegeben, kurz gemischt und bei 42°C fü r 60 min
inkubiert. Die Proben wurden erneut bei 70°C erhitzt und entweder weiter verarbeitet oder
bei -20°C gelagert. Um Kontaminationen mit DNA auszuschließen wurde jedem
Versuchsansatz eine Negativkontrolle mitgefü hrt, wobei die RNA durch ein entsprechendes
Volumen an H2Odest ersetzt wurde.

2 Material und Methoden
34
2.2.2.7 Real-Time quantitative PCR
Die Real-time quantitativer PCR erlaubt neben der Vervielfältigung von Nukleinsäuren
zusätzlich deren Quantifizierung. Die relative Expression der unterschiedlichen Gene wurde
mittels Fluoreszenzfarbstoff (SYBR Green I) analysiert. Nach dem Binden des Farbstoffes an
die doppelsträngige DNA nimmt die Intensität des Farbstoffes um ein Vielfaches zu und
steigt während der Extensionsphase durch das zusätzliche Binden an neu entstandene PCR-
Produkte weiter an. Dieses Fluoreszenzsignal wird anschließend gemessen und ausgewertet.
Die Versuche wurden mit dem LightCycler480 (Roche) und dem SYBR Green I Kit der Firma
Roche durchgefü hrt. Der Reaktionsansatz wurde dabei nach folgendem Schema pipettiert:
H2Odest 6 µl
„ forward“ Primer (25 µM) 1 µl
„ reverse“ Primer (25 µM) 1 µl
Master Mix 10 µl
cDNA (40 ng) 2 µl
Volumen 20 µl
Um potentielle Kontaminationen des Master Mixes bzw. der cDNA auszuschließen wurde
eine Leerprobe (keine DNA) zugefü gt. Die Real-time PCR wurde mit 45 Zyklen durchgefü hrt.
Nach Beendigung der Reaktion konnten die Ct-Werte anhand der Messdaten mit Hilfe der
LightCycler480 Software release (Version 1.5.0) berechnet werden. Alle Proben wurden in
Duplikaten pipettiert und das „house-keeping“ Gen HPRT (Hypoxanthin-Phosphoribosyl-
Transferase) als Kalibrator fü r die Normalisierung verwendet. Die Daten wurden relativ
quantifiziert mittels ΔΔCt-Methode, mittels GraphPad Prism 5 statistisch ausgewertet und
graphisch dargestellt. Alle Exprerimente wurden dreimal wiederholt.
2.2.2.8 Amplifikation von DNA/cDNA-Fragmenten mittels Polymerase-Kettenreaktion
(„polymerase chain reaction“, PCR)
Mittels PCR lassen sich in vitro DNA-Bereiche zwischen zwei bekannten Regionen
amplifizieren. In mehreren Zyklen werden dabei die folgenden Schritte wiederholt:
Denaturierung der DNA-Stränge, Hybridisierung der Oligomere an den Matrizen-Strang und
Synthese des komplementären Stranges durch Elongation der Oligomere. Dadurch wird der

2 Material und Methoden
35
Sequenzbereich zwischen den Oligonukleotiden exponentiell amplifiziert. Der allgemeine
PCR-Ansatz in einem Reaktionsvolumen von 25 µl wurde dabei wie folgt angesetzt:
DreamTaqTM Green PCR Master Mix (Fermentas) 12,5 µl
sense Primer (10 µM) 2 µl
anti-sense Primer (10 µM) 2 µl
DNA (hier: 50 ng) 1 µl
H2Odest 7,5 µl
Volumen 25 µl
Die Synthesereaktion erfolgte in der PCR Maschine (VWR) mittels folgenden
Syntheseprotokolls:
Erste Denaturierung 5 min 95°C
Amplifikation 25-35 Zyklen 1 min 30 sec 95°C (Denaturierung)
1 min 30 sec x°C (Hybridisierung)
2 min 72°C (Elongation)
Finale Elongation 5 min 72°C
Die Hybridisierungstemperatur der einzelnen Oligonukleotide richtete sich dabei nach der
jeweiligen Oligomersequenz. Nach Beendigung der Synthese wurde das PCR-Produkt auf ein
Agarosegel aufgetragen, als Nachweis fü r die gelungene Amplifikation.
2.2.2.9 Agarosegelelektrophorese von DNA
Mittels Gelelektrophorese kö nnen DNA-Fragmente in einem elektrischen Feld abhängig von
ihrer Molekü lgrö ße aufgetrennt werden. Das Agarosegel dient dabei als molekulares Sieb
durch das die negativ geladenen DNA-Fragmente wandern kö nnen. Dabei werden
hochprozentige Agarosegele (1,5- 2%) zur Auftrennung von kleinen DNA-Fragmenten (<500
bp) und niederprozentige Agarosegele (0,5- 1%) zur Auftrennung von großen DNA-
Fragmenten (>1000 bp) verwendet. Fü r die Gelelektrophorese wurden 1%-ige w/v
Agarosegele mit 1x TBE Laufpuffer und 0,5 µg/ml Ethidiumbromid verwendet. Die DNA-
Proben wurden mit 6x Gelladepuffer versehen, auf das Gel aufgetragen und bei einer

2 Material und Methoden
36
Spannung von 50- 100 V fü r ca. 30 min aufgetrennt. Anschließend wurden die Gele unter
einem UV-Transilluminator analysiert und zu Dokumentationszwecken fotografiert.
2.2.2.10 Photometrische Bestimmung von Nukleinsäuren
Die Konzentration von RNA, DNA, cDNA und Plasmid-DNA wurde mit dem Nanodrop 2000
Spectrophotometer (Thermo Scientific) bei 260 nm bestimmt. Die Reinheit wurde mit der
Ratio260/280 bestimmt.
2.2.3 Immunologische Methoden
2.2.3.1 Immunhistochemie (IHC)
Bei der Immunhistochemie kö nnen Proteine in Gewebeschnitten mittels Antikö rper-Antigen-
Reaktion sichtbar gemacht werden. Dabei bindet in einem ersten Schritt ein spezifischer
Antikö rper (Primärantikö rper) gegen ein Epitop, also den bestimmten Bereich eines
Antigens. In einem zweiten Schritt bindet ein Sekundärantikö rper, der wiederrum mit einem
Enzym (hier: Peroxidase) gekoppelt ist, spezifisch an den Primärantikö rper. Durch eine
Enzym-Substrat (hier: Wasserstoffperoxid)-Reaktion wird ein Chromogen (hier DAB= 3,3´-
Diaminobenzidin) durch Oxidation in einen braunen Farbstoff umgesetzt, der im Gewebe
sichtbar ist. Fü r die Immunhistochemie wurden 3,5 µm dicke Schnitte von Formalin fixiertem
und in Paraffin eingebettetem Gewebe erstellt. Anschließend wurden die Schnitte in einem
ca. 40°C warmen Wasserbad geglättet, auf Objektträger aufgezogen und ü ber Nacht bei 37°C
im Inkubator getrocknet. Fü r die Immunhistochemie wurden dabei folgende Schritte im
Einzelnen durchgefü hrt:
1 Deparaffinierung Roticlear 3x 10 min 2 Rehydratation Ethanol 100% 3x 3 min Ethanol 95% 3 min Ethanol 70% 3 min Ethanol 50% 3 min 3 Waschen H2Odest 5 min 4 Hitzeinduzierte Antigendemaskierung 1x Citratpuffer 2x 5 min (600 W) 5 Abkü hlen Raumtemperatur 15- 20 min 6 Waschen 1x TBST 5 min 7 Permeabilisierung 0,5% TritonX100/PBS 5 min 8 Waschen Waschen 5 min 9 Hydrogen Peroxidase Behandlung 3% H2O2 5 min 10 Waschen Aquadest 5 min

2 Material und Methoden
37
11 Blocken 1x TBST/3% BSA 1 h 12 Primärantikö rper AK in 1x TBST/3% BSA ü ber Nacht 13 Waschen 1x TBST 2x 5 min 14 Sekundärantikö rper 1 h 15 Waschen 1x TBST 2x 5 min 16 DAB Reaktion 1-2 min 17 Stoppen der DAB Reaktion in Wasser 18 Hämatoxylin 15 sec 19 Waschen H2Odest 15 min 20 Dehydratation Ethanol 70% 3 min Ethanol 95% 3 min Ethanol 100% 3x 3 min Roticlear 3x 5 min 21 Fixierung Mounting medium
2.2.3.1.1 Quantifizierung der Kif20a Immunhistochemie
Fü r die Analyse der Kif20a Expression mit dem Ü berleben von Patienten mit duktalem
Adenokarzinom wurden Gewebeschnitte immunhistochemisch mit dem polyklonalen Kif20a
Antikö rper angefärbt (siehe 2.2.3.1) und von zwei unabhängigen Personen analysiert (D.S,
M.E.). Die Schnitte wurden entsprechend der Anzahl der Kif20a gefärbten Tumorzellen (0-
30%= 1, 31- 60%= 2, 61- 100%= 3) sowie der Intensität der Kif20a Färbung (schwach= 1,
mittel= 2, stark= 3) ausgewertet. Die Werte der Intensität bzw. der Fläche wurden fü r jeden
Patienten multipliziert und als Kif20a „ tief“ (Wert <6) bzw. Kif20a „hoch“ (Wert ≥ 6)
eingeteilt.
2.2.3.1.2 Quantifizierung der PCNA („Proliferating cell nuclear antigen“)
Immunhistochemie
Fü r die Analyse der PCNA Expression wurden Gewebeschnitte wie unter 2.2.3.1 beschrieben
angefärbt und Bilder in 200-facher Vergrö ßerung angefertigt. Fü nf repräsentative
Bildauschnitte mit je 100 Zellen wurden fü r die Evaluierung ausgewählt und die Gesamtzahl
der PCNA positiv gefärbten Tumorzellen gezählt.
2.2.3.2 Immunfluoreszenz (IF)
Mittels Immunfluoreszenz kö nnen Proteine in Zellen oder Gewebeschnitten durch
Antikö rper-Antigen-Reaktion sichtbar gemacht werden. Dabei bindet in einem ersten Schritt
ein spezifischer Primärantikö rper an ein Epitop des zu untersuchenden Proteins. In einem
zweiten Schritt bindet ein gegen den ersten Antikö rper gerichteter Sekundärantikö rper, der

2 Material und Methoden
38
wiederrum mit einem Fluorochrom gekoppelt ist. Unter dem Immunfluoreszenzmikroskop
kann der Farbstoff sichtbar gemacht werden. Dabei trifft kurzwelliges Anregungslicht auf das
Präparat, dessen gebundenes Fluorochrom angeregt wird und längerwellig fluoresziert. Fü r
die Immunfluoreszenz wurden dabei folgende Schritte durchgefü hrt: Zellen wurden ü ber
Nacht auf Objektträgern ausgesät. Der Objektträger wurde dann mit 1x PBS gewaschen und
die Zellen anschließend fü r 30 min mit 4% Formaldehyd fixiert. Nach einem weiteren 5min
Waschschritt mit 1x PBS wurden die Zellen fü r 15 min mit 0,5% TritonX100 behandelt um
diese zu permeabilisieren. Nach einer 30 min Inkubation mit 1x TBST/3%BSA wurde der in 1x
PBS verdü nnte Primärantikö rper auf die Zellen gegeben und ü ber Nacht bei 4°C inkubiert.
Die Zellen wurden dann mit 1x PBS/0,1%Tween20 gewaschen, um unspezifische
Bindungsstellen abzusättigen, und anschließend mit dem Sekundärantikö rper fü r eine
Stunde im Dunkeln inkubiert. Nach einem finalen Waschschritt mit 1x PBS/0,1%Tween20
wurden die Zellen mit „Fluorescence Mounting Medium“ fixiert. Dabei enthielt das
„Mounting Medium“ das 4´,6-Diamidino-2-phenylindol (DAPI), dass zur Färbung der Kerne
verwendet wurde. Die Präparate wurden unter dem Fluoreszenzmikroskop analysiert und zu
Demonstrationszwecken fotografiert.
Tab. 17: Übersicht der Fluoreszenzfarbstoffe
Fluorochrom
Farbe Extinktion Emission
Alexa488 Grü n 495 519
Alexa594 Orange-Rot 590 617
DAPI Blau 358 461
Probidiumiodid Rot 535 617
2.2.4 Proteinanalytik
2.2.4.1 Herstellung des RIPA Puffers fü r die Proteinextraktion
Mittels RIPA Puffer kö nnen Zellen lysiert und Proteine gelö st werden. Dabei werden die
Proteine weder degradiert noch deren biologische Aktivität zerstö rt. Fü r die Herstellung
eines RIPA-Puffers wurden zunächst 750 mg Tris Baze in 75 ml Wasser gegeben und
anschließend 900 mg NaCl unter ständigem Rü hren darin gelö st. Der pH-Wert wurde dabei
mit 5 M HCL auf 7,4 eingestellt. Nach dem vollständigen Lö sen der Feststoffe wurden 10 ml

2 Material und Methoden
39
10% NP-40 und 2,5 ml 10% Na-deoxycholate unter ständigem Rü hren zur Lö sung gegeben. In
einem finalen Schritt wurden 1 ml 100 mM EDTA zum Ansatz pipettiert und das Volumen auf
100 ml mit destilliertem Wasser eingestellt. Der RIPA-Puffer wurde dann bis zur weiteren
Verwendung bei 2- 8°C gelagert. Direkt vor Gebrauch des RIPA-Puffers wurden diesem noch
folgende Substanzen in einer Konzentration von 1 µg/ml hinzugefü gt: je 100 µl Aprotinin,
Leupeptin, Pepstatin, sowie je 500 µl PMSF, Na3VO4 und NaF. Dabei handelt es sich um
Inhibitoren (Proteinasen, Phosphatasen), welche zum einen die Proteolyse verhindern, und
zum anderen die Phosphorylierung von Proteinen aufrechterhalten sollen.
2.2.4.2 Herstellung von Proteinlysaten
Fü r die Herstellung von Proteinlysaten wurden die Zellen mit 1x PBS gewaschen, frisch
angesetzter RIPA-Puffer dazu gegeben und die adhärenten Zellen mit einem Zellschaber von
der Zellkulturschale abgekratzt. Anschließend wurden die Zellen mit einer 1 ml Insulinspritze
resuspendiert und bei 4°C fü r 15 min bei 10.000 rpm zentrifugiert, um das Zelllysat von den
Zellfragmenten zu trennen. Der Ü berstand wurde weiter verarbeitet oder bei -20°C gelagert.
2.2.4.3 Proteinbestimmung
Die Bestimmung der Proteinkonzentration erfolgte mittels Bradford-Methode. Dazu wurde
eine BSA-Standard Verdü nnungsreihe mit den Konzentrationen 0,125 µg/µl bis 2 µg/µl
hergestellt. Die Reagenzien A und B des „Pierce BCA Protein Assay Kit” wurden anschließend
im Verhältnis 1:50 gemischt und je 200 µl/Loch einer 96-Loch Platte vorgelegt. Je 5 µl des zu
untersuchenden Proteinlysats sowie 5 µl der Standards wurden in Duplikaten zu den „wells“
pipettiert und die Platte im ELISA-Reader bei 595 nm gemessen. Mittels der erstellten BSA-
Standard-Eichkurve konnten anschließend die jeweiligen Proteinkonzentrationen der Proben
berechnet werden.
2.2.4.4 Polyacrylamid-Gelelektrophorese (SDS-PAGE)
Mittels Natriumdodecylsulfat-Polyacrylamid-Gel-Elektrophorese (SDS-PAGE, sodium dodecyl
sulfate polyacrylamid gel electrophoresis) lassen sich Proteine entsprechend ihrer
molekularen Masse auftrennen [137]. Durch die Vorbehandlung des Proteingemisches mit
dem Detergenz SDS wird a) die Sekundärstruktur der Proteine aufgehoben und b) die
Proteine negativ geladen. Dadurch wird die Wandereigenschaft der Proteine vorwiegend
durch ihr Molekulargewicht und nicht durch deren Eigenladung oder

2 Material und Methoden
40
Aminosäurezusammensetzung bestimmt. Fü r die SDS-Page wurden 10%-ige SDS Gele (siehe
Tab. 15) gegossen. Dabei werden in hochprozentigen Gelen kleine Proteine und in
niedrigprozentigen Gelen große Proteine aufgetrennt. Die Proteinproben wurden vor dem
Auftragen auf das SDS-Gel mit 6,25 µl „4x NuPage LDS Sample Buffer“ (Invitrogen) und 2,5 µl
„NuPage Reducing Agent“ (Invitrogen) gemischt und 10 min bei 95°C erhitzt. Anschließend
wurden die Proben 5 min bei 4°C inkubiert, kurz auf Eis gestellt und anschließend auf das
SDS-Gel aufgetragen. In einer vertikalen Gelelektrophoresekammer mit Laufpuffer wurden
die Proben 55 min bei einer Spannung von 200 V aufgetrennt und fü r weitere Western Blot
Experimente eingesetzt.
Tab. 18: Zusammensetzung eines 10%-igen SDS-Gels
Lösung Sammelgel Trenngel
H2Odest 1,5 ml 3,3 ml
30% Bis-Acrylamid 330 µl 2,8 ml
4x Upper Tris (pH 6,8) 625 µl -
4x Lower Tris (pH 8,8) - 2,8 ml
SDS 10% 25 µl 80 µl
APS 10% 20 µl 60 µl
TEMED 2,5 µl 2,5 µl
Volumen 2502,5 µl 9042,5 µl
2.2.4.5 Transfer von Proteinen auf eine Nitrocellulosemembran (Western Blot)
Um die Identität elektrophoretisch aufgetrennter Proteine zu untersuchen wurden diese auf
eine Nitrocellulosemembran transferiert. Die Ü bertragung der Proteine erfolgte in einer
„semi-dry“ Blotting Kammer mit Transferpuffer bei einer Spannung von 30 V fü r 1,5 h. Nach
dem Transfer wurde die Membran fü r Immundetektions-Analysen weiter behandelt.
2.2.4.6 Immundetektion von Proteinen (Immunblotanalyse)
Bei der Immundetektion wurden die auf die Membran transferierten Proteine mittels
Antigen-Antikö rper Reaktion sichtbar gemacht. Dabei bindet in einem ersten Schritt ein
spezifischer Antikö rper die immobilisierten Antigene auf der Membran und in einem zweiten
Schritt wird dieser wiederum von einem Sekundärantikö rper erkannt, der an eine

2 Material und Methoden
41
Meerrettich-Peroxidase gekoppelt ist. Durch diese enzymatische Aktivität werden die
Antigenbanden detektiert. Nach dem Transfer der Proteine auf die Membran wurde diese
zweimal kurz mit 1x TBS gewaschen. Die freien Bindungsstellen wurden durch die Inkubation
mit 5%-iger Milch/TBST Lö sung (Blocking Puffer) fü r 20 min bei Raumtemperatur abgesättigt.
Aufgrund der starken Phosphorylierung vieler Milchproteine wurde bei der Verwendung von
Antikö rpern gegen phosphorylierte Proteine die Membran mit einer 5%-igen BSA/TBST
Lö sung geblockt. Der erste Antikö rper wurde in 5%-iger Milch/TBST- bzw. 5%-iger BSA/TBST
Lö sung verdü nnt und die Membran fü r min 2 h bei Raumtemperatur oder ü ber Nacht bei 4°C
unter ständigem Schü tteln inkubiert. Durch dreimaliges Waschen mit 1x TBST fü r 10 min
wurde der ü berschü ssige Primärantikö rper von der Membran entfernt. Anschließend wurde
der HRPT-gekoppelte Spezies-spezifische Sekundärantikö rper, der ebenfalls mit Blocking-
Puffer verdü nnt wurde, dazu gegeben und die Membran fü r eine Stunde unter ständigem
Schü tteln bei Raumtemperatur inkubiert. Vor der Detektion mit dem „ECL-Kit“ (Invitrogen)
wurde ein dreimaliger Waschschritt mit 1x TBST fü r 10 min durchgefü hrt. Fü r die Detektion
wurden die ECL Reagenzien A und B im Verhältnis 1:1 gemischt und fü r eine Minute auf die
Membran gegeben. Anschließend wurde der Blot luftblasenfrei in eine Klarsichtfolie gelegt
und auf einem Rö ntgenfilm exponiert. Sollte dieselbe Membran mit einem weiteren
Antikö rper inkubiert werden, wurde diese zunächst fü r 10 min mit 0,2 M NaOH-Lö sung bei
Raumtemperatur behandelt, um die bereits gebundenen Antikö rper zu entfernen. Nach
einem dreimaligen Waschschritt fü r 10 min mit 1x TBST konnte die Membran erneut
geblockt und mit einem Antikö rper inkubiert werden.
2.2.5 In vitro Analysen
2.2.5.1 Transfektion mit siRNA
Als Transfektion bezeichnet man das Einbringen genetischen Materials in eukaryotische
Zellen. Es kö nnen hierbei Plasmide fü r die Ü berexpression von Genen oder siRNA („small
interfering RNA“) fü r die Hemmung solcher eingeschleust werden. Die Hemmung der
Genexpression spezifischer Gene durch RNA Molekü le wird als RNA- Interferenz bezeichnet
[138]. siRNAs sind kleine RNA Molekü le, die komplementär an Strukturen der mRNA Binden
und diese durch verschiedenste zelluläre Mechanismen degradieren. Daraus folgert eine
spezifische Unterdrü ckung der Genexpression. Fü r die Transfektion von humanen
Pankreaskarzinomzellen wurden am Tag zuvor Zellen mit einer Dichte von 50x103 Zellen/
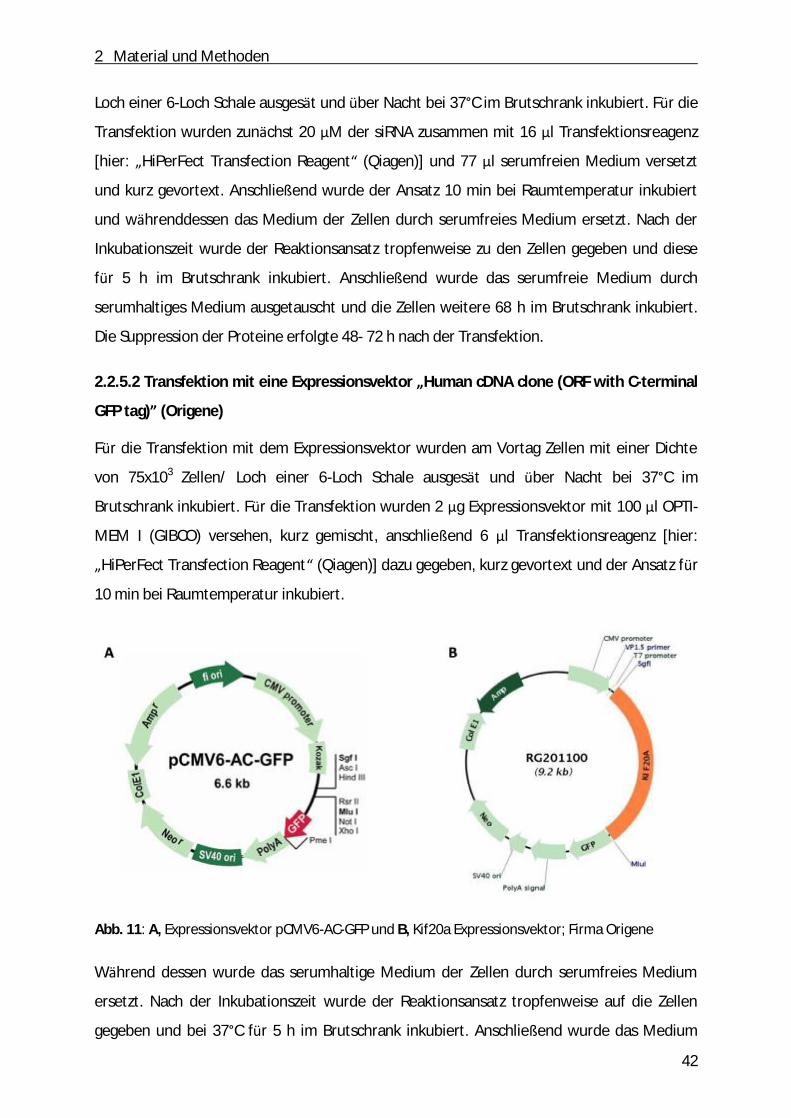
2 Material und Methoden
42
Loch einer 6-Loch Schale ausgesät und ü ber Nacht bei 37°C im Brutschrank inkubiert. Fü r die
Transfektion wurden zunächst 20 µM der siRNA zusammen mit 16 µl Transfektionsreagenz
[hier: „HiPerFect Transfection Reagent“ (Qiagen)] und 77 µl serumfreien Medium versetzt
und kurz gevortext. Anschließend wurde der Ansatz 10 min bei Raumtemperatur inkubiert
und währenddessen das Medium der Zellen durch serumfreies Medium ersetzt. Nach der
Inkubationszeit wurde der Reaktionsansatz tropfenweise zu den Zellen gegeben und diese
fü r 5 h im Brutschrank inkubiert. Anschließend wurde das serumfreie Medium durch
serumhaltiges Medium ausgetauscht und die Zellen weitere 68 h im Brutschrank inkubiert.
Die Suppression der Proteine erfolgte 48- 72 h nach der Transfektion.
2.2.5.2 Transfektion mit eine Expressionsvektor „Human cDNA clone (ORF with C-terminal
GFP tag)” (Origene)
Fü r die Transfektion mit dem Expressionsvektor wurden am Vortag Zellen mit einer Dichte
von 75x103 Zellen/ Loch einer 6-Loch Schale ausgesät und ü ber Nacht bei 37°C im
Brutschrank inkubiert. Fü r die Transfektion wurden 2 µg Expressionsvektor mit 100 µl OPTI-
MEM I (GIBCO) versehen, kurz gemischt, anschließend 6 µl Transfektionsreagenz [hier:
„HiPerFect Transfection Reagent“ (Qiagen)] dazu gegeben, kurz gevortext und der Ansatz fü r
10 min bei Raumtemperatur inkubiert.
Abb. 11: A, Expressionsvektor pCMV6-AC-GFP und B, Kif20a Expressionsvektor; Firma Origene
Während dessen wurde das serumhaltige Medium der Zellen durch serumfreies Medium
ersetzt. Nach der Inkubationszeit wurde der Reaktionsansatz tropfenweise auf die Zellen
gegeben und bei 37°C fü r 5 h im Brutschrank inkubiert. Anschließend wurde das Medium

2 Material und Methoden
43
durch serumhaltiges Medium ersetzt und die Zellen fü r weitere 24- 48 h im Brutschrank bei
37°C inkubiert. Fü r die Versuche wurde der Expressionsvektor pCMV6-AC-GFP der Firma
Origene verwendet, der neben einer Ampicillin-Resistenz und einer GFP-Markierung die
humane Kif20a Sequenz enthält (Abb. 11).
2.2.5.3 Synchronisation von Krebszellen
Um Zellen in verschiedenen Zellzyklusstadien untersuchen zu kö nnen, mü ssen sie in
verschiedenen Stadien synchronisiert werden. Mittels doppeltem Thymidinblock kö nnen
Zellen in der G1-Phase des Zellzyklus arretiert werden. Das Ribonukleotid Thymidin wirkt
dabei als allosterischer Inhibitor auf die Ribunukleotid-Reduktase. Durch einen Ü berschuss
an Thymidin wird die Synthese von Pyrimidinnukleotiden unterbunden. Dadurch wird die
DNA-Synthese gehemmt und die Zellen am G1/S-Checkpoint synchronisiert. Ein doppelter
Thymidinblock wurde durchgefü hrt, um alle Zellen zu erreichen und zu arretieren. Hierfü r
wurden die Zellen fü r 20 h mit 2,5 mM Thymidin inkubiert. Nach zweimaligem Waschen mit
PBS wurden die Zellen fü r 9 h mit Standardmedium versehen. Anschließend erfolgte ein
erneuter Inkubationsschritt mit 2,5 mM Thymidin fü r 16 h. Nach einem weiteren
Waschschritt mit PBS wurden die Zellen bis zur endgü ltigen Analyse mit Standardmedium
versehen.
2.2.6 Biologische Analysen
2.2.6.1 Proliferationsassay (MTT-Assay)
Um den Effekt von Substanzen auf die Proliferation von Zellen zu untersuchen, kann ein
MTT-Assays durchgefü hrt werden. Das Prinzip des Testes beruht dabei auf der Spaltung des
gelben Tetrazoliumsalzes MTT (3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromid)
in das blau-violette, wasserunlö sliche Formazan durch die Succinat Dehydrogenase. Diese
zelluläre Reduktion wird ausschließlich von metabolisch aktiven Zellen durchgefü hrt und
benö tigt die Co-Faktoren NADH und NADHP. Durch die Zugabe von Isopropanol zu den
Formazan-Kristallen werden diese gelö st und die Konzentration der resultierenden gefärbten
Lö sung im ELISA-Reader (Thermo Scientific) bei 570 nm photometrisch gemessen.

2 Material und Methoden
44
2.2.6.2 MTT-Assay nach Transfektion mit siRNA oder Expressionsvektor
Um den Einfluss der Runter- oder Ü berregulierung eines Gens auf die Zellproliferation zu
untersuchen wurden die Zellen, wie unter 3.1.4.3 beschrieben, vorbehandelt. 24 h vor
Inkubationsende wurden die Zellen in Mikrotiterplatten („96-well“) mit einer Zelldichte von
5x103 Zellen/Loch in 100 µl Kulturmedium ausgesät und 24 h bei 37°C und 5% CO2 im
Brutschrank inkubiert. Anschließend wurden 10 µl/Loch der MTT-Lö sung zu den Zellen
gegeben und die Platte fü r 4h bei 37°C im Brutschrank inkubiert. Danach wurde das Medium
abgesaugt und die Platte erneut fü r 30 min bei 37°C getrocknet. In einem finalen Schritt
wurden 100 µl Isopropanol pro Loch pipettiert und die Platte fü r 5 min bei 300 rpm
geschü ttelt. Der Ansatz wurde in einem ELISA-Reader (Thermo Scientific) bei 570 nm
gemessen und ausgewertet.
2.2.6.3 MTT-Assay nach Runter- oder Überregulierung eines Gens mit zusätzlicher
Zytostatika Behandlung (Taxol/Gemcitabine)
In diesen Versuchen wurde der zusätzliche Effekt der Zytostatika Taxol und Gemcitabine auf
bereits transfizierte Zellen (siRNA/ Expressionsvektor) untersucht. Die Zellen wurden
zunächst wie unter 3.1.4.3 beschrieben, vorbehandelt. 24 h vor Inkubationsende wurden die
Zellen gesplittet und auf Mikrotiterplatten („96-well“) mit einer Zelldichte von 5x103
Zellen/Loch ausgesät. Zusätzlich wurde zu den Zellen in Versuch 1: Taxol (2 µM, 20 µM) und
in Versuch 2: Gemcitabine (10 µM, 50 µM, 100 µM, 500 µM, 1000 µM) gegeben und die
Platten fü r 24 h bei 37°C im Brutschrank inkubiert. Die weitere Durchfü hrung und
Auswertung des MTT-Assays erfolgte wie unter 2.2.5.1 beschrieben.
2.2.6.4 Migrationsassay
Mittels Migrationsassay sollte die Auswirkung der Runter- bzw. Ü berregulierung von Kif20a
auf die Migration von Krebszellen in vitro untersucht werden. Fü r die Migrationsversuche
wurden Migrations-Platten („24-Multiwell Insert System“) der Firma BD verwendet und laut
Herstellerprotokoll verfahren. Die Zellen wurden vorab wie unter 2.2.5.1 bzw. 2.2.5.2
beschrieben behandelt.
2.2.6.5 Invasionsassay
Um die Invasionsfähigkeit von Krebszellen nach Transfektion zu untersuchen, wurden
Matrigel beschichtete „ Insert Systems“ („ 24-multiwell insert systems with matrigel“) der

2 Material und Methoden
45
Firma BD verwendet. Dabei handelt es sich um kleine Inserts, die eine PET Membran mit 8
µm großen Poren enthalten. Diese Membran ist mit einer dü nnen Schicht Matrigel
beschichtet, die in vitro als eine rekonstituierte Basalmembran fungiert. Durch die
Beschichtung mit Matrigel werden nicht invasive Zellen davon abgehalten, durch die
Membran zu wandern. Invasive Zellen (maligne und nicht maligne) kö nnen durch das
Matrigel wandern und die Membran passieren. Die Versuche wurden laut
Herstellerprotokoll durchgefü hrt. Vorab wurden die Zellen wie unter Punkt 2.2.5.1
beschrieben, behandelt.
2.2.6.6 Motilitätsanalysen mittels „Time- lapse“-Mikroskopie
Das Prinzip der „ time lapse“-Mikroskopie beruht auf der Aufnahme einer bestimmten Zelle
oder Zellpopulation ü ber einen bestimmten Zeitraum. Dabei wird das Objekt bei gleichen
Einstellungen und gleicher Position in bestimmten Intervallen gescannt. Dadurch kö nnen
dynamische Vorgänge innerhalb einzelner Zellen oder zwischen Zellpopulationen verfolgt
werden. Mittels des „Manual Tracking“ Programms kö nnen bestimmte Zellen ausgewählt
und ü ber einen Zeitraum verfolgt werden. Somit lassen sich mit einem weiteren
„Chemotaxis“-Programm die Geschwindigkeit, die zurü ckgelegte Distanz sowie die
Richtungsabhängigkeit der Zellen berechnen und statistisch auswerten. Hierfü r wurden die
Zellen wie unter 2.2.5.1 beschrieben behandelt und 16 h bevor die Runterregulierung ihr
Maximum (hier: 72 h) erreicht hatte, in einer Dichte von 50.000 Zellen/ Loch einer 6-Loch
Schale ausgesät. Anschließend wurde die Bewegung der Zellen mittels Mikroskop fü r 22 h
aufgenommen. Die Auswertung der Zellen erfolgte mittels „Manual Tracking Program“
(Image J software) sowie einem „Chemotaxis tool“ der Firma Ibidi.
2.2.6.7 Zellzyklusanalysen mittels „Fluorescence activated cell sorting“ (FACS)
Mittels Propidiumiodid (PI)- Färbung lassen sich die einzelnen Zellzyklusstadien, durch die
Bestimmung des DNA-Gehalts der Zellen, genauer untersuchen. Dabei bindet PI
stö chiometrisch an die DNA. Ü ber die Menge des Farbstoffs kann anschließend mittels FACS
Analyse der Gehalt der DNA berechnet werden. Dafü r wurden die behandelten Zellen
trypsiniert, mit PBS gewaschen und anschließend in 300 µl PBS resuspendiert. Anschließend
wurden 700 µl eiskalter Ethanol zu der Zellsuspension gegeben und diese mindestens 4 h bei
4°C fixiert (die Zellen waren nun bis zu 4 Wochen bei 4°C haltbar). Fü r die FACS Analyse
wurden die Zellen 4 min bei 1200 rpm zentrifugiert und der Ü berstand vorsichtig abgesaugt.

2 Material und Methoden
46
Das Pellet wurde mit PBS gewaschen und erneut fü r 4 min bei 1200 rpm zentrifugiert.
Anschließend wurde der Ü berstand verworfen und das Pellet in 400 µl PBS (plus 10 mg/ml
RNAseA) resuspendiert. Durch die Zugabe der RNAseA wurden unspezifische RNA Molek ü le
durch hydrolytische Spaltung der Phosphodiesterbindung abgebaut. Nach dem Inkubieren
der Zellsuspension fü r 30- 45 min bei 60°C wurden die Zellen mit 2 µg/ml PI gefärbt und
mittels FACS-Gerät analysiert.
2.2.6.8 Apoptose Assay
Um lebende Zellen von Zellen zu unterscheiden, die sich in der Apoptose befinden, gibt es
unterschiedliche Mö glichkeiten. In der vorliegenden Arbeit wurden fü r die Bestimmung von
apoptotischen Zellen Immunfluoreszenz- Färbungen mittels „Cleaved Caspase 3“
durchgefü hrt. In lebenden Zellen gibt es sogenannte Initiator-Caspasen (Caspase 8 und 9),
die durch eine Reihe von Enzymkaskaden aktiviert werden und daraufhin Effektor-Caspasen
(Caspase 3, 6, 7) spalten. Durch die Spaltung der Effektor-Caspasen werden diese wiederum
aktiviert und spalten zelleigene Proteine wie Laminin und Aktin. Zudem werden Nukleasen
aktiviert, die im Rahmen der Apoptose nukleäre DNA spaltet. Mittels Immunfluoreszenz lässt
sich die gespaltene Caspase 3 (hier: „Cleaved Caspase 3“) als Färbung sichtbar machen.
Hierfü r wurden die Zellen wie unter Punkt 2.2.3.2 beschrieben behandelt und anschließend
unter dem Mikroskop analysiert.
2.2.6.10 Graphische Darstellung und statistische Analysen
Alle Versuche wurden dreimal unabhängig voneinander wiederholt und statistisch sowie
grafisch mit dem Programm GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA)
ausgewertet. Vergleiche von demographischen und klinischen Parametern zwischen
Gruppen, wurden mittels „Fisher Test“ und „Chi-Quadrat Test“ durchgefü hrt. Die Korrelation
der Kif20a Expression mit dem Ü berleben von Patienten mit duktalem Adenokarzinom
wurde mittels „Kaplan- Meier“ Methode und „Log rank test“ analysiert. Beim Vergleich von
zwei Gruppen wurden Medianwerte als „Cut-off“ Limits verwendet. Die Ergebnisse sind als
Mittelwerte +/- Standardfehler des Mittelwertes („standard error of the mean“, SEM)
dargestellt. Statistisch signifikante Unterschiede wurden mit p ≤ 0,05 akzeptiert und sind mit
einen Stern (*) indiziert.

3 Ergebnisse
47
3 Ergebnisse
3.1 Analyse der Kif20a Expression in humanem Pankreasgewebe und
Karzinomzelllinien
3.1.1 Kif20a Expression in humanem Pankreasgewebe
Die immunhistochemischen Untersuchungen wurden an Formalin-fixierten und in Paraffin
eingebetteten Geweben durchgefü hrt. Fü r die Untersuchungen wurden Schnitte mit
normalem Pankreasgewebe (n= 5), chronischer Pankreatitis (n= 10), interepithelialen
Neoplasien (PanIN) (n= 5), sowie duktalem Adenokarzinom (n= 10) verwendet.
3.1.1.1 Gesunder Pankreas
Die Analysen des gesunden Pankreasgewebes zeigten eine schwache zytoplasmatische
Kif20a Färbung des azinären Gewebes, der duktalen Zellen, sowie der Inselzellen. Gefäße
und Nervenzellen zeigten keinerlei positive Färbung (Abb. 12). Eine Färbung der Nuklei
konnte nicht detektiert werden.
Abb. 12: Immunhistochemische Analysen von Kif20a in gesundem Pankreas. Formalin-fixierte Paraffinschnitte wurden mit einem Antikö rper gegen Kif20a inkubiert und mit Hämatoxylin gegengefärbt. Die histologische Färbung zeigte eine schwache zytoplasmatische Kif20a Lokalisation in allen duktalen sowie azinären Strukturen. Inselzellen wiesen eine etwas stärkere Kif20a Färbung auf. Vergrö ßerung x100
3.1.1.2 Chronische Pankreatitis
Gewebeschnitte mit chronischer Pankreatitis zeigten eine stärkere zytoplasmatische Kif20a
Färbung in Azinus- und duktalen Zellen im Vergleich zu gesundem Gewebe. Die Gefäße und
die Inselzellen waren in den chronischen Pankreatitis Proben deutlich stärker gefärbt. Eine
Färbung der Kerne wurde nicht beobachtet (Abb. 13).

3 Ergebnisse
48
Abb. 13: Immunhistochemische Analysen von Kif20a in chronischer Pankreatitis. Formalin-fixierte Paraffinschnitte wurden mit einem Antikö rper gegen Kif20a inkubiert und mit Hämatoxylin gegengefärbt. Eine stärkere zytoplasmatische Kif20a Färbung konnte sowohl in den Inselzellen als auch in den azinären und duktalen Strukturen detektiert werden. Die Kerne waren dabei nicht gefärbt. Vergrö ßerung x100 (A), x200 (B, C).
3.1.1.3 PanIN Läsionen
In PanIN Läsionen konnte eine deutliche zytoplasmatische Kif20a Färbung beobachtet
werden, die je nach untersuchtem Präparat unterschiedlich stark ausfiel. Circa 80% der
Präparate wiesen eine starke zytoplasmatische Kif20a Färbung der PanIN Läsionen sowie der
Inselzellen auf (Abb. 14A). In weniger als 1% der Zellen konnte zudem eine positive
Kernfärbung detektiert werden (Abb. 14B, rote Pfeile). Ein Fü nftel der angefärbten Schnitte
zeigte eine sehr schwache zytoplasmatische Kif20a Färbung, sowohl in den Bereichen mit
PanIN Läsionen als auch in den Inselzellen. Zellen des umliegenden Stromas wurden nicht
angefärbt (Abb. 14C).
Abb. 14: Immunhistochemische Analysen von Kif20a in verschiedenen PanIN Läsionen des Pankreas. Formalin-fixierte Paraffinschnitte wurden mit einem Kif20a Antikö rper inkubiert und mit Hämatoxylin gegengefärbt. A, eine starke zytoplasmatische Kif20a Expression konnte in den PanIN Läsionen festgestellt werden. B, circa 1% der Kerne waren Kif20a positiv. C, umliegende Zellen des Stromas waren nicht angefärbt. Vergrö ßerung x100 (A, C), x200 (B).
3.1.1.4 Duktales Adenokarzinom
Im Karzinomgewebe konnte eine starke Kif20a Färbung in den Tumorzellen festgestellt
werden, die je nach Präparat leichte Schwankungen der Farbintensität aufwies. Dabei
zeigten 80% der Präparate eine sehr starke Kif20a Färbung der Tumorzellen, die vor allem im

3 Ergebnisse
49
Zytoplasma nachgewiesen werden konnte. Inselzellen sowie Gefäße zeigten ebenfalls eine
stärkere Kif20a Färbung. In diesen Präparaten konnte zudem in circa 5% der Tumorzellen
eine deutliche Kernfärbung beobachtet werden. Zellen des umliegenden Stroma waren je
nach Gewebeprobe schwach oder gar nicht angefärbt (Abb. 15A). Die restlichen 20% der
untersuchten Präparate wiesen eine mittlere bis schwache Kif20a Färbung auf, wobei hier
ebenfalls die Färbung des Zytoplasmas dominierte (Abb. 15B). Lediglich in ungefähr 5% der
Tumorzellen konnte ebenfalls ein Kif20a Färbung der Nuklei identifiziert werden (Abb. 15C).
Abb. 15: Immunhistochemische Analysen von Kif20a im Tumorgewebe des Pankreas. Formalin-fixierte Paraffinschnitte wurden mit einem Kif20a Antikö rper inkubiert und mit Hämatoxylin gegengefärbt. A, eine starke zytoplasmatische Kif20a Färbung wurde in den Tumorzellen sowie den Inselzellen detektiert (roter Pfeil). B, einige Präparate zeigten eine schwache Kif20a Färbung des Zytoplasmas. C, circa 5% der Zellen wiesen zudem eine deutliche Kif20a Färbung des Nukleus auf. Vergrö ßerung x100 (A, B), x400 (C).
3.1.2 Kif20a Expression in humanen Karzinomzelllinien des Pankreas
Fü r die in vitro Studien standen in der vorliegenden Arbeit sechs verschiedene
Pankreaskarzinomzelllinien zur Verfü gung. Dabei handelte es sich im Einzelnen um die drei
duktalen Adenokarzinom (PDAC) -Zelllinien T3M4, SU86.86 und Panc-1, sowie die beiden
neuroendokrinen (NET) Karzinomzelllinien CM und QCP. Im Folgenden wurden diese
Zelllinien mittels QRT-PCR sowie Immunblot Analysen auf ihre Kif20a Expression untersucht.
Als Referenz bzw. fü r die Normalisierung wurde die Zelllinie HPDE verwendet bei der es sich
um eine immortalisierte humane duktale Epithelzelllinie des Pankreas handelt. Als interne
Ladekontrolle wurde GAPDH verwendet. Fü r die QRT-PCR Analysen wurden vorerst Primer-
Effizienz Untersuchungen fü r Kif20a und HRPT, das als interne Referenz verwendet wurde,
durchgefü hrt. Zuerst wurden Qualitätsprü fungen der extrahierten RNA aus den
Karzinomzelllinien ausgefü hrt. Dafü r wurde die extrahierte RNA auf ein Agarosegel
aufgetragen und elektrophoretisch aufgetrennt. Bei einer qualitativ hochwertigen RNA sind
dabei zwei Banden sichtbar. Es handelt sich hierbei um die 28S- und die 18S- ribosomale
RNA. Das Intensitätsverhältnis der beiden Banden sollte dabei 2:1 sein, was deutlich zu
erkennen war (Abb. 16).
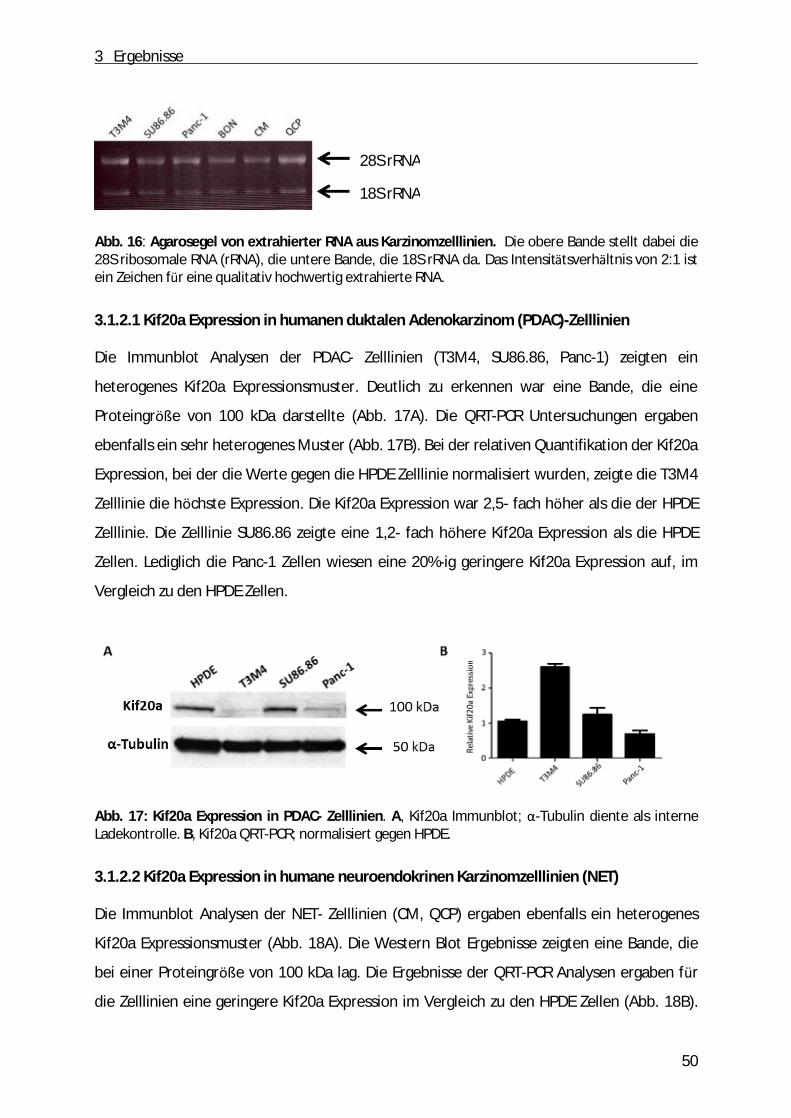
3 Ergebnisse
50
Abb. 16: Agarosegel von extrahierter RNA aus Karzinomzelllinien. Die obere Bande stellt dabei die 28S ribosomale RNA (rRNA), die untere Bande, die 18S rRNA da. Das Intensitätsverhältnis von 2:1 ist ein Zeichen fü r eine qualitativ hochwertig extrahierte RNA.
3.1.2.1 Kif20a Expression in humanen duktalen Adenokarzinom (PDAC)-Zelllinien
Die Immunblot Analysen der PDAC- Zelllinien (T3M4, SU86.86, Panc-1) zeigten ein
heterogenes Kif20a Expressionsmuster. Deutlich zu erkennen war eine Bande, die eine
Proteingrö ße von 100 kDa darstellte (Abb. 17A). Die QRT-PCR Untersuchungen ergaben
ebenfalls ein sehr heterogenes Muster (Abb. 17B). Bei der relativen Quantifikation der Kif20a
Expression, bei der die Werte gegen die HPDE Zelllinie normalisiert wurden, zeigte die T3M4
Zelllinie die hö chste Expression. Die Kif20a Expression war 2,5- fach hö her als die der HPDE
Zelllinie. Die Zelllinie SU86.86 zeigte eine 1,2- fach hö here Kif20a Expression als die HPDE
Zellen. Lediglich die Panc-1 Zellen wiesen eine 20%-ig geringere Kif20a Expression auf, im
Vergleich zu den HPDE Zellen.
Abb. 17: Kif20a Expression in PDAC- Zelllinien. A, Kif20a Immunblot; α -Tubulin diente als interne Ladekontrolle. B, Kif20a QRT-PCR; normalisiert gegen HPDE.
3.1.2.2 Kif20a Expression in humane neuroendokrinen Karzinomzelllinien (NET)
Die Immunblot Analysen der NET- Zelllinien (CM, QCP) ergaben ebenfalls ein heterogenes
Kif20a Expressionsmuster (Abb. 18A). Die Western Blot Ergebnisse zeigten eine Bande, die
bei einer Proteingrö ße von 100 kDa lag. Die Ergebnisse der QRT-PCR Analysen ergaben fü r
die Zelllinien eine geringere Kif20a Expression im Vergleich zu den HPDE Zellen (Abb. 18B).
18S rRNA
28S rRNA
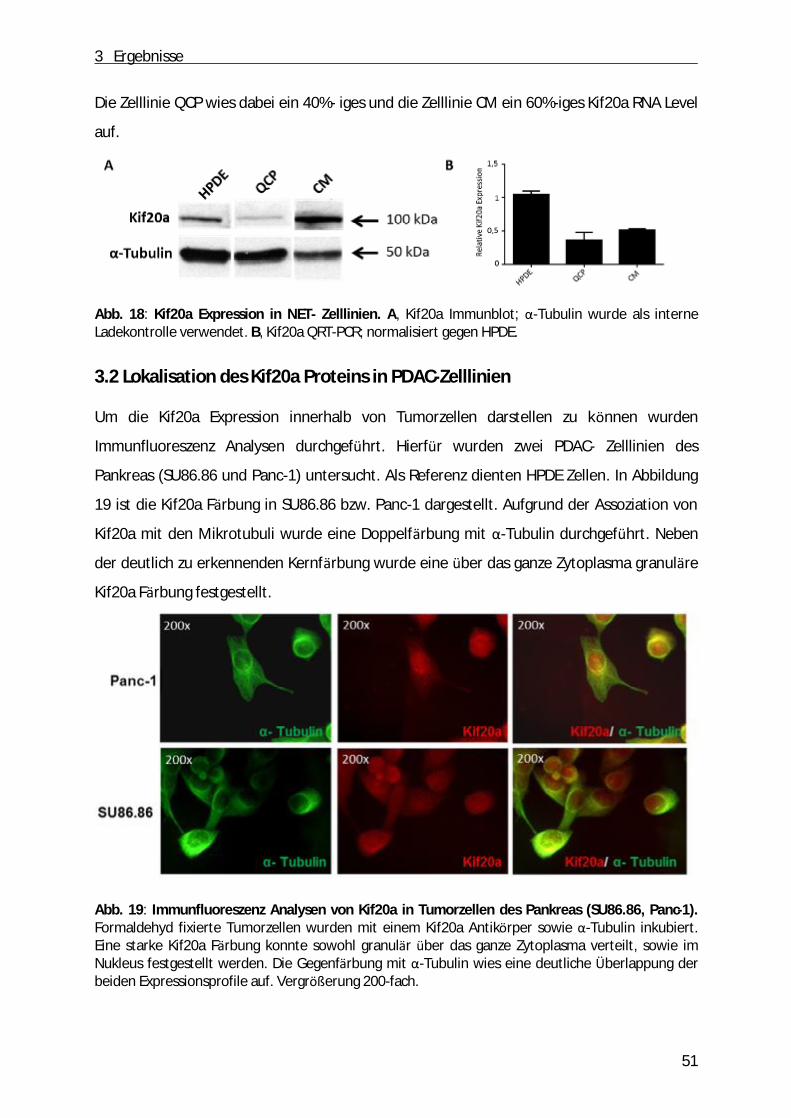
3 Ergebnisse
51
Die Zelllinie QCP wies dabei ein 40%- iges und die Zelllinie CM ein 60%-iges Kif20a RNA Level
auf.
Abb. 18: Kif20a Expression in NET- Zelllinien. A, Kif20a Immunblot; α -Tubulin wurde als interne Ladekontrolle verwendet. B, Kif20a QRT-PCR; normalisiert gegen HPDE.
3.2 Lokalisation des Kif20a Proteins in PDAC-Zelllinien
Um die Kif20a Expression innerhalb von Tumorzellen darstellen zu kö nnen wurden
Immunfluoreszenz Analysen durchgefü hrt. Hierfü r wurden zwei PDAC- Zelllinien des
Pankreas (SU86.86 und Panc-1) untersucht. Als Referenz dienten HPDE Zellen. In Abbildung
19 ist die Kif20a Färbung in SU86.86 bzw. Panc-1 dargestellt. Aufgrund der Assoziation von
Kif20a mit den Mikrotubuli wurde eine Doppelfärbung mit α -Tubulin durchgefü hrt. Neben
der deutlich zu erkennenden Kernfärbung wurde eine ü ber das ganze Zytoplasma granuläre
Kif20a Färbung festgestellt.
Abb. 19: Immunfluoreszenz Analysen von Kif20a in Tumorzellen des Pankreas (SU86.86, Panc-1). Formaldehyd fixierte Tumorzellen wurden mit einem Kif20a Antikö rper sowie α -Tubulin inkubiert. Eine starke Kif20a Färbung konnte sowohl granulär ü ber das ganze Zytoplasma verteilt, sowie im Nukleus festgestellt werden. Die Gegenfärbung mit α -Tubulin wies eine deutliche Ü berlappung der beiden Expressionsprofile auf. Vergrö ßerung 200-fach.

3 Ergebnisse
52
Die Fluoreszenz Analysen der HPDE Zellen ergaben ein ähnliches Ergebnis der Kif20a
Färbung. Deutlich stärker war auch hier der Zellkern im Gegensatz zum Zytoplasma
angefärbt, das ebenfalls granuläre Strukturen aufwies (Abb. 20).
Abb. 20: Immunfluoreszenz Analysen von Kif20a in humanen immortalisierten duktalen Epithelzellen des Pankreas (HPDE). Formaldehyd fixierte HPDEs wurden mit einem Kif20a Antikö rper inkubiert. Der Zellkern wurde mit DAPI angefärbt. Eine deutliche Kif20a Färbung konnte sowohl im Zytoplasma als auch im Kern detektiert werden. Vergrö ßerung 200-fach.
3.3 Etablierung einer temporären Kif20a Suppression in PDAC- Zelllinien
Um den Einfluss der Kif20a Expression auf Proliferation, Migration sowie die Apoptose zu
analysieren, wurde die Methode des transienten Kif20a „knock-downs“ verwendet. Dafü r
wurde in den PDAC- Zelllinien T3M4, SU86.86 und Panc-1 ein validiertes siRNA-Target fü r
Kif20a von der Firma Qiagen verwendet. Zur Ü berprü fung der Spezifität der siRNA fü r Kif20a
wurden die Zelllinien T3M4, SU86.86 und Panc-1 mit dem Konstrukt transfiziert. Nach 24 h,
48 h und 72 h Inkubationszeit wurde jeweils gesamt-RNA aus den Zellen extrahiert, in cDNA
umgeschrieben und in einer QRT-PCR weiter untersucht. Die Ergebnisse der QRT-PCR in
Abbildung 21 zeigen die Reduktion der Kif20a Expression in den unterschiedlichen Zelllinien.
Die beste Kif20a Suppression konnte dabei nach 48h Inkubation detektiert werden. Hier lag
die Kif20a RNA Expression fü r die Zelllinie T3M4 nur noch bei 15% (p= 0,08) und fü r die
Zelllinie SU86.86 bei 33% (p= 0,15). Die grö ßte Suppression wurde in der Zelllinie Panc-1
erzielt. Hier lag die Kif20a RNA Expression nach der Transfektion bei 10% (p= 0,07). Als
Kontrolle wurde eine validierte „AllStar Negative Control siRNA“ der Firma Qiagen
verwendet.
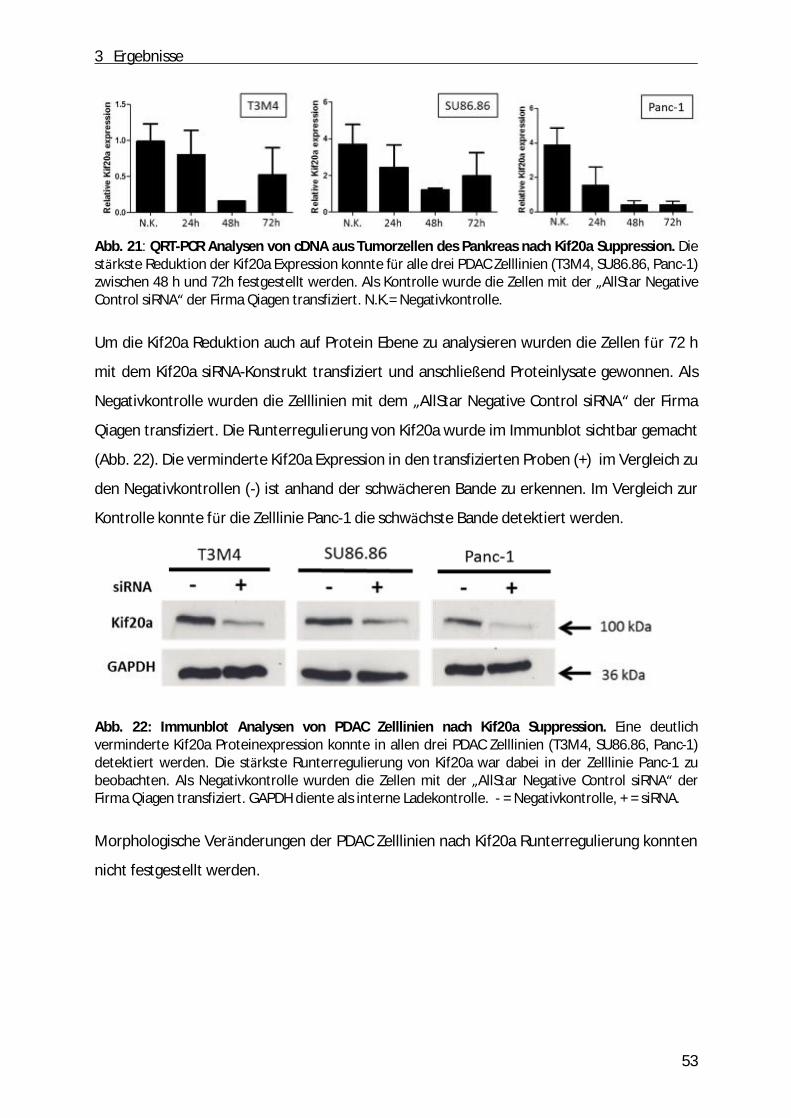
3 Ergebnisse
53
Abb. 21: QRT-PCR Analysen von cDNA aus Tumorzellen des Pankreas nach Kif20a Suppression. Die stärkste Reduktion der Kif20a Expression konnte fü r alle drei PDAC Zelllinien (T3M4, SU86.86, Panc-1) zwischen 48 h und 72h festgestellt werden. Als Kontrolle wurde die Zellen mit der „AllStar Negative Control siRNA“ der Firma Qiagen transfiziert. N.K.= Negativkontrolle.
Um die Kif20a Reduktion auch auf Protein Ebene zu analysieren wurden die Zellen fü r 72 h
mit dem Kif20a siRNA-Konstrukt transfiziert und anschließend Proteinlysate gewonnen. Als
Negativkontrolle wurden die Zelllinien mit dem „AllStar Negative Control siRNA“ der Firma
Qiagen transfiziert. Die Runterregulierung von Kif20a wurde im Immunblot sichtbar gemacht
(Abb. 22). Die verminderte Kif20a Expression in den transfizierten Proben (+) im Vergleich zu
den Negativkontrollen (-) ist anhand der schwächeren Bande zu erkennen. Im Vergleich zur
Kontrolle konnte fü r die Zelllinie Panc-1 die schwächste Bande detektiert werden.
Abb. 22: Immunblot Analysen von PDAC Zelllinien nach Kif20a Suppression. Eine deutlich verminderte Kif20a Proteinexpression konnte in allen drei PDAC Zelllinien (T3M4, SU86.86, Panc-1) detektiert werden. Die stärkste Runterregulierung von Kif20a war dabei in der Zelllinie Panc-1 zu beobachten. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ der Firma Qiagen transfiziert. GAPDH diente als interne Ladekontrolle. - = Negativkontrolle, + = siRNA.
Morphologische Veränderungen der PDAC Zelllinien nach Kif20a Runterregulierung konnten
nicht festgestellt werden.
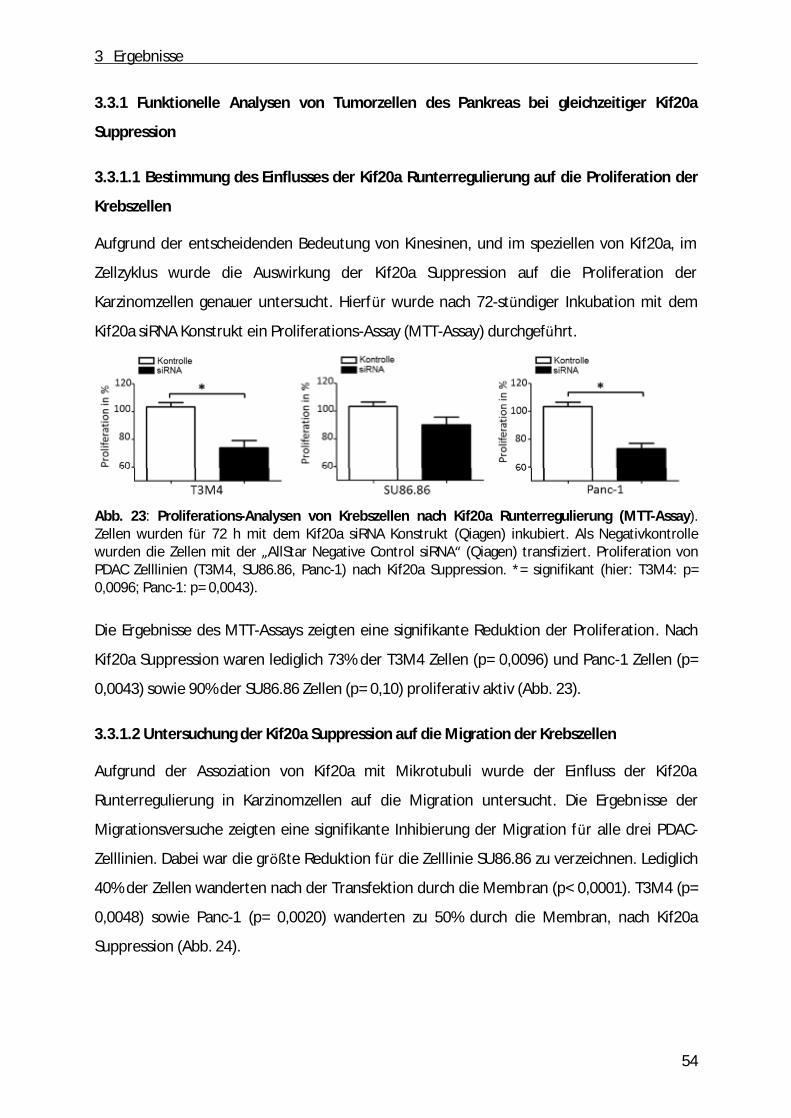
3 Ergebnisse
54
3.3.1 Funktionelle Analysen von Tumorzellen des Pankreas bei gleichzeitiger Kif20a
Suppression
3.3.1.1 Bestimmung des Einflusses der Kif20a Runterregulierung auf die Proliferation der
Krebszellen
Aufgrund der entscheidenden Bedeutung von Kinesinen, und im speziellen von Kif20a, im
Zellzyklus wurde die Auswirkung der Kif20a Suppression auf die Proliferation der
Karzinomzellen genauer untersucht. Hierfü r wurde nach 72-stü ndiger Inkubation mit dem
Kif20a siRNA Konstrukt ein Proliferations-Assay (MTT-Assay) durchgefü hrt.
Abb. 23: Proliferations-Analysen von Krebszellen nach Kif20a Runterregulierung (MTT-Assay). Zellen wurden fü r 72 h mit dem Kif20a siRNA Konstrukt (Qiagen) inkubiert. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ (Qiagen) transfiziert. Proliferation von PDAC Zelllinien (T3M4, SU86.86, Panc-1) nach Kif20a Suppression. *= signifikant (hier: T3M4: p= 0,0096; Panc-1: p= 0,0043).
Die Ergebnisse des MTT-Assays zeigten eine signifikante Reduktion der Proliferation. Nach
Kif20a Suppression waren lediglich 73% der T3M4 Zellen (p= 0,0096) und Panc-1 Zellen (p=
0,0043) sowie 90% der SU86.86 Zellen (p= 0,10) proliferativ aktiv (Abb. 23).
3.3.1.2 Untersuchung der Kif20a Suppression auf die Migration der Krebszellen
Aufgrund der Assoziation von Kif20a mit Mikrotubuli wurde der Einfluss der Kif20a
Runterregulierung in Karzinomzellen auf die Migration untersucht. Die Ergebnisse der
Migrationsversuche zeigten eine signifikante Inhibierung der Migration fü r alle drei PDAC-
Zelllinien. Dabei war die grö ßte Reduktion fü r die Zelllinie SU86.86 zu verzeichnen. Lediglich
40% der Zellen wanderten nach der Transfektion durch die Membran (p< 0,0001). T3M4 (p=
0,0048) sowie Panc-1 (p= 0,0020) wanderten zu 50% durch die Membran, nach Kif20a
Suppression (Abb. 24).
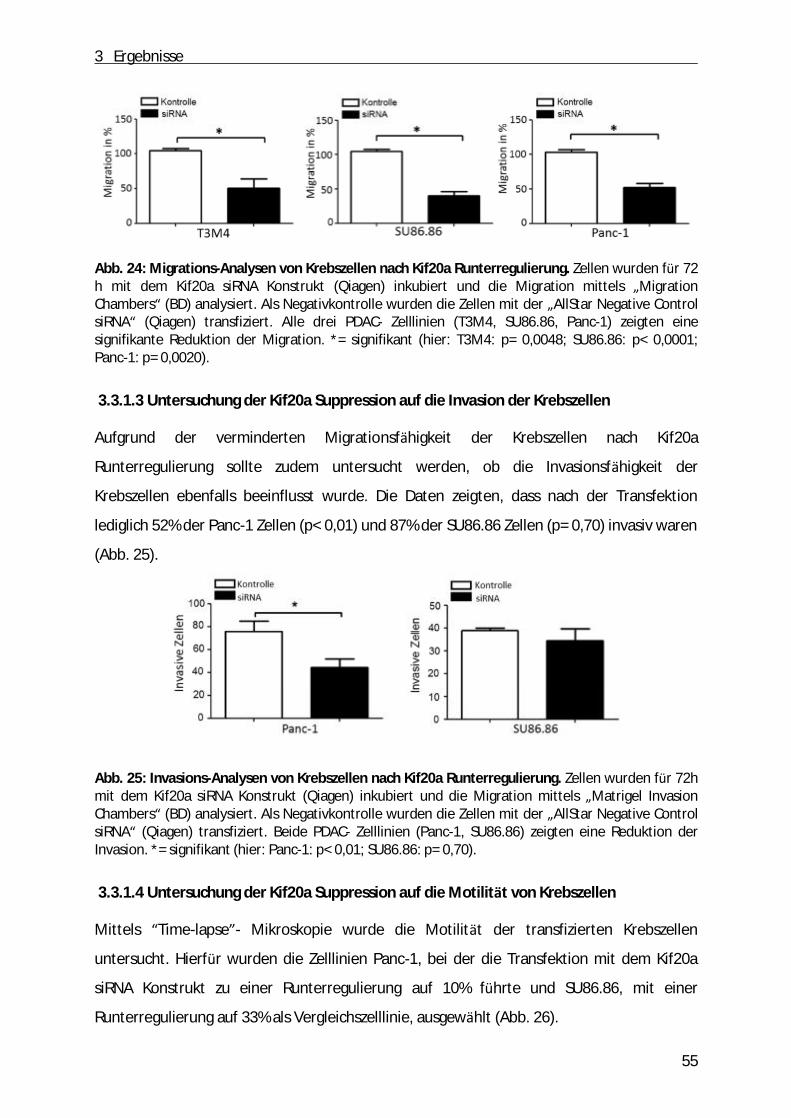
3 Ergebnisse
55
Abb. 24: Migrations-Analysen von Krebszellen nach Kif20a Runterregulierung. Zellen wurden fü r 72 h mit dem Kif20a siRNA Konstrukt (Qiagen) inkubiert und die Migration mittels „Migration Chambers“ (BD) analysiert. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ (Qiagen) transfiziert. Alle drei PDAC- Zelllinien (T3M4, SU86.86, Panc-1) zeigten eine signifikante Reduktion der Migration. *= signifikant (hier: T3M4: p= 0,0048; SU86.86: p< 0,0001; Panc-1: p= 0,0020).
3.3.1.3 Untersuchung der Kif20a Suppression auf die Invasion der Krebszellen
Aufgrund der verminderten Migrationsfähigkeit der Krebszellen nach Kif20a
Runterregulierung sollte zudem untersucht werden, ob die Invasionsfähigkeit der
Krebszellen ebenfalls beeinflusst wurde. Die Daten zeigten, dass nach der Transfektion
lediglich 52% der Panc-1 Zellen (p< 0,01) und 87% der SU86.86 Zellen (p= 0,70) invasiv waren
(Abb. 25).
Abb. 25: Invasions-Analysen von Krebszellen nach Kif20a Runterregulierung. Zellen wurden fü r 72h mit dem Kif20a siRNA Konstrukt (Qiagen) inkubiert und die Migration mittels „Matrigel Invasion Chambers“ (BD) analysiert. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ (Qiagen) transfiziert. Beide PDAC- Zelllinien (Panc-1, SU86.86) zeigten eine Reduktion der Invasion. *= signifikant (hier: Panc-1: p< 0,01; SU86.86: p= 0,70).
3.3.1.4 Untersuchung der Kif20a Suppression auf die Motilität von Krebszellen
Mittels “Time-lapse” - Mikroskopie wurde die Motilität der transfizierten Krebszellen
untersucht. Hierfü r wurden die Zelllinien Panc-1, bei der die Transfektion mit dem Kif20a
siRNA Konstrukt zu einer Runterregulierung auf 10% fü hrte und SU86.86, mit einer
Runterregulierung auf 33% als Vergleichszelllinie, ausgewählt (Abb. 26).

3 Ergebnisse
56
Abb. 26: “Time-lapse” Mikroskopie (Richtungsabhängigkeit, Distanz, Geschwindigkeit). A, Vergleich von Kif20a Runterregulierten Panc-1 Zellen mit Kontrollzellen. B, Vergleich von Kif20a supprimierten SU86.86 Zellen mit Kontrollzellen. Kontrollzellen wurden mit dem “AllStars Neg. Control siRNA” Konstrukt (Qiagen) transfiziert. *= signifikant (hier: Panc-1: p< 0,001).
Nach Runterregulierung des Kif20a Gens konnte eine Reduzierung der zurü ckgelegten
Distanz der Panc-1 Zellen festgestellt werden. Im Vergleich zu den Kontrollzellen wanderten
die Zellen nur noch ein Drittel so weit (p< 0,001). Ebenso die Geschwindigkeit der Zellen
reduzierte sich um 75% (p< 0,001). Durch die reduzierte Streckendistanz, sowie die
verminderte Geschwindigkeit konnte eine Erhö hung der Richtungsabhängigkeit um 50% (p=
nicht signifikant) beobachtet werden (Abb. 26A). Bei der Untersuchung der SU86.86 Zellen
konnte kein Unterschied zwischen den transfizierten Zellen und den Kontrollzellen
beobachtet werden. Sowohl Richtungsabhängigkeit, zurü ckgelegte Distanz als auch
Geschwindigkeit waren annähernd gleich (Abb. 26B).
3.3.1.5 Zellzyklusanalysen von Krebszellen nach Kif20a Runterregulierung
Aufgrund der reduzierten Proliferation nach Runterregulierung von Kif20a wurden zusätzlich
Apoptose Färbungen durchgefü hrt. Bereits die DAPI Färbungen zeigten eine Veränderung
der Morphologie der Zellkerne von Kif20a runterregulierten Panc-1 Zellen. Anstatt der
normalen runden Morphologie waren die Zellkerne bohnenfö rmig gekrü mmt (Abb. 27).
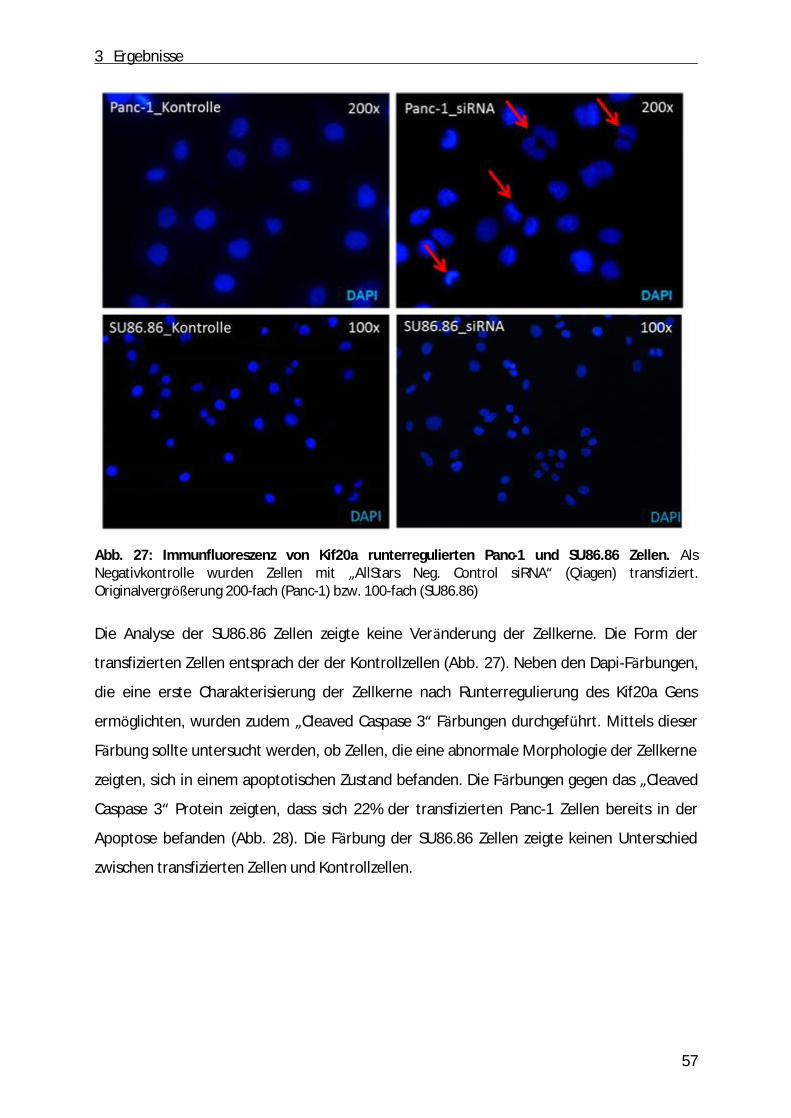
3 Ergebnisse
57
Abb. 27: Immunfluoreszenz von Kif20a runterregulierten Panc-1 und SU86.86 Zellen. Als Negativkontrolle wurden Zellen mit „AllStars Neg. Control siRNA“ (Qiagen) transfiziert. Originalvergrö ßerung 200-fach (Panc-1) bzw. 100-fach (SU86.86)
Die Analyse der SU86.86 Zellen zeigte keine Veränderung der Zellkerne. Die Form der
transfizierten Zellen entsprach der der Kontrollzellen (Abb. 27). Neben den Dapi-Färbungen,
die eine erste Charakterisierung der Zellkerne nach Runterregulierung des Kif20a Gens
ermö glichten, wurden zudem „Cleaved Caspase 3“ Färbungen durchgefü hrt. Mittels dieser
Färbung sollte untersucht werden, ob Zellen, die eine abnormale Morphologie der Zellkerne
zeigten, sich in einem apoptotischen Zustand befanden. Die Färbungen gegen das „Cleaved
Caspase 3“ Protein zeigten, dass sich 22% der transfizierten Panc-1 Zellen bereits in der
Apoptose befanden (Abb. 28). Die Färbung der SU86.86 Zellen zeigte keinen Unterschied
zwischen transfizierten Zellen und Kontrollzellen.
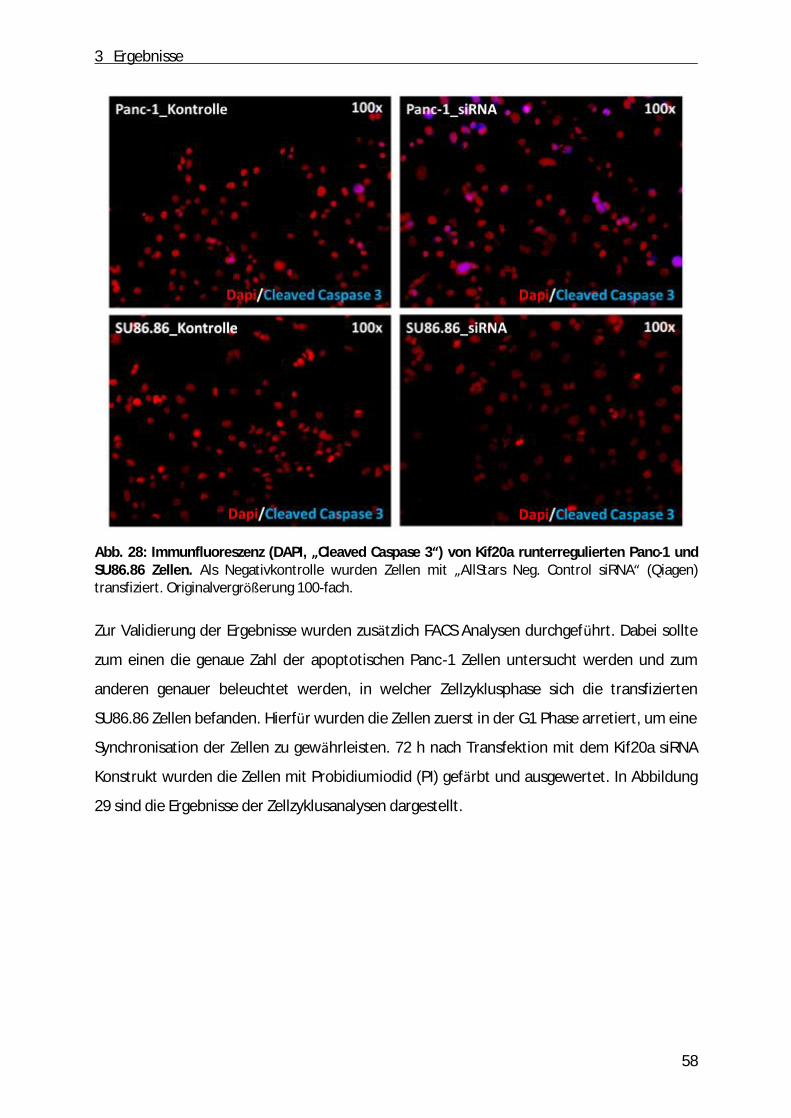
3 Ergebnisse
58
Abb. 28: Immunfluoreszenz (DAPI, „Cleaved Caspase 3“) von Kif20a runterregulierten Panc-1 und SU86.86 Zellen. Als Negativkontrolle wurden Zellen mit „AllStars Neg. Control siRNA“ (Qiagen) transfiziert. Originalvergrö ßerung 100-fach.
Zur Validierung der Ergebnisse wurden zusätzlich FACS Analysen durchgefü hrt. Dabei sollte
zum einen die genaue Zahl der apoptotischen Panc-1 Zellen untersucht werden und zum
anderen genauer beleuchtet werden, in welcher Zellzyklusphase sich die transfizierten
SU86.86 Zellen befanden. Hierfü r wurden die Zellen zuerst in der G1 Phase arretiert, um eine
Synchronisation der Zellen zu gewährleisten. 72 h nach Transfektion mit dem Kif20a siRNA
Konstrukt wurden die Zellen mit Probidiumiodid (PI) gefärbt und ausgewertet. In Abbildung
29 sind die Ergebnisse der Zellzyklusanalysen dargestellt.
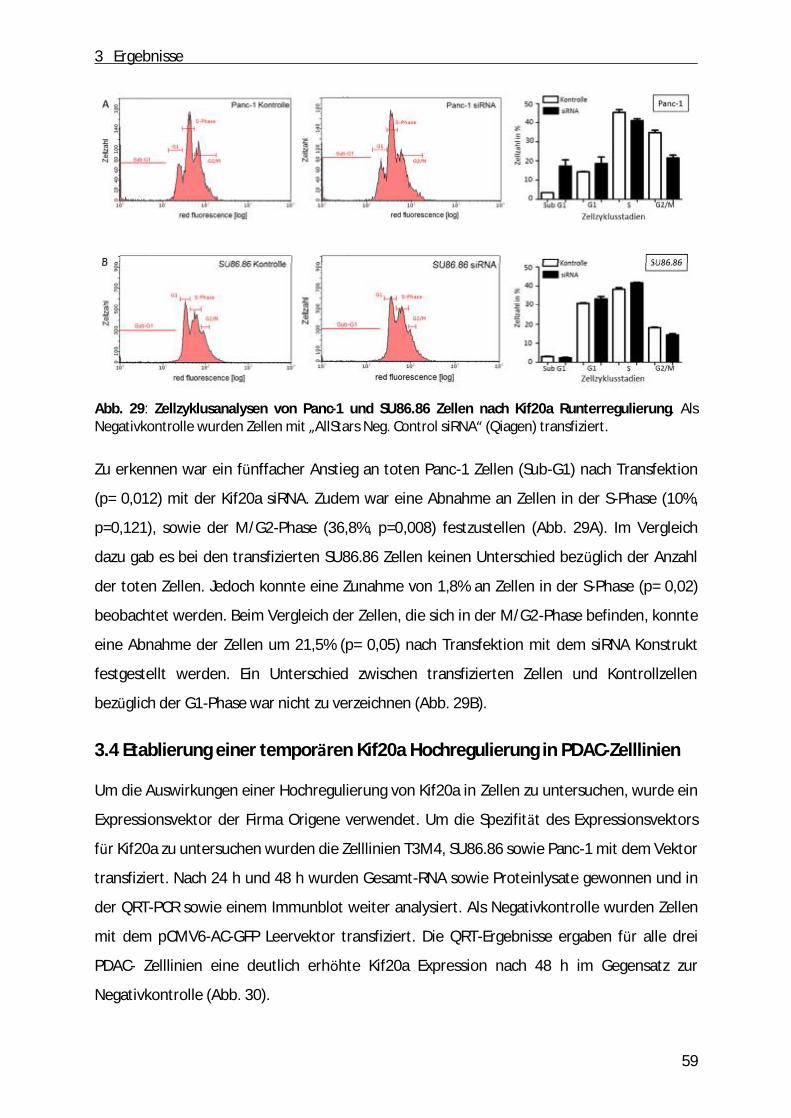
3 Ergebnisse
59
Abb. 29: Zellzyklusanalysen von Panc-1 und SU86.86 Zellen nach Kif20a Runterregulierung. Als Negativkontrolle wurden Zellen mit „AllStars Neg. Control siRNA“ (Qiagen) transfiziert.
Zu erkennen war ein fü nffacher Anstieg an toten Panc-1 Zellen (Sub-G1) nach Transfektion
(p= 0,012) mit der Kif20a siRNA. Zudem war eine Abnahme an Zellen in der S-Phase (10%,
p=0,121), sowie der M/G2-Phase (36,8%, p=0,008) festzustellen (Abb. 29A). Im Vergleich
dazu gab es bei den transfizierten SU86.86 Zellen keinen Unterschied bez ü glich der Anzahl
der toten Zellen. Jedoch konnte eine Zunahme von 1,8% an Zellen in der S-Phase (p= 0,02)
beobachtet werden. Beim Vergleich der Zellen, die sich in der M/G2-Phase befinden, konnte
eine Abnahme der Zellen um 21,5% (p= 0,05) nach Transfektion mit dem siRNA Konstrukt
festgestellt werden. Ein Unterschied zwischen transfizierten Zellen und Kontrollzellen
bezü glich der G1-Phase war nicht zu verzeichnen (Abb. 29B).
3.4 Etablierung einer temporären Kif20a Hochregulierung in PDAC-Zelllinien
Um die Auswirkungen einer Hochregulierung von Kif20a in Zellen zu untersuchen, wurde ein
Expressionsvektor der Firma Origene verwendet. Um die Spezifität des Expressionsvektors
fü r Kif20a zu untersuchen wurden die Zelllinien T3M4, SU86.86 sowie Panc-1 mit dem Vektor
transfiziert. Nach 24 h und 48 h wurden Gesamt-RNA sowie Proteinlysate gewonnen und in
der QRT-PCR sowie einem Immunblot weiter analysiert. Als Negativkontrolle wurden Zellen
mit dem pCMV6-AC-GFP Leervektor transfiziert. Die QRT-Ergebnisse ergaben fü r alle drei
PDAC- Zelllinien eine deutlich erhö hte Kif20a Expression nach 48 h im Gegensatz zur
Negativkontrolle (Abb. 30).
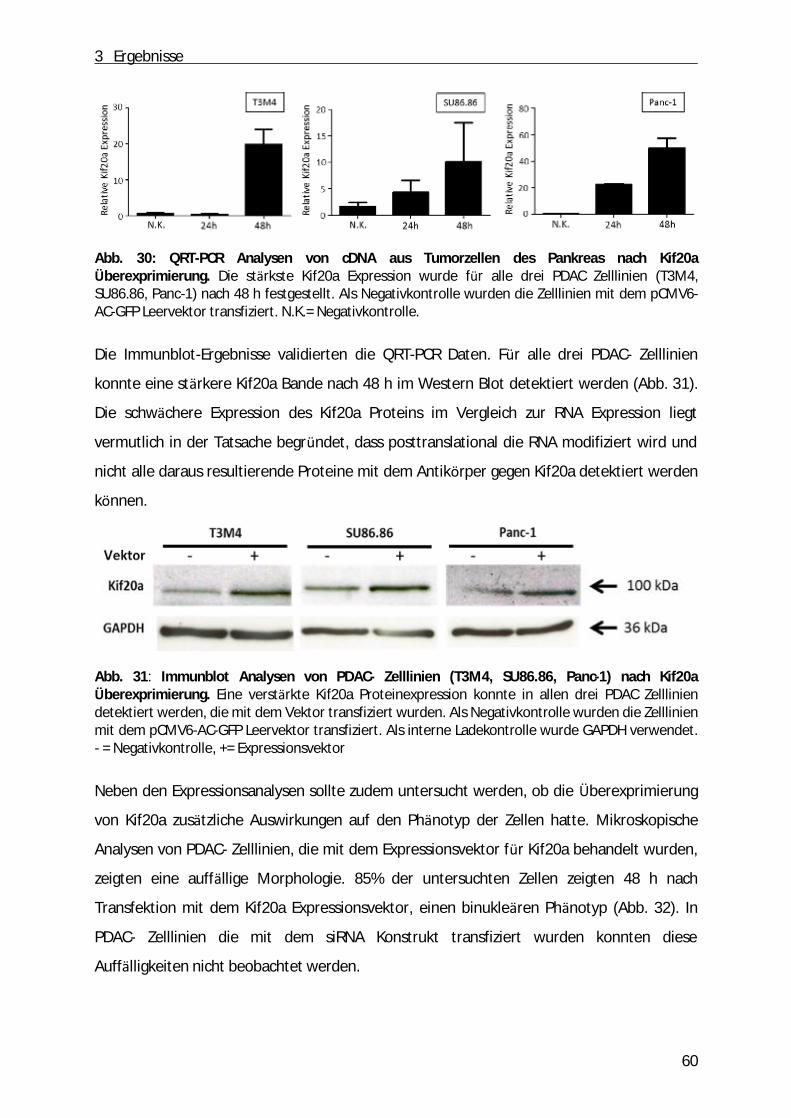
3 Ergebnisse
60
Abb. 30: QRT-PCR Analysen von cDNA aus Tumorzellen des Pankreas nach Kif20a Überexprimierung. Die stärkste Kif20a Expression wurde fü r alle drei PDAC Zelllinien (T3M4, SU86.86, Panc-1) nach 48 h festgestellt. Als Negativkontrolle wurden die Zelllinien mit dem pCMV6-AC-GFP Leervektor transfiziert. N.K.= Negativkontrolle.
Die Immunblot-Ergebnisse validierten die QRT-PCR Daten. Fü r alle drei PDAC- Zelllinien
konnte eine stärkere Kif20a Bande nach 48 h im Western Blot detektiert werden (Abb. 31).
Die schwächere Expression des Kif20a Proteins im Vergleich zur RNA Expression liegt
vermutlich in der Tatsache begrü ndet, dass posttranslational die RNA modifiziert wird und
nicht alle daraus resultierende Proteine mit dem Antikö rper gegen Kif20a detektiert werden
kö nnen.
Abb. 31: Immunblot Analysen von PDAC- Zelllinien (T3M4, SU86.86, Panc-1) nach Kif20a Überexprimierung. Eine verstärkte Kif20a Proteinexpression konnte in allen drei PDAC Zelllinien detektiert werden, die mit dem Vektor transfiziert wurden. Als Negativkontrolle wurden die Zelllinien mit dem pCMV6-AC-GFP Leervektor transfiziert. Als interne Ladekontrolle wurde GAPDH verwendet. - = Negativkontrolle, += Expressionsvektor
Neben den Expressionsanalysen sollte zudem untersucht werden, ob die Ü berexprimierung
von Kif20a zusätzliche Auswirkungen auf den Phänotyp der Zellen hatte. Mikroskopische
Analysen von PDAC- Zelllinien, die mit dem Expressionsvektor fü r Kif20a behandelt wurden,
zeigten eine auffällige Morphologie. 85% der untersuchten Zellen zeigten 48 h nach
Transfektion mit dem Kif20a Expressionsvektor, einen binukleären Phänotyp (Abb. 32). In
PDAC- Zelllinien die mit dem siRNA Konstrukt transfiziert wurden konnten diese
Auffälligkeiten nicht beobachtet werden.

3 Ergebnisse
61
Abb. 32: Phänotyp von PDAC- Zelllinien (T3M4, SU86.86, Panc-1) nach Kif20a Überexprimierung. Sowohl in T3M4, SU86.86 als auch Panc-1 konnten 48 h nach der Transfektion mit dem Kif20a Expressionsvektor Zellen mit einer binukleären Morphologie festgestellt werden. Vergrö ßerung 400-fach.
3.4.1 Funktionelle Analysen von Tumorzellen des Pankreas bei gleichzeitiger Kif20a
Hochregulierung
3.4.1.1 Effekt der Kif20a Hochregulierung auf die Proliferation der Krebszellen
Um die Auswirkung der Hochregulierung von Kif20a in Karzinomzellen auf die Proliferation
der Zellen zu untersuchen wurden die Zelllinien T3M4, SU86.86 sowie Panc-1, 24 h bzw. 48 h
mit dem Kif20a Expressionsvektor behandelt und anschließend ein MTT-Assay durchgefü hrt.
Die Ergebnisse der Proliferations-Analysen sind in Abbildung 33 dargestellt. Sowohl fü r T3M4
als auch fü r SU86.86 konnte eine signifikante Reduktion der Proliferation 48 h nach der
Transfektion beobachtet werden. Lediglich 70% der SU86.86 Zellen (p= 0,0006) sowie 80%
der T3M4 Zellen (p= 0,0058) proliferierten nach der Transfektion mit dem Kif20a
Expressionsvektor. Die Analyse der Panc-1 Zellen ergab eine 90%-ige Proliferation der Zellen
nach Transfektion und war nicht signifikant. Zellen die 24 h mit dem Kif20a
Expressionsvektor transfiziert wurden, zeigten in den MTT-Assays keine inhibierende
Wirkung. Dieses Ergebnis korreliert wiederum mit der zu schwachen Hochregulierung von
Kif20a, 24 h nach Transfektion (vgl. Abb. 30). Lediglich die transfizierten T3M4 Zellen
proliferierten zu 90% im Vergleich zu Kontrollzellen (p= 0,15).
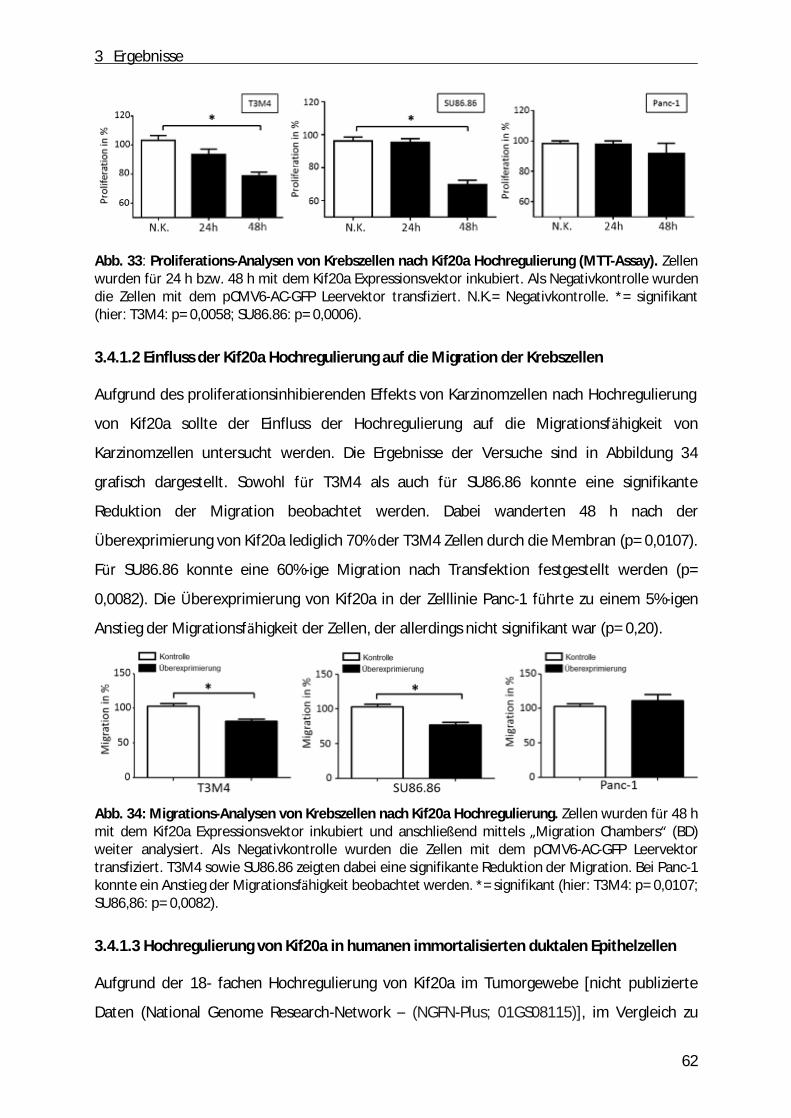
3 Ergebnisse
62
Abb. 33: Proliferations-Analysen von Krebszellen nach Kif20a Hochregulierung (MTT-Assay). Zellen wurden fü r 24 h bzw. 48 h mit dem Kif20a Expressionsvektor inkubiert. Als Negativkontrolle wurden die Zellen mit dem pCMV6-AC-GFP Leervektor transfiziert. N.K.= Negativkontrolle. *= signifikant (hier: T3M4: p= 0,0058; SU86.86: p= 0,0006).
3.4.1.2 Einfluss der Kif20a Hochregulierung auf die Migration der Krebszellen
Aufgrund des proliferationsinhibierenden Effekts von Karzinomzellen nach Hochregulierung
von Kif20a sollte der Einfluss der Hochregulierung auf die Migrationsfähigkeit von
Karzinomzellen untersucht werden. Die Ergebnisse der Versuche sind in Abbildung 34
grafisch dargestellt. Sowohl fü r T3M4 als auch fü r SU86.86 konnte eine signifikante
Reduktion der Migration beobachtet werden. Dabei wanderten 48 h nach der
Ü berexprimierung von Kif20a lediglich 70% der T3M4 Zellen durch die Membran (p= 0,0107).
Fü r SU86.86 konnte eine 60%-ige Migration nach Transfektion festgestellt werden (p=
0,0082). Die Ü berexprimierung von Kif20a in der Zelllinie Panc-1 fü hrte zu einem 5%-igen
Anstieg der Migrationsfähigkeit der Zellen, der allerdings nicht signifikant war (p= 0,20).
Abb. 34: Migrations-Analysen von Krebszellen nach Kif20a Hochregulierung. Zellen wurden fü r 48 h mit dem Kif20a Expressionsvektor inkubiert und anschließend mittels „Migration Chambers“ (BD) weiter analysiert. Als Negativkontrolle wurden die Zellen mit dem pCMV6-AC-GFP Leervektor transfiziert. T3M4 sowie SU86.86 zeigten dabei eine signifikante Reduktion der Migration. Bei Panc-1 konnte ein Anstieg der Migrationsfähigkeit beobachtet werden. *= signifikant (hier: T3M4: p= 0,0107; SU86,86: p= 0,0082).
3.4.1.3 Hochregulierung von Kif20a in humanen immortalisierten duktalen Epithelzellen
Aufgrund der 18- fachen Hochregulierung von Kif20a im Tumorgewebe [nicht publizierte
Daten (National Genome Research-Network – (NGFN-Plus; 01GS08115)], im Vergleich zu

3 Ergebnisse
63
gesundem Pankreasgewebe, sollte eine immortalisierte duktale Epithelzelllinie des Pankreas
(HPDE), die annähernd einen normal Phänotyp hat, mit dem Kif20a Expressionsvektor
transfiziert werden. Die Transfektion der HPDE Zellen fü hrte zu einer signifikanten
Ü berexprimierung von Kif20a (Abb. 35). 24 h bis 48 h nach der Transfektion starben jedoch
alle Zellen, wodurch weitere funktionelle Analysen dieser Zelllinie nicht mö glich waren.
Abb. 35: Überexprimierung von Kif20a in immortalisierten humanen duktalen Epithelzellen (HPDE).
3.5 Funktionelle Analysen von PDAC- Zelllinien nach Taxolbehandlung bei
gleichzeitiger Kif20a Suppression
Durch das irreversible Binden von Paclitaxel an die Untereinheiten des Tubulin kommt es zur
Anordnung der Mikrotubuli, was zu abnormalen mitotischen Spindelformationen f ü hrt. Als
Folge kö nnen Chromosomenbrü che, sowie eine Inhibierung der Zellreplikation bzw.
Migration auftreten. In den folgenden Versuchen sollte analysiert werden, welchen Effekt
die zusätzliche Runterregulierung von Kif20a bei gleichzeitiger Behandlung mit Paclitaxel auf
die Proliferation bzw. die Migrationsfähigkeit der Tumorzellen hat. Da Kif20a als Kinesin-
Motorprotein mit Mikrotubuli assoziiert ist, und eine Suppression von Kif20a zu einem
abnormalem Zellzyklus fü hrt (siehe Vorversuche), sollte diese kombinierte Behandlung
genauer untersucht werden.
Dafü r wurden zuerst die alleinige Wirkung von Taxol auf die Proliferation von PDAC-
Zelllinien (T3M4, SU86.86, Panc-1) untersucht. Hierfü r wurden die Zelllinien fü r 24 h mit
Paclitaxel in den Konzentrationen 2 µM und 20 µM behandelt. Anschließend wurde ein
Proliferationstest (MTT-Assay) durchgefü hrt. Die Ergebnisse zeigen, dass in allen drei
Zelllinien die Behandlung mit 2 µM Paclitaxel den grö ßten inhibierenden Effekt auf die
Proliferation der Zellen bewirkte (Abb. 36). Nach der Behandlung waren 25% der T3M4
Zellen (p< 0,0001), 70% der SU86.86 Zellen (p= 0,0070) sowie 40% der Panc-1 Zellen (p=
0,0003) proliferativ. Die Behandlung mit 20 µM Paclitaxel ergab ebenfalls eine deutliche
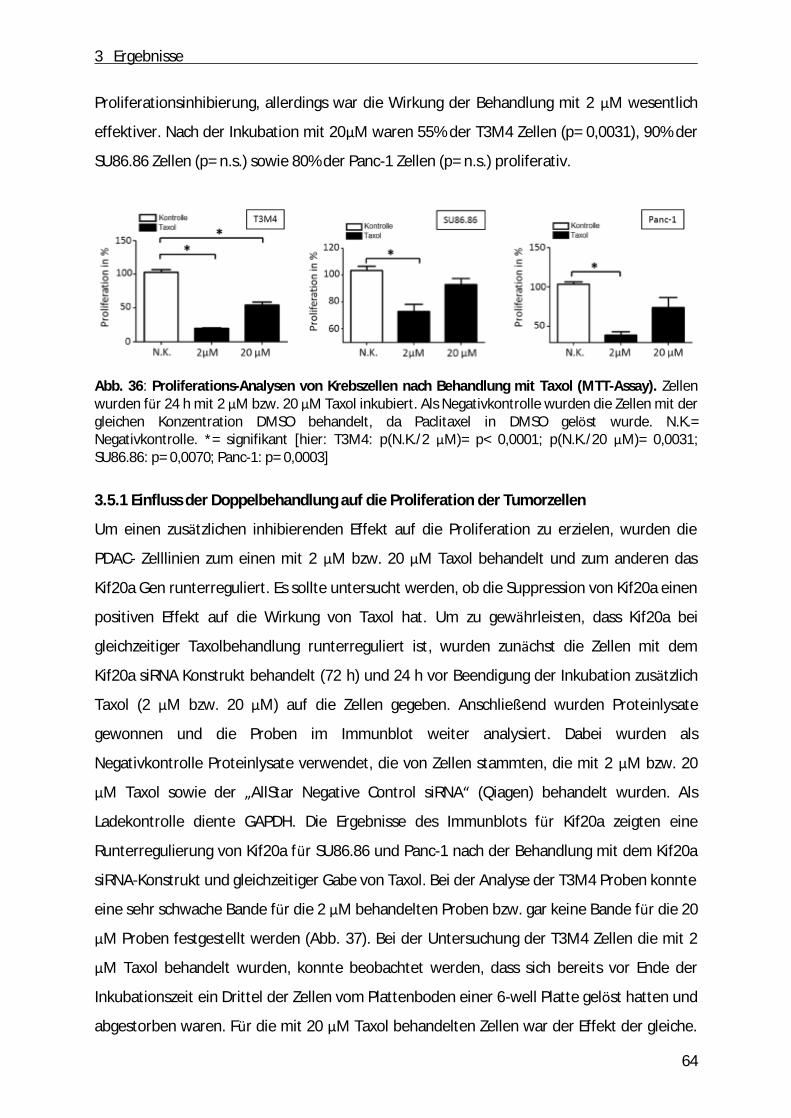
3 Ergebnisse
64
Proliferationsinhibierung, allerdings war die Wirkung der Behandlung mit 2 µM wesentlich
effektiver. Nach der Inkubation mit 20µM waren 55% der T3M4 Zellen (p= 0,0031), 90% der
SU86.86 Zellen (p= n.s.) sowie 80% der Panc-1 Zellen (p= n.s.) proliferativ.
Abb. 36: Proliferations-Analysen von Krebszellen nach Behandlung mit Taxol (MTT-Assay). Zellen wurden fü r 24 h mit 2 µM bzw. 20 µM Taxol inkubiert. Als Negativkontrolle wurden die Zellen mit der gleichen Konzentration DMSO behandelt, da Paclitaxel in DMSO gelö st wurde. N.K.= Negativkontrolle. *= signifikant [hier: T3M4: p(N.K./2 µM)= p< 0,0001; p(N.K./20 µM)= 0,0031; SU86.86: p= 0,0070; Panc-1: p= 0,0003]
3.5.1 Einfluss der Doppelbehandlung auf die Proliferation der Tumorzellen
Um einen zusätzlichen inhibierenden Effekt auf die Proliferation zu erzielen, wurden die
PDAC- Zelllinien zum einen mit 2 µM bzw. 20 µM Taxol behandelt und zum anderen das
Kif20a Gen runterreguliert. Es sollte untersucht werden, ob die Suppression von Kif20a einen
positiven Effekt auf die Wirkung von Taxol hat. Um zu gewährleisten, dass Kif20a bei
gleichzeitiger Taxolbehandlung runterreguliert ist, wurden zunächst die Zellen mit dem
Kif20a siRNA Konstrukt behandelt (72 h) und 24 h vor Beendigung der Inkubation zusätzlich
Taxol (2 µM bzw. 20 µM) auf die Zellen gegeben. Anschließend wurden Proteinlysate
gewonnen und die Proben im Immunblot weiter analysiert. Dabei wurden als
Negativkontrolle Proteinlysate verwendet, die von Zellen stammten, die mit 2 µM bzw. 20
µM Taxol sowie der „AllStar Negative Control siRNA“ (Qiagen) behandelt wurden. Als
Ladekontrolle diente GAPDH. Die Ergebnisse des Immunblots fü r Kif20a zeigten eine
Runterregulierung von Kif20a fü r SU86.86 und Panc-1 nach der Behandlung mit dem Kif20a
siRNA-Konstrukt und gleichzeitiger Gabe von Taxol. Bei der Analyse der T3M4 Proben konnte
eine sehr schwache Bande fü r die 2 µM behandelten Proben bzw. gar keine Bande fü r die 20
µM Proben festgestellt werden (Abb. 37). Bei der Untersuchung der T3M4 Zellen die mit 2
µM Taxol behandelt wurden, konnte beobachtet werden, dass sich bereits vor Ende der
Inkubationszeit ein Drittel der Zellen vom Plattenboden einer 6-well Platte gelö st hatten und
abgestorben waren. Fü r die mit 20 µM Taxol behandelten Zellen war der Effekt der gleiche.

3 Ergebnisse
65
Hier waren mehr als 90% der Zellen vor Ende der Inkubationszeit nicht mehr vital. Aus
diesem Grund wurde in den folgenden Analysen die Zelllinie T3M4, bei gleichzeitiger
Behandlung des Kif20a siRNA Konstrukts und 20 µM Taxol, ausgeschlossen.
Abb. 37: Immunblot Analysen von PDAC- Zelllinien nach Kif20a Suppression und gleichzeitiger Taxol Behandlung (2 µ M/ 20 µ M). Eine deutlich verminderte Kif20a Proteinexpression konnte in allen drei PDAC- Zelllinien (T3M4, SU86.86, Panc-1) sowohl fü r die Behandlung mit 2 µM als auch fü r 20 µM detektiert werden. T3M4 zeigte fü r die Behandlung mit 20 µM keinerlei Banden aufgrund des Absterbens der Mehrheit der Zellen während der Behandlung. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ (Qiagen) sowie 2 µM/ 20 µM Taxol inkubiert. GAPDH diente als interne Kontrolle. - = Negativkontrolle, + = siRNA.
Die Ergebnisse des anschließenden Proliferations-Assays sind in Abbildung 45 dargestellt. Fü r
die Zelllinie SU86.86 konnte dabei der grö ßte Effekt festgestellt werden. Zellen die
ausschließlich mit Taxol behandelt wurden (2 µM) zeigten eine 10- 15%-ige Inhibierung der
Proliferation im Gegensatz zur Behandlung von 2 µM Taxol plus dem Kif20a siRNA Konstrukt.
Hier lag die Inhibierung doppelt so hoch bei 30- 35% (p= 0,0058). Nach der Behandlung mit
20 µM Taxol wurde ebenfalls eine 10%-ige Reduktion der Proliferation detektiert im
Gegensatz zu 60- 65% nach der zusätzlichen Gabe des Kif20a Konstrukts (p= 0,0212). Die
Auswirkungen der Doppelbehandlung auf die Zelllinie Panc-1 zeigten einen schwächeren
Effekt bezü glich der Proliferationsinhibierung. Die Behandlung der Zellen mit 2 µM Taxol
fü hrte zu einer Wachstumsinhibierung von 50- 55%. Die zusätzliche Gabe des Kif20a siRNA-
Konstrukts erhö hte den Inhibierenden Effekt auf 60- 65% (Abb. 38A). Die Durchfü hrung des
Versuches mit einer Taxol-Konzentration von 20 µM zeigte nahezu das gleiche Ergebnis. Bei
der Behandlung von T3M4 mit 2 µM Taxol zeigte sich bereits eine sehr starke Inhibierung der
Proliferation (80- 85%). Bei der zusätzlichen Runterregulierung von Kif20a wurde der Effekt
nicht gesteigert (85%) (Abb. 38B).

3 Ergebnisse
66
Abb. 38: Proliferations-Analysen von Krebszellen nach Behandlung mit Taxol und Kif20a siRNA (MTT-Assay). Als Negativkontrolle wurden die Zellen mit der gleichen Konzentration DMSO behandelt sowie der „AllStar Negative Control siRNA“ (Qiagen). N.K. = Negativkontrolle. A, MTT-Assay nach Behandlung mit 2 µM Taxol/ Kif20a siRNA. *= signifikant [hier: SU86.86: p(2 µM/2 µM+siRNA)= 0,0058]. B, MTT-Assay nach Behandlung mit 20 µM Taxol /Kif20a siRNA. *= signifikant [hier: SU86.86: p(20 µM/20 µM+siRNA)= 0,0212].
Ausgehend von diesen Daten wurden fü r die folgenden Versuche Taxolkonzentrationen von
2 µM verwendet, da diese den grö ßten inhibierenden Effekt auf das Wachstum der
Karzinomzellen zeigten.
3.5.2 Effekt der Doppelbehandlung auf die Migration der Tumorzellen
Aufgrund der teilweise starken Auswirkungen der Runterregulierung von Kif20a und
gleichzeitiger Taxol Behandlung auf die Proliferation, sollte zudem analysiert werden,
welchen Einfluss die kombinierte Behandlung auf die Migrationsfähigkeit der Zellen hatte.
Als Vergleichskontrolle wurden dabei Zellen ausschließlich mit 2 µM Taxol behandelt. In
Abbildung 39 sind die Ergebnisse des Migrations-Assays grafisch dargestellt. Man konnte
eine Reduktion der Migrationsfähigkeit sowohl fü r die Zelllinien SU86.86 als auch fü r Panc-1
feststellen. Dabei wanderten im Einzelnen nach der Doppelbehandlung lediglich 4,5% der
SU86.86 Zellen (p< 0,0001) und 30% der Panc-1 Zellen (p< 0,0001) durch die Membran. Wie
die Vorversuche zeigten, reagierten T3M4 Zellen auf die Doppelbehandlung des Kif20a siRNA
Konstrukts mit Taxol sehr sensibel. Auch nach mehrmaligem Wiederholen der
Migrationsanalysen konnte kein Ergebnis der T3M4 Zellen erlangt werden, da diese während
der Versuche abstarben.

3 Ergebnisse
67
Abb. 39: Migrations-Analysen von Krebszellen nach Kif20a Runterregulierung und gleichzeitiger Behandlung mit 2 µ M Taxol. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ (Qiagen) transfiziert. Sowohl SU86.86 als auch Panc-1 zeigten eine signifikante Migrationsinhibierung nach Doppelbehandlung. *= signifikant (hier: SU86.86: p< 0,0001; Panc-1: p< 0,0001).
3.6 Funktionelle Analysen von PDAC- Zelllinien nach Gemcitabinebehandlung
bei gleichzeitiger Kif20a Suppression
Seit 1997 gilt Gemcitabine (2`, 2`-difluoro 2`-deoxycytidin, dFdC) als der Goldstandard fü r die
Behandlung von Patienten mit fortgeschrittenem Pankreaskarzinom. Allerdings erhö hten
auftretende Resistenzen gegenü ber Gemcitabine die Lebensrate der behandelten Patienten
daher lediglich um ca. 6 Monate. In den folgenden in vitro Versuchen sollte die kombinierte
Gabe von Gemcitabine bei gleichzeitiger Kif20a Suppression untersucht werden.
Insbesondere sollte dabei die Wirkung auf die Proliferation sowie Migration der Tumorzellen
untersucht werden.
3.6.1 Effekte der Doppelbehandlung auf die Proliferation der Tumorzellen
In diesem Versuch sollte untersucht werden, ob die Runterregulierung von Kif20a einen
zusätzlichen inhibierenden Effekt auf die Proliferation von Karzinomzellen hat, die bereits
mit Gemcitabine behandelt wurden. Als Kontrollen dienten hierfü r Zellen, die ausschließlich
mit Gemcitabine in unterschiedlichen Konzentrationen (10 µM, 50 µM, 100 µM, 500 µM,
1000 µM) fü r 24 h sowie der „AllStar Negative Control siRNA“ (Qiagen) behandelt wurden.
Die Ergebnisse zeigen fü r die Zelllinie SU86.86 einen schwachen zusätzlichen Effekt auf die
Proliferationsinhibierung. Nur Zellen, die mit 10 µM Gemcitabine behandelt wurden wiesen
eine Wachstumsreduktion von zusätzlich 9% auf. Fü r hö here Konzentrationen konnte keine
inhibierende Wirkung festgestellt werden (Daten nicht gezeigt). Fü r die Zelllinie Panc-1
konnte fü r alle Konzentrationen eine zusätzliche Verstärkung der Proliferationsinhibierung
detektiert werden. Je nach Konzentration lag diese bei zusätzlichen 1- 10% im Vergleich zu
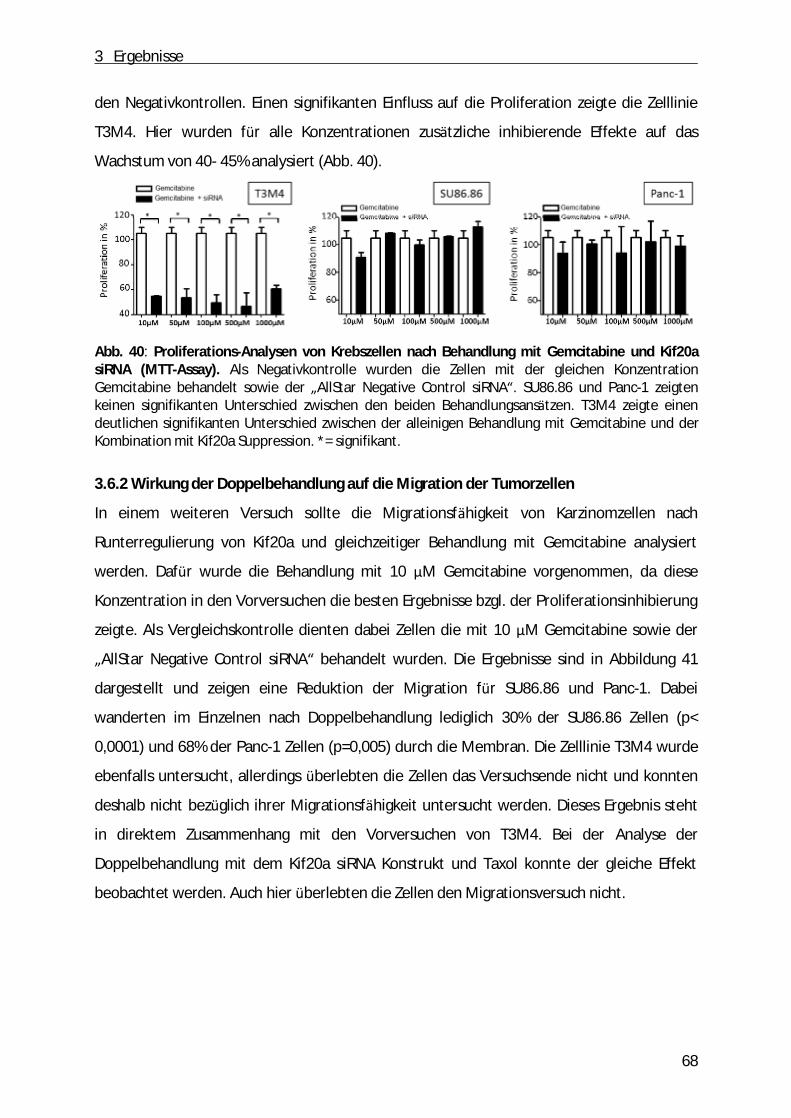
3 Ergebnisse
68
den Negativkontrollen. Einen signifikanten Einfluss auf die Proliferation zeigte die Zelllinie
T3M4. Hier wurden fü r alle Konzentrationen zusätzliche inhibierende Effekte auf das
Wachstum von 40- 45% analysiert (Abb. 40).
Abb. 40: Proliferations-Analysen von Krebszellen nach Behandlung mit Gemcitabine und Kif20a siRNA (MTT-Assay). Als Negativkontrolle wurden die Zellen mit der gleichen Konzentration Gemcitabine behandelt sowie der „AllStar Negative Control siRNA“. SU86.86 und Panc-1 zeigten keinen signifikanten Unterschied zwischen den beiden Behandlungsansätzen. T3M4 zeigte einen deutlichen signifikanten Unterschied zwischen der alleinigen Behandlung mit Gemcitabine und der Kombination mit Kif20a Suppression. *= signifikant.
3.6.2 Wirkung der Doppelbehandlung auf die Migration der Tumorzellen
In einem weiteren Versuch sollte die Migrationsfähigkeit von Karzinomzellen nach
Runterregulierung von Kif20a und gleichzeitiger Behandlung mit Gemcitabine analysiert
werden. Dafü r wurde die Behandlung mit 10 µM Gemcitabine vorgenommen, da diese
Konzentration in den Vorversuchen die besten Ergebnisse bzgl. der Proliferationsinhibierung
zeigte. Als Vergleichskontrolle dienten dabei Zellen die mit 10 µM Gemcitabine sowie der
„AllStar Negative Control siRNA“ behandelt wurden. Die Ergebnisse sind in Abbildung 41
dargestellt und zeigen eine Reduktion der Migration fü r SU86.86 und Panc-1. Dabei
wanderten im Einzelnen nach Doppelbehandlung lediglich 30% der SU86.86 Zellen (p<
0,0001) und 68% der Panc-1 Zellen (p=0,005) durch die Membran. Die Zelllinie T3M4 wurde
ebenfalls untersucht, allerdings ü berlebten die Zellen das Versuchsende nicht und konnten
deshalb nicht bezü glich ihrer Migrationsfähigkeit untersucht werden. Dieses Ergebnis steht
in direktem Zusammenhang mit den Vorversuchen von T3M4. Bei der Analyse der
Doppelbehandlung mit dem Kif20a siRNA Konstrukt und Taxol konnte der gleiche Effekt
beobachtet werden. Auch hier ü berlebten die Zellen den Migrationsversuch nicht.

3 Ergebnisse
69
Abb. 41: Migrations-Analysen von Krebszellen nach Kif20a Runterregulierung und gleichzeitiger Behandlung mit 10 µ M Gemcitabine. Als Negativkontrolle wurden die Zellen mit der „AllStar Negative Control siRNA“ (Qiagen) sowie 10 µM Gemcitabine inkubiert. Sowohl SU86.86 als auch Panc-1 zeigten eine signifikante Migrationsinhibierung nach Doppelbehandlung. *= signifikant (hier: SU86.86: p< 0,0001; Panc-1: p=0,005).
3.7 Funktionelle Analysen von PDAC- Zelllinien nach Behandlung mit
Paprotrain
In den bereits beschriebenen Vorversuchen konnte gezeigt werden, dass die
Runterregulierung des Kinesin Motor Proteins Kif20a deutliche Auswirkungen auf die
Proliferation sowie die Migration von PDAC- Zelllinien hatte. Auch fü r die
Kombinationstherapie mit Taxol bzw. Gemcitabine scheint Kif20a ein interessantes Zielgen
zu sein. Tcherniuk et al. analysierten bereits 8900 kleine Molekü le, die die ATPase Aktivität
des Kinesin Motor Proteins Kif20a inhibieren. Dabei fanden sie ein (Z)-2-(1H-indol-3-yl)-3-
(pyridin-3-yl)acrylonitril, das den Namen Paprotrain (Passenger PROtein TRAnsport Inhibitor)
trägt. Dieser reversible Inhibitor ist unkompetitiv mit ATP und nicht-kompetitiv mit
Mikrotubuli. Zudem ist er sehr spezifisch und interagiert nicht mit anderen Mitgliedern der
Kinesin-6 Superfamilie. Die Versuche konnten zudem zeigen, dass die Behandlung von HeLa
Zellen mit 10- 50 µM des Inhibitors zu einer Inhibierung der Proliferation sowie zu Apoptose
fü hrt. Aus diesem Grund sollte in den folgenden Analysen der Effekt von Paprotrain auf die
Proliferation von PDAC- Zelllinien genauer untersucht werden.
3.7.1 Auswirkung der Paprotrain Behandlung auf die Proliferation der Karzinomzellen
Fü r den folgenden Versuch wurden PDAC- Zellen 24 h lang mit unterschiedlichen Paprotrain
Konzentrationen (1 µM, 10 µM, 15 µM, 20 µM, 22 µM, 50 µM) behandelt und anschließend
ein MTT-Assay durchgefü hrt. Dabei konnte man fü r die Zelllinie T3M4 eine Reduktion der
Proliferation fü r alle Konzentrationen beobachten. Die stärkste Inhibierung erzielte dabei
eine Paprotrain Konzentration von 22µM. Nach der Behandlung waren lediglich 43,2% der
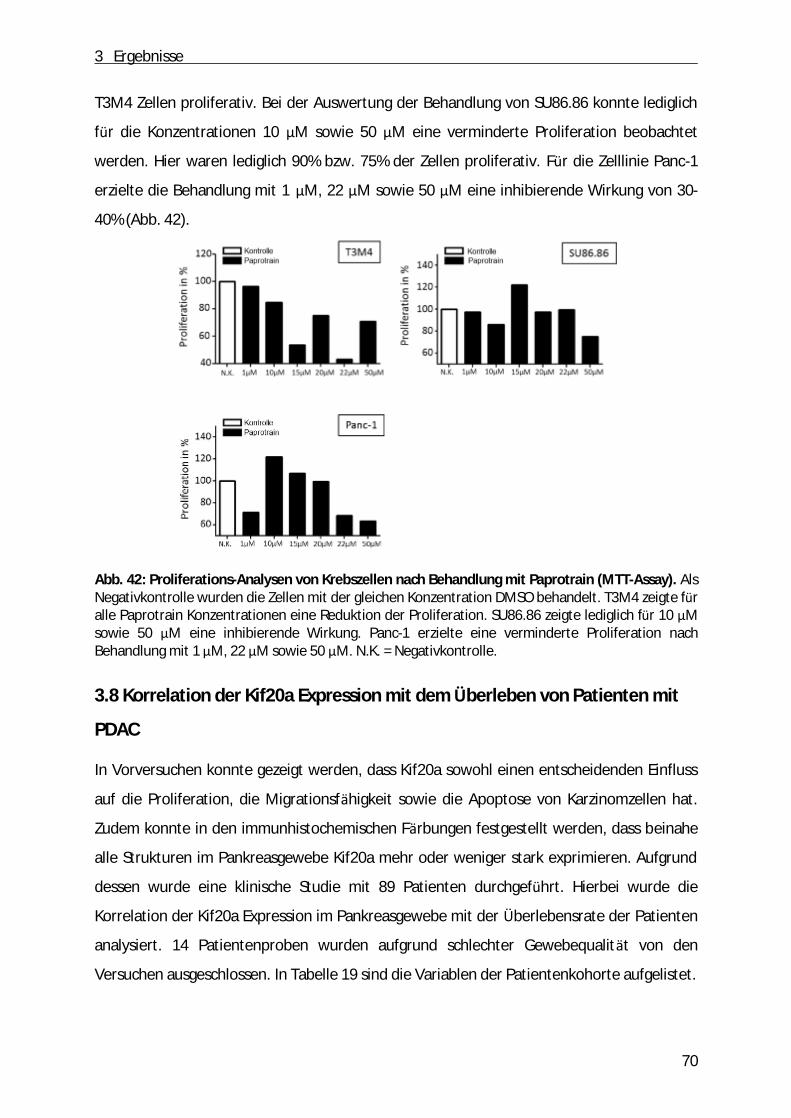
3 Ergebnisse
70
T3M4 Zellen proliferativ. Bei der Auswertung der Behandlung von SU86.86 konnte lediglich
fü r die Konzentrationen 10 µM sowie 50 µM eine verminderte Proliferation beobachtet
werden. Hier waren lediglich 90% bzw. 75% der Zellen proliferativ. Fü r die Zelllinie Panc-1
erzielte die Behandlung mit 1 µM, 22 µM sowie 50 µM eine inhibierende Wirkung von 30-
40% (Abb. 42).
Abb. 42: Proliferations-Analysen von Krebszellen nach Behandlung mit Paprotrain (MTT-Assay). Als Negativkontrolle wurden die Zellen mit der gleichen Konzentration DMSO behandelt. T3M4 zeigte fü r alle Paprotrain Konzentrationen eine Reduktion der Proliferation. SU86.86 zeigte lediglich fü r 10 µM sowie 50 µM eine inhibierende Wirkung. Panc-1 erzielte eine verminderte Proliferation nach Behandlung mit 1 µM, 22 µM sowie 50 µM. N.K. = Negativkontrolle.
3.8 Korrelation der Kif20a Expression mit dem Überleben von Patienten mit
PDAC
In Vorversuchen konnte gezeigt werden, dass Kif20a sowohl einen entscheidenden Einfluss
auf die Proliferation, die Migrationsfähigkeit sowie die Apoptose von Karzinomzellen hat.
Zudem konnte in den immunhistochemischen Färbungen festgestellt werden, dass beinahe
alle Strukturen im Pankreasgewebe Kif20a mehr oder weniger stark exprimieren. Aufgrund
dessen wurde eine klinische Studie mit 89 Patienten durchgefü hrt. Hierbei wurde die
Korrelation der Kif20a Expression im Pankreasgewebe mit der Ü berlebensrate der Patienten
analysiert. 14 Patientenproben wurden aufgrund schlechter Gewebequalität von den
Versuchen ausgeschlossen. In Tabelle 19 sind die Variablen der Patientenkohorte aufgelistet.
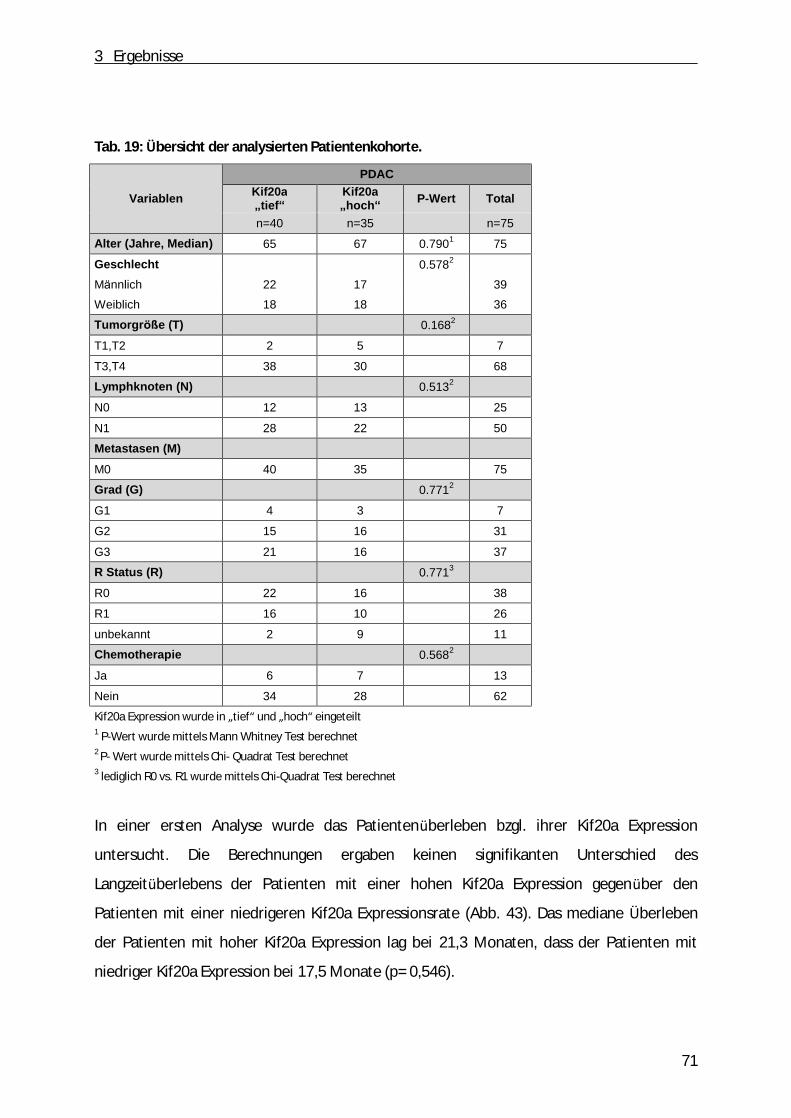
3 Ergebnisse
71
Tab. 19: Übersicht der analysierten Patientenkohorte.
Variablen
PDAC Kif20a „ tief“
Kif20a „ hoch“ P-Wert Total
n=40 n=35 n=75
Alter (Jahre, Median) 65 67 0.7901 75
Geschlecht 0.5782 Mä nnlich 22 17 39
Weiblich 18 18 36
Tumorgröße (T) 0.1682 T1,T2 2 5 7
T3,T4 38 30 68
Lymphknoten (N) 0.5132 N0 12 13 25
N1 28 22 50
Metastasen (M) M0 40 35 75
Grad (G) 0.7712 G1 4 3 7
G2 15 16 31
G3 21 16 37
R Status (R) 0.7713 R0 22 16 38
R1 16 10 26
unbekannt 2 9 11
Chemotherapie 0.5682 Ja 6 7 13
Nein 34 28 62
Kif20a Expression wurde in „ tief“ und „hoch“ eingeteilt 1 P-Wert wurde mittels Mann Whitney Test berechnet 2 P- Wert wurde mittels Chi- Quadrat Test berechnet 3 lediglich R0 vs. R1 wurde mittels Chi-Quadrat Test berechnet
In einer ersten Analyse wurde das Patientenü berleben bzgl. ihrer Kif20a Expression
untersucht. Die Berechnungen ergaben keinen signifikanten Unterschied des
Langzeitü berlebens der Patienten mit einer hohen Kif20a Expression gegenü ber den
Patienten mit einer niedrigeren Kif20a Expressionsrate (Abb. 43). Das mediane Ü berleben
der Patienten mit hoher Kif20a Expression lag bei 21,3 Monaten, dass der Patienten mit
niedriger Kif20a Expression bei 17,5 Monate (p= 0,546).
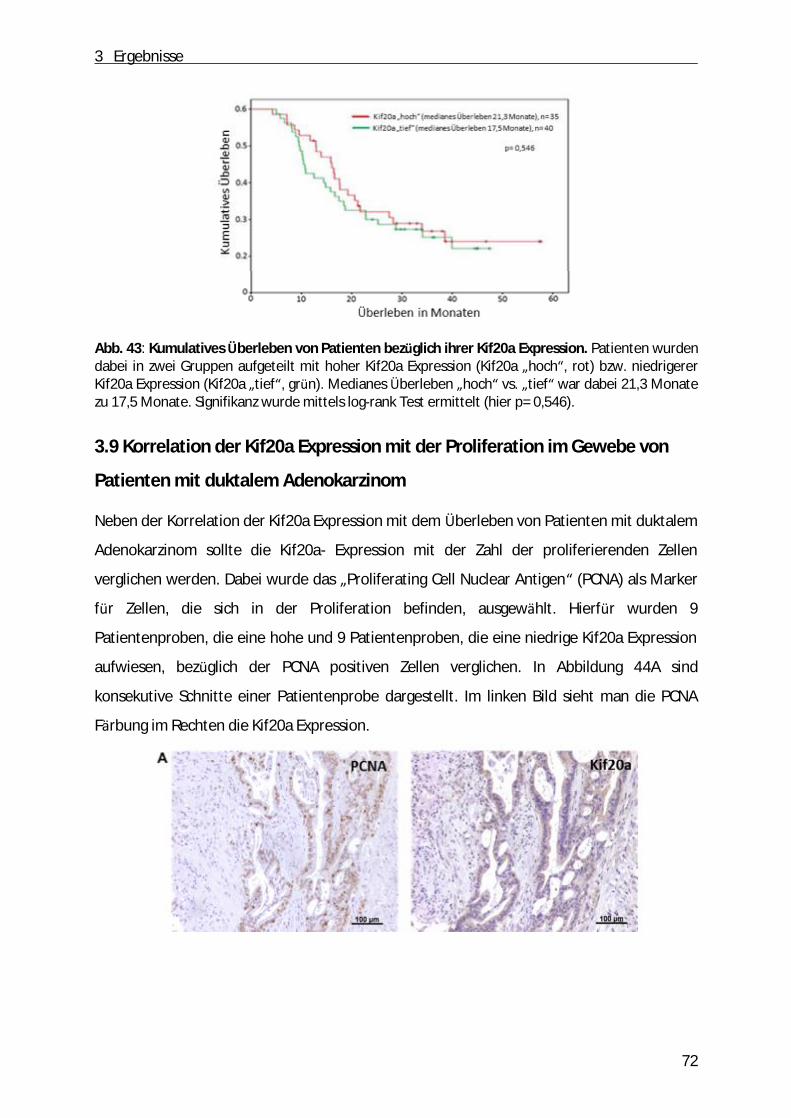
3 Ergebnisse
72
Abb. 43: Kumulatives Überleben von Patienten bezü glich ihrer Kif20a Expression. Patienten wurden dabei in zwei Gruppen aufgeteilt mit hoher Kif20a Expression (Kif20a „hoch“, rot) bzw. niedrigerer Kif20a Expression (Kif20a „ tief“, grü n). Medianes Ü berleben „hoch“ vs. „ tief“ war dabei 21,3 Monate zu 17,5 Monate. Signifikanz wurde mittels log-rank Test ermittelt (hier p= 0,546).
3.9 Korrelation der Kif20a Expression mit der Proliferation im Gewebe von
Patienten mit duktalem Adenokarzinom
Neben der Korrelation der Kif20a Expression mit dem Ü berleben von Patienten mit duktalem
Adenokarzinom sollte die Kif20a- Expression mit der Zahl der proliferierenden Zellen
verglichen werden. Dabei wurde das „Proliferating Cell Nuclear Antigen“ (PCNA) als Marker
fü r Zellen, die sich in der Proliferation befinden, ausgewählt. Hierfü r wurden 9
Patientenproben, die eine hohe und 9 Patientenproben, die eine niedrige Kif20a Expression
aufwiesen, bezü glich der PCNA positiven Zellen verglichen. In Abbildung 44A sind
konsekutive Schnitte einer Patientenprobe dargestellt. Im linken Bild sieht man die PCNA
Färbung im Rechten die Kif20a Expression.
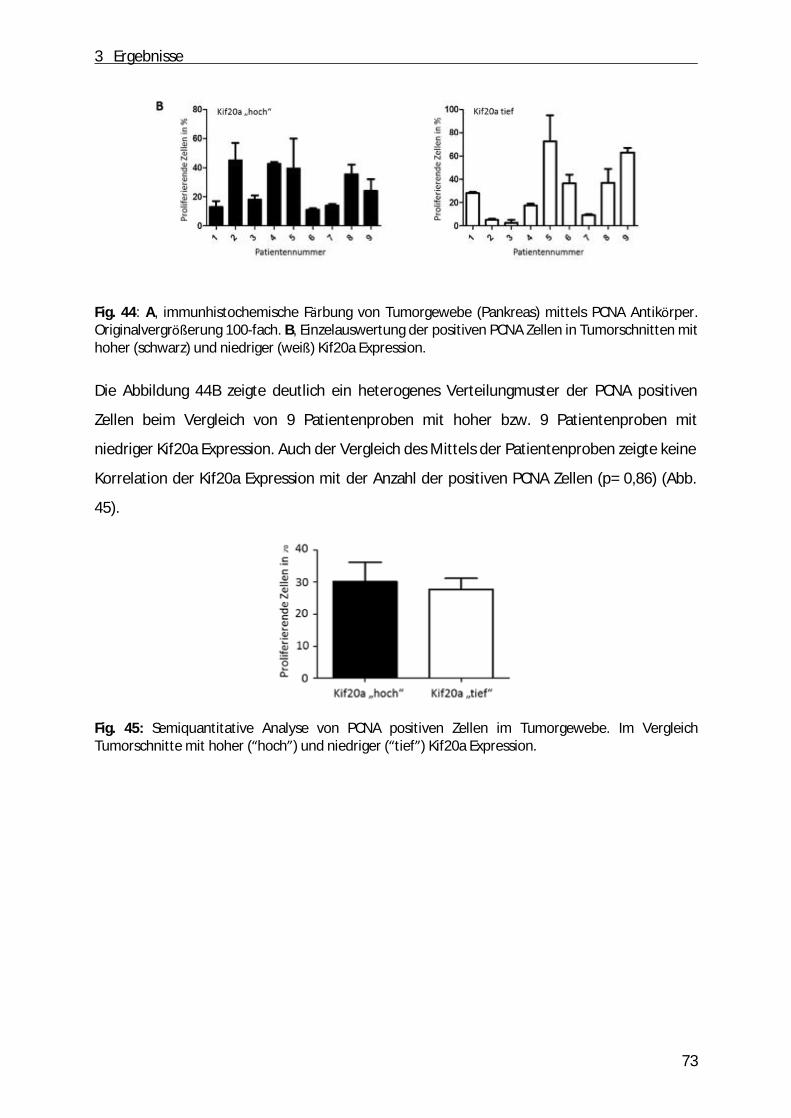
3 Ergebnisse
73
Fig. 44: A, immunhistochemische Färbung von Tumorgewebe (Pankreas) mittels PCNA Antikö rper. Originalvergrö ßerung 100-fach. B, Einzelauswertung der positiven PCNA Zellen in Tumorschnitten mit hoher (schwarz) und niedriger (weiß) Kif20a Expression.
Die Abbildung 44B zeigte deutlich ein heterogenes Verteilungmuster der PCNA positiven
Zellen beim Vergleich von 9 Patientenproben mit hoher bzw. 9 Patientenproben mit
niedriger Kif20a Expression. Auch der Vergleich des Mittels der Patientenproben zeigte keine
Korrelation der Kif20a Expression mit der Anzahl der positiven PCNA Zellen (p= 0,86) (Abb.
45).
Fig. 45: Semiquantitative Analyse von PCNA positiven Zellen im Tumorgewebe. Im Vergleich Tumorschnitte mit hoher (“hoch” ) und niedriger (“tief” ) Kif20a Expression.

4 Diskussion
74
4 Diskussion
Das Pankreaskarzinom stellt die fü nft häufigste Todesursache in westlichen Ländern dar
[139]. Diese schlechte Prognose ist auf das späte Erkennen des Tumors sowie das teilweise
schlechte Ansprechen auf Chemotherapien zurü ckzufü hren. Obwohl die Resektion das
Langzeitü berleben drastisch steigern kann, sind lediglich 15- 20% der detektierten Tumoren
resizierbar [44]. Intrinsische und erworbene Resistenzen gegenü ber Gemcitabine, das als
Standardtherapie bei Pankreaskarzinompatienten eingesetzt wird, sind zudem Faktoren, die
die Prognose verschlechtern. Da die 5- Jahres- Ü berlebensrate mit Pankreaskarzinom bei 5%
liegt ist es wichtig neue therapeutische Ansätze und Strategien zu entwickeln [140-144].
Meta-Analysen verschiedener „high-throughput“ Untersuchungen von Tumorgewebe und
Zelllinien des Pankreas zeigten einen signifikanten Unterschied der Kif20a Expression im
Tumorgewebe gegenü ber gesunden Pankreasgewebe (National Genome Research-Network
– (NGFN-Plus; 01GS08115). Dabei war die Kif20a Expression im Tumorgewebe 18- fach hö her
als im normalen Pankreas [Daten nicht verö ffentlicht (National Genome Research-Network –
(NGFN-Plus; 01GS08115)]. Auch in kleinzelligem Lungenkarzinom, Blutkrebs und Brustkrebs
konnten Forschergruppen eine Ü berexprimierung des Kinesin Motor Proteins Kif20a
feststellen [136, 145, 146]. Im normalen Gewebe ist Kif20a ausschließlich in großen Mengen
in der fö talen Leber, dem erwachsenen Knochenmark sowie dem Thymus exprimiert.
Geringe Mengen konnten zudem in der Plazenta und dem Herzen detektiert werden. In der
Milz und den Lymphknoten ist Kif20a nur in sehr geringem Maße vorhanden und in
Bauchspeicheldrü se, Lunge, Gehirn, Leber, Niere sowie Skelettmuskulatur konnte keinerlei
Kif20a Expression beobachtet werden [125]. Erste Analysen des Kinesin Motor Proteins
Kif20a aus dem Jahr 1998 konnten das Protein erstmals zytoplasmatisch in der Nähe des
Golgi Apparats lokalisieren [123]. Hier interagiert es als Motor mit der Guanosintriphosphat
(GTP) gebunden Form von RAB6A sowie RAB6B. Diese beiden Proteine sind kleine GTPasen,
die die Assoziation bzw. Dissoziation von Kif20a an Mikrotubuli regulieren. Dabei bindet die
konservierte Motordomäne von Kif20a an die Mikrotubuli und generiert mechanische
Energie durch die Hydrolyse von ATP. Mittels dieser freigewordenen Energie kö nnen nun
Golgimembranen, Vesikel und Organelle retrograd durch das Zytoplasma der Zelle
transportiert werden [123] [129] [147]. Kif20a ist zudem an diversen Prozessen des Zellzyklus
beteiligt, wie der Formation der mitotischen Spindeln, der Chromosomen Separierung sowie
während der Zytokinese [108]. Aufgrund seiner unterschiedlichen Funktionen innerhalb der

4 Diskussion
75
Zelle und während des Zellzyklusses, sowie seiner Ü berexprimierung im Krebsgewebe ist
Kif20a ein interessantes Zielgen fü r die Forschung. In der vorliegenden Doktorarbeit sollte
die Expression des Kinesin Motor Proteins in der Bauchspeicheldrü se sowie in PDAC- und
NET- Tumorzellen untersucht werden (Kapitel 3.1-3.2). Zudem sollte mittels funktioneller
Analysen der Einfluss von Kif20a auf die Proliferation, die Migration, die Invasion, die
Motilität sowie die Apoptose von PDAC-Zelllinien erforscht werden. Es wurden zudem
Zellzyklusanalysen durchgefü hrt, um festzustellen, in welchen Stadien sich Tumorzellen nach
Kif20a Suppression befinden (Kapitel 3.3-3.4). Des Weiteren wurden Analysen zur
Doppelbehandlung durchgefü hrt. Hierbei sollte der Effekt von Chemotherapeutika wie Taxol
und Gemcitabine bei gleichzeitiger Kif20a Suppression auf die Proliferation, sowie die
Migration untersucht werden (Kapitel 3.5-3.6). In Proliferationsanalysen mit Tumorzellen
sollte zudem der kommerziell erwerbbare Kif20a Inhibitor „Paprotrain“ untersucht werden
(Kapitel 3.7). Zusätzlich wurde die Kif20a Expression mit dem Ü berleben von 89 Patienten
mit duktalem Adenokarzinom korreliert. Dafü r wurden die Kohorte in zwei Gruppen
eingeteilt: Patienten mit einer hohen Kif20a Expression, und Patienten mit einer niedrigen
Kif20a Expression (Kapitel 3.8). Des Weiteren wurden die Korrelation der Kif20a Expression
mit der Expression des PCNA („Proliferating Cell Nuclear Antigen“) Proteins untersucht
(Kapitel 3.9).
4.1 Kif20a Expression im Pankreasgewebe und Tumorzellen des Pankreas
Fü r die Untersuchung der Kif20a Expression wurden Gewebeschnitte des gesunden
Pankreas, Gewebe mit chronischer Pankreatitis, PanIN Läsionen sowie Tumorgewebe
immunhistochemisch analysiert (Abb. 12- 15). Dabei konnte keine bis sehr schwache Kif20a
Expression in allen duktalen sowie azinären Zellen des gesunden Pankreas beobachtet
werden (Abb. 12). Im Vergleich dazu waren Schnitte mit chronischer Pankreatitis etwas
stärker angefärbt. Sowohl gesundes Pankreasgewebe als auch Gewebe mit chronischer
Pankreatitis zeigten ausschließlich eine zytoplasmatische Kif20a Färbung (Abb. 13). Eine
Kif20a Expression in den Kernen konnte nicht detektiert werden. Bei der Untersuchung der
Gewebeproben mit PanIN Läsionen bzw. Tumorgewebe war eine sehr starke Kif20a Färbung
des Zytoplasmas zu verzeichnen (Abb. 14, 15). In circa 5% der positiv gefärbten Zellen der
Vorläuferläsionen konnte zudem eine deutliche nukleäre Färbung beobachtet werden. Bei
der Analyse der Tumorpräparate stieg die Zahl der positiven Kerne sogar auf 5% an. Die

4 Diskussion
76
entscheidende Rolle von Kif20a während des Zellzyklus ist mö glicherweise der Grund,
warum lediglich in Gewebe mit PanIN Läsionen bzw. Tumorzellen Kif20a positive Kerne
detektiert werden konnten [109, 127, 130]. Studien haben gezeigt, dass Kif20a vor allem in
der M-Phase des Zellzyklus hochreguliert ist [127, 130, 132]. Aufgrund der hohen Expression
in mitotisch aktiven Zellen sollte die Kif20a Expression in verschiedenen Zelllinien des
Pankreas untersucht werden. Dafü r wurden drei duktale Adenokarzinomzelllinien (PDAC-
Zelllinien) und zwei neuroendokrine Tumorzelllinien (NET- Zelllinien) analysiert. Die Kif20a
Expressionsprofile zeigten fü r alle fü nf Zelllinien eine sehr heterogene Verteilung (Abb. 17,
18). Dabei wurde in der QRT-PCR fü r T3M4 die hö chste Kif20a Rate festgestellt gefolgt von
SU86.86 und Panc-1 (Abb. 17). In beiden NET-Zelllinien lag die Kif20a Expression unter der
der HPDE Zellen, die fü r die Normalisierung herangezogen wurden (Abb. 18). Da HPDE Zellen
immortalisierte duktale Epithelzellen des Pankreas sind, wäre es mö glich, das aus diesem
Grund die Kif20a Expression geringer ist als bei den PDAC Zelllinien [148]. Da es sich bei den
NET Zelllinien nicht um duktale Zellen handelt sind HPDE Zellen mö glicherweise nicht die
richtige Zelllinie fü r die Normalisierung der NET Zelllinien. Die Proteinanalyse erbrachte
ebenfalls ein sehr heterogenes Kif20a Expressionsprofil. Studien mit anderen Zelllinien des
Pankreas wie PK8, PK9, PK45H, PK45P, PK59, KLM1, MIA-Paca2 sowie Hs700T zeigten
ebenfalls ein sehr heterogenes Kif20a Expressionsmuster [136]. Ü ber die Expression von
Kif20a ist zum jetzigen Stand nicht viel bekannt, manche Studien gehen allerdings davon aus,
dass die sehr heterogene Verteilung im direkten Zusammenhang mit genomischer
Instabilität steht [149]. Aufgrund der unterschiedlichen Aufgabe von Kif20a innerhalb der
Zelle wurde in der vorliegenden Arbeit die Lokalisation von Kif20a in Tumorzellen
untersucht. Die Immunfluoreszenzergebnisse zeigten eine starke Kif20a Expression im
Nukleus sowie eine ü ber das ganze Zytoplasma verteilte granuläre Färbung (Abb. 19). Diese
Ergebnisse bekräftigen wiederum die Aufgaben von Kif20a sowohl im Zellkern während der
Mitose als auch innerhalb des Zytoplasmas, wo es am Transport der Golgimembranen sowie
Vesikeln und Organellen beteiligt ist [109, 127, 129, 130]. Die Doppelfärbung mit α -Tubulin
zeigte die Assoziation von Kif20a mit den Mikrotubuli (Abb. 19). Dabei bindet das Kinesin
Motor Protein allerdings nicht direkt an die Untereinheiten der Mikrotubuli sondern ü ber
zwei kleine GTPase, RAB6A und RAB6B [123, 131]. Die Vergleichsfärbungen der HPDE Zellen
zeigten eine ähnliche Kif20a Lokalisation innerhalb der Zelle. Aufgrund der sehr kleinen

4 Diskussion
77
HPDE Zellen war es schwierig, die Kif20a Verteilung innerhalb des Zytoplasmas genauer zu
untersuchen (Abb. 20).
4.2 Funktionelle Analysen von PDAC Zelllinien nach Kif20a Suppression
Analysen haben ergeben, dass Kif20a in normalem Gewebe kaum bzw. in geringen Mengen
exprimiert wird [125]. Im Tumorgewebe steigt die Expression dagegen drastisch an. Sowohl
in kleinzelligem Lungenkarzinom, Blutkrebs, Brustkrebs als auch im
Bauchspeicheldrü senkrebs konnte dieser Anstieg beobachtet werden [132, 136, 145, 146].
Aus diesem Grund sollte in der vorliegenden Arbeit sowohl der Effekt der Runterregulierung
als auch der Effekt der Hochregulierung von Kif20a in Tumorzellen untersucht werden. Dabei
wurden verschiedene biologische Analysen wie Proliferations-, Migrations-, Invasions-,
Motilitäts- und Apoptose- Assays durchgefü hrt. Nach Kif20a Suppression konnte fü r alle drei
PDAC Zelllinien eine deutliche Reduktion der Proliferation beobachtet werden (Abb. 23).
Nach Kif20a Suppression waren lediglich 73% der T3M4 sowie Panc-1 Zellen und 90% der
SU86.86 Zellen proliferativ. Beim Vergleich der QRT-PCR Ergebnisse nach der Behandlung
mit dem Kif20a siRNA Konstrukt konnte festgestellt werden, dass die Kif20a Expression fü r
die Zelllinie T3M4 nur noch bei 15% und bei der Zelllinie Panc-1 bei 10% lag. Im Vergleich
dazu lag die Kif20a Expression in der Zelllinie SU86.86 bei 33%. (Abb. 21, 22). Aufgrund der
hö heren Kif20a Rate fü r SU86.86 lässt sich vermutlich die weniger starke Reduktion der
Proliferation fü r SU86.86 erklären. Aufgrund des Einflusses von Kif20a während der Mitose
ist erklärbar, warum eine Suppression des Kif20a Gens, eine Verminderung der Proliferation
zur Folge hat. Morphologische Veränderungen zeigten die transfizierten Zellen im Vergleich
zur Kontrolle jedoch nicht. Dies lässt darauf schließen, dass die Inhibierung der Proliferation
nicht alleine durch das Ausbleiben der Zytokinese begrü ndet ist und dass andere Kinesine die
mitotische Funktion von Kif20a mö glicherweise ü bernehmen. Taniuchi et. al konnte in
seinen Studien ein ähnliches Ergebnis erzielen. Nach der Transfektion von MIA-Paca2 und
PK59 Zellen konnte eine Wachstumsinhibierung festgestellt werden. Multinukleäre Zellen
wurden auch dort nicht beobachtet [132]. Auch Experimente mit der Brustkrebszelllinie
MCF7 zeigten keine multinukleären Zellen. Sie konnten zudem beweisen, dass die Kif20a
supprimierten Zellen in der G1 Phase akkumulieren und anschließend durch lysosomalem
Zelltod absterben [150]. Studien mit HeLa Zellen, die aus einem Zervixkarzinom isoliert
wurden, konnten jedoch zeigen, dass Transfektionsversuche mit dem Kif20a siRNA Konstrukt

4 Diskussion
78
zu binukleären Zellen fü hrten. Bereits 24 h nach der Transfektion konnte eine
Akkumulierung der Zellen mit doppeltem Nukleus beobachtet werden [128, 151]. Ebenso in
Leberkarzinomzellen wurden binukleäre Zellen nach der Transfektion mit dem Kif20a siRNA
Konstrukt beobachtet, das auf einen Fehler während der Zytokinese zurü ckzufü hren ist
[149].
Fü r die Migration von Zellen ist das Zusammenspiel von Aktin, Mikrotubuli sowie den damit
assoziierten Proteinen und Motorproteinen von entscheidender Bedeutung [152]. In
unterschiedlichen Studien wurde bereits die Rolle von Kinesin Motor Proteinen in
Migrationsprozessen untersucht. Im kolorektalen Karzinom fü hrte die Suppression von
Kif18A zu einer verminderten Migrationsfähigkeit der Tumorzellen [153]. Ebenso die
Runterregulierung des Kinesin Motor Proteins Kif14 in Lebertumorzellen reduzierte die
Migration der Zellen [154]. Neben dem Einfluss auf die Proliferation sollte deshalb zudem die
Wirkung der Kif20a Runterregulierung auf die Migration untersucht werden (Abb. 24). Dabei
konnte beobachtet werden, dass fü r alle drei PDAC- Zelllinien die Migration vermindert war.
Fü r die T3M4 sowie Panc-1 Zelllinien konnte eine 50%-ige Reduktion festgestellt werden.
Den grö ßten Effekt hatte die Kif20a Suppression auf die Zelllinie SU86.86. Lediglich 40% der
Zellen wanderten nach Kif20a Suppression durch die Membran. Odrowaz et. al erzielte in
seinen Kif20a Suppressionsstudien ebenfalls ähnliche Ergebnisse. Die Migration von MCF10A
Brustkrebszellen konnte auch hier minimiert werden [155]. Studien ü ber die genaue
Funktionsweise von Kif20a während der Zellmigration gibt es allerdings noch nicht.
Mö glicherweise sind Motorproteine von entscheidender Bedeutung, um die Zellform zu
vergrö ßern, notwenige Organelle durch die Zelle zu transportieren und letzten Endes die
Zellform erneut wiederherzustellen. Aufgrund des Effekts der Kif20a Suppression auf die
Migration der Tumorzellen sollte zudem untersucht werden, ob die Runterregulierung
ebenfalls Einfluss auf die Invasionseigenschaft der Tumorzellen hatte. Die Ergebnisse
zeigten, dass lediglich 52% der Panc-1 Zellen und 87% der SU86.86 Zellen nach der
Transfektion invasiv waren (Abb. 25). Der Grund fü r die stärkere Reduktion fü r Panc-1
kö nnte in der stärkeren Runterregulierung von Kif20a liegen.
Neben der eingeschränkten Migrations- und Invasionsfähigkeit der Zellen nach Kif20a
Suppression, sollte untersucht werden, ob die Runterregulierung Einfluss auf die Motilität
der Zellen hatte. Bislang gibt es keine Daten, die den Einfluss von Kif20a auf die Motilität von
Zellen, beschreiben. In der vorliegenden Arbeit wurden dafü r „Time-lapse“- Analysen

4 Diskussion
79
durchgefü hrt. Die Ergebnisse zeigten, dass transfizierte Panc-1 Zellen eine deutlich
niedrigere Bewegungsgeschwindigkeit (75%) aufwiesen. Auch die zurü ckgelegte Distanz war
im Vergleich zu den Kontrollzellen deutlich verringert (67%) (Abb. 26A). Auffällig war, dass
sich die Zellen weniger stark im Raum bewegten, was durch einen Anstieg der
Richtungsabhängigkeit (50%) der Zellen gezeigt werden konnte. Als weiteres Beispiel wurde
auch hier die Zelllinie SU86.86 analysiert um festzustellen, ob eine geringere Kif20a
Suppression, ein anderes Ergebnis erzielte. Die Ergebnisse zeigten jedoch keinen
Unterschied zwischen transfizierten Zellen und Kontrollzellen (Abb. 26B).
Neben Analysen, die die Bewegungsfähigkeit von Krebszellen nach Kif20a Suppression
beschrieben, sollte mittels Zellzyklusanalysen festgestellt werden, ob sich die Zellen nach der
Runterregulierung des Kif20a Gens, in einer bestimmten Zellzyklusphase arretieren oder ob
die Zellen bereits apoptotisch waren. Hierfü r wurden die Zelllinien Panc-1 und SU86.86
verwendet, um eine Zelllinie mit hö herer Kif20a Suppression und eine Zelllinie mit niedriger
Kif20a Suppression zu vergleichen. Mittels Apoptose Färbungen sollte die Vermutung, dass
Panc-1 Zellen nach Kif20a Transfektion apoptotisch werden, ü berprü ft werden. Bereits die
Färbung der Zellkerne mittels DAPI zeigt eine Veränderung der Panc-1 Zellkerne nach
Transfektion mit dem Kif20a siRNA Konstrukt. Die ursprü nglich runden Zellkerne waren
anschließend bohnenfö rmig gekrü mmt (Abb. 27). Dies lässt mö glicherweise auf eine
Fehlfunktion des Zellzyklus und im Speziellen auf eine abnormale Zytokinese schließen. Die
SU86.86 Zellkernen hingegen zeigten keinen Unterschied in ihrer Morphologie nach Kif20a
Suppression (Abb. 27). Als Apoptose Marker wurde ein Antikö rper gegen das Protein
„Cleaved Caspase 3“ verwendet, das in gesunden Zellen in seiner inaktiven Form als
Procaspase (32kDa) vorliegt und in apoptotischen Zellen in zwei Fragmente gespalten wird
(p12, p17). Die Färbungen in Abbildung 28 zeigten einen Anstieg an apoptotischen Panc-1
Zellen nach Transfektion mit dem Kif20a siRNA Konstrukt. Im Vergleich dazu war keinerlei
positive Färbung der transfizierten SU86.86 Zellen zu beobachten. Experimente mit
Leberkarzinomzellen ergaben ebenfalls eine Reduktion der Proliferation der Zellen nach
Kif20a Runterregulierung, nicht aber eine Induzierung der Apoptose [149]. Groth-Pedersen
konnte in seinen Studien mit MCF7 Brustkrebszellen belegen, dass Kif20a supprimierte
Zellen absterben. Allerdings konnten diese Analysen verdeutlichen, dass es sich dabei nicht
um den apoptotischen Zelltod, sondern um einen lysosomalen Zelltod handelt. Um die
Ergebnisse zu verifizieren, wurden FACS Analysen durchgefü hrt. Die Ergebnisse zeigten, dass

4 Diskussion
80
ein 5-facher Anstieg an toten Panc-1 Zellen nach Transfektion mit der Kif20a siRNA zu
verzeichnen war. Die Zahl der Zellen die sich in der G2/M- Phase befanden, war dagegen
vermindert (Abb. 29A). Die Runterregulierung des Kif20a Gens fü hrte scheinbar, schon bevor
die Zelle in die Mitose eintraten, zum Zelltod. Während des Zellzyklusses ü bernimmt Kif20a
unterschiedliche Aufgaben, wie die Formation der mitotischen Spindelfasern, der
Separierung der Zentrosomen, der Chromosomen Aufteilung sowie der Zytokinese [109, 127,
130]. Auch wenn die genaue Ursache der Apoptose hier nicht ganz geklärt werden konnte,
geht man davon aus, dass Bereiche dieser Aufgaben während des Zellzyklusses gestö rt oder
verändert sind, und damit die Zelle unweigerlich in Apoptose geht. Bei der Untersuchung der
SU86.86 Zellen konnte jedoch ein anderes Ergebnis erzielt werden. Die Kif20a
Runterregulierung bewirkte hier keinen Anstieg an toten Zellen, dafü r nahm die Zahl der sich
in der S-Phase befindlichen Zellen zu (hier: 1,8%). Hier konnte eine Arretierung der Zellen in
der S-Phase beobachtet werden (Abb. 29B). Der Vergleich der beiden Zelllinien lässt
vermuten, dass eine starke Kif20a Expression zu einem Zellzyklus Arrest in der S-Phase fü hrt,
und die Funktion von Kif20a durch andere Proteine kompensiert werden kann, wohingegen
eine schwache Kif20a Expression zum Tod der Zelle fü hrt. Yan et al. [156] konnte zeigen,
dass Kif20a unter anderem eine Rolle bei der Arretierung von Magenkarzinomzellen in der
G2/M Phase spielt. Warum die Zellen allerdings in der G2/M Phase arretiert waren, konnte
nicht geklärt werden.
4.3 Funktionelle Analysen von PDAC Zelllinien nach Kif20a Hochregulierung
Neben den funktionellen Analysen nach Kif20a Runterregulierung sollte der Einfluss der
Kif20a Hochregulierung auf die Proliferation und Migration untersucht werden. Bislang gibt
es keine Studien ü ber Analysen von Krebszellen, in denen Kif20a zusätzlich ü berexprimiert
wurde. Dafü r wurde PDAC Zellen mit einem Kif20a Expressionsvektor transfiziert und
anschließend analysiert (Abb. 30). Eine deutliche Kif20a Ü berexprimierung konnte fü r alle
drei Zelllinien nach 48 h beobachtet werden (Abb. 30, 31). Bereits 24 h nach der Transfektion
konnten binukleäre Zellen unter dem Mikroskop entdeckt werden, deren Zahl sich nach
weiteren 24 h deutlich erhö hte (Abb. 32). Anscheinend hat die Ü berexprimierung des
Kinesin Motor Proteins eine entscheidende Auswirkung auf die Zytokinese. Mö glicherweise
ist nach Ü berexprimierung die Menge an Kif20a so stark erhö ht, dass nicht alle Kif20a
Proteine durch PLK1 phosphoryliert werden kö nnen und somit die Funktion währen der

4 Diskussion
81
Zytokinese behindert ist. Bei den Suppressionsversuchen konnten dagegen keine
binukleären Zellen festgestellt werden. Proliferationsanalysen ergaben, dass lediglich 70%
der SU86.86 Zellen sowie 80% der T3M4 Zellen proliferierten nach der Transfektion mit dem
Kif20a Expressionsvektor. Die Analyse der Panc-1 Zellen ergab eine 90%-ige Proliferation der
Zellen nach Transfektion und war nicht signifikant (Abb. 33). Interessanterweise scheint die
Inhibierung des Wachstums mit der Kif20a Expression zu korrelieren. Beim Vergleich der
QRT-PCR Analysen mit den Ergebnissen des MTT-Assays ließ sich folgendes Muster
erkennen: Je hö her die Kif20a Expression, desto geringer die Wachstumsinhibierung.
Krebszellen, die eine sehr starke Kif20a Expression aufweisen waren somit vitaler und
aggressiver. Dies kö nnte von entscheidender Bedeutung fü r mö gliche in vivo Studien sein.
Aus diesem Grund sollte zudem untersucht werden, ob sich ebenfalls eine Korrelation bzgl.
der Migration feststellen ließ (Abb. 34). Dabei wanderten 48 h nach der Ü berexprimierung
von Kif20a lediglich 70% der T3M4 Zellen und 60% der SU86.86 Zellen durch die Membran.
Neben der Hochregulierung in Tumorzellen wurde zudem die Transfektion der Zelllinie HPDE
untersucht (Abb. 35). Dabei handelt es sich um eine immortalisierte duktale Zelllinie, die in
der Literatur als Kontrollzelllinie bzw. als annähernd normale Pankreaszelllinie beschrieben
wird [148]. Allerdings fü hrte die Ü berexprimierung von Kif20a in HPDE Zellen nach 24- 48 h
zum Zelltod.
4.4 Funktionelle Analysen von PDAC Zelllinien nach Kombinationsbehandlung
von Chemotherapeutika bei gleichzeitiger Kif20a Suppression
Neueste Studien in der Krebstherapie beschäftigen sich mit der Inhibierung von Proteinen,
die an regulatorischen Prozessen des Zellzyklusses beteiligt sind. Taxane werden zum
Beispiel bei der Therapie von verschiedenen soliden Tumoren als adjuvante, neoadjuvante
oder Sekundärtherapie eingesetzt [73-78]. Die Wirkungsweise dieser mitotischen Inhibitoren
beruht dabei auf folgendem Prinzip: Nach der Bindung von Taxol an die β-Untereinheit der
Mikrotubuli werden diese stabilisiert und dadurch die Depolymerisierung verhindert. Es
kommt in Folge dessen zu einer verminderten Dynamik sowie einer Bü ndelung der
Mikrotubuli in der Interphase, was Zellen in einen mitotischen Arrest versetzt und zum
anderen die Apoptose einleitet [157, 158]. Erworbene Resistenzen gegenü ber Taxol
beeinträchtigen jedoch die klinische Effektivität [159-161]. Aus diesem Grund sollte in der
vorliegenden Arbeit der Effekt der Doppelbehandlung von Taxol bei gleichzeitiger

4 Diskussion
82
Suppression des Kinesin Motor Proteins Kif20a untersucht werden. Daf ü r wurden in
Vorversuchen zwei unterschiedliche Konzentrationen an Taxol (2 µM, 20 µM) getestet, um
die optimale Inhibierung der Proliferation herauszufinden. Die Ergebnisse des MTT-Assays
zeigten, dass fü r alle drei Zelllinien eine Taxol Konzentration von 2 µM fü r 24 h am
wirksamsten war (Abb. 36). Die Proliferation der T3M4 Zellen lag bei 25%, bei SU86.86 Zellen
bei 70% und bei der Zelllinie Panc-1 bei 40%. Fü r die Behandlung mit 20 µM Taxol konnte ein
Wachstum von 55% fü r T3M4, 90% fü r SU86.86 sowie 80% fü r Panc-1 beobachtet werden.
Nach der Doppelbehandlung von Taxol (2 µM, 20 µM) bei gleichzeitiger Kif20a Suppression
konnte eine Runterregulierung von Kif20a im Immunblot festgestellt werden (Abb. 37). Bei
der Doppelbehandlung von T3M4 mit dem Kif20a siRNA Konstrukt und 20 µM Taxol konnten
jedoch keine Banden im Immunblot sichtbar gemacht werden. Da GAPDH unter anderem mit
Mikrotubuli assoziierten Proteinen, wie zum Beispiel tau oder MAP1B („microtubuli
associated protein 1B“) interagiert, kö nnte es sein, dass GAPDH zudem mit dem Kinesin
Motor Protein Kif20a interagiert [162-164]. Aus diesem Grund kö nnte vermutlich bei der
Kombination von Taxol und Kif20a siRNA die GAPDH Bande im Immunblot verschwinden.
Studien ü ber eine mö gliche Verbindung gibt es bislang nicht, ebenso liegen keine Analysen
bzgl. der Beziehung zwischen der GAPDH-Tubulin Interaktion und der Bindung von
Mikrotubulitoxinen wie Taxol vor. Aus diesem Grund wurde die Zelllinie T3M4 von weiteren
funktionellen Analysen nach Doppenbehandlung (Kif20a siRNA und 20 µM Taxol)
ausgeschlossen. Die folgenden Proliferationsanalysen der SU86.86 und Panc-1 Zelllinie
wurden sowohl mit 2 µM als auch mit 20 µM Taxol sowie dem Kif20a siRNA Konstrukt
durchgefü hrt. Es sollte analysiert werden, ob die zusätzliche Runterregulierung von Kif20a
einen zusätzlichen inhibierenden Effekt auf das Wachstum der PDAC Zelllinien hatte (Abb.
38). Fü r die Zelllinien Panc-1 sowie T3M4 konnte dabei kein signifikanter Anstieg der
Inhibierung beobachtet werden. Einzig fü r die Zelllinie SU86.86 konnte fü r eine
Konzentration von 2 µM eine doppelt so hohe Reduktion des Wachstums festgestellt
werden, nach zusätzlicher Kif20a Runterregulierung. Ebenso bei der Behandlung mit 20 µM
und dem siRNA Konstrukt konnte die Inhibierung von 10% auf ü ber 60% gesteigert werden.
Warum SU86.86 gegenü ber den anderen Zelllinien eine so starke Reduktion zeigt, muss in
weiteren Analysen gezeigt werden. Die Vorversuche zeigten, dass sowohl T3M4 als auch
Panc-1 sensitiver auf Taxol reagierten als SU86.86 und damit eventuell die Proliferation
dieser beiden Zelllinien nicht zusätzlich durch die Suppression von Kif20a gesteigert werden

4 Diskussion
83
konnte. Somit wäre fü r Tumorzellen, die eine weniger starke Sensitivität fü r Taxol haben, die
Suppression von Kif20a eine gute Alternative zur Reduktion der Proliferation. Neben den
Analysen zur Proliferationsinhibierung sollte zudem der Einfluss auf die Migration der PDAC
Zellen untersucht werden (Abb. 39). Dafü r wurde ausschließlich eine Konzentration von 2
µM Taxol verwendet, da diese Konzentration bereits die besten Erfolge in den Vorversuchen
erzielte. Die Ergebnisse der Versuche zeigten, dass lediglich 4,5% der doppelt behandelten
SU86.86 Zellen und 30% der behandelten Panc-1 Zellen durch die Membran wanderten, im
Vergleich zu den Kontrollzellen, die aussschließlich mit Taxol behandelt wurden. Fü r die
Zelllinie T3M4 konnten keine Ergebnisse erzielt werden, da die Zellen die Doppelbehandlung
und den Migrationsversuch nicht ü berlebten. Es ist anzunehmen, dass zwei entscheidende
Mechanismen diesen enormen Effekt ausmachten. Zum einen wird durch die Gabe von Taxol
die Dynamik der Mikrotubuli vermindert und zum anderen kann Kif20a durch die
Suppression nicht mehr in ausreichendem Maße an den Mikrotubuli entlangwandern [157].
Als Motor Protein hat es zudem die Aufgabe des Organelltransports, dass während der
Migration der Zellen ebenso von entscheidender Bedeutung ist [129, 130]. Eine
Runterregulierung wü rde zudem einen verminderten Transport von wichtigen Molekü len
während der Migration inhibieren. Analysen des Kinesin Motor Proteins Kif18A, bei dem es
sich ebenfalls um ein Mitglied der N-Kinesine handelt, zeigte ebenfalls eine deutliche
Reduktion der Migration nach Kif18A Suppression [153]. Studien ü ber Kif20a und den
Einfluss auf die Migration bei gleichzeitiger Taxolbehandlung gibt es bislang noch nicht.
In einem zweiten Abschnitt sollte der Einfluss der Doppelbehandlung von Gemcitabine und
Kif20a Suppression auf die Proliferation sowie Migration untersucht werden. Fü r die
Proliferationsanalysen wurden dabei unterschiedliche Konzentrationen an Gemcitabine (10
µM, 50 µM, 100 µM, 500 µM, 1000 µM) eingesetzt (Abb. 40). Die Ergebnisse zeigten fü r die
Zelllinien SU86.86 sowie Panc-1 lediglich fü r die Konzentration von 10 µM eine leichte
Reduktion des Wachstums bei gleichzeitiger Kif20a Runterregulierung (SU86.86 = 9%; Panc-1
= 7%). Fü r die Zelllinie T3M4 konnte fü r alle Konzentrationen eine signifikante
Proliferationsinhibierung von bis zu 50% nach Doppelbehandlung verzeichnet werden.
Bereits die Taxolversuche konnten eine hö here Sensitivität der T3M4 Zelllinie auf die
Doppelbehandlung zeigen im Gegensatz zu SU86.86 und Panc-1. Fü r die Migrationsversuche
wurde anschließend eine Konzentration von 10 µM Gemcitabine verwendet, da diese
Konzentration den grö ßten inhibierenden Effekt auf die Proliferation erzielte. Ä hnlich wie bei

4 Diskussion
84
den Taxolversuchen ü berlebten die T3M4 Zellen die Behandlung jedoch nicht. Nach
Doppelbehandlung wanderten nur noch 30% der SU86.86 Zellen und 68% der Panc-1 Zellen
durch die Membran im Vergleich zu den Kontrollzellen, die ausschließlich mit 10 µM
Gemcitabine behandelt wurden (Abb. 41). Studien ü ber Kif20a Suppression bei gleichzeitiger
Gemcitabinebehandlung wurden bislang nicht verö ffentlicht. Zusammenfassend lässt sich
sagen, dass die Kombinationstherapie von Taxol oder Gemcitabine bei gleichzeitiger Kif20a
Suppression ein sehr heterogenes Ergebnis bzgl. der Reduktion von Proliferation und
Migration lieferte.
4.5 Wirkung von Paprotrain auf die Proliferation von PDAC Zelllinien
Vergleicht man die Ergebnisse der Proliferations- und Migrationsversuche sowohl nach
Kif20a Suppression als auch nach Kif20a Ü berexprimierung lässt sich ein direkter
Zusammenhang zwischen der Kif20a Expression und der Vitalität bzw. Mobilität der
Krebszellen erkennen. Je hö her die Kif20a Expression war, desto vitaler und mobiler waren
die Tumorzellen. Eine deutliche Reduktion der Migration und Proliferation konnte in den
Zellen beobachtet werden, die eine geringere Kif20a Expression aufwiesen. Aus diesem
Grund sollte in einer weiteren Analyse der Effekt des Kif20a Inhibitors Paprotrain auf die
Proliferation der Tumorzellen untersucht werden. Tscherniuk et al. analysierten einen Pool
von 8900 kleinen Molekü len, die alle die Wirkungsweise der ATPase Aktivität des Kif20a
Proteins unterdrü cken sollten. Dabei wurde das (Z)-2-(1H-indol-3-yl)-3-(pyridin-3-
yl)acrylonitril, das den Namen Paprotrain (Passenger PROtein TRAnsport Inhibitor) trägt, als
effektivster Inhibitor identifiziert. Proliferationsanalysen mit Paprotrain konnten erste
Erfolge in HeLa Zellen dokumentieren. Erste Ergebnisse in PDAC Zellen zeigten eine deutliche
Reduktion der Proliferation. Dabei konnte man fü r die Zelllinie T3M4 eine Reduktion der
Proliferation fü r alle Konzentrationen beobachten. Die stärkste Inhibierung erzielte dabei
eine Paprotrain Konzentration von 22µM. Nach der Behandlung waren lediglich 43,2% der
T3M4 Zellen proliferativ. Bei der Auswertung der Behandlung von SU86.86 konnte lediglich
fü r die Konzentrationen 10 µM sowie 50 µM eine verminderte Proliferation beobachtet
werden. Hier waren lediglich 90% bzw. 75% der Zellen proliferativ. Fü r die Zelllinie Panc-1
erzielte die Behandlung mit 1 µM, 22 µM sowie 50 µM eine inhibierende Wirkung von 30-
40% (Abb. 42). Diese Ergebnisse liefern erste Analysen ü ber die Wirksamkeit von Paprotrain
in Pankreaskarzinomzellen und mü ssen in weiteren Versuchen auf ihre Genauigkeit

4 Diskussion
85
ü berprü ft werden. Bislang belegen unterschiedliche Studien jedoch einen Effekt auf die
Proliferation bzw. die Apoptose von verschiedenen Krebszellen nach Behandlung mit Kinesin
Inhibitoren. Neben Einzelbehandlung mit Inhibitoren wie K5Is [165], Dimethylenastron [165]
oder Monastrol [166] konnten auch Kombinationsstudien mit Chemotherapeutika
vielversprechende Effekte in vitro erzielen. In einer Studie von Basso et al. konnte ein Kinesin
Motor Protein Inhibitor (SCH 2047069) antitumorale Effekte sowohl in der Einzeltherapie als
auch in Kombination mit Paclitaxel, Gemcitabine oder Vincristine erzeugen [167].
Nichtdestotrotz scheint die Wirkungsweise der Kinesin Inhibitoren in klinischen Studien oft
weniger effektiv [168-170]. Studien ü ber einen Kif20a Inhibitor oder Paprotrain im speziellen
wurden bislang nicht publiziert, sind aber aufgrund der vorliegenden Daten sehr
vielversprechend.
4.6 Korrelation der Kif20a Expression mit dem Überleben von Patienten mit
duktalem Adenokarzinom
In Vorversuchen konnte gezeigt werden, dass die Kif20a Expression einen Einfluss auf die
Proliferation sowie die Migration von PDAC Zelllinien hatte. Auch die
immunhistochemischen Analysen zeigten eine stärkere Kif20a Expression in
Vorläuferläsionen sowie im Tumorgewebe im Vergleich zum gesunden Pankreasgewebe. Aus
diesem Grund sollte die Korrelation der Kif20a Expression mit dem Ü berleben von Patienten
mit duktalem Adenokarzinom genauer untersucht werden. Dafü r wurden Gewebeproben
von 89 Patienten immunhistochemisch gefärbt und ausgewertet. Mittels Kaplan-Meier Test
wurden die Ü berlebenskurven der beiden Gruppen verglichen (Abb. 43). Dabei konnte fü r
Patienten mit einer hohen Kif20a Expression (Kif20a“hoch“) ein medianes Ü berleben von
21,3 Monaten ermittelt werden im Vergleich zu Patienten mit einer niedrigen Kif20a
Expression (Kif20a“tief“). Hier lag das mediane Ü berleben bei 17,5 Monaten. Die Ergebnisse
sind jedoch nicht signifikant gewesen (p= 0,546). Analysen der Kif18A Expression und dem
Ü berleben von Patienten mit kolorektalem Karzinom zeigten ein entgegengesetztes Muster.
Hier hatten Patienten mit einer hohen Kif18A Expression eine deutlich schlechtere Prognose
im Vergleich zu Patienten mit einer niedrigeren Kif18A Expression (p=0,0007)[153]. Studien
an Patienten mit einem hepatozellulären Karzinom zeigten, dass eine erhö hte Kif14
Expression mit einer schlechteren Prognose korrelierte (p< 0,001)[154]. Auch fü r Eg5 zeigten
sich schlechtere Prognosen bei Patienten mit Nierenkarzinom wenn die Kinesin Expression

4 Diskussion
86
erhö ht war (p= 0,003) [171]. In der vorliegenden Arbeit wurden erstmals Analysen von
Patienten mit PDAC hinsichtlich ihrer Kif20a Expression und dem Ü berleben vorgestellt.
Bislang gibt es keine Verö ffentlichungen ü ber den Zusammenhang der Expression von
Kinesinen und dem Ü berleben von Patienten mit Pankreastumoren. Die Analysen zeigten
deutlich, dass es keinen signifikanten Unterschied gab bzgl. der Kif20a Expression und dem
medianen Ü berleben. Dieses hier vorgestellte in vitro Modell ist schwierig, da es sich in der
Zellkultur lediglich um eine Art von Zellen handelt, bei der äußere Einflü sse fehlen und
Gewebeproben einen Pool von unterschiedlichsten Zellen darstellen. Um eine bessere
Aussage treffen zu kö nnen mü sste eine grö ßere Patientenkohorte analysiert werden. Des
Weiteren ist die Immunhistochemie eine schwierige Methode die Korrelation zwischen
Ü berleben und Kif20a Expression zu berechnen, da Kif20a in unterschiedlichsten Strukturen
des Pankreas exprimiert wird.
In einem letzten Schritt wurde die Korrelation der Kif20a Expression mit der Expression des
PCNA („Proliferating Cell Nuclear Antigen“) Proteins untersucht. Die Ergebnisse zeigten, dass
die Intensität der Kif20a Expression nicht mit der Anzahl der PCNA positiv gefärbten
Tumorzellen korreliert (Abb. 44, 45).
4.7 Ausblick
Aufgrund der Hochregulierung von Kif20a im Pankreaskarzinom und seinen Einflü ssen auf
die Proliferation sowie Migration der Tumorzellen ist Kif20a ein gutes Ausgangsgen f ü r neue
Therapieansätze. Zudem konnte bewiesen werden, dass eine Kombinationstherapie von
Chemotherapeutika sowie einer Suppression des Kinesin Motor Proteins eine Reduktion der
Proliferation sowie Migration/ Invasion zeigte. Dieser Ansatz ist mö glicherweise fü r
zukü nftige Kombinationstherapie bei Patienten von entscheidender Bedeutung.
Verschiedene Studien konnten bereits zeigen, dass Kfi20a ein gutes Kandidatengen fü r die
Immuntherapie ist. Patienten wurde mit einem Kif20a Vakzin behandelt und erlangten
dadurch eine bessere Prognose [172-174]. Kommende Untersuchungen werden sich zudem
mit einem Kif20a-knock out Mausmodell beschäftigen, in dem das Tumorwachstum genauer
erforscht werden soll.

5 Literaturverzeichnis
87
5 Literaturverzeichnis
1. Vaupel, Physiologie des Menschens: Funktionen des Magen Darm Kanals.1995: Springer Verlag. 2. Bardeesy, N. and R.A. DePinho, Pancreatic cancer biology and genetics. Nat Rev
Cancer, 2002. 2(12): p. 897-909. 3. Schiebler, Anatomie. 9. Auflage ed: Springer Verlag. 4. Rauch, L.-. Taschenlehrbuch Histologie2003: Georg Thieme Verlag. 5. Anatomy, G.s., Lehrbuch der Anatomie2005: Urban& Fischer. 6. Klimstra, D.S., et al., Acinar cell carcinoma of the pancreas. A clinicopathologic
study of 28 cases. Am J Surg Pathol, 1992. 16(9): p. 815-37. 7. Klimstra, D.S., et al., Pancreatoblastoma. A clinicopathologic study and review of the
literature. Am J Surg Pathol, 1995. 19(12): p. 1371-89. 8. Ohike, N., M. Kosmahl, and G. Kloppel, Mixed acinar-endocrine carcinoma of the
pancreas. A clinicopathological study and comparison with acinar-cell carcinoma. Virchows Arch, 2004. 445(3): p. 231-5.
9. Chen, M., et al., Molecular pathology of pancreatic neuroendocrine tumors. J Gastrointest Oncol, 2012. 3(3): p. 182-8.
10. Jones, S., et al., Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science, 2008. 321(5897): p. 1801-6.
11. Yachida, S., et al., Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature, 2010. 467(7319): p. 1114-7.
12. Campbell, P.J., et al., The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature, 2010. 467(7319): p. 1109-13.
13. Brugge, W.R., et al., Cystic neoplasms of the pancreas. N Engl J Med, 2004. 351(12): p. 1218-26.
14. Maitra, A., et al., Precursors to invasive pancreatic cancer. Adv Anat Pathol, 2005. 12(2): p. 81-91.
15. Hruban, R.H., R.E. Wilentz, and A. Maitra, Identification and analysis of precursors to invasive pancreatic cancer. Methods Mol Med, 2005. 103: p. 1-13.
16. Hruban, R.H., et al., Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol, 2001. 25(5): p. 579-86.
17. Kloppel, G. and J. Luttges, WHO-classification 2000: exocrine pancreatic tumors. Verh Dtsch Ges Pathol, 2001. 85: p. 219-28.
18. Hruban, R.H., A. Maitra, and M. Goggins, Update on pancreatic intraepithelial neoplasia. Int J Clin Exp Pathol, 2008. 1(4): p. 306-16.
19. Chang, D.K., N.D. Merrett, and A.V. Biankin, Improving outcomes for operable pancreatic cancer: is access to safer surgery the problem? J Gastroenterol Hepatol, 2008. 23(7 Pt 1): p. 1036-45.
20. Feldmann, G., et al., Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg, 2007. 14(3): p. 224-32.
21. Maitra, A. and R.H. Hruban, Pancreatic cancer. Annu Rev Pathol, 2008. 3: p. 157-88. 22. Institut, R.K., Krebs in Deutschland2013. 23. Coughlin, S.S., et al., Predictors of pancreatic cancer mortality among a large cohort
of United States adults. Cancer Causes Control, 2000. 11(10): p. 915-23. 24. Qiu, D., et al., Overview of the epidemiology of pancreatic cancer focusing on the
JACC Study. J Epidemiol, 2005. 15 Suppl 2: p. S157-67.

5 Literaturverzeichnis
88
25. Yun, Y.H., et al., Cigarette smoking and cancer incidence risk in adult men: National Health Insurance Corporation Study. Cancer Detect Prev, 2005. 29(1): p. 15-24.
26. Yun, Y.H., et al., Relative and absolute risks of cigarette smoking on major histologic types of lung cancer in Korean men. Cancer Epidemiol Biomarkers Prev, 2005. 14(9): p. 2125-30.
27. Arslan, A.A., et al., Anthropometric measures, body mass index, and pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan). Arch Intern Med, 2010. 170(9): p. 791-802.
28. Bagnardi, V., et al., Alcohol consumption and the risk of cancer: a meta-analysis. Alcohol Res Health, 2001. 25(4): p. 263-70.
29. Bagnardi, V., et al., A meta-analysis of alcohol drinking and cancer risk. Br J Cancer, 2001. 85(11): p. 1700-5.
30. Everhart, J. and D. Wright, Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA, 1995. 273(20): p. 1605-9.
31. Shikata, K., T. Ninomiya, and Y. Kiyohara, Diabetes mellitus and cancer risk: Review of the epidemiological evidence. Cancer Sci, 2013. 104(1): p. 9-14.
32. La Vecchia, C., et al., Tea consumption and cancer risk. Nutr Cancer, 1992. 17(1): p. 27-31.
33. Michaud, D.S., et al., Coffee and alcohol consumption and the risk of pancreatic cancer in two prospective United States cohorts. Cancer Epidemiol Biomarkers Prev, 2001. 10(5): p. 429-37.
34. Silverman, D.T., Risk factors for pancreatic cancer: a case-control study based on direct interviews. Teratog Carcinog Mutagen, 2001. 21(1): p. 7-25.
35. Michaud, D.S., et al., Dietary meat, dairy products, fat, and cholesterol and pancreatic cancer risk in a prospective study. Am J Epidemiol, 2003. 157(12): p. 1115-25.
36. Hruban, R.H., et al., Genetics of pancreatic cancer. From genes to families. Surg Oncol Clin N Am, 1998. 7(1): p. 1-23.
37. Huang, L., et al., Identification of common variants in BRCA2 and MAP2K4 for susceptibility to sporadic pancreatic cancer. Carcinogenesis, 2013.
38. Goggins, M., et al., Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res, 1996. 56(23): p. 5360-4.
39. Barton, C.M., et al., Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer, 1991. 64(6): p. 1076-82.
40. Scarpa, A., et al., Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol, 1993. 142(5): p. 1534-43.
41. Hruban, R.H., et al., Molecular pathology of pancreatic cancer. Cancer J, 2001. 7(4): p. 251-8.
42. Trede, M., A. Richter, and K. Wendl, Personal observations, opinions, and approaches to cancer of the pancreas and the periampullary area. Surg Clin North Am, 2001. 81(3): p. 595-610.
43. Richter, A., et al., Long-term results of partial pancreaticoduodenectomy for ductal adenocarcinoma of the pancreatic head: 25-year experience. World J Surg, 2003. 27(3): p. 324-9.
44. Varadhachary, G.R., Preoperative therapies for resectable and borderline resectable pancreatic cancer. J Gastrointest Oncol, 2011. 2(3): p. 136-42.
45. Burris, H.A., 3rd, et al., Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol, 1997. 15(6): p. 2403-13.
46. Di Marco, M., et al., Metastatic pancreatic cancer: is gemcitabine still the best standard treatment? (Review). Oncol Rep, 2010. 23(5): p. 1183-92.

5 Literaturverzeichnis
89
47. Philip, P.A., et al., Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol, 2010. 28(22): p. 3605-10.
48. Kindler, H.L., et al., Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol, 2010. 28(22): p. 3617-22.
49. Moore, M.J., et al., Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol, 2007. 25(15): p. 1960-6.
50. Conroy, T., et al., FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med, 2011. 364(19): p. 1817-25.
51. Rothenberg, M.L., et al., A phase II trial of gemcitabine in patients with 5-FU-refractory pancreas cancer. Ann Oncol, 1996. 7(4): p. 347-53.
52. Mini, E., et al., Cellular pharmacology of gemcitabine. Ann Oncol, 2006. 17 Suppl 5: p. v7-12.
53. Nakano, Y., et al., Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer, 2007. 96(3): p. 457-63.
54. Li, D., et al., Pancreatic cancer. Lancet, 2004. 363(9414): p. 1049-57. 55. Berlin, J.D., et al., Phase III study of gemcitabine in combination with fluorouracil
versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J Clin Oncol, 2002. 20(15): p. 3270-5.
56. Rocha Lima, C.M., et al., Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol, 2004. 22(18): p. 3776-83.
57. Louvet, C., et al., Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol, 2005. 23(15): p. 3509-16.
58. Oettle, H., et al., A phase III trial of pemetrexed plus gemcitabine versus gemcitabine in patients with unresectable or metastatic pancreatic cancer. Ann Oncol, 2005. 16(10): p. 1639-45.
59. Abou-Alfa, G.K., et al., Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancer. J Clin Oncol, 2006. 24(27): p. 4441-7.
60. Bramhall, S.R., et al., A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer, 2002. 87(2): p. 161-7.
61. Van Cutsem, E., et al., Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol, 2004. 22(8): p. 1430-8.
62. Van Cutsem, E., et al., Lessons from the adjuvant bevacizumab trial on colon cancer: what next? J Clin Oncol, 2011. 29(1): p. 1-4.
63. Philip, P.A., Improving treatment of pancreatic cancer. Lancet Oncol, 2008. 9(1): p. 7-8.
64. Cascinu, S., et al., Cetuximab plus gemcitabine and cisplatin compared with gemcitabine and cisplatin alone in patients with advanced pancreatic cancer: a randomised, multicentre, phase II trial. Lancet Oncol, 2008. 9(1): p. 39-44.

5 Literaturverzeichnis
90
65. Heinemann, V., et al., Increased survival using platinum analog combined with gemcitabine as compared to single-agent gemcitabine in advanced pancreatic cancer: pooled analysis of two randomized trials, the GERCOR/GISCAD intergroup study and a German multicenter study. Ann Oncol, 2007. 18(10): p. 1652-9.
66. Sultana, A., et al., Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer. J Clin Oncol, 2007. 25(18): p. 2607-15.
67. Huanwen, W., et al., Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol Cancer, 2009. 8: p. 125.
68. Zalatnai, A. and J. Molnar, Review. Molecular background of chemoresistance in pancreatic cancer. In Vivo, 2007. 21(2): p. 339-47.
69. Hiddemann, B., Die Onkologie. 2. Auflage ed: Springer Verlag. 70. Wani, M.C., et al., Plant antitumor agents. VI. The isolation and structure of taxol, a
novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc, 1971. 93(9): p. 2325-7.
71. Singla, A.K., A. Garg, and D. Aggarwal, Paclitaxel and its formulations. Int J Pharm, 2002. 235(1-2): p. 179-92.
72. Appendino, G., The phytochemistry of the yew tree. Nat Prod Rep, 1995. 12(4): p. 349-60.
73. Cantu, M.G., et al., Randomized controlled trial of single-agent paclitaxel versus cyclophosphamide, doxorubicin, and cisplatin in patients with recurrent ovarian cancer who responded to first-line platinum-based regimens. J Clin Oncol, 2002. 20(5): p. 1232-7.
74. Paclitaxel and docetaxel in breast and ovarian cancer. Drug Ther Bull, 1997. 35(6): p. 43-6.
75. Eisenhauer, E.A., et al., European-Canadian randomized trial of paclitaxel in relapsed ovarian cancer: high-dose versus low-dose and long versus short infusion. J Clin Oncol, 1994. 12(12): p. 2654-66.
76. Gore, M.E., et al., Platinum-Taxol non-cross resistance in epithelial ovarian cancer. Br J Cancer, 1995. 71(6): p. 1308-10.
77. Trimble, E.L., et al., Paclitaxel for platinum-refractory ovarian cancer: results from the first 1,000 patients registered to National Cancer Institute Treatment Referral Center 9103. J Clin Oncol, 1993. 11(12): p. 2405-10.
78. Bishop, J.F., et al., Initial paclitaxel improves outcome compared with CMFP combination chemotherapy as front-line therapy in untreated metastatic breast cancer. J Clin Oncol, 1999. 17(8): p. 2355-64.
79. Bonomi, P., et al., Comparison of survival and quality of life in advanced non-small-cell lung cancer patients treated with two dose levels of paclitaxel combined with cisplatin versus etoposide with cisplatin: results of an Eastern Cooperative Oncology Group trial. J Clin Oncol, 2000. 18(3): p. 623-31.
80. McGuire, W.P., et al., Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med, 1996. 334(1): p. 1-6.
81. Piccart, M.J., et al., Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: three-year results. J Natl Cancer Inst, 2000. 92(9): p. 699-708.
82. Shukuya, T., et al., Weekly Paclitaxel after failure of gemcitabine in pancreatic cancer patients with malignant ascites: a retrospective study. Jpn J Clin Oncol, 2010. 40(12): p. 1135-8.

5 Literaturverzeichnis
91
83. Maeda, S., et al., Paclitaxel as second-line chemotherapy in patients with gemcitabine-refractory pancreatic cancer: a retrospective study. Int J Clin Oncol, 2011. 16(5): p. 539-45.
84. Kim, Y.J., et al., Phase II study of 5-fluorouracil and paclitaxel in patients with gemcitabine-refractory pancreatic cancer. Cancer Chemother Pharmacol, 2009. 63(3): p. 529-33.
85. Eisenhauer, E.A. and J.B. Vermorken, The taxoids. Comparative clinical pharmacology and therapeutic potential. Drugs, 1998. 55(1): p. 5-30.
86. Schiff, P.B., J. Fant, and S.B. Horwitz, Promotion of microtubule assembly in vitro by taxol. Nature, 1979. 277(5698): p. 665-7.
87. Hamel, E., et al., Interactions of taxol, microtubule-associated proteins, and guanine nucleotides in tubulin polymerization. J Biol Chem, 1981. 256(22): p. 11887-94.
88. Parness, J. and S.B. Horwitz, Taxol binds to polymerized tubulin in vitro. J Cell Biol, 1981. 91(2 Pt 1): p. 479-87.
89. Schiff, P.B. and S.B. Horwitz, Taxol assembles tubulin in the absence of exogenous guanosine 5'-triphosphate or microtubule-associated proteins. Biochemistry, 1981. 20(11): p. 3247-52.
90. Rowinsky, E.K., L.A. Cazenave, and R.C. Donehower, Taxol: a novel investigational antimicrotubule agent. J Natl Cancer Inst, 1990. 82(15): p. 1247-59.
91. Spencer, C.M. and D. Faulds, Paclitaxel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs, 1994. 48(5): p. 794-847.
92. Rowinsky, E.K., et al., Clinical pharmacology and metabolism of Taxol (paclitaxel): update 1993. Ann Oncol, 1994. 5 Suppl 6: p. S7-16.
93. Schnitzer, M.J. and S.M. Block, Kinesin hydrolyses one ATP per 8-nm step. Nature, 1997. 388(6640): p. 386-90.
94. Hua, W., et al., Coupling of kinesin steps to ATP hydrolysis. Nature, 1997. 388(6640): p. 390-3.
95. Asbury, C.L., A.N. Fehr, and S.M. Block, Kinesin moves by an asymmetric hand-over-hand mechanism. Science, 2003. 302(5653): p. 2130-4.
96. Yildiz, A., et al., Kinesin walks hand-over-hand. Science, 2004. 303(5658): p. 676-8. 97. Woehlke, G. and M. Schliwa, Walking on two heads: the many talents of kinesin. Nat
Rev Mol Cell Biol, 2000. 1(1): p. 50-8. 98. Zhu, C., E. Bossy-Wetzel, and W. Jiang, Recruitment of MKLP1 to the spindle
midzone/midbody by INCENP is essential for midbody formation and completion of cytokinesis in human cells. Biochem J, 2005. 389(Pt 2): p. 373-81.
99. Vale, R.D., T.S. Reese, and M.P. Sheetz, Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell, 1985. 42(1): p. 39-50.
100. Lawrence, C.J., et al., A standardized kinesin nomenclature. J Cell Biol, 2004. 167(1): p. 19-22.
101. Miki, H., et al., All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci U S A, 2001. 98(13): p. 7004-11.
102. Vale, R.D. and R.J. Fletterick, The design plan of kinesin motors. Annu Rev Cell Dev Biol, 1997. 13: p. 745-77.
103. Case, R.B., et al., The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell, 1997. 90(5): p. 959-66.
104. Endow, S.A. and K.W. Waligora, Determinants of kinesin motor polarity. Science, 1998. 281(5380): p. 1200-2.
105. Henningsen, U. and M. Schliwa, Reversal in the direction of movement of a molecular motor. Nature, 1997. 389(6646): p. 93-6.

5 Literaturverzeichnis
92
106. Romberg, L., D.W. Pierce, and R.D. Vale, Role of the kinesin neck region in processive microtubule-based motility. J Cell Biol, 1998. 140(6): p. 1407-16.
107. Sablin, E.P., et al., Direction determination in the minus-end-directed kinesin motor ncd. Nature, 1998. 395(6704): p. 813-6.
108. Verhey, K.J. and J.W. Hammond, Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol, 2009. 10(11): p. 765-77.
109. Hirokawa, N., et al., Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol, 2009. 10(10): p. 682-96.
110. Hirokawa, N., R. Nitta, and Y. Okada, The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol, 2009. 10(12): p. 877-84.
111. Gennerich, A. and R.D. Vale, Walking the walk: how kinesin and dynein coordinate their steps. Curr Opin Cell Biol, 2009. 21(1): p. 59-67.
112. Mayr, M.I., et al., The human kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression. Curr Biol, 2007. 17(6): p. 488-98.
113. Wordeman, L., Microtubule-depolymerizing kinesins. Curr Opin Cell Biol, 2005. 17(1): p. 82-8.
114. Gupta, M.L., Jr., et al., Plus end-specific depolymerase activity of Kip3, a kinesin-8 protein, explains its role in positioning the yeast mitotic spindle. Nat Cell Biol, 2006. 8(9): p. 913-23.
115. Varga, V., et al., Yeast kinesin-8 depolymerizes microtubules in a length-dependent manner. Nat Cell Biol, 2006. 8(9): p. 957-62.
116. Hirokawa, N., et al., Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell, 1989. 56(5): p. 867-78.
117. Kanai, Y., N. Dohmae, and N. Hirokawa, Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron, 2004. 43(4): p. 513-25.
118. Diefenbach, R.J., et al., The C-terminal region of the stalk domain of ubiquitous human kinesin heavy chain contains the binding site for kinesin light chain. Biochemistry, 1998. 37(47): p. 16663-70.
119. Wolf, F.W., et al., vab-8 is a key regulator of posteriorly directed migrations in C. elegans and encodes a novel protein with kinesin motor similarity. Neuron, 1998. 20(4): p. 655-66.
120. Miki, H., Y. Okada, and N. Hirokawa, Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol, 2005. 15(9): p. 467-76.
121. Hizlan, D., et al., Structural analysis of the ZEN-4/CeMKLP1 motor domain and its interaction with microtubules. J Struct Biol, 2006. 153(1): p. 73-84.
122. Mishima, M., S. Kaitna, and M. Glotzer, Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity. Dev Cell, 2002. 2(1): p. 41-54.
123. Echard, A., et al., Interaction of a Golgi-associated kinesin-like protein with Rab6. Science, 1998. 279(5350): p. 580-5.
124. Neef, R., et al., Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J Cell Biol, 2003. 162(5): p. 863-75.
125. Lai, F., et al., cDNA cloning, expression pattern, genomic structure and chromosomal location of RAB6KIFL, a human kinesin-like gene. Gene, 2000. 248(1-2): p. 117-25.
126. Horrevoets, A.J., et al., Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood, 1999. 93(10): p. 3418-31.

5 Literaturverzeichnis
93
127. Fontijn, R.D., et al., The human kinesin-like protein RB6K is under tight cell cycle control and is essential for cytokinesis. Mol Cell Biol, 2001. 21(8): p. 2944-55.
128. Tcherniuk, S., et al., Relocation of Aurora B and survivin from centromeres to the central spindle impaired by a kinesin-specific MKLP-2 inhibitor. Angew Chem Int Ed Engl, 2010. 49(44): p. 8228-31.
129. Hirokawa, N., Y. Noda, and Y. Okada, Kinesin and dynein superfamily proteins in organelle transport and cell division. Curr Opin Cell Biol, 1998. 10(1): p. 60-73.
130. Hill, E., M. Clarke, and F.A. Barr, The Rab6-binding kinesin, Rab6-KIFL, is required for cytokinesis. EMBO J, 2000. 19(21): p. 5711-9.
131. White, J., et al., Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J Cell Biol, 1999. 147(4): p. 743-60.
132. Taniuchi, K., et al., Down-regulation of RAB6KIFL/KIF20A, a kinesin involved with membrane trafficking of discs large homologue 5, can attenuate growth of pancreatic cancer cell. Cancer Res, 2005. 65(1): p. 105-12.
133. Opdam, F.J., et al., The small GTPase Rab6B, a novel Rab6 subfamily member, is cell-type specifically expressed and localised to the Golgi apparatus. J Cell Sci, 2000. 113 ( Pt 15): p. 2725-35.
134. Barton, N.R. and L.S. Goldstein, Going mobile: microtubule motors and chromosome segregation. Proc Natl Acad Sci U S A, 1996. 93(5): p. 1735-42.
135. Goldstein, L.S. and A.V. Philp, The road less traveled: emerging principles of kinesin motor utilization. Annu Rev Cell Dev Biol, 1999. 15: p. 141-83.
136. Imai, K., et al., Identification of HLA-A2-restricted CTL epitopes of a novel tumour-associated antigen, KIF20A, overexpressed in pancreatic cancer. Br J Cancer, 2011. 104(2): p. 300-7.
137. Laemmli, U.K., Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 1970. 227(5259): p. 680-5.
138. Fire, A., et al., Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 1998. 391(6669): p. 806-11.
139. Jemal, A., et al., Global cancer statistics. CA Cancer J Clin, 2011. 61(2): p. 69-90. 140. Eloubeidi, M.A., et al., Prognostic factors for survival in pancreatic cancer: a
population-based study. Am J Surg, 2006. 192(3): p. 322-9. 141. Goonetilleke, K.S. and A.K. Siriwardena, Systematic review of carbohydrate antigen
(CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur J Surg Oncol, 2007. 33(3): p. 266-70.
142. Jemal, A., et al., Cancer statistics, 2007. CA Cancer J Clin, 2007. 57(1): p. 43-66. 143. Erkan, M., et al., The role of stroma in pancreatic cancer: diagnostic and therapeutic
implications. Nat Rev Gastroenterol Hepatol, 2012. 9(8): p. 454-67. 144. Hidalgo, M. and D.D. Von Hoff, Translational therapeutic opportunities in ductal
adenocarcinoma of the pancreas. Clin Cancer Res, 2012. 18(16): p. 4249-56. 145. Ho, J.R., et al., Deregulation of Rab and Rab effector genes in bladder cancer. PLoS
One, 2012. 7(6): p. e39469. 146. Kikuchi, T., et al., Expression profiles of non-small cell lung cancers on cDNA
microarrays: identification of genes for prediction of lymph-node metastasis and sensitivity to anti-cancer drugs. Oncogene, 2003. 22(14): p. 2192-205.
147. Seog, D.H., D.H. Lee, and S.K. Lee, Molecular motor proteins of the kinesin superfamily proteins (KIFs): structure, cargo and disease. J Korean Med Sci, 2004. 19(1): p. 1-7.
148. Ouyang, H., et al., Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am J Pathol, 2000. 157(5): p. 1623-31.

5 Literaturverzeichnis
94
149. Gasnereau, I., et al., KIF20A mRNA and its product MKlp2 are increased during hepatocyte proliferation and hepatocarcinogenesis. Am J Pathol, 2012. 180(1): p. 131-40.
150. Groth-Pedersen, L., et al., Identification of cytoskeleton-associated proteins essential for lysosomal stability and survival of human cancer cells. PLoS One, 2012. 7(10): p. e45381.
151. Neef, R., U. Gruneberg, and F.A. Barr, Assay and functional properties of Rabkinesin-6/Rab6-KIFL/MKlp2 in cytokinesis. Methods Enzymol, 2005. 403: p. 618-28.
152. Ridley, A.J., et al., Cell migration: integrating signals from front to back. Science, 2003. 302(5651): p. 1704-9.
153. Nagahara, M., et al., Kinesin 18A expression: clinical relevance to colorectal cancer progression. Int J Cancer, 2011. 129(11): p. 2543-52.
154. Yang, T., X.B. Zhang, and Z.M. Zheng, Suppression of KIF14 expression inhibits hepatocellular carcinoma progression and predicts favorable outcome. Cancer Sci, 2013.
155. Odrowaz, Z. and A.D. Sharrocks, The ETS Transcription Factors ELK1 and GABPA Regulate Different Gene Networks to Control MCF10A Breast Epithelial Cell Migration. PLoS One, 2012. 7(12): p. e49892.
156. Yan, G.R., et al., Genistein-induced mitotic arrest of gastric cancer cells by downregulating KIF20A, a proteomics study. Proteomics, 2012. 12(14): p. 2391-9.
157. Jordan, M.A. and L. Wilson, Microtubules as a target for anticancer drugs. Nat Rev Cancer, 2004. 4(4): p. 253-65.
158. Wilson, L., D. Panda, and M.A. Jordan, Modulation of microtubule dynamics by drugs: a paradigm for the actions of cellular regulators. Cell Struct Funct, 1999. 24(5): p. 329-35.
159. Orr, G.A., et al., Mechanisms of Taxol resistance related to microtubules. Oncogene, 2003. 22(47): p. 7280-95.
160. Gottesman, M.M. and I. Pastan, Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem, 1993. 62: p. 385-427.
161. Kavallaris, M., et al., Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest, 1997. 100(5): p. 1282-93.
162. Wang, Q., et al., Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J, 2005. 19(7): p. 869-71.
163. Cueille, N., et al., Microtubule-associated protein 1B binds glyceraldehyde-3-phosphate dehydrogenase. J Proteome Res, 2007. 6(7): p. 2640-7.
164. Durrieu, C., F. Bernier-Valentin, and B. Rousset, Microtubules bind glyceraldehyde 3-phosphate dehydrogenase and modulate its enzyme activity and quaternary structure. Arch Biochem Biophys, 1987. 252(1): p. 32-40.
165. Liu, M., et al., Validating the mitotic kinesin Eg5 as a therapeutic target in pancreatic cancer cells and tumor xenografts using a specific inhibitor. Biochem Pharmacol, 2008. 76(2): p. 169-78.
166. Kapoor, T.M., et al., Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol, 2000. 150(5): p. 975-88.
167. Basso, A.D., et al., SCH 2047069, a novel oral kinesin spindle protein inhibitor, shows single-agent antitumor activity and enhances the efficacy of chemotherapeutics. Mol Cancer Ther, 2010. 9(11): p. 2993-3002.
168. Infante, J.R., et al., A Phase I study to assess the safety, tolerability, and pharmacokinetics of AZD4877, an intravenous Eg5 inhibitor in patients with advanced solid tumors. Cancer Chemother Pharmacol, 2012. 69(1): p. 165-72.

5 Literaturverzeichnis
95
169. Khoury, H.J., et al., A phase 1 dose-escalation study of ARRY-520, a kinesin spindle protein inhibitor, in patients with advanced myeloid leukemias. Cancer, 2012. 118(14): p. 3556-64.
170. Kimura, H., et al., ALK fusion gene positive lung cancer and 3 cases treated with an inhibitor for ALK kinase activity. Lung Cancer, 2012. 75(1): p. 66-72.
171. Sun, D., et al., The expression of Eg5 predicts a poor outcome for patients with renal cell carcinoma. Med Oncol, 2013. 30(1): p. 476.
172. Asahara, S., et al., Phase I/II clinical trial using HLA-A24-restricted peptide vaccine derived from KIF20A for patients with advanced pancreatic cancer. J Transl Med, 2013. 11: p. 291.
173. Suzuki, N., et al., A phase I clinical trial of vaccination with KIF20A-derived peptide in combination with gemcitabine for patients with advanced pancreatic cancer. J Immunother, 2014. 37(1): p. 36-42.
174. Tomita, Y., et al., Identification of promiscuous KIF20A long peptides bearing both CD4+ and CD8+ T-cell epitopes: KIF20A-specific CD4+ T-cell immunity in patients with malignant tumor. Clin Cancer Res, 2013. 19(16): p. 4508-20.

6 Tabellenverzeichnis
96
6 Tabellenverzeichnis
Tab. 1: Übersicht der Kinesin Superfamilien
Tab. 2: Chemikalien und Reagenzien
Tab. 3: Geräte
Tab. 4: Verbrauchsmaterialien
Tab. 5: Puffer, Lösungen und Medien
Tab. 6: Zellkulturmedien und Zusätze
Tab. 7: Bakterien
Tab. 8: Zelllinien
Tab. 9: Eingesetzte Oligonukleotide
Tab. 10: Vektoren und siRNA
Tab. 11: Primäre Antikörper
Tab. 12: Sekundäre Antikörper
Tab. 13: Kits und DNA/Protein Leiter
Tab. 14: DNA und Protein Leiter
Tab. 15: Software
Tab. 16: Übersicht der verwendeten Zelllinien
Tab. 17: Übersicht der Fluoreszenzfarbstoffe
Tab. 18: Zusammensetzung eines 10%-igen SDS-Gels
Tab. 19: Übersicht der analysierten Patientenkohorte

7 Abbildungsverzeichnis
97
7 Abbildungsverzeichnis
Abb. 1: Schematische Darstellung des Pankreas.
Abb. 2: Tumorprogressionsmodell des duktalen Adenokarzinoms sowie damit verbundene genetische Aberrationen in zeitlicher Abfolge.
Abb. 3: Prozentualer Anteil der häufigsten Tumorlokalisationen aller Krebsneuerkrankungen in Deutschland 2010 [22].
Abb. 4: Vergleich der relativen 5-Jahres-Überlebensraten, nach Lokalisation und Geschlecht, Deutschland 2009 - 2010 (Periodenanalyse) [22].
Abb. 5: Chemische Struktur von Gemcitabine.
Abb. 6: Chemische Struktur von Paclitaxel (5β ,20-Epoxy-1,2α ,4,7β ,13α -hexahydroxytax-11-en-9-one4, 10-diacetate2-benzoate13-ester mit (2R,3S)-N-benzoyl-3-phenyllisoserine).
Abb. 7: Schematische Darstellung der drei „Prototypen“ von Motorproteinen.
Abb. 8: Übersicht der Kinesin Superfamilien und deren Struktur.
Abb. 9: Mechanismus der Kinesin Bewegung entlang von Mikrotubuli.
Abb. 10: Schematische Darstellung des Kinesin Motor Proteins Kif20a.
Abb. 11: A, Expressionsvektor pCMV6-AC-GFP und B, Kif20a Expressionsvektor; Firma Origene.
Abb. 12: Immunhistochemische Analysen von Kif20a in gesundem Pankreas.
Abb. 13: Immunhistochemische Analysen von Kif20a in chronischer Pankreatitis.
Abb. 14: Immunhistochemische Analysen von Kif20a in verschiedenen PanIN Läsionen des Pankreas.
Abb. 15: Immunhistochemische Analysen von Kif20a im Tumorgewebe des Pankreas.
Abb. 16: Agarosegel von extrahierter RNA aus Karzinomzelllinien.
Abb. 17: Kif20a Expression in PDAC- Zelllinien.
Abb. 18: Kif20a Expression in NET- Zelllinien.
Abb. 19: Immunfluoreszenz Analysen von Kif20a in Tumorzellen des Pankreas (SU86.86, Panc-1).
Abb. 20: Immunfluoreszenz Analysen von Kif20a in humanen immortalisierten duktalen Epithelzellen des Pankreas (HPDE).
Abb. 21: QRT-PCR Analysen von cDNA aus Tumorzellen des Pankreas nach Kif20a Suppression.

7 Abbildungsverzeichnis
98
Abb. 22: Immunblot Analysen von PDAC Zelllinien nach Kif20a Suppression.
Abb. 23: Proliferations-Analysen von Krebszellen nach Kif20a Runterregulierung (MTT-Assay).
Abb. 24: Migrations-Analysen von Krebszellen nach Kif20a Runterregulierung.
Abb. 25: Invasions-Analysen von Krebszellen nach Kif20a Runterregulierung.
Abb. 26: “Time-lapse” Mikroskopie (Richtungsabhängigkeit, Distanz, Geschwindigkeit).
Abb. 27: Immunfluoreszenz von Kif20a runterregulierten Panc-1 und SU86.86 Zellen.
Abb. 28: Immunfluoreszenz (DAPI, „Cleaved Caspase 3“) von Kif20a runterregulierten Panc-1 und SU86.86 Zellen.
Abb. 29: Zellzyklusanalysen von Panc-1 und SU86.86 Zellen nach Kif20a Runterregulierung.
Abb. 30: QRT-PCR Analysen von cDNA aus Tumorzellen des Pankreas nach Kif20a Überexprimierung.
Abb. 31: Immunblot Analysen von PDAC- Zelllinien (T3M4, SU86.86, Panc-1) nach Kif20a Überexprimierung.
Abb. 32: Phänotyp von PDAC- Zelllinien (T3M4, SU86.86, Panc-1) nach Kif20a Überexprimierung.
Abb. 33: Proliferations-Analysen von Krebszellen nach Kif20a Hochregulierung (MTT-Assay) .
Abb. 34: Migrations-Analysen von Krebszellen nach Kif20a Hochregulierung.
Abb. 35: Überexprimierung von Kif20a in immortalisierten humanen duktalen Epithelzellen (HPDE).
Abb. 36: Proliferations-Analysen von Krebszellen nach Behandlung mit Taxol (MTT-Assay).
Abb. 37: Immunblot Analysen von PDAC- Zelllinien nach Kif20a Suppression und gleichzeitiger Taxol Behandlung (2 µ M/ 20 µ M).
Abb. 38: Proliferations-Analysen von Krebszellen nach Behandlung mit Taxol und Kif20a siRNA (MTT-Assay).
Abb. 39: Migrations-Analysen von Krebszellen nach Kif20a Runterregulierung und gleichzeitiger Behandlung mit 2 µ M Taxol.
Abb. 40: Proliferations-Analysen von Krebszellen nach Behandlung mit Gemcitabine und Kif20a siRNA (MTT-Assay).
Abb. 41: Migrations-Analysen von Krebszellen nach Kif20a Runterregulierung und gleichzeitiger Behandlung mit 100 µ M Gemcitabine.
Abb. 42: Proliferations-Analysen von Krebszellen nach Behandlung mit Paprotrain (MTT-Assay)
Abb. 43: Kumulatives Überleben von Patienten bezü glich ihrer Kif20a Expression.
Fig. 44: Immunhistochemische Färbung von Tumorgewebe (Pankreas) mittels PCNA Antikörper.
Fig. 45: Semiquantitative Analyse von PCNA positiven Zellen im Tumorgewebe.

8 Danksagung
99
8 Danksagung
An dieser Stelle mö chte ich allen danken, die zum Gelingen der Arbeit beigetragen haben.
Die vorliegende Arbeit wurde am Klinikum rechts der Isar, der TU Mü nchen, in der Abteilung
Chirurgie (Pankreasforschungslabor) von Prof. Jö rg Kleeff in der Zeit vom 01.10.2009 bis
31.04.2014 unter Anleitung von PD. Erkan angefertigt.
Besonderer Dank gilt meinem Doktorvater PD. Dr. med. Mert Erkan sowie meinem
Arbeitsgruppenleiter Prof. Dr. med. Jö rg Kleeff. Auch PD. Dr. med. Christoph Michalski
mö chte ich danken fü r die jahrelange Unterstü tzung. Ihre ständige Diskussionsbereitschaft
und konstruktiven Arbeitsvorschläge haben mir während der Durchfü hrung meiner
Doktorarbeit stets wertvolle Hilfe geleistet.
Des Weiteren mö chte ich Prof. Dr. rer. nat. Bernhard Kü ster danken, fü r die Ü bernahme des
Zweitgutachtens und die wertvollen Tipps während des „Thesis Commitee Meetings“.
Fü r die technische Unterstü tzung und das tolle Arbeitsklima mö chte ich allen Kollegen aus
der AG Kleeff danken. Mit euch verging die Zeit (fast) wie im Flug!
Besonderer Dank gilt auch Carsten, Kathi, Manja, Simone, Susanne, Sylvia und Timo fü r die
jahrelange Unterstü tzung und schö nen Momente nicht nur im Labor, sondern auch im
Privaten.
Hiermit mö chte ich im Speziellen auch allen Danken, die nicht namentlich aufgezählt
wurden!
Mein grö ßter Dank gebü hrt meinen Eltern, Geschwistern, Felix und meinen Freunden, die
mich bei Hö hen und Tiefen immer begleitet und unterstü tzt haben.





![~ld~~Jil;i ,J,.L;~~J~~ t f ''J -c · Ahmed, nr. 3034) kıyyüddin el-Fasl'nin İbn Rafi'in eserin den seçerek meydana getirdiği el-Mün tel]abü '1-mul]tar el-mü~eyyel bihi 'ala](https://static.fdocuments.nl/doc/165x107/5f6827af11a2800f36032666/ldjili-jlj-t-f-j-c-ahmed-nr-3034-kyyddin-el-faslnin-bn.jpg)











![Kanter portfolio.PPT [Kompatibilitätsmodus] - Kanter Media ...Kanter Media - mehr Qualität in der Bildung 3 Bildungsinstitute S h d D l t h i tit t Mü hSprachen und Dolmetscherinstitut](https://static.fdocuments.nl/doc/165x107/6056ea3d803b870fd35ea4d1/kanter-kompatibilittsmodus-kanter-media-kanter-media-mehr-qualitt.jpg)