
Single-event microkinetische modellering van de ...
96
Faculteit Ingenieurswetenschappen Vakgroep Chemische Proceskunde en Technische Chemie Laboratorium voor Petrochemische Techniek Directeur: Prof. Dr. Ir. G. B. Marin Single-event microkinetische modellering van de hydrogenering van benzeen op platina Bart Matheeussen Promotor: Prof. Dr. Ir. G. B. Marin Prof. Dr. Ir. J. Thybaut Begeleider: Prof. Dr. Ir. J. Thybaut Master thesis ingediend tot het behalen van de graad van burgerlijk scheikundig ingenieur Aacademiejaar 2005-2006
Transcript of Single-event microkinetische modellering van de ...

Faculteit Ingenieurswetenschappen
Vakgroep Chemische Proceskunde en Technische Chemie
Laboratorium voor Petrochemische Techniek Directeur: Prof. Dr. Ir. G. B. Marin
Single-event microkinetische modellering van de hydrogenering van benzeen op platina
Bart Matheeussen
Promotor: Prof. Dr. Ir. G. B. Marin Prof. Dr. Ir. J. Thybaut Begeleider: Prof. Dr. Ir. J. Thybaut
Master thesis ingediend tot het behalen van de graad van burgerlijk scheikundig ingenieur
Aacademiejaar 2005-2006


Faculteit Ingenieurswetenschappen
Vakgroep Chemische Proceskunde en Technische Chemie
Laboratorium voor Petrochemische Techniek Directeur: Prof. Dr. Ir. G. B. Marin
Single-event microkinetische modellering van de hydrogenering van benzeen op platina
Bart Matheeussen
Promotor: Prof. Dr. Ir. G. B. Marin Prof. Dr. Ir. J. Thybaut Begeleider: Prof. Dr. Ir. J. Thybaut
Master thesis ingediend tot het behalen van de graad van burgerlijk scheikundig ingenieur
Aacademiejaar 2005-2006

Dankwoord In de eerste plaats wil ik graag professor Marin en professor Thybaut bedanken voor de kans
die ze mij gegeven hebben om deze master thesis tot stand te laten komen.
Heel veel dank aan professor Thybaut voor de begeleiding gedurende het jaar. De praktische
kennis en hulp is onmisbaar geweest.
Mijn medestudenten kan ik zeker niet vergeten in dit dankwoord. Meer specifiek wil ik Filip,
Tom en Wouter bedanken voor hun steun en relativeringsvermogen. Merci.
Tenslotte een dankwoord aan mijn ouders, zus en vriendin voor hun morele steun en
financiële bijdrage gedurende de opleiding.
Bart Matheeussen
Juni 2006

Opleidingscommissie Scheikunde
Verklaring in verband met de toegankelijkheid van de scriptie
Ondergetekende, Bart Matheeussen
afgestudeerd aan de UGent in het academiejaar 2005 - 2006 en auteur van de
scriptie met als titel:
Single-event microkinetische modellering van de hydrogenering van benzeen op platina
verklaart hierbij:
1. dat hij/zij geopteerd heeft voor de hierna aangestipte mogelijkheid in verband met
de consultatie van zijn/haar scriptie:
o de scriptie mag steeds ter beschikking gesteld worden van elke aanvrager
o de scriptie mag enkel ter beschikking gesteld worden met uitdrukkelijke,
schriftelijke goedkeuring van de auteur of de promotoren
o de scriptie mag ter beschikking gesteld worden van een aanvrager na een
wachttijd van jaar
o de scriptie mag nooit ter beschikking gesteld worden van een aanvrager
2. dat elke gebruiker te allen tijde gehouden is aan een correcte en volledige
bronverwijzing
Gent, (datum)
(Handtekening)
FACULTEIT INGENIEURSWETENSCHAPPEN
Chemische Proceskunde en Technische ChemieLaboratorium voor Petrochemische Techniek
Directeur: Prof. Dr. Ir. Guy B. Marin

Overzicht In hoofdstuk 1 wordt de relevantie van het schatten van modelparameters binnen het single-
event concept beschreven. Het hydrogeneren van benzeen op Pt kadert binnen het
hydrokraken. Voorafgaand aan de doelstellingen van het eindwerk wordt het hydrokraken en
relevante studies uit de literatuur kort toegedicht. De beschrijving van de experimentele
opstelling en de functies van de programmacode komt aan bod in hoofdstuk 2. De
experimentele dataset en het effect van de instelvariabelen op de reactiesnelheid wordt
weergegeven in hoofdstuk 3. In hoofdstuk 4 wordt uitgebreid ingegaan op het single-event
concept en hoe de resultaten van de kwantumchemische berekeningen in dit model kunnen
verwerkt worden. Door regressie van het SEMK model worden de modelparameters uit het
SEMK model geschat. De significantie en fysische relevantie van de modelparameters wordt
begroot in hoofdstuk 5. In het tweede deel van hoofdstuk 5 worden progressiemogelijkheden
tot verdere verfijning van het fundamenteel model uitgewerkt en geëvalueerd binnen het
single-event concept. Met een algemene conclusie van de resultaten en mogelijke
verbeteringen wordt in hoofdstuk 6 de thesis afgesloten.

Inhoudsopgave
Symbolenlijst Extended abstract Hoofdstuk 1 Inleiding 1
1.1 Hydrokraken 1
1.1.1 Kinetiek van de hydrogenering van tolueen op PT/ZSM-22 (Thybaut et al.,2002) 6
1.1.2 Kinetisch model voor de hydrogenering van tolueen gebaseerd op elementaire
principles (Saeys et al., 2005) 10
1.2 Doel eindwerk 14
Hoofdstuk 2 Procedures 16
2.1 Beschrijving van de experimentele opstelling 16
2.1.1 Voedingssectie 16
2.1.2 Reactiesectie 16
2.1.3 Uitlaatsectie 17
2.1.4 Analysesctie 18
2.2 Katalysator 21
2.3 Uitvoering van de experimenten 21
2.4 Berekeningsmethoden 22
2.4.1 Verwerking van de experimentele resultaten 22
2.4.2 Parameterschattingen en sequentieel experimenteel ontwerp 24
2.4.2.1 Reactorsimulatie - TWCFUN 24
2.4.2.2 Parameterschattingen - TWMARS 26
2.4.2.3 Sequentieel ontwerp van experimenten voor precieze
parameterschattingen - BLHSR 29
2.4.2.3.1 Minimumvolumecriterium 31
2.4.2.3.2 Vormcriterium 31

Inhoudsopgave
Hoofdstuk 3 Benzeenhydrogeneringsexperimenten op Pt/ZSM-22 33 3.1 Experimentele instelcondities 33
3.2 Effect van procesparameters op de hydrogenering van benzeen 33
3.2.1 Effect van de temperatuur 34
3.2.2 Effect van de waterstofpartieeldruk 36
3.2.3 Effect van de benzeenpartieeldruk 37
3.2.4 Effect van de totaaldruk 38
3.3 Besluit 39
Hoofdstuk 4 Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten 40
4.1 SEMK model voor de benzeenhydrogenering 42
4.2 Parameters in het SEMK model voor de hydrogenering van benzeen 45
4.2.1 Parameterschattingen voor de hydrogenering van benzeen 45
4.2.2 Thermodynamische consistentie 47
4.3 Reparameterisatie 48
Hoofdstuk 5 SEMK model regressie voor de hydrogenering van
benzeen op Pt/ZSM-22 51
5.1 Parameterschattingen 51
5.2 Modeladequaatheid en globale significantie van het SEMK model 57
5.3 Progressiemogelijkheden tot verdere verfijning van het huidige fundamenteel model
binnen het single-event concept 59
5.4 Semi-competitieve chemisorptie 61
5.5 Besluit 65
Hoofdstuk 6 Algemeen besluit 67
Appendix A: Transportlimitaties 69
Appendix B: Hydrogeneringsexperimenten op benzeen 72

Inhoudsopgave
Appendix C: Kalibrering van de massadebietregelaars van
waterstof en stikstof en inwendige standaard 73
Appendix D: Kalibratiefactoren van de gaschromatograaf 75
Referentielijst

Symbolenlijst
Romeinse symbolen * actief centrum op de katalysator
* radicalaire positie in de gasfase-equivalent van de geadsorbeerde molecule
A aromaat, piekoppervlakte in chromatogram, pre-exponentiële factor
A* gereparameteriseerde pre-exponentiële factor
AHn aromatische component met n H-addities
b modelparametervector
c0 concentratie van het aantal actieve centra mol m-3
c concentratie
ct totale concentratie van actieve centra
CF kalibreringsfactor
EA activeringsenergie J mol-1
∆ H enthalpieverschil J mol-1
F molair debiet mol s-1
F F-waarde voor de significantie van de regressie
h constante van Planck 6.62 10-34 J s
H waterstofatoom
H2 waterstofmolecule
i reactie-intermediair
J jacobiaan
k reactiesnelheidscoëfficiënt mol kgkat-1
s-1
KA chemisorptie-evenwichtscoëfficiënt van aromaat J/mol
kB constante van Boltzmann 1.83 10-23 J K-1
Ksurf oppervlakevenwichtscoëfficiënt J/mol
k0, K0 pre-exponentiële factor kgkat mol-1
mf massafractie mol kgkat-1
s-1
MW moleculair gewicht kg mol-1
n aantal experimenten
nc aantal koolstofatomen
ne aantal single-events, aantal herhalingsexperimenten

Symbolenlijst
pA partieeldrukvan component A, aantal parameters Pa
Q moleculaire partitiefunctie
R netto vomingsnelheid mol kgkat-1
s-1
R gasconstante 8.31 J mol-1 K-1
s standaardafwijking
∆ S entropieverschil J/mol
t student t-waarde voor de significantie van individuele parameter
T temperatuur K
V(b)i,j element van covariantiematrix op rij i en kolom j
W katalysatormassa kg
X conversie
y molfractie
Griekse symbolen α competitiefactor voor chemisorptie van een molecule
α Evans-Polanyi coëfficiënt
β echte paramatervector
θ bedekkingsgraad van het katalysatoroppervlak
ρi,j binaire correlatiecoëfficiënt tussen parameter i en j
σ symmetriegetal
Subscripts ≠ geactiveerd complex
_ vector
A aromaat, benzeen
act activering
AH6 gehydrogeneerd aromaat
B benzeen
ber berekend
chem chemisorptie
deh dehydrogenering
e uitlaat reactor
exp experimenteel
ext extern

Symbolenlijst
gem rekenkundig gemiddelde
glob globaal
int intern
HC koolwaterstoffen
hyd hydrogenering
r reactie
ref referentie
rot rotatie
surf oppervlak
t totaal
trans translatie
vib vibratie
Superscripts
A reactie-orde benzeen
App schijnbare
B reactie-orde waterstof Comp samengesteld
N aantal actieve centra betrokken bij snelheidsbepalende stap
N gemiddelde
~ uitlaat mixer
~ single-event
transitietoestand
^ modelberekend
0 inlaat reactor
0 aanduiding pre-exponentiële factor
Afkortingen BALc balans van de koolwaterstoffen
BALm totale balans
FID flame ionization detector
H waterstofatoom
H2 waterstofmolecule
HPLC high pressure liquid chromatography
≠

Symbolenlijst
LH Langmuir-Hinshelwood
LHHW Langmuir-Hinshelwood-Hougen-Watson
nob aantal uitgevoerde experimenten
SBS snelheidsbepalende stap
SEMK single-event micorkinetisch model

Single-event microkinetic modeling of the hydrogenation of benzene on Pt
Bart Matheeussen
Promoter(s): Prof. dr. ir. G.B. Marin, Prof. dr. ir. J. Thybaut
Abstract The study of the gas phase benzene hydrogenation kinetics over a 0.5 wt% Pt/ZSM-22 catalyst is based on an experimental database consisting of 43 measurements. A qualitive picture of the benzene hydrogenation over a Pt catalyst [1], based on quantumchemical calculations serves as the basis for the construction of a Single-Event MicroKinetics (SEMK) model. Single-event rate coefficients, k(m,n), are defined depending on the saturation state of the nearest neighbours (m) and the degreee of branching of the carbon atom to which the H-atom is added (n). The number of parameters is reduced to 7 due to the calculation of the pre-exponential factors ([1], [2]) and the thermodynamic consistency.The estimation of all parameters results in an F-value of 5 103. The coverage dependent chemisorption enthalpy of hydrogen amounts to -60.0 ± 11.3 kJ/mol and corresponds with a surface coverage of 25%. A value of -64.4 ± 6.4 kJ/mol for the chemisorption enthalpy of benzene is estimated. Under typical experimental conditions the surface coverages of all carbon species amounts to 60%. There is no dominant carbon species present on the catalyst surface. The remaining 15 % are free. Keywords hydrogenation, benzene, single-event
microkinetic modeling
I. INTRODUCTION The hydrogenation of benzene is studied as part
of the hydrocracking process. The hydrogenation or saturation of aromatic components is of increasing interest due to the more stringent environmental legislation. Moreover, the removal of aromatic components is benificial for the diesel’s quality as the cetane number increases with decreasing aromatic content.
The kinetic modeling of aromatic hydrogenation reactions based on steady-state data without in situ characterization of the surface cannot make use of direct information concerning the surface intermediates. This has lead to a variety of possible kinetic models presented for the hydrogenation of aromatic model components. However, recently a reaction path analysis for the catalyzed benzene hydrogenation based on quantumchemical calculations [1] has lead to the construction of more fundamental kinetic models ([2], [3]). Unlike in the previous Langmuir-Hinshelwood models ([2], [3]),
no dominant reaction pad is considered in the current kinetic model. This emplies that the entire network has to be considered. The benzene hydrogenation to cyclohexane was found to follow a Horiuti-Polanyi-type mechanism in wich hydrogen atoms add sequentially to the adsorbed benzene molecule.
A Single-Event MicroKinetic model (SEMK) has been constructed [4]. A judicious definition of the single-event hydrogenation rate coefficients is required to obtain reliable parameter estimates. The hydrogenation rate coefficient for an elementary step is written as the product of the number of single-events and the single-event reaction rate coefficients, which take into account the saturation state of the nearest neighbours (m= 0,1 or 2) and the degree of branching of the reacting C-atom (n=2). Microkinetic models constructed from the fundamental kinetics of elementary reaction steps offer the ability to cover a broader range of process conditions, along with improved accuracy.
II. EXPERIMENTAL RESULTS The experiments were performed in a gas phase
continuous stirred tank reactor at temperatures in the range of 423–498 K, H2 inlet partial pressures of 100–600 kPa and benzene inlet partial pressures of 10–60 kPa. The experiments were selected based on the criteria of sequential design. The effect of the operating conditions on the hydrogenation rate is consistent with the literature [2]. The benzene hydrogenation rate shows a maximum as function of the temperature. At a certain temperature, the increase of the hydrogenation rate coefficient with the temperature is overcompensated by the decrease of the hydrocarbons surface concentrations. The maximum shifts to higher temperatures if the total pressure is increased. A higher temperature is then necessary to decrease the hydrocarbons surface concentrations, more specifically, the one of benzene (Figure 1). The kinetic model simulates accurately the experimental trends. An increase in the hydrogen inlet partial pressure enhances the cyclohexane outlet flow rate. A n opposite effect is observed for the effect of the benzene inlet partial pressure. The single-event model provides

information on all hydrocarbons surface concentrations and that of hydrogen. The kinetic model simulates that the catalyst surface is practically saturated with the hydrocarbon species. At higher temperatures, the saturation of the metal surface by hydrocarbon species is no longer maintained. Sites that were previously occupied by benzene are now available for H2 chemisorption, explaining the competitive character of H2 and benzene chemisorption (section III).
0
5
10
15
20
25
30
400 420 440 460 480 500 520
temperature (K)
reac
tion
rate
(m
mol
kg k
at-1
s-1
)
sim 10 bar
sim 20 bar
sim 30 bar
Figure 1 reaction rate as function of the tempeture (xHC = 0.02, H2/HC = 5, W/F = 23.10 kgkat s mol-1), (●): p = 10 bar; (■): p = 20 bar; (▲): p = 30 bar
III. SINGLE-EVENT MICROKINETIC MODEL The following model assumptions for the
benzene hydrogenation are made: 1. competitive dissociative H2 and molecular
benzene chemisorption on identical sites 2. H2, benzene and cyclohexane chemisorption are
quasi-equilibrated 3. no dehydrogenated surface species are
considered [1] 4. no rate-determing step or dominant reaction
path is assumed 5. each reaction step of the netwerk is considered
to be reversibel 6. the steady state hypothesis is applied for the
partially hydrogenated surface intermediates
IV. SEMK MODELING RESULTS The parameter estimates are performed using a
Levenberg-Marquardt algorithm. The sum of squared residuals between the observed and calculated outlet cyclohexane flow rates is minimized by adjusting the model parameter vector b.
min)FF(SSQ b2cyclohex
nob
1jcyclohex →−=∑
=
(1)
Several sets of values for the pre-exponential factors were tested, each corresponding with different assumptions concerning the surface mobility. The parameter estimates and pre-exponential factors are shown in Table 1. The coverage dependent chemisorption enthalpy of hydrogen amounts to -60.0 ± 11.3 kJ/mol and is an intermediate value of the quantumchemical calculated range of 40–90 kJ/mol [1]. The
chemisorption enthalpy of benzene, -64.4 ± 6.4 kJ/mol, is in close agreement with the quantumchemical calculated value of -71 kJ/mol [1]. The chemisorption of cyclohexane, which is calculated via the thermodynamic constraint, is not physically relevant. However it confirms the fast and irreversible chemisorption of cyclohexane([2], [3]). The pre-exponential factor amounts to 1.2 10-
25 and the chemisorption enthalpy is slightly exotherm -4.7 kJ/mol.
Due to the difference in size of both reagens molecules a constant limited number of sites exists which are available for the dissociative H2 chemisorption. The semi competitive chemisorption is presented as a model hypothese to obtain a physically relevant value for the chemisorption coefficient of cyclohexane. Table 1 fixed and estimated parameter values with their 95% significanca interval
(0,2) (1,2) (2,2) C6H6 H2
EA (kJ/mol)
62.0 ± 2.6
48.1 ± 46.7
66.5 ± 3.3
∆H (kJ/mol)
2.3 ± 6.2 4.4 *
6.4 ± 7.2
-64.4 ± 6.4
-60.0 ± 11.3
A 1.0E+16 1.0E+16 1.0E+16
Exp(∆S/R) 1 1 * 1 1.0E-12 1.0E-11
The surface coverage under typical experimental
conditions with parameters as reported in Table 1 corresponds to 60% of carbon species, 25 % of hydrogen and the remaining part is free. The model simulations result in a surface coverage with no dominant carbon species. The contribution of each elementary hydrogenation stap is important. The catalyst coverage values are consistent with the other reported values in the literature [2].
05
101520253035404550
0 5 10 15 20 25 30 35 40 45 50Fexp (µmol s-1)
Fber (µmol s-1)
Figure 2 parity diagram for the cyclohexane outlet flow rate; line: experimental, dots: calculated based on the SEMK model with model parameters from Table 1
The good agreement between the calculated and the experimental cyclohexane outlet flow rates results in a F-value of 5 103 (Figure 2) for the global significance of the regression.

REFERENCES [1] M. Saeys, M.-F. Reyniers, G.B. Marin, J. Phys. Chem. B,
106, 7489-7498 (2002) Density Functional Study of benzene adsorption on Pt(111)
[2] J. Thybaut, M. Saeys en G.B. Marin, Chem. Eng. J., 90, 117-229 (2002) Hydrogenation kinetics of toluene on Pt/ZSM-22
[3] M. Saeys, M.-F. Reyniers, J. Thybaut, M. Neurock, G.B.
Marin, J. Catal., 236, 129-138 (2005) First-principles based kinetic model for the hydrogenation of toluene
[4] V. Vangenende, Laboratorium voor Petrochemische Techniek Universiteit Gent, 2003-2004 Development of a single-event model for the catalytic hydrogenation of aromatic compounds

1
Hoofdstuk 1 Inleiding
1.1 Hydrokraken
Ruwe aardolie wordt in een raffinaderij omgezet in bruikbare fracties. In een eerste stap wordt
aan de hand van een destillatie een scheiding in verscheidene fracties gerealiseerd, zie Figuur
1-1 ‘Atmospheric Distillation’. De lichtere fracties, voorgesteld door stromen 2 tot en met 6,
ondergaan nog enkele veredelingsprocessen alvorens als eindproduct aangewend te worden.
De zwaardere fracties, voorgesteld door stromen 7 tot en met 15, worden onderworpen aan
veredelings- en conversieprocessen waarbij deze stromen worden gekraakt tot lichtere
componenten. De steeds zwaardere voedingen die opgepompt en geraffineerd moeten worden
tonen het belang aan van het kraken als conversieproces.
Figuur 1-1 schematisch overzicht van raffinaderij

Inleiding
2
‘Hydrotreating’ en ‘hydroconversion’ zijn termen die gebruikt worden om verschillende
raffinageprocessen te beschrijven waarbij onzuiverheden worden verwijderd, het gemiddeld
moleculair gewicht van de voeding verlaagd wordt en/of de viscositeit van de zwaardere
fracties gereduceerd wordt in aanwezigheid van waterstof. ‘Hydrotreating’ en
‘hydroconversion’ zullen aan belang winnen ten gevolge van de uitputting van de lichtere
ruwe aardoliën en het bijgevolg steeds groter wordende aandeel van zware fracties in de te
behandelen ruwe oliën. Heden is hydrokraken in deze context wellicht de belangrijkste
technologie binnen deze types van processen. Een belangrijk kenmerk is dat simultaan met de
verlaging van het gemiddeld moleculair gewicht de H/C verhouding van de oliefracties
verhoogt. Dit leidt tot een betere kwaliteit van de verbrandingseigenschappen van de
dieselfracties geproduceerd via hydrokraken.
Het hydrokraken van zwaardere fracties werd een concurrentieel alternatief van het
katalytisch kraken op het ogenblik dat katalytische reforming zijn intrede deed. In een
katalytische reformer wordt immers waterstof, dat als reagens gebruikt wordt bij hydrokraken,
als nevenproduct gevormd. Beide kraakprocessen beogen een verhoging van de H/C
verhouding van de te behandelen fractie. Bij het katalytisch kraken is deze verhoging het
gevolg van het onttrekken van koolstofatomen aan de voeding. De cokesafzetting op de
katalysator leidt tot de vorming van onverzadigde, aromatische koolwaterstoffen. Dit heeft
een positief effect op de benzinekwaliteit. Bij het hydrokraken is de verhoging van de H/C
verhouding een gevolg van de sterk hydrogenerende reactiecondities. De kraking gebeurt in
aanwezigheid van waterstof en leidt tot verzadigde eindproducten. Het cetaangetal van diesel
wordt daarbij positief beïnvloed.
Het hydrokrakingsproces verloopt volgens een bifunctioneel reactiemechanisme dat
metallische en zure centra omvat, zie Figuur 1-2. Verzadigde reactanten fysisorberen in de
katalysatorporiën. Een dehydrogeneringsreactie treedt op na chemisorptie op het metallisch
centrum. Het gevormde alkeen verlaat het metallisch centrum en migreert naar een zuur
centrum waar het geprotoneerd wordt tot een carbenium ion. Deze carbenium ionen
ondergaan isomerisatiereacties, zoals alkylverschuivingen of PCP-vertakkingen, en
krakingsreacties (beta-scissies). Isomeren en gekraakte producten verlaten de zure centra. Na
migratie naar het metallisch centrum ondergaan de onverzadigde species een
hydrogeneringsreactie.

Inleiding
3
Figuur 1-2 bifunctioneel reactiemechanisme bij hydrokraken (Thybaut et al., 2002)
Moderne, industriële hydrokrakers zijn eenheden met een twee-stap-configuratie namelijk een
eerste stap waarin voornamelijk ontzwaveling, ontstikstoffing en hydrogenering van aromaten
optreedt op een sulfidekatalysator en een tweede stap waarin een doorgedreven hydrokraking
uitgevoerd wordt, in sommige gevallen zelfs op een edelmetaalkatalysator. De zwavel- en
stikstofverwijdering is in dit geval van groot belang wegens de gevoeligheid van de
edelmetaalkatalysatoren aan deze contaminanten. In aanwezigheid van waterstof worden
waterstofsulfide en ammoniak gevormd. Een typevoorbeeld van een twee-stap-configuratie
voor een industriële hydrokraker is weergegeven in Figuur 1-3. De potentie om beide stappen
individueel te sturen verhoogt de flexibiliteit en de mogelijkheid tot optimalisatie van het
volledige proces.
a) H2 verwarmer; b) reactor 1ste stap (hydrotreating); c) reactor 2de stap (hydrocracking); d) hoge druk scheiding;
e) H2 compressor; f ) lage druk scheiding; g) scheiding in fracties
Figuur 1-3 twee-stap-configuratie voor een industriële hydrokraker
gas phase
physisorption
(de)-hydrogenation
(de)-protonationalkyl-shift PCP-branching
ß-scission
zeolite
metal sites
acid sites
+
+
+
+
+
+
Inleiding
4
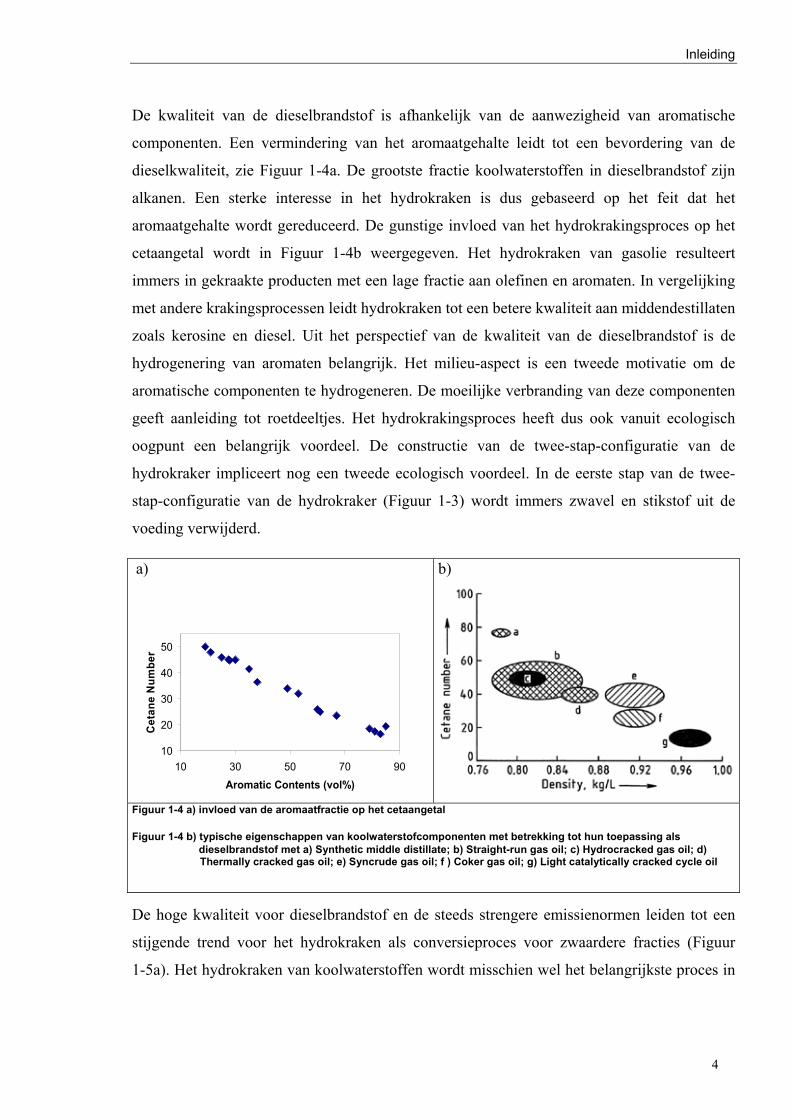
De kwaliteit van de dieselbrandstof is afhankelijk van de aanwezigheid van aromatische
componenten. Een vermindering van het aromaatgehalte leidt tot een bevordering van de
dieselkwaliteit, zie Figuur 1-4a. De grootste fractie koolwaterstoffen in dieselbrandstof zijn
alkanen. Een sterke interesse in het hydrokraken is dus gebaseerd op het feit dat het
aromaatgehalte wordt gereduceerd. De gunstige invloed van het hydrokrakingsproces op het
cetaangetal wordt in Figuur 1-4b weergegeven. Het hydrokraken van gasolie resulteert
immers in gekraakte producten met een lage fractie aan olefinen en aromaten. In vergelijking
met andere krakingsprocessen leidt hydrokraken tot een betere kwaliteit aan middendestillaten
zoals kerosine en diesel. Uit het perspectief van de kwaliteit van de dieselbrandstof is de
hydrogenering van aromaten belangrijk. Het milieu-aspect is een tweede motivatie om de
aromatische componenten te hydrogeneren. De moeilijke verbranding van deze componenten
geeft aanleiding tot roetdeeltjes. Het hydrokrakingsproces heeft dus ook vanuit ecologisch
oogpunt een belangrijk voordeel. De constructie van de twee-stap-configuratie van de
hydrokraker impliceert nog een tweede ecologisch voordeel. In de eerste stap van de twee-
stap-configuratie van de hydrokraker (Figuur 1-3) wordt immers zwavel en stikstof uit de
voeding verwijderd. a) b)
Figuur 1-4 a) invloed van de aromaatfractie op het cetaangetal Figuur 1-4 b) typische eigenschappen van koolwaterstofcomponenten met betrekking tot hun toepassing als
dieselbrandstof met a) Synthetic middle distillate; b) Straight-run gas oil; c) Hydrocracked gas oil; d) Thermally cracked gas oil; e) Syncrude gas oil; f ) Coker gas oil; g) Light catalytically cracked cycle oil
De hoge kwaliteit voor dieselbrandstof en de steeds strengere emissienormen leiden tot een
stijgende trend voor het hydrokraken als conversieproces voor zwaardere fracties (Figuur
1-5a). Het hydrokraken van koolwaterstoffen wordt misschien wel het belangrijkste proces in
10
20
30
40
50
10 30 50 70 90Aromatic Contents (vol%)
Cet
ane
Num
ber

Inleiding
5
de raffinage van petroleum. Omwille van deze voordelen wordt verwacht dat de interesse in
het hydrokraken in de toekomst nog zal toenemen (Figuur 1-5b).
a)
b)
Figuur 1-5 a) trend hydrokraken – katalytisch kraken Figuur 1-5 b) capaciteitsverandering in Europa (2005 – 2010); CDU: crude oil distillation unit; VDU: vacuum distillation unit; FCC: fluid catalytic cracking; RCC: residual catalytic cracking; HDC: hydrocracking
Kinetische modellen voor de katalytische hydrogenering van aromaten zijn uitvoerig
bestudeerd (Van Meerten et al., 1977; Rahaman en Vannice, 1991; Lin en Vannice, 1993;
Rousset et al., 2001; Thybaut et al., 2002; Salmi et al., 2004). Als modelcomponent wordt
vooral in minder recente studies gebruik gemaakt van benzeen. Tegenwoordig worden ook
andere modelcomponenten onderzocht zoals tolueen, xyleen isomeren, ethylbenzeen,
naftaleen. De aromaathydrogenering is voor een verscheidenheid aan metalen of
metaalcombinaties zoals Ni, Pt, Pd, PtPd, etc. geanalyseerd aangezien het effect van de
katalysator op de hydrogeneringssnelheid aanzienlijk kan zijn. De kinetische modellen
werden opgesteld op basis van data in stationaire toestand zonder in situ karakterisatie van het
oppervlak. De afwezigheid van informatie omtrent de oppervlakintermediairen heeft geleid tot
een waaier van mogelijke kinetische modellen. Veelal werd gebruik gemaakt van Langmuir-
Hinshelwood modellen. Verschillende modellen onderscheiden zich op basis van de gemaakte
onderstellingen. Lin en Vannice (1993) beschouwden 4 categorieën. Het onderscheid tussen
de verschillende categorieën was gebaseerd op de aanwezigheid van een snelheidsbepalende
stap en de atomaire H- of moleculaire H2-additie.
Gebaseerd op de kwantumchemische berekeningen werden fundamenteel kinetische modellen
geconstrueerd voor de hydrogenering van tolueen door Thybaut et al. (2002) en Saeys et al.

Inleiding
6
(2005). Het model van Thybaut et al. (2002) ziet af van de aanname van een
snelheidsbepalende stap, maar onderstelt identieke snelheidscoëfficiënten voor de eerste vier
H-atoomaddities en identieke chemisorptiecentra voor waterstofatomen en aromatische
koolwaterstoffen. Het in rekening brengen van de potentiële verschillen van de
snelheidscoëfficiënten voor de verschillende H-atoomadditities leidt tot een microkinetisch
model, zie verder. Een analoog Langmuir-Hinshelwood model wordt door Saeys et al. (2005)
opgesteld met de vijfde H-additie als snelheidsbepalende stap.
Een verschuiving van Langmuir-Hinshelwood modellen naar microkinetische modellen is
merkbaar. Microkinetische modellen werden reeds ontwikkeld door Verstraete (1997) en Van
Engelandt (1998), maar werden bij de uiteindelijke toepassing zodanig gereduceerd dat de
door deze auteurs gebruikte modellen voor hydrogenering equivalent waren met Langmuir-
Hinshelwood-Hougen-Watson modellen met een snelheidsbepalende stap. Voor de
hydrogenering van aromaten is er momenteel een vernieuwde interesse in de ontwikkeling en
toepassing van deze single-event microkinetische modellen. De voorgestelde single-event
methodiek (Vagenende, 2004) leidt tot de constructie van een gedetailleerd microkinetisch
model dat de oppervlakconcentraties van de verschillende koolwaterstofintermediairen in
rekening brengt. Het volledige reactienetwerk wordt in het SEMK model in rekening gebracht
waarbij de concentraties van de verschillende koolwaterstofintermediairen op het
katalysatoroppervlak worden bepaald via de pseudo-stationaire toestandshypothese. Deze
verfijning wordt verwacht de accuraatheid van het model te vergroten.
1.1.1 Kinetiek van de hydrogenering van tolueen op PT/ZSM-22 (Thybaut et al., 2002)
Kwantumchemische berekeningen (Saeys et al., 2002) bieden inzicht in de
hydrogeneringsreactie van aromaten. In combinatie met beschikbare literatuurgegevens van
de hydrogenering van aromatische componenten en met een fysicochemische studie van de
interactie van aromatische componenten en overeenkomstige gehydrogeneerde producten met
het metaaloppervlak is een alternatief model geconstrueerd (Figuur 1-6).

Inleiding
7
Figuur 1-6 enthalpieniveau’s van de componenten betrokken bij de hydrogenering van benzeen
Deze informatie geeft aanleiding tot een aantal hypothesen waarop het algemene model
steunt:
1. de eerste vier H-atoom addities zijn niet in quasi-evenwicht in tegenstelling
tot de vijfde en de zesde H-atoom additie
2. H2 (dissociatieve chemisorptie) en aromatische reactanten chemisorberen
competitief
3. (a) chemisorptie van H2 en aromatische reactanten is in quasi-evenwicht
(b) desorptie van gehydrogeneerd product is snel en irreversibel
4. geen gedehydrogeneerd species is op het oppervlak aanwezig
In het algemene model worden de reactiesnelheidscoëfficiënten van de eerste vier H-atoom
addities en de evenwichtscoëfficiënten van de vijfde en de zesde H-atoom additie aan elkaar
gelijkgesteld. Een limietgeval van dit algemeen model is een model met de vierde H-
atoomadditie als snelheidsbepalende stap. Voor het algemene model kan een expliciete
reactiesnelheidsvergelijking opgesteld worden.
2
23
23
AAHH
HHAA3
surft)g(AH
)1BBB()1B2B3B4(pK)pK1(
pKpKBkCR
22
22
6
+++
+++++
= ( 1-1)
met 22 HHsurf**Hsurf pKK/KB =θθ= ( 1-2)
Eén van de modelveronderstellingen steunt op de hypothese van competitieve chemisorptie.
De meeste andere modellen gaan echter uit van niet competitieve chemisorptie tussen

Inleiding
8
waterstof en de aromatische component. Algemeen kan gesteld worden dat repulsieve
interacties tussen twee gechemisorbeerde species op naburige actieve centra bestaan. Een
zekere vorm van competitie tussen het gechemisorbeerde waterstof en de aromatische species
kan optreden zelfs wanneer verschillende types van actieve centra betrokken zijn. Het
waarnemen van het optreden van competitieve of niet-competitieve chemisorptie blijkt
afhankelijk te zijn van de experimentele condities. Bij lage temperaturen is het
metaaloppervlak volledig verzadigd met aromatische species. Een beperkt aantal actieve
centra blijft beschikbaar voor de dissociatieve chemisorptie van waterstof. De
bedekkingsgraad van het katalysatoroppervlak door aromaten zal afnemen indien de
temperatuur stijgt. Bij een zekere temperatuur is het oppervlak niet langer volledig verzadigd
met aromatische species. Deze actieve centra worden eveneens beschikbaar voor waterstof.
Deze verschillende fenomenen kunnen het optreden van niet-competitieve chemisorptie bij
lagere temperaturen en van competitieve chemisorptie bij hogere temperaturen verklaren. De
‘transitietemperatuur’ wordt bepaald door de aangewende partieeldrukken. Nog hogere
temperaturen leiden tot lage bedekkingsgraden van zowel aromaten als waterstof en zorgen
ervoor dat geen competitieve effecten meer worden waargenomen tussen beide species.
In Figuur 1-7 wordt de reactiesnelheid voor de hydrogenering van tolueen weergegeven als
functie van de temperatuur. Er wordt een maximum waargenomen. Dit fenomeen is reeds
eerder waargenomen voor tolueen en andere aromatische componenten op typische
hydrogeneringsmetalen zoals Pt, Pd, Ni, etc (Lin en Vannice, 1993; Smeds et al., 1996;
Thybaut et al., 2002). Het wordt algemeen aangenomen dat de effecten van de
bedekkingsgraad aan de oorsprong liggen van het maximum. De stijging van de
reactiesnelheid wordt bij een bepaalde temperatuur overgecompenseerd door de daling van de
concentratie van de aromatische reactie-intermediairen.

Inleiding
9
Figuur 1-7 reactiesnelheid voor de hydrogenering van tolueen in functie van de temperatuur
Indien enkel de inlaatpartieeldruk van waterstof wordt verhoogd en alle andere experimentele
variabelen onveranderd blijven, stijgt de reactiesnelheid (Figuur 1-8).
Figuur 1-8 reactiesnelheid voor de hydrogenering van tolueen in functie van de inlaatpartieeldruk van waterstof
Een tegengesteld effect wordt waargenomen indien enkel de inlaatpartieeldruk van tolueen
wordt verhoogd, zie Figuur 1-9. Tolueen heeft een inhiberend effect op de reactiesnelheid.
Figuur 1-9 reactiesnelheid voor de hydrogenering van tolueen in functie van de inlaatpartieeldruk van tolueen

Inleiding
10
De parameters in het algemene model corresponderend met de beste regressie duiden aan dat
de koolwaterstofspecies relatief immobiel zijn op het katalysatoroppervlak in tegenstelling tot
de H-atomen die een hogere mobiliteit bezitten. Typische experimentele condities resulteren
in een oppervlak dat voor 60% bedekt wordt door tolueen en voor 20% door H-atomen. De
overige actieve centra zijn vrij. De chemisorptie–enthalpie van tolueen en H2 worden door
regressie van het algemeen kinetisch model significant geschat. De respectievelijke waarden
zijn -70 kJ/mol en -42 kJ/mol. Uit de geschatte waarde voor de samengestelde
activeringsenergie wordt een waarde voor de echte activeringsenergie verkregen in een gebied
van 40 – 50 kJ/mol. De waarden die door regressie van het model geschat worden zijn in
overeenstemming met de kwantumchemisch berekende waarden, zie paragraaf 1.1.2
1.1.2 Kinetisch model voor de hydrogenering van tolueen gebaseerd op elementaire principes (Saeys et al., 2005)
De constructie van dit fundamenteel kinetisch model voor de hydrogenering van tolueen op Pt
is gebaseerd op kwantumchemische berekeningen. De kwantumchemische berekeningen voor
de hydrogenering van benzeen suggereren dat het reactienetwerk is opgebouwd uit atomaire
waterstofaddities Bij de experimentele condities van de gebruikte dataset heeft de
dehydrogenering in dit reactienetwerk geen significante bijdrage zowel omwille van
thermodynamische als van kinetische redenen. Een ab initio analyse van het reactiepad voor
de hydrogenering van benzeen op Pt(111) resulteert in een dominant reactiepad. De
sequentiële additie van de waterstofatomen in de eerste drie reactiestappen van het dominant
reactiepad wordt telkens gerealiseerd in de meta-positie ten opzichte van elkaar. De
activeringsenergieën van de volgende reactiestappen zijn significant hoger omdat er geen
atomaire additie van waterstof meer mogelijk is in de meta-positie. Op basis van deze
kwantumchemische berekeningen wordt aangetoond dat gechemisorbeerd benzeen twee
stabiele configuraties heeft, namelijk de hollow- en bridgeconfiguratie (Figuur 1-10). Enkel
de hollow chemisorptieconfiguratie wordt in het model beschouwd aangezien dit de meer
reactieve gechemisorbeerde vorm is. Bovendien is dit de vorm waarin benzeenchemisorptie
preferentieel optreedt bij hogere bedekkingsgraden van het oppervlak door benzeen.

Inleiding
11
Figuur 1-10 illustratie van het geadsorbeerd benzeen op de Pt22 cluster; bridge configuratie (links) en hollow
configuratie (rechts)
Aangezien de hydrogenering van een oppervlakintermediair op een ‘hollow site’ kinetisch en
thermodynamisch met 20 kJ/mol of meer bevoordeeld is, wordt enkel het gehydrogeneerd
product van het geadsorbeerde benzeen op de hollow configuratie in het model in rekening
gebracht voor de additie van het tweede waterstofatoom. Saeys (2002) onderscheidt voor de
tweede waterstofadditiestap, namelijk deze aan het cyclohexadienyl species, vijf potentiële
reactiestappen. De mogelijke reactiepaden voor de tweede waterstofadditie en de
corresponderende activeringsenergieën en oppervlakreactie-enthalpieën worden weergegeven
in Figuur 1-11. Een verschil van 19 kJ/mol voor de activeringsenergie van het dominant pad
wordt berekend in vergelijking met alternatieve reacties.
Figuur 1-11 verschillende mogelijke reacties voor de tweede hydrogenatiestap, de activeringsenergieën en reactie-
enthalpieën zijn weergegeven in kJ/mol Een dominant reactiepad voor de volledige hydrogenering van de aromatische ring kan
voorgesteld worden aangezien de kwantumchemische berekeningen voor de volgende
waterstofaddities tot analoge bemerkingen leiden. Elke elementaire stap van het dominant
reactiepad heeft een activeringsenergie die minimaal 18 kJ/mol lager is dan een alternatieve
elementaire reactiestap. De reactiestap die het verst verwijderd is van het evenwicht is deze
voor de vorming van cyclohexyl met een berekende activeringsenergie van 104 kJ/mol

Inleiding
12
(Figuur 1-12). Uitgaande van zijn kwantumchemische berekeningen van de
activeringsenergieën is door Saeys et al. (2005) een model opgesteld. De
modelveronderstelling dat de vijfde waterstofadditie als snelheidsbepalende stap volgt
rechtstreeks uit de ab initio reactiepad analyse.
Figuur 1-12 overzicht van het reactienetwerk met de berekende activeringsenergieën (kJ/mol) voor het dominante reactiepad voor de hydrogenering benzeen in vet (Saeys et al.,2002) De activeringsenergieën, adsorptie- en reactie-enthalpieën in het model zijn
kwantumchemisch berekend. De pre-exponentiële factoren worden berekend op basis van
statistische thermodynamica, waarbij veronderstellingen zijn afgeleid gebaseerd op
kwantumchemische berekeningen (Saeys et al., 2002). De kwantumchemisch berekende
adsorptie-enthalpie voor cyclohexaan is -27 kJ/mol waardoor de desorptie van cyclohexaan
snel en irreversibel kan verondersteld worden. Experimenteel gerapporteerde waarden voor de
adsorptie-enthalpie van cyclohexaan bedragen ongeveer -58 kJ/mol (Bussel et al., 1992).
Echter de desorptie van cyclohexaan wordt nog steeds snel en irreversibel verondersteld
aangezien deze experimentele waarde laag is in vergelijking met de adsorptie-enthalpie van
benzeen.
In het kinetisch model is de waterstofchemisorptie-enthalpie een belangrijke parameter en de
enige die wordt geschat. De waterstofchemisorptie-enthalpie is immers sterk afhankelijk van
de bedekkingsgraad ten gevolge van repulsieve interacties. Bij een lage bedekkingsgraad
bedraagt deze -60 kJ/mol tot -90 kJ/mol. Deze waarde wordt sterk minder negatief, tot -40
kJ/mol, bij hogere bedekkingsgraden. Het belang van deze parameter wordt aangeduid in
Figuur 1-13. De reactie-enthalpie van de elementaire hydrogeneringsstappen wordt in sterke

Inleiding
13
mate beïnvloed door de waterstofchemisorptie-enthalpie. In deze figuur is de
activeringsenergie voor de hydrogenering onafhankelijk van de waterstofadsorptie-enthalpie.
In werkelijkheid resulteert, in overeenstemming met de Polanyi-Evans relatie, een lagere H-Pt
bindingsenergie corresponderend met een lagere chemisorptie-enthalpie van waterstof in een
lagere activeringsenergie voor de hydrogeneringsreactie.
Figuur 1-13 DFT berekend energie profiel voor dominant reactiepad in Figuur 1-12
( ,2H,adsH∆ = - 94.3 kJ/mol, ---,
2H,adsH∆ = - 45.0kJ/mol)
De kwantumchemische berekeningen vormen de basisonderstelling voor een Langmuir-
Hinshelwood-Hougen-Watson (LHHW) kinetisch model voor de katalytische hydrogenering
van tolueen op Pt. Het LHHW model wordt verondersteld om de experimentele data enkel
kwalitatief te reproduceren aangezien de modelveronderstellingen uitgaan van
vereenvoudigingen. Het model voor de katalytische hydrogenering van tolueen is immers
gebaseerd op de kwantumchemische berekeningen voor benzeen. De accuraatheid van de
modelhypothesen gebaseerd op ab initio reactiepad analyse kan getest worden.
1. competitieve dissociatieve chemisorptie van H2 en moleculaire chemisorptie van
aromaten op identieke chemisorptiecentra
2. desorptie van het gehydrogeneerde product is snel en irreversibel
3. dominant pad met de vijfde waterstofadditie snelheidsbepalend
4. stap 1 – 4 zijn in quasi -evenwicht
5. oppervlakconcentraties van de partieel gehydrogeneerde species worden verwaarloosd
Een LHHW snelheidsvergelijking wordt opgesteld:

Inleiding
14
( )2HHAA
25HA
25HA
4
1jj5t
22
22
pKpK1
ppKKKkC
r++
=
∏=
( 1-3)
Het belang van de waterstofchemisorptie-enthalpie in het kinetisch model is aanzienlijk
(Figuur 1-13) en wordt geschat op -54.0 ± 10 kJ/mol. De sterke afhankelijkheid van deze
parameter van de bedekkingsgraad resulteert in een intermediaire geschatte waarde voor de
waterstofchemisorptie-enthalpie die consistent is met een gemiddelde gesimuleerde
waterstofbedekkingsgraad van 61%.
1.2 Doel eindwerk
Met dit eindwerk wordt een stap verder gezet in de toepassing van een fundamenteel, single-
event microkinetisch model voor de hydrogenering van aromaten. Een model voor de
hydrogenering van aromaten is bijvoorbeeld een wezenlijk onderdeel in de modellering van
het hydrokrakingsproces, omwille van de sterke exothermiciteit van het proces dat tot lokale
temperatuursverhogingen kan leiden. In het verleden zijn deze hydrogeneringsreacties reeds
uitvoerig bestudeerd. Veel gebruikte modellen houden slechts in beperkte mate rekening met
de onderliggende chemie, wat hun bruikbaarheid beperkt. De ontwikkeling van fundamenteel
kinetische modellen die wel rekening houden met de onderliggende chemie, laat toe om
simulatiemodellen voor industriële eenheden te ontwikkelen die toepasbaar zijn in meer
uitgebreide werkingsgebieden.
Een analyse van het reactiepad voor de hydrogenering van benzeen op basis van
kwantumchemische berekeningen (Saeys, 2002) resulteert in de constructie van meer
fundamenteel kinetische modellen dan tot dan toe het geval was. In paragraaf 1.1.1 en 1.1.2
werden twee dergelijke fundamenteel kinetische modellen, ontwikkeld door respectievelijk
Thybaut et al. (2002) en Saeys et al. (2005), beschreven. De single-event methodologie voor
de hydrogenering van aromaten werd door Vagenende (2004) ontwikkeld. Dit model maakt
abstractie van een dominant reactiepad en beschouwt aldus elk mogelijk intermediair. Elke
elementaire reactiestap kan bijdragen tot de reactiesnelheid. De voorgestelde elementaire
stappen voor de hydrogenering van aromaten worden zodanig gedefinieerd dat de
overeenkomstige snelheidscoëfficiënten onafhankelijk zijn van de beschouwde reagentia.
Debruyne (2005) maakte gebruik van het SEMK model voor de katalytische hydrogenering

Inleiding
15
van mono-aromaten om parameters te schatten voor de benzeenhydrogenering. Niet alle
parameters uit het SEMK model konden significant geschat worden.
Dit eindwerk heeft als doel het stel experimentele gegevens van de hydrogenering van
benzeen op Pt/ZSM-22 zodanig uit te breiden dat de verschillende modelparameters op een
significante manier kunnen geschat worden. Daartoe wordt gebruik gemaakt van de
methodiek van de sequentiële planning van experimenten ter verkrijging van meer precieze
parameterschattingen. Indien de regressie van het model significant is, kan het stel van single-
event reactiesnelheidscoëfficiënten bepaald worden. Een fysische interpretatie volgt uit de
resultaten van het modelleringswerk. De nieuwe benzeengegevens kunnen getoetst worden
aan andere opgestelde modellen voor de hydrogenering van mono-aromaten door Thybaut et
al. (2002) en Saeys et al. (2005). Tenslotte kunnen uit de interpretatie van het
modelleringswerk suggesties voortkomen ter verbetering van het kinetisch model.

16
Hoofdstuk 2 Procedures
2.1 Beschrijving van de experimentele opstelling
2.1.1 Voedingssectie
De voeding bestaat uit benzeen (vloeibaar), waterstof (gas) en stikstof (gas), elk met hoge
zuiverheid. De debieten van de gasvormige voeding H2 en N2 wordt geregeld via twee
onafhankelijke thermische massadebietsregelaars (4). De kalibratiecurve van de regelaars
voor deze voedingscomponenten wordt in Appendix C weergegeven. Het waterstofdebiet
wordt ingesteld tussen 9.3 10-5 mol/s en 5.7 10-4 mol/s. Stikstof wordt toegevoegd om de
partieeldrukken van benzeen en waterstof zo te realiseren dat intrinsieke kinetiek kan gemeten
worden. De ingestelde stikstofdebieten bevinden zich in een bereik van 7.2 10-4 mol/s tot 5.2
10-3 mol/s. In de voorverwarmer/verdamper (6) wordt de gasstroom gemengd met vloeibaar
benzeen. Het vloeibare benzeen wordt vanuit een gesloten recipiënt (1) gevoed via een HPLC
– pomp (High Pressure Liquid Chromatography) (2) tussen de 1.9 10-5 en 5.6 10-5 mol/s. Een
elektronische balans voorziet de mogelijkheid om de correcte werking van de pomp te
verifiëren. In de voorverwarmer/verdamper zal het vloeibare benzeen verdampen en worden
de gasvormige reactanten opgewarmd tot de gewenste temperatuur. Het gasvormige
reactantenmengsel bereikt de reactor via een opening in de reactorwand net boven de
schoepen van de roerder (11), zie sectie 2.1.2. Een kleine fractie van de gasstroom, bestaande
uit stikstof en waterstof, stroomt rechtstreeks de reactor binnen via de magnetische roerder.
Deze stroom vermijdt lokale condensatie van koolwaterstoffen onderin de opstelling en voert
de warmte af die wordt veroorzaakt door de werking van de roerder.
2.1.2 Reactiesectie
De koolwaterstoffen worden gehydrogeneerd in een Berty – type reactor (8). Het volkomen
vermengde karakter van de reactor wordt gerealiseerd door een roerder (11). Het toerental van
de roerder bedraagt 1500 toeren per minuut en genereert een interne recycle stroom. De
aandrijving van de roerder is magnetisch waardoor dichtingsproblemen vermeden worden.

Procedures
17
Temperatuursgradiënten in de reactor zijn afwezig ten gevolge van de intense convectie. Een
CSTR massabalans voldoet als modelvergelijking voor een Berty reactor.
In deze gasfase volkomen vermengde reactor stromen de reactanten binnen ter hoogte van de
roerder. Het vooropgestelde stromingsprofiel voorziet dat de gassen opwaarts stromen tussen
de reactorwand (8) en het katalysatormandje (9) en neerwaarts over het katalysatorbed (10).
De stromingsweerstand over het katalysatorbed is minimaal. Het effluent verlaat de reactor
onder de schoepen van de roerder. Het katalysatorbed waarover de gassen stromen, is in lagen
opgebouwd. Op het draagnet van het katalysatormandje wordt een laag inerte sferen
aangebracht met intermediaire diameter. Een inerte laag met dezelfde diameter als de
katalysatorkorrels wordt afwisselend met een katalysatorlaag verdeeld over het
katalysatorbed. Vervolgens wordt een laag inerte sferen met intermediaire diameter en een
laag grote sferen verspreid over het katalysatorbed. Om de activiteit op lange termijn te
waarborgen wordt een termijn van 22 uur tussen de experimenten gerespecteerd. Een
waterstofstroom wordt over het katalysatorbed gestuurd met een debiet van 4.0 10-4 mol/s. In
de gasfase volkomen vermengde reactor wordt de druk ingesteld aan de hand van een
tegendrukregelaar (18). De druk in de reactor wordt gevarieerd tussen 10 en 30 bar. Via een
verwarmingskap (12) rond het reactorblok wordt de temperatuur geregeld in een bereik van
423K tot 498K. De reactor wordt voorzien van 2 thermokoppels (13) respectievelijk boven en
onder het katalysatormandje.
Door middel van het aanschroeven van 8 bouten wordt het reactordeksel aangebracht op het
reactorlichaam (8). De dichting wordt gerealiseerd aan de hand van een aluminium
dichtingsring. Aluminium is een ductiel materiaal waardoor het zich aanpast aan het te
dichten oppervlak bij het aanschroeven.
2.1.3 Uitlaatsectie
In de gasmengkamer wordt aan het reactoreffluent een inwendige standaard toegevoegd.
Methaan wordt gebruikt als inwendige standaard. In Appendix C wordt de kalibratiecurve van
deze gasstroom weergegeven. De inwendige structuur van de gasmenger bestaat uit zeven
schotten. Op deze manier wordt een goede menging gerealiseerd tussen de reactieproducten
en methaan. Het methaandebiet gelegen tussen 1.9 10-5 en 5.6 10-5 mol/s wordt ingesteld via
een thermische massadebietregelaar (5). De gasstroom die de gasmenger verlaat wordt
opgesplitst in 2 fracties. In een koeler/condensor (17) worden de aanwezige koolwaterstoffen

Procedures
18
gecondenseerd. De lichtere componenten stromen doorheen de tegendrukregelaar. Een
kleinere fractie stroomt doorheen de zeswegkraan (16). Oven en menger worden op
temperatuur gehouden om condensatie van koolwaterstoffen te vermijden.
2.1.4 Analysesctie
Via de zeswegkraan (6) kan een monster (20 µl) genomen worden van de reactieproducten en
de inwendige standaard. De samenstelling wordt geanalyseerd aan de hand van een
gaschromatograaf (Hewlett Packard Series II ) (21) met een FID detector (Flame Ionization
Detector). In de capillaire kolom met een 0.25 µm polydimethylsiloxaanfilm wordt de
scheiding gerealiseerd. De mogelijkheid om een betere scheiding te verwezenlijken door
cryogene koeling met vloeibare stikstof is aanwezig. De gaschromatograaf is uitgerust met
een kolom met een lengte van 50 m en een inwendige diameter 0.25 mm. De FID detector
analyseert het effluent. Het principe van een flame ionization detector is gebaseerd op de
productie van ionen in een vlam. De waterstof/lucht vlam wordt gestabiliseerd door het
toevoegen van stikstof. Een collector elektrode is gelocaliseerd boven de vlam. Het
resulterende signaal is een maat voor het aantal koolstofatomen dat per tijdseenheid op de
detector verschijnt. Het elektrisch signaal wordt versterkt en verwerkt door het
softwarepakket X-chrom. Deze techniek laat toe de meeste organische componenten te
detecteren.
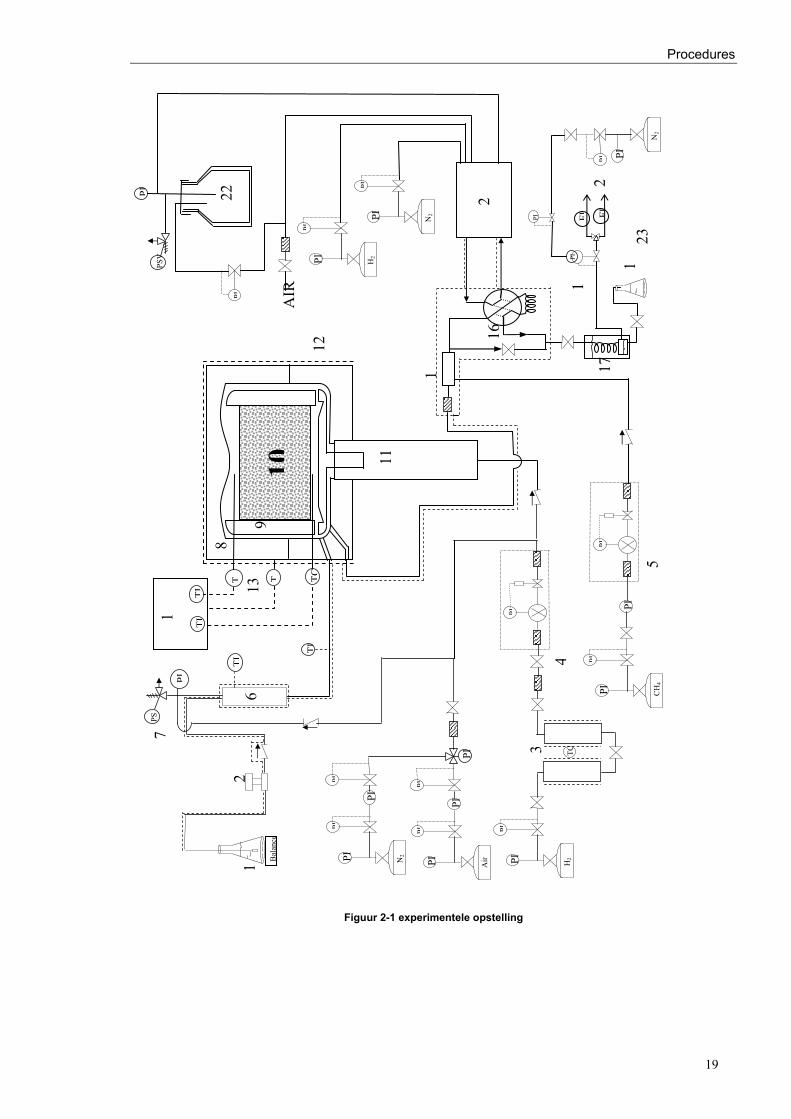
Procedures
19
Figuur 2-1 experimentele opstelling
PI
H
2 PI
PI
N
2 PI
PI
A
ir PI
PI
PI
PI PI
PI
PI
PI
C
H4
PI
PI
PI
PI
TI
TI
FIPI
N
2
PI
PI
H
2 PI
PI N
2 PI
PI
AIR
PI
PI
PSPS
V
TCT
TI
T
FI
PI
12
TC 3
4
5
6
7
8
9 10 11
12
13
1
1
16
17
1
1
2
222
TI
Bal
ance
23

Procedures
20
TC
PI Drukindicatie
TI Temperatuurindicatie
FI Debietindicatie
Afsluiter
PSV Veiligheidsklep Verwarmde leiding
Terugslagklep
Filter
Thermokoppel Controlelijn
Figuur 2-2 betekenis symbolen gebruikt bij Figuur 2-1
2.1.1 De voedingssectie 1 Recipiënt koolwaterstoffen en balans 2 Voedingspomp 3 Waterstofvoorbehandeling 4 Thermische massadebietregelaar H2 5 Thermische massadebietregelaar CH4 2.1.2 De reactiesectie 6 Verdamper/voorverwarmer 7 Veiligheidsklep 8 Reactorlichaam 9 Katalysatorhouder 10 Katalysatorbed 11 Magnetische roerder 12 Verwarmingskap 13 Thermokoppels 14 Temperatuurregelaar 2.1.3– 2.1.4 De uitlaat- en analysesectie
15 Gasmengkamer 16 Monsternamekraan (zeswegkraan) 17 Koeler/condensor 18 Tegendrukregelaar 19 Afscheider/collector - aftap vloeibaar effluent 20 Afvoer van gassen via natte gas debietmeters 21 Gaschromatograaf 22 Droging perslucht 23 Natte gas debietmeters
Figuur 2-3 legende bij Figuur 2-1

Procedures
21
2.2 Katalysator
De hydrogenering van de modelcomponent benzeen werd bestudeerd met een Pt/ZSM-22
katalysator. Pt (0.5 gew%) bevindt zich op het uitwendig oppervlak van het zeoliet. Wegens
de vormselectiviteit worden zuur gekatalyseerde volgreacties slechts in verwaarloosbare mate
waargenomen, namelijk <1%. De poedervormige katalysator wordt in tabletten geperst,
gebroken en gezeefd. Katalysatorkorrels met een diameter van 0.5 tot 0.7 mm worden
geselecteerd. Het droge gewicht van de katalysator ( ± 1.3 g) wordt exact bepaald door
weging. Alvorens de experimentele data uit te breiden wordt de poedervormige katalysator in
het reactorbed gereduceerd. De reductie wordt gerealiseerd bij 623 K en atmosfeerdruk
gedurende 4 uur.
2.3 Uitvoering van de experimenten
Bij aanvang van een set van experimenten wordt de Berty-reactor geladen met een gewogen
hoeveelheid katalysator ( ± 1.3 g). Het katalysatormandje wordt in de reactor geplaatst. De
temperatuurregeling wordt voorzien door 2 thermokoppels die worden aangebracht. De
reactor wordt gesloten door het kruislings aanschroeven van het reactordeksel op het
reactorlichaam. De katalysator wordt gedurende 4 uur op een temperatuur van 623 K
gereduceerd. Vervolgens wordt de reactor afgekoeld en de gewenste temperatuur wordt
ingesteld. De bouten van de reactor worden na afkoeling opnieuw aangeschroefd zodat de
reactor op druk kan worden gebracht. Deze procedure is éénmalig voor een hele reeks
experimenten tot de katalysatoractiviteit afneemt.
Elk experiment vereist het instellen van de gewenste druk, temperatuur en de debieten van de
verschillende componenten. De temperatuur wordt ingesteld via de 2 thermokoppels. Bij het
aanleggen van de verwarming van de reactor wordt de waterkoeling van de roerder aangezet.
Deze koeling verhindert dat de warmte van de reactor via conductie naar de roerder zou gaan
en tot oververhitting van de koolstoflagers zou leiden. Op deze manier wordt ook de
demagnetisering van de magnetische koppeling bij hoge temperatuur vermeden. Het
koelwater wordt verder in de opstelling gebruikt om het reactoreffluent te condenseren. De
druk in de reactor wordt bereikt via een tegendrukregelaar. De druk wordt verhoogd in
stappen van 5 bar tot de gewenste druk bereikt is. Hierbij wordt ook de druk op de gasflessen
geleidelijk verhoogd zodat het drukverschil tussen de fles en de reactor minstens 10 à 15 bar

Procedures
22
bedraagt. Bij een kleiner drukverschil is er geen gasstroom van de voedingsflessen naar de
reactor door de drukval over de leidingen en de debietmeters. Als de druk is opgebouwd,
worden de massadebietregelaars voor waterstof, stikstof en de inwendige standaard methaan
ingesteld zoals vereist voor het experiment. De magnetische roerder wordt in werking gesteld
(1500 omwentelingen per minuut). En het gewenste benzeendebiet wordt aan de hand van een
HPLC pomp ingesteld. Na enige tijd wordt de stabiliteit van het ingestelde debiet van benzeen
verzekerd (stabiel gebied: 0 – 25). Het debiet kan gecontroleerd worden aan de hand van een
balans. De stabilisatietijd van de opstelling na het instellen van de experimentele condities
bedraagt ongeveer 1 uur. Een monster van het experiment wordt genomen via een
zeswegkraan ongeveer 1u25 na het instellen van de experimentele condities. Als controle
wordt na 1u45 een tweede monster naar de GC gestuurd. Na het uitvoeren van het experiment
moet condensatie van benzeen in de installatie vermeden worden. De HPLC pomp van
benzeen wordt om die reden eerst uitgeschakeld. Gedurende 22 uur wordt een waterstroom
over de katalysator gestuurd om vermindering van de activiteit van de katalysator op langere
termijn te vermijden. De reactor wordt daarbij op een lichte overdruk van 2 – 4 bar gehouden.
De lekdichtheid van de installatie kan op deze manier gemakkelijk gecontroleerd worden.
2.4 Berekeningsmethoden
2.4.1 Verwerking van de experimentele resultaten
De samenstelling van het reactiemengsel wordt geanalyseerd aan de hand van een
gaschromatograaf met een FID detector. De integraal (V.s) van een piek in het chromatogram
is recht evenredig met de totaal geïnjecteerde stofhoeveelheid. De FID detector is echter niet
even gevoelig voor alle componenten. De relatieve piekoppervlakte van een bepaalde
component kan dus niet rechtstreeks gelijkgesteld worden aan de fractie van deze component.
Uit een mengsel met gekende samenstelling kunnen de kalibratiefactoren berekend worden.
De massafractie in het effluent wordt berekend uit onderstaande formule.
∑=
= n
1j)j(CF)j(A
)i(CF)i(A)i(mf
( 2-1)
De verschillende debieten in de experimentele opstellingen worden schematisch weergegeven
in Figuur 2-4.

Procedures
23
Figuur 2-4 definiëring van debieten in experimentele opstelling
De bepaling van het uitlaatdebiet wordt gerealiseerd door gebruik te maken van een
inwendige standaard. In de gasmenger wordt het methaan samengevoegd met het effluent dat
de reactor verlaat. Het gekende molaire debiet van methaan en de concentratie na de
gasmenger laten toe het molaire koolwaterstofdebiet te bepalen. De samenstelling van het
reactoreffluent wordt via de volgende vergelijking bepaald.
4
4
CH
0CHe
HC y~F
F~ = ( 2-2)
Het molair debiet van de verschillende koolstofcomponenten in het reactoreffluent wordt
berekend via:
eHCi
ei F~y~F~ = ( 2-3)
De koolstof- en massabalansen over de reactor kunnen met het gebruik van een inwendige
standaard uitgerekend worden. De verhouding van de debieten over de reactor wordt bepaald.
De controle van de koolstof- en massabalans laat toe de kwaliteit van het uitgevoerde
experiment na te gaan. Een afwijking van minder dan 5% wordt getolereerd. Het experiment
wordt opnieuw uitgevoerd indien de afwijking meer dan 5% bedraagt.
( )( ) %100
s/molinbietkoolstofdes/moluitbietkoolstofde(%)BALc ×= ( 2-4)
%100
yCF
yCFBALc
c
c
n
1i
0ii
0HC
n
1i
eii
eHC
×=
∑
∑
=
= ( 2-5)
( )( ) %100
s/kgetinlaatdebitotales/kgietuitlaatdebtotale(%)BALm ×= ( 2-6)
Reactor Mixer
0i
0HC y,F
0H2
F
ei
eH
eHC y,F,F
2
0CH4
F
ei
eH
eHC y~,F~,F~
2

Procedures
24
%100
yMWFFMW
yMWFFMW(%)BALm
c
22
c
22
n
1i
0ii
0HC
0HH
n
1i
eii
eHC
0HH
×
+
+
=
∑
∑
=
= ( 2-7)
De inwendige standaard wordt in BALc en BALm niet beschouwd. Een meer gevoelige test
wordt verkregen.
2.4.2 Parameterschattingen en sequentieel experimenteel ontwerp
Het gebruikte softwareprogramma bevat essentieel 2 subroutines, die op hun beurt gebruik
maken van onderliggende subroutines en functies. De subroutine TWMARS schat de
parameters via de Levenberg-Marquardt methode. Het sequentieel ontwerp voor preciezere
parameterschatting wordt gerealiseerd met behulp van de tweede subroutine BLHSR. De data
met de preliminaire experimenten en de beginschattingen van de parameters wordt ingelezen
uit het gegevensbestand ‘sdestm.da’. Het programma kan uitgevoerd worden in een
simulatiemode, waarbij enkel de dieperliggende subroutine TWCFUN aangeroepen wordt ter
simulatie van de experimentele gegevens met de huidige parameterwaarden. Daarnaast kan
het programma uitgevoerd worden in een parameterschattingsmode en planningsmode. De
communicatie van de resultaten van het programma naar de gebruiker toe verloopt via een
resultatenbestand ‘sdestm.li’. In het geval van parameterschattingen worden de resultaten van
de verschillende statistische toetsen, die het model en de modelparameters beoordelen,
weergegeven in dit bestand. Daarnaast bevat hetzelfde resultatenbestand in de planningsmode
ook het resultaat van het sequentieel ontwerp van experimenten. Het experiment dat het meest
bijdraagt tot meer precieze parameterschattingen op basis van het volume- en het
vormcriterium wordt voorgesteld.
2.4.2.1 Reactorsimulatie - TWCFUN
De experimentele instelcondities en de parameterwaarden van het model zijn de
noodzakelijke gegevens om de modelberekende afhankelijk variabele namelijk het
effluentdebiet van cyclohexaan te berekenen. De afhankelijke variabele wordt berekend aan
de hand van het reactormodel (CSTR) en het reactiemodel (single-event microkinetisch
model). De respectievelijke subroutines DNSQE en KINETICS worden aangeroepen in
TWCFUN. In DNSQE worden tevens algebraïsche vergelijkingen opgelost die opgesteld zijn
op basis van de pseudo-stationaire toestandshypothese. Deze leiden tot de benodigde

Procedures
25
oppervlakconcentraties van de reactie-intermediairen voor de berekening van de
reactiesnelheden.
Omwille van de afwezigheid van concentratie-, druk- en temperatuurgradiënten kan de Berty-
reactor gemodelleerd worden als een continue reactor met volkomen vermenging, zoals eerder
vermeld. De accumulatieterm in de CSTR-balans voor cyclohexaan kan geëlimineerd worden
ten gevolge van de stationaire toestand. Er wordt geen cyclohexaan gevoed zodat geldt:
WRF cyclohexcyclohex = ( 2-8)
Het uitlaatdebiet wordt iteratief berekend zodanig dat aan de molaire balans wordt voldaan.
In KINETICS wordt de hydrogeneringskinetiek berekend waarbij elke elementaire stap in het
reactienetwerk reversibel is. De netto-productiesnelheid van de verschillende componenten
wordt berekend. Zoals eerder vermeld, worden de concentraties van partieel gehydrogeneerde
intermediairen verkregen via de pseudostationaire toestandshypothese. De chemisorptie van
waterstof, benzeen en cyclohexaan op het katalysatoroppervlak wordt in quasi-evenwicht
verondersteld.
H2 (g) + 2∗ 2H∗ 2H,chemK =
2H2*
2H
pcc ∗ ( 2-9)
C6H6 (g) + ∗ C6H6∗ 66HC,chemK =
66
66
HC
*HC
pc
c
∗
( 2-10)
C6H6 (g) + ∗ C6H6∗ 126HC,chemK =
126
126
HC
*HC
pc
c
∗
( 2-11)
∑=
++++=12
2i*i*HC*HC*H*t CCCCCC
12666 ( 2-12)
Dit leidt tot de berekening van de concentraties op het oppervlak van benzeen, waterstof,
cyclohexaan en de vrije centra. Het single-event model voor hydrogenering geeft dus inzicht
in de concentraties van de verschillende koolwaterstoffen op het oppervlak. De
partieeldrukken in de bovenstaande vergelijkingen volgen uit de kennis van het berekende
uitlaatdebiet van een cyclohexaan. Het uitlaatdebiet van cyclohexaan volstaat om via atomaire
balansen het uitlaatdebiet van benzeen en waterstof te kennen. De reactiesnelheid is dus
functie van de temperatuur, de totaaldruk en het berekende uitlaatdebiet van cyclohexaan.

Procedures
26
2.4.2.2 Parameterschattingen - TWMARS
De routine TWMARS schat de modelparameters. De afhankelijke variabele nodig voor het
kleinste kwadratencriterium wordt berekend door TWCFUN. TWCFUN is de
simulatieroutine in het programma en wordt aangeroepen als onderdeel van TWMARS. Het
single-event microkinetisch model is een niet-lineair model in de parameters. Algemeen kan
een niet lineair model voorgesteld worden door:
iii ),x(fy ε+β= ( 2-13)
De modelparameters worden geschat door het minimaliseren van het kleinste
kwadratencriterium.
[ ]∑=
→β−=βn
1i
2ii min),x(fy)(S ( 2-14)
Specifiek in dit eindwerk is de doelfunctie opgebouwd uit de residuele kwadratensom over
alle i experimenten van het experimenteel waargenomen en het modelberekende uitlaatdebiet
van cyclohexaan.
( ) MinFF bnob
1i
2ii →−∑
=
( 2-15)
De minimalisatie van de kwadratensom leidt tot de modelparametervector b die de echte
parametervector β benadert. Het oplossen van een stelsel niet lineaire normaalvergelijkingen
stuit op numerische problemen. De Levenberg-Marquardt techniek (Marquardt, 1963) is
daarentegen gebaseerd op de linearisatie van de modelvergelijking met betrekking tot de
parameters aan de hand van een Taylorreeksontwikkeling rond de preliminaire schattingen
0b . De resulterende set van waarnemingsvergelijkingen is lineair voor alle jb∆ :
j
bβ
p
1j j
i0ii b∆.
β
)β,x(f)b,x(f)β,x(f
0==∑ ∂
∂+≅ ( 2-16)
Statistische testen zijn uitgewerkt voor modellen lineair in de parameters op basis van de
normaalverdeling van de experimentele fout. Aan de hand van een Taylorreeksontwikkeling
rond de geconvergeerde parameterschattingen worden de statistische testen ook toegepast op
modellen die niet lineair zijn in de parameters.
Om de kwaliteit van het model en de modelparameters te evalueren wordt gebruik gemaakt
van verscheidene statistische testen. De modeladequaatheid wordt getest op basis van de ‘lack
of fit’ kwadratensom en de ‘pure error’ kwadratensom. Een schatting van de experimentele
fout wordt berekend na het uitvoeren van ne herhalingsexperimenten.

Procedures
27
1n
)yy(
se
2n
1jj
2e
e
−
−
=∑= ( 2-17)
met y het gemiddelde van de waargenomen waarden tijdens de herhalingsexperimenten.
De F-test voor de modeladequaatheid is gebaseerd op de volgende verhouding Fc, die wordt
vergeleken met de corresponderende getabelleerde waarde:
)α1;1n,1n-p-F(n~
1n
)yy(
1npn
)yy()yy(
F ee
e
2n
1ii
e
n
1i
2n
1ii
2ii
ce
e
−−+
−
−
+−−
−−−
=
∑
∑ ∑
=
= =
( 2-18)
Indien de berekende F-waarde de getabelleerde waarde overschrijdt dan is het model
inadequaat met een probabiliteit van α−1 wegens een tekort aan aanpassingsvermogen.
Een andere F-test voor de significantie van de regressie kan toegepast worden en gaat na of
niet alle parameters simultaan gelijk aan nul kunnen zijn. De verhouding van de regressie
kwadratensom en de residuele kwadratensom is een naar F(p,n-p) verdeelde veranderlijke.
)α1; p-n,F(p~
pn
)yy(
p
y
F2
n
1iii
n
1i
2i
c −
−
−
=
∑
∑
=
=
( 2-19)
Een berekende waarde die groter is dan )α1; p-n,F(p − impliceert dat de regressie significant
is. Een berekende F-waarde die beduidend groter is dan de getabelleerde F-waarde komt
overeen met een sterke verwerping van de nulhypothese. De regressie is significant of m.a.w.
het model is in staat om een groot deel van de totale kwadratensom te verklaren.
De significantie van de individuele parameterschattingen kan ook onderworpen worden aan
een statistische analyse. Aangezien )b( jσ onbekend is word de standaardafwijking geschat:
[ ] j,jj )b(V)b(s = ( 2-20)
met de covariantiematrixpn)b(S
.)JJ()b(V 1T
−= − ( 2-21)

Procedures
28
Via een t distributie wordt op basis van een tweezijdige t-test geverifieerd of de
parameterschatting jb significant verschilt van nul.
)2
1; p-nt(~)b(s
0bt
j
jc
α−
−= ( 2-22)
Indien de getabelleerde waarde )2
1; p-nt( α− kleiner is dan de berekende waarde dan wordt
hypothese dat jβ gelijk is aan nul verworpen. Uit de bovenstaande formule kunnen de
individuele betrouwbaarheidsintervallen berekend worden. De waarden binnen de limieten
van het betrouwbaarheidsinterval wijken niet significant af van de optimale
parameterschatting jb met een probabiliteit van α−1 .
)b(s)2α1;pn(tbβ)b(s)
2α1;pn(tb jjjjj −−+≤≤−−− ( 2-23)
De correlatie tussen de geschatte parameterwaarden is gebaseerd op de binaire lineaire
correlatiecoëfficiënten: ( )
( ) ( ) j,ji,i
j,ij,i
bVbV
bVρ = ( 2-24)
De correlatiecoëfficiënt geeft de lineaire afhankelijkheid weer tussen 2 parameters. Een hoge
binaire correlatiecoëfficient tussen 2 parameters betekent dat deze door 1 parameter kan
vervangen worden. Het omgekeerde is niet noodzakelijk waar. Een lage binaire
correlatiecoëfficient betekent niet dat de 2 parameters noodzakelijk zijn in het model. Dit is
enkel af te leiden uit de significantie van de individuele parameters. Absolute waarden van j,iρ
die 1 benaderen, wijzen op een sterk lineair verband tussen de geschatte waarden voor de
corresponderende parameters i en j. De waarden van de parameter i bepalen de waarde van
parameter j, evenredig als j,iρ ~ 1 of omgekeerd evenredig als j,iρ ~ -1.
Het gezamenlijke betrouwbaarheidsinterval wordt verkregen door het simultaan variëren van
alle parameters. Alle parameterwaarden β die voldoen aan de volgende vergelijking:
−−
−+= )α1;pn,p(F
pnp1).b(S)β(S ( 2-25)
begrenzen het oppervlak waarbinnen alle parametercombinaties niet significant verschillen
van de optimale parameterschattingen b met een probabiliteit van α−1 .

Procedures
29
2.4.2.3 Sequentieel ontwerp van experimenten voor precieze parameterschattingen - BLHSR
Het programma sdestm.f (Hosten,1974) werd door Debruyne (2005) aangepast tot
sdestm_single_event.f om het sequentieel ontwerp voor meer preciese parameterschattingen
toe te laten. De eerste stap in het sequentieel ontwerp is het vastleggen van het experimenteel
gebied van de onafhankelijke variabelen waarin intrinsieke kinetiek (Appendix A) kan
gemeten worden, met een bijhorend rooster met mogelijke instelcondities, zie paragraaf 3.1.
Ieder knooppunt in het rooster correspondeert met een mogelijk uit te voeren experiment. De
mogelijke experimenten worden ingelezen in TWGRID. In de subroutine BLHSR worden de
roosterpunten getoetst aan het minimumvolume- en het vormcriterium. Om het effect van de
procesparameters (paragraaf 3.2) op de reactiesnelheid vast te leggen is er een beperkte keuze
aan experimenten mogelijk. De selectie tussen de experimenten gebeurt op basis van het
sequentieel ontwerp. Het geselecteerde criterium evalueert ieder knooppunt. De
instelcondities van het volgende experiment corresponderen met het roosterpunt dat
aanleiding geeft tot de optimale waarde van het criterium voor het sequentieel ontwerp voor
precieze parameterschattingen. De meest nauwkeurige parameterschattingen worden
verkregen door experimenten te selecteren in de gebieden waar het model de grootste
onzekerheid vertoont. Deze gebieden corresponderen meestal met de randen van het
experimenteel gebied om zo systematisch het gezamenlijk betrouwbaarheidsgebied van de
parameters te verkleinen (Atkinson en Hunter, 1968).
Meer precieze parameterschattingen zijn het resultaat van het selecteren van experimentele
instelcondities op basis van geactualiseerde informatie. De grootheid waarop gesteund wordt
om de experimenten te selecteren, is de covariantiematrix van de parameterschattingen:
( ) ( ) 21T σXXbV−
= ( 2-26)
In lineaire modellen is de covariantiematrix ( )bV slechts afhankelijk van de foutvariantie 2σ
en van de matrix X , namelijk de instelwaarden van de onafhankelijk variabelen. De
waarnemingsuitslagen van de afhankelijk veranderlijke hebben geen invloed. Voor een niet-
lineair model wordt de covariantiematrix benaderd gegeven door ( ) 21T JJ σ− waarin J de
matrix is met de partiële afgeleiden van de antwoordvergelijking naar de parameters,
genomen bij de waarden die de kwadratensom van de residuelen minimaliseren. Hier spelen
de waarnemingsuitslagen wel een zekere rol aangezien ze de parameterschattingen

Procedures
30
beïnvloeden en bijgevolg ook de elementen van J . De invloed is echter slechts indirect en in
vele gevallen zijn de criteria weinig sensitief ten opzichte van de parameterwaarden althans
binnen bepaalde grenzen. Deze onzekerheid wordt op een optimale manier behandeld door
een volledig sequentieel ontwerp. Een maximale benutting van de aanwezige experimentele
informatie bij het sequentieel ontwerp voor meer precieze parameterschattingen wordt
gerealiseerd door na ieder experiment alle informatie opnieuw te analyseren. In de betrekking
(2.26) is de experimentele fout 2σ niet onder controle van de experimenteerder. In
tegenstelling tot de instelwaarden van de onafhankelijke veranderlijke die vrij te kiezen zijn
binnen bepaalde grenzen. De precisiematrix met betrekking tot de parameterschattingen is
gekend alvorens de experimenten werkelijk worden uitgevoerd. In de precisiematrix ( ) 1T XX−
is alle informatie met betrekking tot de betrouwbaarheid van de parameterschattingen vervat.
Een particulier stel van experimenten wordt aan de hand van criteria gebaseerd op de
precisiematrix ( ) 1T XX− geselecteerd om de parameters met de grootst mogelijke
betrouwbaarheid te schatten.
Op basis van het gezamenlijk betrouwbaarheidsgebied van de parameters worden de twee
voornaamste globale criteria afgeleid. Deze criteria kunnen betrekking hebben op een
bepaalde parameter of parametergroep. Het principe van het sequentieel ontwerp van
parameterschattingen is dus gebaseerd op het effect van mogelijke experimenten van het
experimenteel rooster op de ( ) 1T XX− matrix. De precisiematrix zit immers vervat in de formule
voor het gezamenlijk betrouwbaarheidsgebied β . Een globaal criterium voor het
experimenteel ontwerp van experimenten met betrekking tot meer precieze
parameterschattingen is het verkleinen van het gezamenlijk betrouwbaarheidsinterval. Dit
gebied beschrijft immers de variabiliteit van alle parameters simultaan. De vergelijking van
dit betrouwbaarheidsgebied β is gegeven door:
( ) ( ) δ=−β−β bXXb TT ( 2-27)
De precisiematrix is een positief definiete matrix zodat het linkerlid in de bovenstaande
vergelijking een hyperellipsoïde voorstelt. Door een orthogonale transformatie wordt de
standaardvergelijking verkregen van een hyperellipsoïde in p dimensies:
( ) ( ) ( )
1zzz
2
p
p
2
2
2
2
1
1 =
λδ++
λδ+
λδK ( 2-28)

Procedures
31
waarbij iλ de eigenwaarde van XXT is en λδ de lengte van een halve as is.
2β
b1 1β
b2
vorm criterium
volume criterium it i
gebied voor optimalisatie
Figuur 2-5 gezamenlijk betrouwbaarheidsgebied van de parameterschattingen met betrekking tot de globale
experimentele ontwerpcriteria voor preciezere parameterschattingen
2.4.2.3.1 Minimumvolumecriterium
Het principe van het minimumvolumecriterium is gebaseerd op het selecteren van de
experimentele voorwaarden die het volume van de betrouwbaarheidsellipsoïde minimaliseren.
∏=
≈=p
1kTk
XXdet
1λδ.constVol ( 2-29)
Het volume is omgekeerd evenredig met XXdet T . De experimentele voorwaarden worden
bijgevolg zo gekozen dat XXdet T gemaximaliseerd wordt en het volume van de
betrouwbaarheidsellipsoïde geminimaliseerd wordt.
2.4.2.3.2 Vormcriterium
Het vormcriterium is een interessant alternatief wegens het minimaliseren van de correlaties
tussen de parameters. De betrouwbaarheidsellipsoïde wordt zo bolvormig mogelijk gemaakt
zonder expliciet rekening te houden met het volume van het betrouwbaarheidsgebied.
Aangezien het voornamelijk de langste as van de ellipsoïde is, die verantwoordelijk is voor de
grootste afwijking t.o.v. de sfeer, dient deze langste as zoveel mogelijk verkort te worden. Uit
vergelijking (2-28) volgt dat de lengte van de halve assen omgekeerd evenredig is met de
wortel van de eigenwaarden van XXT . Het criterium selecteert nu de experimentele
combinaties die de kleinste eigenwaarde van XXT maximaliseren. Dit criterium garandeert
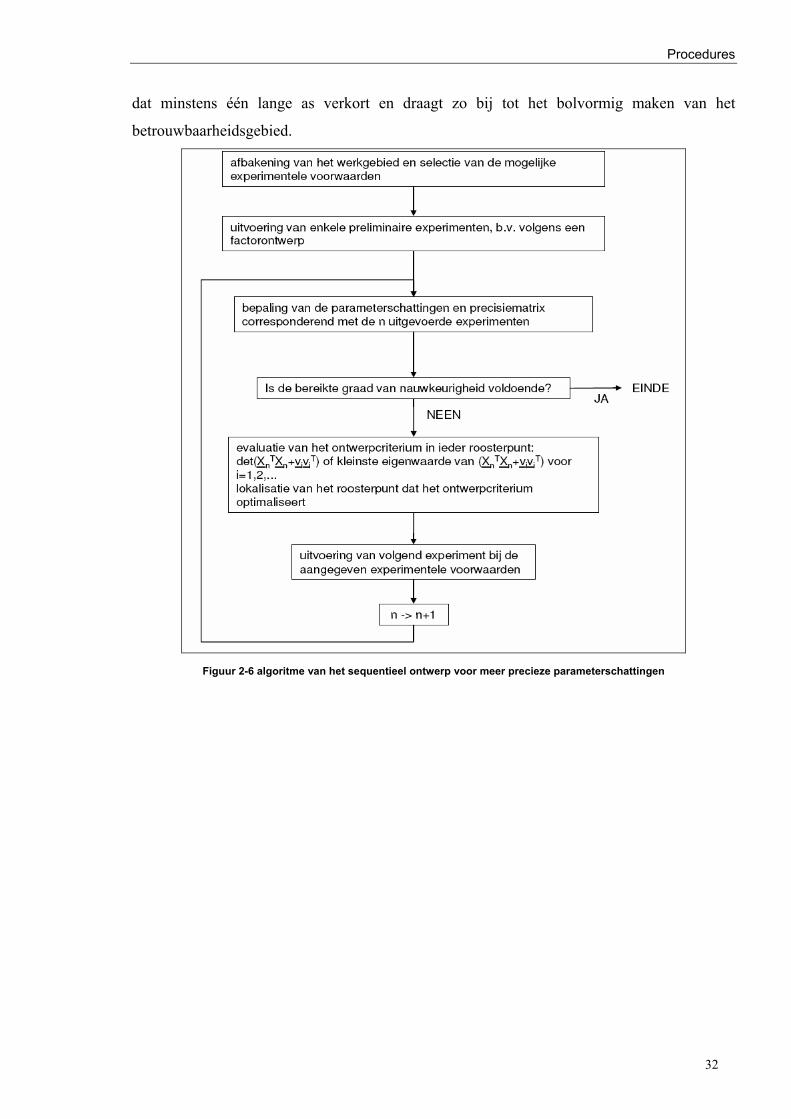
Procedures
32
dat minstens één lange as verkort en draagt zo bij tot het bolvormig maken van het
betrouwbaarheidsgebied.
Figuur 2-6 algoritme van het sequentieel ontwerp voor meer precieze parameterschattingen

33
Hoofdstuk 3 Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
3.1 Experimentele instelcondities
Verscheidene instelvariabelen worden gevarieerd om hun effect op de reactiesnelheid te
begroten. Het bereik van experimentele instelcondities wordt weergegeven in Tabel 3-1. De
experimenteel verkregen conversies en reactiesnelheden met de bijhorende instelcondities
worden weergegeven in appendix B. Tabel 3-1 experimentele instelcondities van uitgevoerde experimenten
ptot (bar) T (K) W/FHC,0 (kgkat s mol-1)66HCp (kPa)
2Hp (kPa)
10 - 30 423 - 498 23.0 – 69.2 10-60 100 – 600
De intrinsieke kinetiek van de hydrogenering van benzeen wordt onderzocht in het
experimentele gebied met afwezigheid van diffusielimitaties, zie Appendix A. De molfractie
benzeen in de gasvormige inlaatstroom varieert van 0.02 – 0.01. Om de conversie van
benzeen te beperken en de intrinsieke hydrogeneringsreactiesnelheid van benzeen waar te
nemen wordt de molverhouding van waterstof op benzeen beperkt tot 10. Een stijgende
partieeldruk van waterstof heeft immers een positieve invloed op de reactiesnelheid, zie
paragraaf 3.2.2. Deze beperking heeft de afwezigheid van diffusielimitaties tot gevolg. Om
totaaldrukken tot 30 bar te realiseren zonder de partieeldruk van benzeen en waterstof te
verhogen wordt een inert gas namelijk stikstof toegevoegd. De experimentele instelcondities
maken een grondige studie van de intrinsieke kinetiek van de hydrogenering van benzeen
mogelijk. De reproduceerbaarheid van de experimenten is nagegaan alvorens de
experimentele dataset is opgebouwd.
3.2 Effect van procesparameters op de hydrogenering van benzeen
De adequaatheid van een model kan kwalitatief onderzocht worden door de
modelvoorspellingen te toetsen aan de experimenteel waargenomen trends. Een
overeenstemming van het effect van de procesparameters op de experimentele reactiesnelheid
en de modelberekende reactiesnelheden moet waargenomen worden. Een fysische

Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
34
interpretatie kan volgen uit de mogelijkheid van het single-event microkinetisch model om de
concentraties van de verschillende koolwaterstoffen en waterstof op het katalysatoroppervlak
te berekenen.
3.2.1 Effect van de temperatuur
De reactiesnelheid voor de hydrogenering van benzeen vertoont een maximum in functie van
de temperatuur. Dit fenomeen is reeds vastgesteld voor andere aromaten. Het maximum wordt
verklaard doordat de stijging van de reactiesnelheidscoëfficiënt bij hogere temperaturen wordt
overgecompenseerd door de daling van de bedekkingsgraad van de reactie-intermediairen.
Het single-event concept laat toe om de concentraties van de verschillende geadsorbeerde
koolwaterstoffen en het atomair geadsorbeerd waterstof op het katalysatoroppervlak te
berekenen. Kwalitatief kan bovenvermeld fenomeen echter ook verklaard worden gebaseerd
op een eenvoudiger kinetisch model van de vorm:
nBBHH
bH
aBHB
)pKpK1(
ppKkKr
22
22
++= ( 3-1)
met n = het aantal actieve centra betrokken bij de snelheidsbepalende stap (n≥1).
Lage temperaturen resulteren in een hoge bedekkingsgraad van de actieve centra door
benzeenmoleculen. De reactiesnelheid wordt dan herleid.
bH
na
BH
n1
B 22ppKkKr = ( 3-2)
met bijhorende schijnbare activeringsenergie:
2HBact
appact HH)n1(EE ∆+∆+= ( 3-3)
De reactiesnelheid bij hogere temperaturen kan, in overeenstemming met een dalend
percentage van de actieve centra dat door koolwaterstofspecies wordt bezet bij een stijgende
temperatuur, vereenvoudigd worden.
bH
aBHB 22ppKkKr = ( 3-4)
met bijhorende schijnbare activeringsenergie:
2HBact
appact HHEE ∆+∆+= ( 3-5)
De bijdrage van de chemisorptie-enthalpie (∆HB<0) van benzeen tot de waarde van de
schijnbare activeringsenergie is in het geval van lage temperaturen 0 voor n=1 of positief voor
n>1. Indien de schijnbare activeringsenergie appactE positief is dan stijgt de reactiesnelheid met
toenemende temperatuur. Bij hogere temperaturen bevat de schijnbare activeringsenergie een
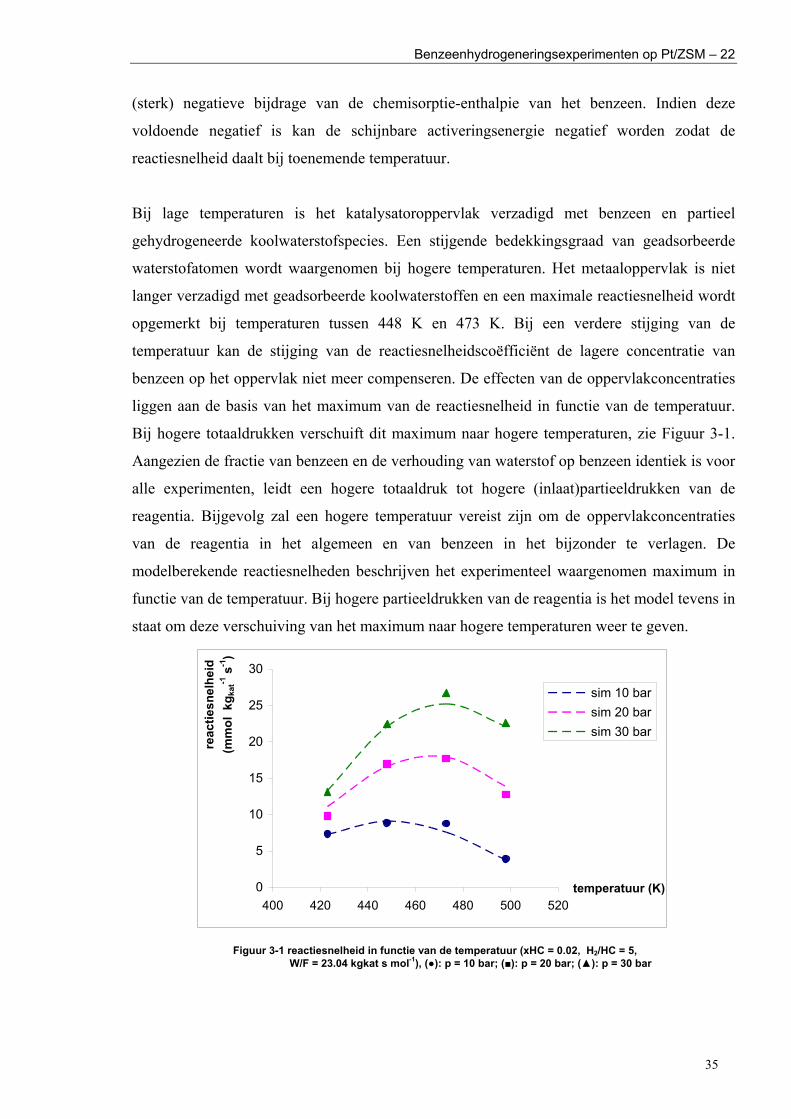
Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
35
(sterk) negatieve bijdrage van de chemisorptie-enthalpie van het benzeen. Indien deze
voldoende negatief is kan de schijnbare activeringsenergie negatief worden zodat de
reactiesnelheid daalt bij toenemende temperatuur.
Bij lage temperaturen is het katalysatoroppervlak verzadigd met benzeen en partieel
gehydrogeneerde koolwaterstofspecies. Een stijgende bedekkingsgraad van geadsorbeerde
waterstofatomen wordt waargenomen bij hogere temperaturen. Het metaaloppervlak is niet
langer verzadigd met geadsorbeerde koolwaterstoffen en een maximale reactiesnelheid wordt
opgemerkt bij temperaturen tussen 448 K en 473 K. Bij een verdere stijging van de
temperatuur kan de stijging van de reactiesnelheidscoëfficiënt de lagere concentratie van
benzeen op het oppervlak niet meer compenseren. De effecten van de oppervlakconcentraties
liggen aan de basis van het maximum van de reactiesnelheid in functie van de temperatuur.
Bij hogere totaaldrukken verschuift dit maximum naar hogere temperaturen, zie Figuur 3-1.
Aangezien de fractie van benzeen en de verhouding van waterstof op benzeen identiek is voor
alle experimenten, leidt een hogere totaaldruk tot hogere (inlaat)partieeldrukken van de
reagentia. Bijgevolg zal een hogere temperatuur vereist zijn om de oppervlakconcentraties
van de reagentia in het algemeen en van benzeen in het bijzonder te verlagen. De
modelberekende reactiesnelheden beschrijven het experimenteel waargenomen maximum in
functie van de temperatuur. Bij hogere partieeldrukken van de reagentia is het model tevens in
staat om deze verschuiving van het maximum naar hogere temperaturen weer te geven.
0
5
10
15
20
25
30
400 420 440 460 480 500 520temperatuur (K)
reac
tiesn
elhe
id
(mm
ol k
g kat
-1 s
-1)
sim 10 barsim 20 barsim 30 bar
Figuur 3-1 reactiesnelheid in functie van de temperatuur (xHC = 0.02, H2/HC = 5,
W/F = 23.04 kgkat s mol-1), (●): p = 10 bar; (■): p = 20 bar; (▲): p = 30 bar

Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
36
3.2.2 Effect van de waterstofpartieeldruk
Een verhoging van de H2-partieeldruk bij constante partieeldruk van het benzeen leidt tot een
toename van de reactiesnelheid. De reactie-orde van waterstof kan bepaald worden uit de
volgende snelheidsvergelijking:
bH
aB 2
ppkr = ( 3-6)
In Figuur 3-2 wordt aangetoond dat de reactie-orde van waterstof positief is.
10
15
20
25
30
35
40
200 300 400 500 600 700pH2 (kPa)
reac
tiesn
elhe
id
(mm
ol k
g kat
-1 s
-1)
sim T = 423 Ksim T = 448 Ksim T = 473 Ksim T = 498 K
Figuur 3-2 reactiesnelheid in functie van de inlaat waterstof partieeldruk (pbenz = 60 kPa, W/F = 23.04 kgkat s mol-1), (●): T = 423 K; (■): T = 448 K; (▲): T = 473 K; (× ): T = 498 K
Afhankelijk van de procescondities kan de chemisorptie van de reagentia competitief of niet-
competitief optreden. De aanname in het model dat het aromaat en waterstof competitief
chemisorberen wordt voor de gebruikte procescondities bevestigd in Figuur 3-3a en Figuur
3-3b. Op hetzelfde type van actief centrum kan zowel een aromaat als waterstof
chemisorberen. Bij lage temperaturen is het oppervlak volledig bezet door benzeen en partieel
gehydrogeneerde producten. Slechts een beperkt aantal interstitiële actieve centra resteert
voor de kleinere molecule waterstof. Het competitieve karakter van waterstof- en
aromaatchemisorptie wordt het sterkst waargenomen bij de temperaturen 448 K tot 473 K.
Vanaf een bepaalde temperatuur wordt de bedekkingsgraad van het oppervlak voldoende laag
zodat geen competitie meer wordt waargenomen. Een verdere stijging van de temperatuur
leidt tot een groter aantal vrije centra.

Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
37
a)
0.0E+00
1.0E-04
2.0E-04
3.0E-04
4.0E-04
5.0E-04
6.0E-04
7.0E-04
8.0E-04
9.0E-04
1.0E-03
200 300 400 500 600 700
pH2 (kPa)
bede
kkin
gsgr
aad
HC
b)
0.0E+00
1.0E-04
2.0E-04
3.0E-04
4.0E-04
5.0E-04
6.0E-04
7.0E-04
8.0E-04
9.0E-04
1.0E-03
200 300 400 500 600 700
pH2 (kPa)
bede
kkin
gsgr
aad
H 2
Figuur 3-3 a) modelberekende bedekkingsgraad van geadsorbeerde koolwaterstoffen op het katalysatoroppervlak in functie van de partieeldruk van waterstof (pbenz = 60 kPa, W/F = 23.04 kgkat s mol-1), (●) : T = 423 K; (■) : T = 448 K; (▲): T = 473 K; (× ): T = 498 K Figuur 3-3 b) modelberekende bedekkingsgraad van atomair geadsorbeerd waterstof op het katalysatoroppervlak in functie van de partieeldruk van waterstof (pbenz = 60 kPa, W/F = 23.04 kgkat s mol-1), (●) : T = 423 K; (■) : T = 448 K; (▲): T = 473 K; (× ): T = 498 K
3.2.3 Effect van de benzeenpartieeldruk
In Figuur 3-4 wordt het effect van een toenemende partieeldruk van benzeen weergegeven
waarbij alle andere instelcondities ongewijzigd blijven.
10
15
20
25
30
0 10 20 30 40 50 60 70 80pbenz (kPa)
reac
tiesn
elhe
id(m
mol
kg k
at-1
s-1
) sim T = 423 Ksim T = 448 Ksim T = 473 Ksim T = 498 K
Figuur 3-4 reactiesnelheid in functie van de inlaat benzeen partieeldruk (pH2 = 300 kPa,
W/F = 32.28 kgkat s mol-1), (●): T = 423 K; (■): T = 448 K; (▲): T = 473 K; (× ): T = 498 K
Bij alle temperaturen wordt een daling van de reactiesnelheid waargenomen. De reactie-orde
van benzeen is negatief. Het inhiberend effect van de inlaatpartieeldruk van benzeen op de

Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
38
reactiesnelheid voor de hydrogenering van benzeen wordt zowel experimenteel waargenomen
als gesimuleerd door het model, zie Figuur 3-4.
De berekende bedekkingsgraad van de koolwaterstoffen en waterstof op het
katalysatoroppervlak in functie van de partieeldruk van benzeen wordt respectievelijk in
Figuur 3-5a en Figuur 3-5b uitgezet. Experimenteel wordt in Figuur 3-4 bij hogere
temperaturen een minder sterke daling van de reactiesnelheid waargenomen. Het inhiberend
effect van benzeen wordt het sterkst waargenomen bij een temperatuur van 423 K. De
bedekkingsgraad van de koolwaterstoffen is bij deze temperatuur immers maximaal. Bij een
hogere inlaatpartieeldruk van benzeen neemt de oppervlakconcentratie van koolwaterstoffen
op de katalysator verder toe. Ook het model simuleert dat het inhiberend effect van benzeen
vermindert naarmate de temperatuur stijgt.
a)
0.0E+00
1.0E-04
2.0E-04
3.0E-04
4.0E-04
5.0E-04
6.0E-04
7.0E-04
8.0E-04
9.0E-04
1.0E-03
0 10 20 30 40 50 60 70 80
pbenz (kPa)
bede
kkin
gsgr
aad
HC
b)
0.0E+00
1.0E-04
2.0E-04
3.0E-04
4.0E-04
5.0E-04
6.0E-04
7.0E-04
8.0E-04
9.0E-04
1.0E-03
0 10 20 30 40 50 60 70 80
pbenz (kPa)
bede
kkin
gsgr
aad
H 2
Figuur 3-5 a) modelberekende bedekkingsgraad van geadsorbeerde koolwaterstoffen op het katalysatoroppervlak in functie van de partieeldruk van benzeen (pH2 = 300kPa, W/F = 32.28 kgkat s mol-1), (●) : T = 423 K; (■) : T = 448 K; (▲): T = 473 K; (× ): T = 498 K Figuur 3-5 b) modelberekende bedekkingsgraad van atomair geadsorbeerd waterstof op het katalysatoroppervlak in functie van de partieeldruk van benzeen (pH2 = 300kPa, W/F = 32.28 kgkat s mol-1), (●) : T = 423 K; (■) : T = 448 K; (▲): T = 473 K; (× ): T = 498 K
3.2.4 Effect van de totaaldruk
In Figuur 3-6 wordt de reactiesnelheid weergegeven in functie van de totaaldruk met als
parameter de temperatuur. De fractie benzeen en de verhouding van de hoeveelheid benzeen
op waterstof is constant. De partieeldrukken van de reagentia variëren dus evenredig met de
totaaldruk. Het waargenomen effect is een samengesteld effect van de partieeldruk van
waterstof en van benzeen, zie paragrafen 3.2.2 en 3.2.3. Uit het effect van de stijgende

Benzeenhydrogeneringsexperimenten op Pt/ZSM – 22
39
totaaldruk wordt afgeleid dat het inhiberend effect van benzeen overgecompenseerd wordt
door het positieve effect van waterstof. Experimenteel wordt waargenomen dat de
reactiesnelheid minder gevoelig is aan de totaaldruk bij een lage temperatuur (423 K). Bij
deze temperatuur is een grote fractie van het katalysatoroppervlak ingenomen door
koolwaterstoffen. Het positieve effect van de toenemende partieeldruk van waterstof is bij
deze temperatuur beduidend minder in vergelijking met hogere temperaturen.
0
5
10
15
20
0 10 20 30ptot (bar)
reac
tiesn
elhe
id(m
mol
kg k
at-1
s-1
) sim T = 423 Ksim T = 448 Ksim T = 473 Ksim T = 498 K
Figuur 3-6 reactiesnelheid in functie van de totaaldruk (xHC = 0.02, H2/HC = 5, W/F = 23.04 kgkat s mol-1), (●): T = 423 K; (■): T = 448 K; (▲): T = 473 K; (× ): T = 498 K
3.3 Besluit
De effecten van de verschillende procesparameters beantwoorden aan de verwachtingen uit
vorige studies in verband met de hydrogenering van aromaten. Een stijging van atomair
geadsorbeerd waterstof heeft een positief effect op de reactiesnelheid. Een tegengesteld effect
wordt waargenomen voor de invloed van de bedekkingsgraad van koolwaterstoffen op de
reactiesnelheid. Beide effecten zijn aanwezig bij een toename van de totaaldruk. Het
inhiberend effect van benzeen wordt overgecompenseerd door het positief effect van
waterstof op de reactiesnelheid. Het temperatuurseffect op de reactiesnelheid resulteert in een
maximum en wordt verklaard door de oppervlakconcentraties.

40
Hoofdstuk 4 Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
De hydrogenering van aromaten is in het verleden uitvoerig bestudeerd. Kinetische modellen
werden opgesteld op basis van data in stationaire toestand zonder in situ karakterisatie van het
oppervlak. De afwezigheid van informatie omtrent de oppervlakintermediairen heeft geleid tot
een waaier van mogelijke kinetische modellen. Veelal werd gebruik gemaakt van Langmuir-
Hinshelwood modellen die uitgaan van één, niet nader gespecifieerd reactiepad met een
snelheidsbepalende stap. Verschillende modellen onderscheiden zich op basis van de
gemaakte onderstellingen. Een analyse van het reactiepad aan de hand van
kwantumchemische berekeningen vormt de basis van de meer fundamentele modellering
gevolgd in dit werk, met name het single-event microkinetisch model (SEMK). Dit
modelconcept ziet af van vereenvoudigende veronderstellingen als de aanwezigheid van een
dominant pad of een snelheidsbepalende stap in het reactienetwerk. Dit impliceert dat het
volledige reactienetwerk moet opgesteld worden en dat de reactiesnelheid wordt bepaald door
de contributie van elke elementaire stap van het netwerk. H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
HH
H H
H
H
H
H
**
*
*
**
*
* *
*
*
*
H
H
H
H
H
H
H
H
H
H
H
*1
3 6
2 4
5
7
8
9
10
11
12 13
Figuur 4-1 elementair reactienetwerk voor de hydrogenering van benzeen

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
41
Indien aan elk van deze elementaire reacties een individuele reactiesnelheidscoëfficiënt wordt
toegekend kan het aantal parameters, afhankelijk van de grootte van het reactienetwerk, sterk
toenemen. In plaats van aan elke elementaire reactie een snelheidscoëfficiënt toe te kennen
kan het aantal parameters gereduceerd worden door verbanden tussen de
snelheidscoëfficiënten van verwante reacties. In deze context kan gebruik gemaakt worden
van lineaire-vrije-energie relaties die een lineair verband tussen de activeringsenergie en de
reactie-enthalpie vooropstellen. Een andere methode is gebaseerd op single-events. Op basis
van bepaalde criteria kunnen elementaire reacties gegroepeerd worden in reactiefamilies.
Reacties die behoren tot een familie hebben dezelfde single-event reactiesnelheidscoëfficiënt
( k~ ). De reactiesnelheid van de elementaire stap wordt bepaald door het product van de
single-event snelheidscoëfficiënt en het aantal single-events.
In dit eindwerk wordt het verkrijgen van fysisch significante modelparameters voor de
katalytische hydrogenering van benzeen door middel van regressie van het reeds opgestelde
SEMK model voor de hydrogenering van mono-aromaten beoogd. De parameters in het
SEMK model worden geschat op basis van het bestaande programma
hydrogenation_CSTR_network_simulation.f (Vagenende, 2004). Het programma sdestm.f
(Hosten, 1974) werd door Debruyne (2005) aangepast om een sequentieel ontwerp van
experimenten voor precieze parameterschattingen toe te laten (Debruyne, 2005). Met behulp
van dit programma, sdestm_single_event.f, kunnen experimentele instelcondities geselecteerd
worden die toelaten de parameters met de grootst mogelijke betrouwbaarheid te schatten, zie
paragraaf 2.4.2.3. Een nieuwe set van experimenten, zie appendix B, werd gebruikt om
significante parameterschattingen te verkrijgen. De te schatten parameters bepalen de single-
event reactiesnelheidcoëfficiënten en de reactie- en chemisorptie-evenwichtscoëfficiënten van
het SEMK model. Deze coëfficiënten bepalen de concentraties van de
oppervlakintermediairen en de reactiesnelheid van de hydrogenering van benzeen tot
cyclohexaan. Om het SEMK model voor de hydrogenering van aromaten, en meer specifiek
voor benzeen, te schetsen, worden de modelveronderstellingen, zie paragraaf 4.1
weergegeven met de bijhorende reactiesnelheidsvergelijking.

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
42
4.1 SEMK model voor de benzeenhydrogenering
Een single-event microkinetisch model voor de katalytische hydrogenering van mono-
aromaten werd reeds opgesteld (Vagenende, 2004). Het model gaat uit van een aantal
gefundeerde onderstellingen.
1. competitieve dissociatieve chemisorptie van H2 en moleculaire chemisorptie van
aromaten op identieke chemisorptiecentra
2. waterstof-, gehydrogeneerd product- en aromaatchemisorptie zijn in quasi-evenwicht
3. op het katalysatoroppervlak wordt geen gedehydrogeneerd species beschouwd (Saeys
et al., 2004)
4. afwezigheid van een snelheidsbepalende stap of dominant reactiepad
5. alle elementaire stappen uit het reactienetwerk worden reversibel beschouwd
6. de pseudo-stationaire toestandshypothese wordt toegepast voor de bepaling van de
concentraties van de partieel gehydrogeneerde intermediairen
Bij het toepassen van het single-event concept worden de snelheidscoëfficiënten opgesplitst in
2 factoren, namelijk het aantal single-events, ne, en de single-event snelheidscoëfficiënt k~ .
Zonder verdere veronderstellingen in verband met de (single-event) snelheidscoëfficiënten,
zou hun aantal gelijk zijn aan het aantal elementaire H-additiestappen. Los van het exacte
aantal coëfficiënten, zou het in elk geval moeilijk zijn alle verschillende parameters zonder
onderlinge correlatie te schatten aangezien slechts 2 van alle koolwaterstofspecies op het
oppervlak ook in de gasfase waargenomen worden, namelijk het reagens, benzeen, en het
product, cyclohexaan. Om die reden worden, gebaseerd op de configuratie van het reactief
centrum tijdens H-addities (Saeys et al, 2002), bepaalde single-event snelheidscoëfficiënten
aan elkaar gelijk gesteld (Vagenende, 2004). Het effect van de configuratie van het in de
hydrogeneringsstap betrokken C-atoom wordt in rekening gebracht door de aard van dit C-
atoom en door de verzadigingstoestand van de naaste buur C atomen. De C-Pt binding wordt
verwacht in sterke mate beïnvloed te worden door de verzadigingstoestand van de naaste buur
C-atomen. De sterische hindering is een tweede factor die de single-event
snelheidscoëfficiënten kan bepalen en wordt door middel van de vertakkingsgraad van het
reagerende C-atoom in rekening gebracht. De elementaire (de)hydrogenering single-event
reactiesnelheidscoëfficiënt k~ wordt dus gekarakteriseerd door het aantal onverzadigde
dichtste buren en de vertakkingsgraad van het reagerende C-atoom.

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
43
De rechtvaardiging is respectievelijk gebaseerd op het effect van de bindingsterkte van de C-
Pt binding en de sterische hindering op de activeringsenergie. Het verschil in
activeringsenergie tussen de verschillende reactiestappen wordt bevestigd door
kwantummechanische berekeningen (Saeys et al., 2004).
De single-event snelheidscoëfficiënten )n,m(k~ worden dus gekarakteriseerd door de
verzadigingsgraad (m) van de naaste buren en de vertakkingsgraad (n) van het C-atoom, dat
een waterstofadditie of waterstofabstractie ondergaat. Per definitie zijn de single-event
snelheidscoëfficiënten onafhankelijk van de beschouwde reagentia. De elementaire
snelheidscoëfficiënten worden bepaald uit de single-event snelheidscoëfficiënten en het aantal
single-events. Het aantal single-events is afhankelijk van de symmetrie van het reactant en het
geactiveerd complex. Dit impliceert dat de elementaire snelheidscoëfficiënten )n,m(k niet
invariant zijn. Een vergelijking tussen benzeen en tolueen als reagentia toont aan dat het
verschil in hydrogeneringssnelheid tussen de elementaire reacties kan geïnterpreteerd worden
als een verschil in aantal single-events. De H-additie wordt beschouwd als een enkelvoudige
gebeurtenis. Benzeen kan op 6 mogelijke C-atomen een H-additie ondergaan waarbij
hetzelfde product gevormd wordt. Bij tolueen wordt het aantal mogelijkheden gereduceerd tot
hoogstens 2 op voorwaarde dat de atomaire waterstof in de ortho- of metapositie addeert . H
*k(2,2) k(2,2)
CH3CH3
H
*
Figuur 4-2 H-additie bij benzeen en tolueen
Uit de transitietoestandstheorie kan het single-event concept rechtstreeks afgeleid worden. De
elementaire reactiesnelheidscoëfficiënt wordt uitgedrukt als:
≠= Kh
Tkk B ( 4-1)
De evenwichtscoëfficiënt ≠K kan weergegeven worden in functie van de
standaardactiverings-enthalpie ≠∆ 0H en -entropie ≠∆ 0S voor de vorming van de
transitietoestand van het reactant. De standaardentropie ≠∆ 0S van een molecule kan
opgesplitst worden in verschillende contributies. De bijdragen tot de standaardentropie zijn de
translatie van de molecule, de externe rotatie van de ganse molecule, de interne rotatie rond
een binding van de molecule en de verschillende vibratiemodes van de bindingen in de
molecule. De rotationele contributie tot de standaardentropie bevat een bijdrage gerelateerd

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
44
aan de symmetrie van de molecule. In deze uitdrukking worden extσ en intσ beschouwd als
het extern en intern symmetriegetal van de molecule. ext,rotS~ en int,rotS~ zijn de intrinsieke
externe en interne rotatie-entropie.
vibrint,rotext,rottrans SSSSS +++= ( 4-2)
extext,rotext,rot σlnRS~S −= ( 4-3)
intint,rotint,rot σlnRS~S −= ( 4-4)
Een symmetriegetal wordt gedefinieerd als het totaal aantal onafhankelijke permutaties van
atomen of identieke groepen (Bischop en Laidler, 1965). Het globale symmetriegetal kan
berekend worden uit het aantal mogelijke rotaties zonder dat de molecule wijzigt. De noemer
wordt bepaald door het aantal chirale centra. Het aantal mogelijke enantiomeren bedraagt 2n.
n
intextglob
2σσ
σ = ( 4-5)
De elementaire snelheidscoëfficiënt k(m,n) kan uitgedrukt worden in functie van de single-
event snelheidscoëfficiënt ( )n,mk~ en het aantal single-events ne. Het aantal single-events is de
verhouding van het globaal symmetriegetal van het reactant op dat van het geactiveerd
complex.
( ) ( ) ( )n,mk~nn,mk~
2
2eeh
Tkn,mk e
,n,extint,
r,nr,extrint,
TRH
RS~
B
,glob
r,glob
00
=σσ
σσ
=σ
σ=
≠≠≠
∆−∆
≠
≠≠
( 4-6)
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
HH
H H
H
H
H
H
**
*
*
**
*
* *
*
*
*
H
H
H
H
H
H
H
H
H
H
H
*1
3 6
2 4
5
7
8
9
10
11
12 13k(2,2) k(2,2)
k(2,2)k(2,2)
k(2,2) k(2,2)k(1,2)
k(1,2)
k(1,2)
k(1,2)
k(1,2)
k(1,2)
k(1,2)
k(1,2)
k(0,2) k(0,2)
k(0,2)k(0,2)
k(0,2) k(0,2)
Figuur 4-3 elementair reactienetwerk van benzeen; dik: dominant pad
Alle elementaire reacties in het reactienetwerk worden als reversibel beschouwd. Het
volledige netwerk wordt in beschouwing genomen en elke H-additiestap kan de

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
45
productiesnelheid van cyclohexaan beïnvloeden. De netto-productiesnelheid van component j
wordt berekend aan de hand van:
∑ →→ −=i
hyd/dehij
deh/hydjij rrR ( 4-7)
( ) ( )[ ]( ) ( )[ ]∑
+−
+=
∗→
∗→
i jiidehHiihydij,e
iiidehHiihydji,ej
CCn,mk~Cn,mk~n
CCn,mk~Cn,mk~nR
*
* ( 4-8)
Er gelden geen beperkingen in verband met de oppervlakintermediairen. Het volledige
reactienetwerk wordt in beschouwing genomen. Het reactienetwerk bestaat uit 13
intermediairen met 20 H-addities en 20 H-substracties, zie Figuur 4-3. De single-event
snelheidscoëfficiënten brengen de verzadigingsgraad en de vertakkingsgraad in rekening en
zijn per definitie onafhankelijk van de beschouwde reagentia. Het single-event model geeft
inzicht in de concentraties van alle koolwaterstoffen op het katalysatoroppervlak en de
snelheden van alle individuele H-additiestappen. In het bijzonder kan het eventuele optreden
van een meest voorkomend oppervlakintermediair of een dominant reactiepad geverifieerd
worden. Een breed toepassingsgebied voor het SEMK model is dus gegarandeerd.
4.2 Parameters in het SEMK model voor de hydrogenering van benzeen
4.2.1 Parameterschattingen voor de hydrogenering van benzeen
De parameters in het SEMK model, zie Tabel 4-1, zijn drie single-event
snelheidscoëfficiënten, drie single-event evenwichtscoëfficiënten en drie chemisorptie-
eventwichtscoëfficiënten. De temperatuursafhankelijkheid van de coëfficiënten verdubbelt het
aantal parameters tot 18. Tabel 4-1 reactie – en evenwichtscoëfficiënten
snelheidscoëfficiënten
( )2,0k~hyd ( )2,1k~hyd ( )2,2k~hyd
evenwichtscoëfficiënten
( )2,0K~ ( )2,1K~ ( )2,2K~
chemisorptie-evenwichtscoëfficiënten
benzeenK waterstofK ncyclohexaaK

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
46
Het aantal te schatten parameters kan gereduceerd worden tot 7 door gebruik te maken van de
thermodynamische consistentie (zie paragraaf 4.2.2) en het berekenen van de pre-
exponentiële factoren gebaseerd op de transitietoestandstheorie en statistische
thermodynamica (Thybaut et al., 2002). De grenzen van het bereik van de pre-exponentiële
factoren worden berekend op basis van de veronderstelde mobiliteit van de corresponderende
species. Een bereik wordt vooropgesteld voor de pre-exponentiële factoren van de
oppervlaksnelheidscoëfficiënt, oppervlakevenwichtscoëfficiënt en chemisorptie-
evenwichtscoëffciënt van benzeen en waterstof, zie Tabel 4-2. Tabel 4-2 pre-exponentiële factoren gebaseerd op de transitietoestandstheorie en statistische thermodynamica
( ) ( )10hyd s2,mk − ( )2,mK0 ( )10
B,chem PaK − ( )10H,chem PaK
2
−
berekend bereik
(Thybaut,2002)
108 - 1013 1 10-5 – 1 10-10 - 10-13 10-8 - 10-13
Thybaut (2002) 1015 10-2 10-12 10-10
Saeys (2005) 511 0.5 7.4 10-12 2.7 10-11
De reactiesnelheidscoëfficiënt voor de hydrogeneringsreactie van aromaten kan geschreven
worden in functie van moleculaire partitiefuncties. De moleculaire partitiefunctie Q is het
product van de contributies van de translationele, rotationele en vibrationele vrijheidsgraden.
)Tk
Eexp(QQ
Qh
Tkk
B
0
HA
HABHA
m−= −
− ( 4-9)
Een analoge uitdrukking kan opgesteld worden voor de evenwichtscoëfficiënten. De pre-
exponentiële factor in het geval van de chemisorptie-evenwichtscoëfficiënt is een maat voor
de mobiliteit van het geadsorbeerde species op het oppervlak. Een meer uitgebreid bereik
wordt verondersteld voor de chemisorptie van waterstof ten gevolge van het dissociatieve
karakter van de waterstofchemisorptie (Thybaut et al., 2002).
Het model van Thybaut (2002) met als modelveronderstelling gelijke snelheidscoëfficiënten
voor de eerste vier H-addities en quasi-evenwicht voor de vijfde en de zesde H-additie wordt
geselecteerd op basis van modelflexibiliteit, zie paragraaf 1.1.1. Verschillende stellen van pre-
exponentiële factoren zijn uitgetest. Uit de pre-exponentiële factoren van het optimale stel 1 De berekende pre-exponentiële factoren voor de reactiesnelheidscoëfficiënten op het oppervlak zijn gelegen tussen 108 en 1013. Dit bereik komt overeen met waarden waarbij alle species (de reactanten en de transitietoestanden) respectievelijk mobiel of immobiel zijn. Het berekende bereik kan uitgebreid worden indien species met andere oppervlakmobiliteit betrokken zijn in de reacties. (Thybaut et al., 2002)

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
47
parameters wordt aangetoond dat voor de H-atomen een hogere mobiliteit op het
katalysatoroppervlak kan verondersteld worden. De reactant- en productkoolwaterstofspecies
op het katalysatoroppervlak zijn eerder immobiel. De optimale pre-exponentiële factoren
beantwoorden ook aan het feit dat de transitietoestand een hogere oppervlakmobiliteit bezit
dan de overige koolwaterstofspecies.
In het kinetisch model voor de hydrogenering van tolueen door Saeys (2005) wordt de vijfde
stap als snelheidsbepalende beschouwd, zie paragraaf 1.1.2. De pre-exponentiële factoren in
kinetisch model van Saeys (2005) verschillen ten opzichte van de pre-exponentiële factoren
voorgesteld door Thybaut (2002). Een hogere mobiliteit voor de aromatische reactanten ten
opzichte van Thybaut (2002) wordt uit DFT berekeningen afgeleid. In vorig werk werd
immers een lage mobiliteit voor de aromatische reactanten verondersteld. De pre-exponentiële
factor voor de chemisorptie-evenwichtscoëfficiënt van benzeen in het model van Saeys (2005)
komt overeen met een hogere mobiliteit van het geadsorbeerde benzeen in de hollow
configuratie. Waterstof is erg mobiel terwijl de geadsorbeerde gehydrogeneerde
intermediairen minder mobiel zijn. Voor de transitietoestand wordt een intermediaire
mobiliteit verondersteld.
4.2.2 Thermodynamische consistentie
Een SEMK model voor de hydrogenering van benzeen dat gebruik maakt van de
vooropgestelde pre-exponentiële factoren gebaseerd op de transitietoestandstheorie en
statistische thermodynamica (Thybaut et al., 2002) bevat 9 te schatten parameters. De single-
event reactiesnelheidscoëfficiënt voor dehydrogenering )n,m(k~dehydr wordt immers berekend
aan de hand van:
)n,m(K~)n,m(k~
)n,m(k~
hydr
hydrdehydr = ( 4-10)
Omwille van de thermodynamische consistentie kan het aantal te schatten parameters
gereduceerd worden tot 7. Een eerste Born-Haber cyclus wordt gemaakt op het
katalysatoroppervlak. De consecutieve waterstofadditie in de ortho en metha positie van het
gehydrogeneerde C-atoom kan gerealiseerd worden via 2 parallelle reactiepaden, zoals
geïllustreerd in Figuur 4-4.

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
48
H
H
H
H
H
H
H
H
**
*
3 6
2 4
( )2,1~K
( )2,1~K
( )2,0~K
( )2,2~K
Figuur 4-4 hydrogenering van benzeen naar 1,2,3 trihydrobenzeen
Beide reactiepaden resulteren in hetzelfde partieel gehydrogeneerde oppervlaktespecies
namelijk 1,2,3 trihydrobenzeen. De modelparameters moeten voldoen aan de volgende
thermodynamische consistentie:
(0,2)K~ (2,2).K~ (1,2)K~ (1,2).K~ = ( 4-11)
De parameter ( )2,1H∆ wordt gebaseerd op de thermodynamische consistentie op het
katalysatoroppervlak berekend.
( ) ( ) ( )2
2,2H∆2,0H∆2,1H∆ += ( 4-12)
Een tweede Born-Haber cyclus omvat de volledige katalytische cyclus. Het resultaat hiervan
is dat de reactie-enthalpieën en -entropieën van de katalytische hydrogenering op het
metaaloppervlak gelijk moet zijn aan de reactie-enthalpie gas,rH∆ en reactie-entropie gas,rS∆
van de gasfasehydrogenering die kan berekend worden gebaseerd op getabelleerde
thermodynamische grootheden.
( ) ( ) CH,chemrrH,chemB,chemgas,r H∆2,0H∆32,2H∆3H∆3H∆H∆2
−+++= ( 4-13)
De chemisorptie-enthalpie van cyclohexaan wordt berekend uit de globale thermodynamische
consistentie.
( ) ( ) gas,rrrH,chemB,chemCH,chem H∆2,0H∆32,2H∆3H∆3H∆H∆2
−+++= ( 4-14)
Het stelsel algebraïsche vergelijkingen dat gebruikt wordt om de thermodynamische
consistentie te garanderen (Vagenende, 2004) wordt voorgesteld door vergelijkingen 4-12 en
4-14. De thermodynamische consistentie wordt ook gerespecteerd voor de berekende pre-
exponentiële factoren.
4.3 Reparameterisatie
In het SEMK model voor de hydrogenering van mono-aromaten worden de pre-exponentiële
factoren berekend. De thermodynamische consistentie en de berekende pre-exponentiële

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
49
factoren in het SEMK model voor de hydrogenering van benzeen op basis van de
transitietoestandstheorie en de statistische thermodynamica reduceren het aantal te schatten
parameters tot 7. De grenzen van het bereik van de pre-exponentiële factoren worden
berekend op basis van de veronderstelde mobiliteit van de corresponderende species, zie
paragraaf 4.2.1. De kwantumchemisch berekende pre-exponentiële factoren in het kinetisch
model van Saeys et al. (2005) corresponderen met een verschillende mobiliteit van de species
op het katalysatoroppervlak ten opzichte van het model van Thybaut et al. (2002).
In het algemene geval dat de beide parameters uit het Arrheniusverband geschat worden, kan
een reparameterisatie uitgevoerd worden om een hoge correlatie tussen de pre-exponentiële
factor en de activeringsenergie of entropie te vermijden. Immers door de hoge correlatie
tussen de parameters A en E is de -1T )JJ( matrix slecht geconditioneerd en bovendien bijna
singulier. Doordat het bereik waarover de temperatuur kan gevarieerd worden eerder beperkt
is, zal het optimalisatieproces zeer traag verlopen of zelfs niet tot waarden voor de
parameterschattingen convergeren. Door reparameterisatie wordt de sterke correlatie tussen A
en E voor de reactiesnelheidscoëfficiënt verbroken en treden deze problemen in mindere mate
op. De matrix is beter geconditioneerd en de parameters zullen beter geschat kunnen worden.
Het model gedraagt zich meer als een lineair model door de beter geconditioneerde J-matrix.
k=
−−− )
T1
T1(
REexp)
RTEexp(A
gemgem ( 4-15)
=
−
− )T
1T1(
REexpA
gem
* ( 4-16)
Na substitutie correspondeert de pre-exponentiële factor *A met refTk waarbij de
referentietemperatuur het rekenkundig gemiddelde van de temperatuur voor alle experimenten
is. De correlatie tussen *A en E is beduidend minder. Een wijziging van *A kan moeilijker
gecompenseerd worden door E te variëren. Een lagere correlatiecoëfficiënt is het gevolg en
een sterker uitgesproken minimum in de doelfunctie wordt gerealiseerd. Dit geeft aanleiding
tot betere convergentie. Bovendien gedraagt de parameter *A zich als een meer lineaire
parameter dan A. De individuele parameter A is meestal niet significant verschillend van nul
in tegenstelling tot de gereparameteriseerde pre-exponentiële factor *A . Een analoge
redenering kan opgebouwd worden voor de evenwichtscoëfficiënten. Door gebruik te maken
van een gereparameteriseerde uitdrukking voor de evenwichtscoëfficiënten kan de correlatie
tussen xH∆ en xS∆ verminderd worden.

Single-Event MicroKinetische (SEMK) model voor de hydrogenering van mono-aromaten
50
Ondanks het gebruik van berekende pre-exponentiële factoren, blijft het uitdrukken van de
reactiesnelheidscoëfficiënten en de evenwichtscoëfficiënten in gereparameteriseerde vorm in
het parameterschattingsprogramma zinvol, met als doel de accuraatheid van de berekende pre-
exponentiële factoren te verifiëren.

51
Hoofdstuk 5 SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
De parameters in het SEMK model voor de hydrogenering van benzeen op Pt/ZSM-22
worden geschat op basis van de methode van de ongewogen kleinste kwadraten. De
doelfunctie, gedefinieerd als de som van de kwadraten van de niet waarneembare fouten,
wordt geminimaliseerd ter bepaling van een optimale parameterset. De opgebouwde
experimentele dataset voor de hydrogenering van benzeen, bestaande uit 43 experimenten, is
weergegeven in appendix B. Er werd in de eerste plaats gecontroleerd of de verwachte
effecten met betrekking tot de invloed van de procesparameters worden waargenomen, zie
Hoofdstuk 3. Om de single-event microkinetische parameters uit het model voor de
hydrogenering van benzeen op een significante manier te kunnen schatten, was een
uitgebreider stel aan experimentele gegevens nodig. Tot op heden konden immers niet alle
parameters uit het SEMK model significant geschat worden (Debruyne, 2005). Een verdere
uitbreiding van de experimentele gegevens is volledig gebaseerd op het sequentieel ontwerp
zoals opgesteld door Debruyne (2005) zodat de experimenten die het meeste bijdragen tot
meer precieze parameterschattingen worden geselecteerd. Een maximale benutting van de
aanwezige experimentele informatie bij het sequentieel ontwerp wordt gerealiseerd door na
ieder experiment alle informatie opnieuw te analyseren. De parameters in het model worden
geschat onder gereparameteriseerde vorm, zie paragraaf 4.3.
5.1 Parameterschattingen
De literatuur getuigt van intensief onderzoek met betrekking tot de hydrogenering van mono-
aromaten. Op basis van gerapporteerde waarden uit de literatuur kunnen de in dit werk
verkregen parameterwaarden geïnterpreteerd worden. In paragrafen 1.1.1 en 1.1.2 worden de
parameterwaarden en de overeenkomstige conclusies met betrekking tot de fysische relevantie
van beide modellen opgesteld door Thybaut et al. (2002) en Saeys et al. (2005) voor de
hydrogenering van mono-aromaten beschreven. Aangezien de parameters in het model een
fysische betekenis hebben, is niet enkel de significantie van de regressie en de bijhorende
parameters een aandachtspunt maar ook de fysische relevantie van de parameterwaarden. In

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
52
het model wordt de thermodynamische consistentie, zie paragraaf 4.2.2, steeds in acht
genomen. Meer specifiek worden de pre-exponentiële factor en de enthalpie voor de
chemisorptie van cyclohexaan berekend uit andere parameterwaarden.
( ) ( ) gas,rrrH,chemB,chemCH,chem H∆2,0H∆32,2H∆3H∆3H∆H∆2
−+++= ( 5-1)
( ) ( )[ ] ( )[ ]31.85.384
30r
30r
30H,chem
0B,chem0
CH,cheme
2,0K2,2KKKK 2
−= ( 5-2)
Het belang van de chemisorptie van waterstof komt naar voren in beide uitdrukkingen. Ook
het effect van de oppervlakevenwichtscoëfficiënten voor de hydrogenering van benzeen op de
thermodynamisch consistent berekende parameterwaarden is niet onbelangrijk.
Tot op heden werden in de kinetische modellen voor de hydrogenering van aromaten
ontwikkeld aan het LPT nooit meer dan drie modelparameters simultaan geschat. Met het
huidige model en de beschikbare dataset is dit wel mogelijk. De thermodynamische
consistentie en de berekende pre-exponentiële factoren in het SEMK model voor de
hydrogenering van benzeen op basis van de transitietoestandstheorie en de statistische
thermodynamica reduceren het aantal te schatten parameters tot 7. Een simultane regressie
van de activeringsenergieën, de enthalpieën voor de oppervlakreacties en de chemisorptie-
enthalpieën voor benzeen en waterstof wordt uitgevoerd. De enthalpieën voor de
oppervlakreacties, namelijk ∆H(0,2) en ∆H(2,2), worden echter niet significant geschat. De
activeringsenergie EA(1,2) ligt in de omgeving van de getabelleerde t-waarde. De berekende t-
waarden voor de overige simultaan geschatte parameters bevestigen de individuele
significantie van deze parameters (Tabel 5-1 tot Tabel 5-3).
Tussen de geschatte parameterwaarden moeten correlaties vermeden worden. Een hoge
binaire lineaire correlatiecoëfficiënt impliceert immers dat de ene parameter in direct verband
kan gebracht worden met de andere. Indien het ganse reactienetwerk wordt beschouwd zijn
echter correlaties tussen de geschatte parameters moeilijk te vermijden gezien de reeds eerder
vermelde discrepantie tussen het aantal koolwaterstofspecies op het oppervlak en het aantal
dat waargenomen wordt in de gasfase. Een simultane regressie van de 7 single-event
microkinetisch parameters resulteert in een hoge correlatiecoëfficiënt (>0.95) tussen de
chemisorptie-enthalpie voor waterstof en de enthalpieën voor de oppervlakreacties namelijk
∆H(0,2) en ∆H(2,2). Een minder negatieve waarde van de chemisorptie-enthalpie van
waterstof, corresponderend met een stijging van de concentratie van de koolwaterstoffen op

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
53
het katalysatoroppervlak, wordt gecompenseerd door een daling van de oppervlakreactie-
enthalpieën.
Verscheidene parametersets, verkregen vertrekkend van verschillende beginschattingen,
worden geëvalueerd op basis van de significantie van de regressie en de fysische relevantie
van de parameterwaarden. Een optimale parameterset wordt door een simultane regressie van
de 7 single-event microkinetische parameters onder gereparameteriseerde vorm gerealiseerd
en wordt weergegeven in Tabel 5-1 tot Tabel 5-3. Het single-event microkinetisch model met
de huidige parameterwaarden is in staat om de effecten van de temperatuur, inlaatpartieeldruk
van benzeen en waterstof en de invloed van de totaaldruk te beschrijven ( Hoofdstuk 3). Een
vergelijkende studie met de parameterwaarden uit de literatuur leidt globaal tot een
aanvaardbare tot goede overeenkomst. Tabel 5-1 geschatte activeringsenergieën en berekende pre-exponentiële factoren voor de reactiesnelheidscoëfficiënt in het SEMK model
(0,2) (1,2) (2,2)
A 1.0E+16 1.0E+16 1.0E+16
EA (kJ/mol) 62.0± 2.6 48.1 ± 46.7 66.5 ± 3.3
De kwantumchemische berekeningen van Saeys et al. (2002) leiden tot de veronderstelling dat
een dominant reactiepad aanwezig is, zie paragraaf 1.1.2. De sequentiële additie van de
waterstofatomen in de eerste drie reactiestappen van het dominant reactiepad wordt telkens
gerealiseerd in de preferentiële meta-positie ten opzichte van elkaar. De corresponderende
activeringsenergie in het SEMK model bedraagt 66.5 kJ/mol (Tabel 5-1). De
activeringsenergieën van de volgende reactiestappen zijn volgens de kwantumchemische
berekeningen significant hoger omdat er geen atomaire additie van waterstof meer mogelijk is
in de meta-positie. De geschatte waarde voor de corresponderende laatste 3
hydrogenatiestappen van het kwantumchemisch berekend reactiepad is echter 62.0 kJ/mol
(Tabel 5-1). Uitgaande van de betrouwbaarheidsintervallen wordt er dus geen significant
verschil waargenomen tussen de activeringsenergieën in het volgens de kwantumchemische
berekeningen verkregen dominante reactiepad. Bovendien volgt uit dit dominant reactiepad
dat de activeringsenergie voor de overige waterstofadditiestappen, namelijk EA(1,2)
significant hoger zou zijn dan de overige activeringsenergieën. De geschatte waarde voor
EA(1,2) is hiermee niet in overeenstemming, hoewel het corresponderende individuele 95%
betrouwbaarheidsinterval heel breed is. De hogere significantie van EA(2,2) en EA(0,2) wordt
niet verklaard door het belangrijker zijn van de corresponderende elementaire stappen in het
SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
54
parallelle deel van het reactienetwerk, zie Figuur 4-3, maar de invloed van deze parameters op
de vormingsnelheid van cyclohexaan via de eerste en de laatste hydrogenatiestap is
aanzienlijk. Algemeen wordt vastgesteld dat de geschatte activeringsenergieën lager liggen
dan de kwantumchemisch berekende waarden. Echter de optimale parameterset in het model
van Thybaut et al. (2002) resulteert in activeringsenergieën in het bereik van 40 tot 50 kJ/mol.
De schattingen voor de activeringsenergieën resulteren in intermediaire waarden tussen beide
modellen zoals voorgesteld in paragraaf 1.1.1 en 1.1.2. Consistente waarden met de huidige
geschatte parameterwaarden voor de activeringsenergieën worden gerapporteerd door
Vagenende (2004) en Debruyne (2005).
De geschatte enthalpieën en de berekende pre-exponentiële factoren voor de
evenwichtscoëfficiënt van de oppervlakreacties in het SEMK model wordt in Tabel 5-2
weergegeven. De geschatte parameterwaarden zijn niet significant geschat, zoals eerder
vermeld. Tabel 5-2 geschatte enthalpieën en berekende pre-exponentiële factoren voor de evenwichtscoëfficiënt van de
oppervlakreacties in het SEMK model, (*) = berekend via thermodynamische consistentie
(0,2) (1,2) (2,2)
exp(∆S/R) 1 1 * 1
∆H (kJ/mol) 2.2±6.2 4.3 * 6.4±7.2
Volgens de kwantumchemische berekeningen van Saeys et al. (2005) voor de hydrogenering
van benzeen op Pt bedraagt de chemisorptie-enthalpie van benzeen voor de meest reactieve
species, namelijk de hollow configuratie, -71 kJ/mol. Het Langmuir-Hinshelwood van
Thybaut et al. (2002), zie paragraaf 1.1.1, rapporteert een chemisorptie-enthalpie van -70
kJ/mol voor de modelcomponent tolueen. De chemisorptie-enthalpie van benzeen, geschat
door regressie van het SEMK model, is -64.4 kJ/mol (Tabel 5-3) en ligt binnen het bereik
zoals gerapporteerd in de literatuur. Wegens de sterke afhankelijkheid van de
bedekkingsgraad ligt de chemisorptie-enthalpie van waterstof binnen een kwantumchemisch
berekend bereik van -40 tot -90 kJ/mol. Een chemisorptie-enthalpie voor waterstof van -42
kJ/mol wordt door regressie van het model van Thybaut et al. (2002) geschat. De geschatte
chemisorptie-enthalpie van waterstof resulteert in een intermediaire waarde van -60.0 kJ/mol
(Tabel 5-3).

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
55
Tabel 5-3 geschatte enthalpieën en berekende pre-exponentiële factoren voor de chemisorptie-evenwichtscoëfficiënt in het SEMK model, (*) = berekend via thermodynamische consistentie
C6H6 H2 C6H12
∆H (kJ/mol) -64.4 ± 6.4 -60.0 ± 11.3 -4.8 *
exp(∆S/R) 1.0E-12 1.0E-11 1.2E-25 *
De oppervlakconcentraties hebben een grote invloed op de uitlaatdebieten van het
reactieproduct namelijk cyclohexaan. In deze context wordt door het single-event concept het
volledige reactienetwerk beschouwd. Het laat toe om de concentraties van de verschillende
geadsorbeerde koolwaterstoffracties te berekenen. Gebaseerd op de parameterwaarden, zoals
gerapporteerd in Tabel 5-1 tot Tabel 5-3, wordt
het oppervlak voor 60% bedekt door de som van
de verschillende koolwaterstofspecies en een
fractie van 25% wordt ingenomen door
waterstof. De rest van het oppervlak is vrij. Door
een stijging van de temperatuur neemt de
modelberekende fractie van waterstof en vrije
centra toe, in tegenstelling tot de fractie van het
oppervlak dat bezet wordt door de verschillende
koolwaterstofspecies. De verdeling van de
verschillende fracties van de koolwaterstoffen op
het oppervlak is gelijkmatig (Tabel 5-1). De
berekende concentratie van cyclohexaan op het
oppervlak is verwaarloosbaar ten gevolge van de lage chemisorptie-evenwichtscoëfficiënt. De
oppervlakconcentraties berekent door het SEMK model met de parameterwaarden uit Tabel
5-1 tot Tabel 5-3 kennen een sterke overeenkomst met de modelberekende
oppervlakconcentraties van Thybaut et al. (2002), zie paragraaf 1.1.1. De bedekkingsgraad
van tolueen en waterstof bedraagt respectievelijk 60% en 20%. De overige actieve centra zijn
niet bedekt. De concentraties van de partieel gehydrogeneerde species in het model van
Thybaut et al. (2002) zijn verwaarloosbaar. De aanwezigheid van gechemisorbeerd tolueen en
waterstof als de grootste fractie op het katalysatoroppervlak is gemeenschappelijk met het
2 De nummering van de reactie-intermediairen correspondeert met de nummering van Figuur 4-3
Tabel 5-4 berekende fracties van verschillende koolwaterstofspecies (p = 10 bar, xHC = 0.02, H2/HC = 5, T = 448 K, W/F = 23.04 kgkat s mol-1 )
geadsorbeerd benzeen 9.89 %
reactie-intermediair 22 14.71 %
reactie-intermediair 3 12.84 %
reactie-intermediair 4 7.24 %
reactie-intermediair 5 3.62 %
reactie-intermediair 6 11.19 %
reactie-intermediair 7 12.71 %
reactie-intermediair 8 1.21 %
reactie-intermediair 9 9.75 %
reactie-intermediair 10 5.58 %
reactie-intermediair 11 2.79 %
reactie-intermediair 12 8.48 %

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
56
model van Saeys et al. (2005). Dit is echter niet in overeenstemming met het berekende
fracties zoals weergegeven in Tabel 5-4. De berekende concentratie van benzeen is niet
significant hoger dan de concentraties van de reactie-intermediairen. Echter uitgaande van de
geschatte activeringsenergieën uit Tabel 5-1 zijn de oppervlakintermediairen, die betrekking
hebben op k(1,2), de meest aanwezige koolwaterstofspecies.
Bij de parameterschattingen is steeds voldaan aan de thermodynamische consistentie, in het
bijzonder deze met betrekking tot de gehele hydrogeneringscyclus. De pre-exponentiële factor
voor de chemisorptie-evenwichtscoëfficiënt van cyclohexaan (Tabel 5-3), berekend via de
thermodynamische consistentie, wijkt echter af ten opzichte van deze berekend via statistische
thermodynamica. Een uiterst lage, fysisch niet-relevante waarde wordt berekend gebruik
makend van de huidige parameterwaarden (Tabel 5-1 tot Tabel 5-3). Deze waarde is echter in
overeenstemming met de modelhypothese van een snelle, irreversibele chemisorptie van
cyclohexaan, zoals die door beide modellen in paragraaf 1.1.1 en 1.1.2 wordt gebruikt. De
fysische niet-relevantie van de thermodynamisch berekende chemisorptie-enthalpie van
cyclohexaan werd eerder al gerapporteerd door Vagenende (2004) en Debruyne (2005). Het
berekende bereik voor de pre-exponentiële factoren, zoals weergegeven in Tabel 4-2 in
combinatie met vergelijking 5.1, leidt tot de conclusie dat aan een fysisch relevante waarde
voor de chemisorptie-evenwichtscoëfficiënt van cyclohexaan enkel kan voldaan worden
indien de pre-exponentiële factoren van de oppervlakevenwichtscoëfficiënten en van de
chemisorptie-evenwichtscoëfficiënten van benzeen en waterstof zich bevinden tegen de
uiteinden van het berekend bereik. Deze stijging van de pre-exponentiële factoren, om aan de
fysische relevantie te voldoen van de thermodynamisch berekende parameters, moet
gecompenseerd worden door het dalen van de chemisorptie-enthalpieën van waterstof en
benzeen. De thermodynamische consistentie (5.1) resulteert in dat geval echter in een minder
negatieve of zelfs positieve chemisorptie-enthalpie voor cyclohexaan. De stijging van de
chemisorptie-enthalpie van cyclohexaan kan door een daling van de oppervlakreactie-
enthalpieën worden gecompenseerd. Indien de oppervlakreactie-enthalpieën dalen om aan de
thermodynamische consistentie (5.1) te voldoen dan resulteert dit in een minder negatieve
waarde van de chemisorptie-enthalpie van waterstof, omwille van de corresponderende
stijging van de koolwaterstofspecies. Het effect van de waterstofchemisorptie op
thermodynamische consistentie is echter beduidend groter waardoor de beoogde fysisch
relevante waarde voor de chemisorptie-enthalpie van cyclohexaan niet bereikt wordt. Het
vermogen van de activeringsenergieën om de experimenteel waargenomen reactiesnelheden

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
57
binnen het perspectief van de voorafgaande redenering te benaderen is ontoereikend.
Verschillende parametersets werden getest op basis van de bovenstaande bemerkingen. Echter
de significantie van de regressie kan niet meer gegarandeerd worden door de
activeringsenergieën aan te passen. De aanwezigheid van correlatie tussen de verschillende
modelparameters komt sterk naar boven wanneer getracht wordt een significant stel
parameters te schatten, die bovendien leiden tot een fysisch relevante waarde voor de
chemisorptie-evenwichtscoëfficiënt voor cyclohexaan.
5.2 Modeladequaatheid en globale significantie van het SEMK model
De adequaatheid van het model kan worden beoordeeld op basis van de afwijkingen tussen de
experimentele waarde en de modelberekende afhankelijke variabele. Een echte schatting van
de foutvariantie onafhankelijk van de adequaatheid van het model wordt berekend op basis
van 4 herhalingsexperimenten (Appendix B) bij instelcondities: p = 10 bar, xHC = 0.02, H2/HC
= 5, T = 448 K, W/F = 23.04 kgkat s mol-1. De F-test, waarbij de modeladequaatheid wordt
getoetst op basis van de verhouding van de ‘lack of fit’ kwadratensom gedeeld door het aantal
vrijheidsgraden en een echte schatting van de foutvariantie, leidt tot het aanvaarden van de
nulhypothese. Het SEMK model is adequaat en vertoont geen systematische afwijkingen of
een tekort aan aanpassingsvermogen.
5)F(33,3;0.9 F2.845)F(40,3;0.92.440.340.84
s1-n-p-n
omkwadratens LOF
F tab2e
eber =<=<=== ( 5-3)
De totale kwadratensom is een maat voor de variantie van de waarnemingen. Deze variantie
kan gedeeltelijk verklaard worden door het model en komt overeen met de
regressiekwadratensom. Het deel dat niet toegedicht kan worden aan het model wordt
weergeven door de residuele kwadratensom. De verhouding van de regressie kwadratensom
op de residuele kwadratensom gedeeld door hun corresponderende vrijheidsgraden is een F-
verdeelde veranderlijke en tevens een maat voor de globale significantie van het model. Het
simultaan schatten van 7 parameters resulteert in een F-waarde van ± 5 103. Het
pariteitsdiagramma corresponderend met de parameterwaarden (Tabel 5-1 tot Tabel 5-3),
geschat door regressie van het SEMK model, wordt weergegeven in Figuur 5-1. De relatieve
afwijking bevindt zich in de meeste gevallen binnen de 5%.

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
58
05
101520253035404550
0 5 10 15 20 25 30 35 40 45 50
Fexp(µmol s-1)
F ber
(µm
ol s
-1)
Figuur 5-1 pariteitsdiagramma voor de uitlaatdebieten van cyclohexaan; lijn: experimenteel, punten: berekend
gebaseerd op het SEMK model voor de katalytische hydrogenering van aromaten op Pt met de parameters van Tabel 5-1 tot Tabel 5-3
De modelberekende waarden benaderen goed de experimenteel waargenomen uitlaatdebieten
van cyclohexaan. De afwijking ten opzichte van de bissectrice is minimaal. In de figuren van
paragraaf 3.2, waar het effect van de verschillende instelvariabelen op de reactiesnelheid is
nagegaan, wordt de temperatuur steeds als parameter uitgezet. Het simuleren van het effect
van de temperatuur op de reactiesnelheid voor de hydrogenering van benzeen is cruciaal
aangezien de experimentele waarneming algemeen wordt verklaard door een combinatie van
de reactiesnelheidscoëfficiënt en de bedekkingsgraad van de reactie-intermediairen en hun
corresponderende temperatuurafhankelijkheid. Indien het verschil tussen het experimenteel en
het modelberekend uitlaatdebiet van cyclohexaan wordt uitgezet in functie van de
temperatuur, wordt er geen systematische fout waargenomen. Het effect van de temperatuur
op de reactiesnelheid wordt door het model accuraat gesimuleerd (Figuur 5-2). Een
gelijkaardige verdeling van het verschil tussen het experimenteel en het modelberekend
uitlaatdebiet van cyclohexaan, namelijk de afwezigheid van een systematische afwijking in
functie van de instelvariabele, wordt opgemerkt voor de andere instelvariabelen. De
afwijkingen zijn dus onafhankelijk van de instelvariabelen.

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
59
-3
-2
-1
0
1
2
3
420 430 440 450 460 470 480 490 500
temperatuur (K)
e i
Figuur 5-2 verschil tussen het experimenteel en het modelberekend uitlaatdebiet van cyclohexaan in functie van
temperatuur
5.3 Progressiemogelijkheden tot verdere verfijning van het huidige fundamenteel model binnen het single-event concept
De berekende oppervlakconcentraties van benzeen en de verschillende reactie-intermediairen
(Tabel 5-4) suggereren dat de contributie van elke elementaire stap tot de globale
reactiesnelheid niet verwaarloosbaar is. Het in beschouwing nemen van het volledige
reactienetwerk lijkt dus zeker zinvol. Echter het SEMK model als een meer fundamenteel
model heeft nog progressiemarge met betrekking tot de graad van detail bij de beschrijving
van de elementaire stappen in het model, in het bijzonder bij de chemisorptiestappen.
De kwantumchemische berekeningen voor de chemisorptie van benzeen op Pt (Saeys et al.,
2002) tonen aan dat er naast de reactieve chemisorptieconfiguratie, de hollow (30)
configuratie, ook een tweede chemisorptietoestand, de bridge (0) configuratie, een effect kan
hebben op de oppervlakconcentraties van de verschillende reactie-intermediairen (Figuur
5-3). De kwantumchemische berekeningen resulteren in chemisorptie-enthalpieën voor de
hollow configuratie en de bridge configuratie van respectievelijk -71 kJ/mol en -102 kJ/mol.
Enkel de configuratie met de minst negatieve chemisorptie-energie wordt in het bestaande
model in beschouwing genomen.

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
60
Figuur 5-3 schematisch overzicht van de verschillende chemisorptieconfiguraties van benzeen op Pt(111) oppervlak
(hollow- fcc30 is niet weergegeven)
Een tweede aspect leidt tot een model, dat naast het in rekening brengen van de preferentiële
chemisorptiestructuren, ook het aantal Pt-atomen van de corresponderende
chemisorptieconfiguraties in beschouwing neemt. Het aantal Pt-atomen dat betrokken is bij de
chemisorptieconfiguratie van de meest reactieve species van benzeen bedraagt drie. Ook voor
de chemisorptieconfiguratie van de reactie-intermediairen is een totaal aantal van drie actieve
centra noodzakelijk.
Een derde argument behelst het in rekening brengen van de afhankelijkheid van de
chemisorptie-enthalpie van waterstof van de totale bedekkingsgraad van het oppervlak
(Podkolzin et al. 2001). De afhankelijkheid van deze bedekkingsgraad is het gevolg van
repulsieve interacties. De industriële condities van het hydrokraken en dus de hydrogenering
van aromaten omvatten hoge waterstofpartieeldrukken. Een minder negatieve chemisorptie-
enthalpie van waterstof wordt, onder industriële condities voor het hydrokraken en de
overeenkomstige hydrogeneringsreacties van aromaten, verwacht ten gevolge van deze hoge
partieeldruk. De implementatie in het model van deze afhankelijkheid bezit waarschijnlijk de
mogelijkheid om de significantie van het model verder te verbeteren. Een lineaire
afhankelijkheid van de bedekkingsgraad kan vooropgesteld worden.
Het onderstellen van semi-competitieve chemisorptie van beide reagentia is een plausibele
mogelijkheid omwille van het verschil in grootte tussen de aromatische component en
waterstof. Het ontbreken van de fysische relevantie voor de chemisorptie-
evenwichtscoëfficiënt van cyclohexaan, berekend via de thermodynamische consistentie,
binnen het huidige model is een aandachtspunt. De implementatie van de semi-competitieve
chemisorptie als modelhypothese is in dat opzicht een mogelijke uitweg. In het huidige model
wordt verondersteld dat beide reagentia chemisorberen op één actief centrum. Het verschil in

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
61
grootte tussen benzeen en waterstof in rekening gebracht worden door de pre-exponentiële
factoren van beide chemisorptie-enthalpieën (Saeys et al., 2005). In de voorafgaande
paragrafen wordt aangehaald dat het bereik van de pre-exponentiële factoren van het huidige
SEMK model moeilijk resulteert in een fysische relevante waarde voor de chemisorptie-
eventwichtscoëfficiënt van cyclohexaan. In deze context kan het in beschouwing nemen van
semi-competitieve chemisorptie leiden tot een significant model met een fysisch relevante
waarde voor de chemisorptie-evenwichtscoëfficiënt van cyclohexaan .
5.4 Semi-competitieve chemisorptie
De beschrijving van de simultane chemisorptie van waterstof en de aromatische component
op het katalysatoroppervlak is van belang voor de formulering van het kinetisch model en
heeft een aanzienlijk effect op de concentraties van de verschillende fracties op het
katalysatoroppervlak én dus op de reactiesnelheid. Het onderstellen van semi-competitieve
chemisorptie van waterstof en de aromatische species blijkt in deze context een plausibele
mogelijkheid omwille van het verschil in grootte tussen beide reagentia.
De hypothese van competitieve chemisorptie tussen benzeen en waterstof is de
modelveronderstelling van het huidig opgestelde SEMK model (Vagenende, 2004). Een
zekere vorm van competitie tussen de gechemisorbeerde aromatische species en waterstof
wordt experimenteel bevestigd. De chemisorptie van benzeen blijkt echter volgens
experimentele waarnemingen en kwantumchemische berekeningen ongeordend (Saeys et al.,
2002; Morin et al., 2003). Wegens het verschil in grootte tussen de aromatische species en
waterstof blijft, indien het oppervlak verzadigd is met de aromatische component, steeds een
beperkt aantal interstitiële actieve centra beschikbaar voor de kleinere molecule waterstof
(Figuur 5-4). Het optreden van competitieve of niet-competitieve chemisorptie blijkt sterk
afhankelijk te zijn van de experimentele instelcondities. Bij lage temperaturen is het
metaaloppervlak volledig verzadigd met aromatische species. Een beperkt aantal actieve
centra blijft steeds beschikbaar voor de dissociatieve chemisorptie van waterstof. De
bedekkingsgraad van het katalysatoroppervlak zal afnemen indien de temperatuur stijgt. Bij
een zekere temperatuur is het oppervlak niet langer volledig verzadigd met aromatische
species. Competitieve chemisorptie wordt waargenomen aangezien de actieve centra
beschikbaar worden voor waterstof. Nog hogere temperaturen leiden tot een lage
bedekkingsgraad zodat geen competitie meer wordt waargenomen tussen beide species.

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
62
Figuur 5-4 semi-competitieve chemisorptie
Een semi-competitief chemisorptiemodel wordt door Salmi et al. (2004) opgesteld om de
chemisorptie op een fysisch aanvaardbare manier te beschrijven. De hypothesen van het
model van Salmi et al. (2004) onderscheiden zich vooral door de onderstelling van semi-
competitieve chemisorptie.
1. dissociatieve H2 chemisorptie
2. semi-competitieve chemisorptie: H2 en het aromaat chemisorberen respectievelijk op i
en m Pt-atomen
3. geen snelheidsbepalende stap: reactiesnelheden tussen gechemisorbeerde species
worden aan elkaar gelijkgesteld
4. op het oppervlak wordt geen partieel gehydrogeneerd species beschouwd
5. waterstof en het aromaat chemisorberen in quasi-evenwicht
6. desorptie van het gehydrogeneerde product is snel en irreversibel
In Figuur 5-5 worden de elementaire reactiestappen van het semi-competitief reactiemodel
van Salmi (2004) weergegeven. De preferentiële chemisorptieconfiguratie van benzeen
bepaalt het aantal Pt-atomen (m) betrokken bij de moleculaire chemisorptie. Waterstof
chemisorbeert dissociatief op twee actieve centra (i=2). Een specifieke balans wordt opgesteld
voor de adsorptiecentra van benzeen. De som van de concentratie van de bezette ( '*Ac ) en
onbezette ( '*c ) benzeenchemisorptiecentra wordt gelijkgesteld aan de verhouding van het
verschil tussen het totale aantal Pt-atomen en het aantal gechemisorbeerde waterstofatomen en
het noodzakelijk aantal actieve centra met betrekking tot de chemisorptieconfiguratie voor
benzeen (m).

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
63
m
)cc(cc *Ht
'*'*A−α
=+ ( 5-4)
In het rechterlid wordt de waarschijnlijkheid dat de Pt-atomen een centrum vormen (α ) in
rekening gebracht.
A + *’ A*’ (*’ = m*)
H2 + i * i H* (i = 2)
A*’ + j H* AHj*’ +j * (j = 6)
AH6*’ AH6 + *’
Figuur 5-5 elementaire reactiestappen bij semi-competitieve chemisorptie (Salmi et al., 2004)
In het oorspronkelijke kinetisch model wordt aangenomen dat benzeen, cyclohexaan en alle
partieel gehydrogeneerde species chemisorberen op één actief centrum van het
metaaloppervlak. Binnen het single-event concept is het een betrachting om de preferentiële
chemisorptieconfiguraties in rekening te brengen. Het schatten van het aantal Pt-atomen van
de betreffende chemisorptiestructuren leidt tot een model met een hogere graad van detail
voor de beschrijving van de chemisorptie. Kwantumchemische berekeningen (Saeys et al.,
2002) rapporteren twee chemisorptiestructuren voor benzeen namelijk de bridge- en
hollowconfiguratie met respectievelijke chemisorptie-enthalpieën: 102 kJ/mol en 71kJ/mol,
zie paragraaf 1.1.2. Beide waarden zijn in overeenstemming met TPD experimenten (Xu et
al.,1994). Het berekend aantal Pt-atomen voor de meest reactieve chemisorptieconfiguratie
(hollow) is drie. De waterstofmolecule chemisorbeert dissociatief op twee Pt-atomen. Een
meer fundamenteel model kan verwezenlijkt worden door beide types van gechemisorbeerd
benzeen te implementeren in het model. Echter in de volgende uitbreiding is enkel de
kinetisch relevante hollowconfiguratie beschouwd.
De preferentiële chemisorptiestructuur en het corresponderend aantal Pt atomen wordt in
rekening gebracht. De berekening van de concentratie van de vrije Pt doubletten en tripletten
is noodzakelijk. Het aantal buren van een actief centrum op een Pt(111) oppervlak bedraagt

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
64
zes (Figuur 5-3) met een kans tot
*
cc dat het actief centrum vrij is. De concentratie van de
doubletten, betrokken bij de dissociatieve chemisorptie van waterstof, wordt via de
onderstaande vergelijking berekend. Het dubbeltellen van het aantal doubletten van de vrije
centra wordt vermeden. Vanuit fysisch standpunt is deze berekening een kunstmatige
benadering, echter ze stelt ons in staat om het effect van de semi-competitieve chemisorptie
van beide reagentia op de parameters te begroten.
tot
2*tot
**
doublet cc3
2
)cc6(c
c == ( 5-5)
Een analoge uitdrukking wordt berekend voor een vrij triplet:
2tot
3*tot
*doublet
tripletcc2
3
)cc
2(cc == ( 5-6)
Het stelsel van vergelijkingen, opgelost door de subroutine DNSQE, wordt uitgebreid met de
volgende vergelijking:
0c3ccc1pK3c
cc2
pK3cc2
pK312
1i*itot*
totHH*2
tot
3*
AHAH2tot
3*
AA 2266=
−−++
+
∑=
( 5-7)
De concentratie van het totaal aantal actieve centra wordt weergegeven door totc en de
concentratie van de reactie-intermediairen op het katalysatoroppervlak wordt gesymboliseerd
door *ic . Een vooropgestelde fractie van 10% vrije centra resulteert in fysisch relevante
oppervlakconcentraties. Uit de concentratie van de vrije centra kunnen rechtstreeks de
respectievelijke oppervlakconcentraties van benzeen, cyclohexaan en waterstof berekend
worden Via:
AA2tot
3*
*A pKcc
2c = ( 5-8)
66*6
AHAH2tot
3*
AH pKcc
2c = ( 5-9)
tot
HH**H c
pK3cc 22= ( 5-10)
De implementatie van de semi-competitieve chemisorptie hypothese drijft de waarden van de
modelparameters in de verkeerde richting en biedt heden weinig perspectief met het oog op
het evolueren naar een fysisch relevante waarde voor de thermodynamisch berekende
chemisorptie-evenwichtscoëfficiënt voor cyclohexaan.

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
65
Echter de kwantumchemische berekeningen (Saeys et al., 2002) voor de chemisorptie van
benzeen suggereren, naast de reactieve chemisorptietoestand van benzeen, een tweede
chemisorptieconfiguratie (bridge) met een belangrijke invloed op de oppervlakconcentraties
van de verschillende reactie-intermediairen. Naar analogie met vergelijkingen 5.5 en 5.6
wordt de concentratie van de vrije Pt kwartetten, die noodzakelijk zijn voor de bridge
chemisorptieconfiguratie van benzeen, bepaald.
3tot
4*
kwartetc2c3
c = ( 5-11)
De balans over de actieve centra kan uitgebreid worden met de bridge configuratie voor de
chemisorptie van benzeen. In de onderstaande vergelijking worden de symbolen met
betrekking tot de bridge configuratie voorgesteld door een accent.
0c3ccc
1pK3ccc2
pK3cc2
pK3c2c3
pK412
1i*itot*
totHH*2
tot
3*
AHAH2tot
3*
AA3tot
4*
A'A 2266=
−−++
+
+
∑=
( 5-12)
De concentratie op het katalysatoroppervlak kan voor de bridge configuratie berekend worden
aan de hand van:
3tot
4*
A'A'Ac2c3
pKc = ( 5-13)
De optimale parameterset voor het model is nog niet gevonden. Een inschatting van het model
en de invloed op de oppervlakconcentratie, met het oog op de fysische relevantie, is een
mogelijke verbetering naar de toekomst toe. Voor de evaluatie van de mogelijke
verbeteringen is de nieuwe dataset immers voldoende groot.
5.5 Besluit
De beschikbare dataset stelde Debruyne (2005) niet in staat om alle parameters uit het SEMK
model opgesteld door Vagenende (2004) significant te schatten. Om de single-event
microkinetische parameters uit het model voor de hydrogenering van benzeen op platina op
een significante manier te kunnen schatten, was een uitgebreider stel aan experimentele
gegevens nodig. In het SEMK model wordt, aan de hand van de thermodynamische
consistentie en de berekende pre-exponentiële factoren voor de hydrogenering van benzeen op
basis van de transitietoestandstheorie en de statistische thermodynamica, het aantal te schatten
parameters gereduceerd tot 7. Tot op heden werden in de kinetische modellen voor de
hydrogenering van aromaten ontwikkeld aan het LPT nooit meer dan drie modelparameters
simultaan geschat. Met het huidige model en de beschikbare dataset is dit wel mogelijk. De
simultane regressie van alle parameters resulteert in significante waarden uitgezonderd voor

SEMK model regressie voor de hydrogenering van benzeen op Pt/ZSM-22
66
de enthalpieën van de oppervlakreacties en EA(1,2). Een vergelijkende studie met de
parameterwaarden uit de literatuur leidt globaal tot een aanvaardbare tot goede overeenkomst.
Uitgaande van de betrouwbaarheidsintervallen wordt er geen significant verschil
waargenomen tussen de eerste drie en de laatste drie geschatte activeringsenergieën
respectievelijk de parameters EA(2,2) en EA(0,2) uit het SEMK model. De chemisorptie-
enthalpie van benzeen, geschat door regressie van het SEMK model, is -64.4 kJ/mol. De
geschatte chemisorptie-enthalpie van waterstof resulteert in een waarde van -60.0 kJ/mol
binnen het kwantumchemisch berekend bereik van -40 tot -90 kJ/mol (Saeys et al., 2005). Op
basis van de parameterwaarden, zoals gerapporteerd in Tabel 5-1 tot Tabel 5-3, wordt het
oppervlak voor 60% bedekt door de som van de verschillende koolwaterstofspecies en een
fractie van 25% wordt ingenomen door waterstof. De rest van het oppervlak is vrij. Op het
katalysatoroppervlak wordt geen dominant koolwaterstofspecies waargenomen. De
contributie van elke elementaire stap tot reactiesnelheid voor de vorming van cyclohexaan is
niet verwaarloosbaar. Het in rekening brengen van het ganse reactienetwerk is dus zinvol.
Een F-waarde van ± 5 103 voor de significantie van de regressie wordt berekend. Het SEMK
model, waarbij bepaalde single-event snelheidscoëfficiënten gebaseerd op de configuratie van
het reactief centrum tijdens de H-addities aan elkaar gelijkgesteld (Saeys et al., 2002) worden
en de contributie van elke elementaire stap in rekening wordt gebracht, is adequaat gebaseerd
op de berekende F-waarde voor de modeladequaatheid. De pre-exponentiële factor voor de
chemisorptie-evenwichtscoëfficiënt van cyclohexaan, berekend via de thermodynamische
consistentie, wijkt af ten opzichte van deze berekend via statistische thermodynamica. Een
uiterst lage, fysisch niet-relevante waarde wordt berekend gebruik makend van de huidige
parameterwaarden (Tabel 5-1 tot Tabel 5-3). Deze waarde is echter in overeenstemming met
de modelhypothese van een snelle, irreversibele chemisorptie van cyclohexaan, zoals die door
beide modellen in paragraaf 1.1.1 en 1.1.2 wordt gebruikt. De beschrijving van de semi-
competitieve chemisorptie van waterstof en benzeen lijkt in deze context een plausibele
mogelijkheid. Een optimale parameterset voor dit model is nog niet gevonden. Met het oog op
de fysische relevantie lijkt in eerste instantie deze modelhypothese weinig verbetering te
brengen.

67
Hoofdstuk 6 Algemeen besluit
Om de single-event microkinetische parameters uit het model voor de hydrogenering van
benzeen op een Pt/ZSM-22 katalysator op een significante manier te kunnen schatten, was een
uitgebreider stel aan experimentele gegevens nodig. In overeenstemming met vroeger werk
werd als functie van de temperatuur een maximum waargenomen van de
hydrogeneringssnelheid. Het maximum verschuift naar hogere temperaturen bij hogere
drukken. Een hogere temperatuur is immers vereist om de oppervlakconcentraties van de
reagentia in het algemeen en van benzeen in het bijzonder te verlagen. Een stijging van
atomair geadsorbeerd waterstof heeft een positief effect op de reactiesnelheid. Een
tegengesteld effect wordt waargenomen voor de invloed van de bedekkingsgraad van
koolwaterstoffen op de reactiesnelheid. Het single-event microkinetisch model voor de
hydrogenering van benzeen is in staat de experimentele gegevens op een adequate manier te
beschrijven. Zowel de effecten van de temperatuur, waterstofpartieeldruk en
benzeenpartieeldruk worden op een voldoende manier gesimuleerd.
Op basis van de configuratie van het reactief centrum tijdens H-addities (Saeys et al, 2002)
worden bepaalde single-event snelheidscoëfficiënten aan elkaar gelijk gesteld. De single-
event snelheidscoëfficiënten )n,m(k~ worden gekarakteriseerd door de verzadigingsgraad (m)
van de naaste buren en de vertakkingsgraad (n) van het C-atoom, dat een waterstofadditie of
waterstofabstractie ondergaat. Pre-exponentiële factoren voor de verschillende snelheids- en
evenwichtscoëfficiënten in het SEMK model worden berekend op basis van de
transitietoestandstheorie en statistische thermodynamica. Thermodynamische consistentie op
het katalysatoroppervlak en voor de gehele thermodynamische cyclus laten toe het aantal te
schatten parameters te reduceren tot 7. Een simultane regressie van alle parameters leidt
globaal tot een aanvaardbare tot goede overeenkomst. De chemisorptie-enthalpie van
benzeen, geschat door regressie van het SEMK model, is -64.4 kJ/mol. De geschatte
chemisorptie-enthalpie van waterstof resulteert in een waarde van -60.0 kJ/mol binnen het
kwantumchemisch berekend bereik van -40 tot -90 kJ/mol (Saeys et al., 2005). De F-waarde
voor de significantie van het model bedraagt ± 5 103. Het oppervlak wordt voor 60% bedekt

68
door de som van de verschillende koolwaterstofspecies en een fractie van 25% wordt
ingenomen door waterstof. De rest van het oppervlak is vrij. Op het katalysatoroppervlak
wordt geen dominant koolwaterstofspecies waargenomen. De contributie van elke elementaire
stap tot reactiesnelheid voor de vorming van cyclohexaan is niet verwaarloosbaar. De pre-
exponentiële factor voor de chemisorptie-evenwichtscoëfficiënt van cyclohexaan, berekend
via de thermodynamische consistentie, is een uiterst lage waarde, hetgeen overeenkomt met
een snelle, irreversibele desorptie. De beschrijving van de semi-competitieve chemisorptie
van waterstof en benzeen, rekening houdend met het verschil in ruimte ingenomen op het
oppervlak door deze moleculen, lijkt in deze context een plausibele mogelijkheid. Een eerste
stap in deze richting werd gezet als onderdeel van dit eindwerk.

69
Appendix A: Transportlimitaties
De studie van de kinetiek van de hydrogenering van benzeen veronderstelt de afwezigheid
van transportlimitaties (EUROKIN). Temperatuur- en concentratiegradiënten moeten
vermeden worden op reactorschaal van de Berty reactor, maar ook in en rondom de
katalysatorkorrels. De intrinsieke kinetiek van reacties wordt in het algemeen gemeten in
laboratoriumopstellingen, waar de reactiecondities goed instelbaar zijn en het
stromingspatroon een éénduidige interpretatie van de gegevens mogelijk maakt. Interne
temperatuurgradiënten zijn in reactor op laboratoriumschaal relatief onbelangrijk wegens de
kleine korreldiameter en de goede warmtegeleiding op de katalysator. Aan het criterium voor
de afwezigheid van externe temperatuurgradiënten is minder snel voldaan. Het optreden van
concentratiegradiënten is eerder kritisch intern dan extern.
1.1 Externe gradiënten Het niet-optreden van externe massatransportlimitaties wordt gegarandeerd indien voldaan
wordt aan (A-1). De waargenomen productiesnelheid mag maximaal 5% afwijken van de
werkelijke productiesnelheid met n de partiële reactie-orde. Daartoe moet gelden dat:
n05.0
C
CC
b,A
s,Ab,A<
− (A-1)
Het criterium wordt omgevormd tot alle grootheden waarneembaar of berekenbaar zijn:
n05.0
Cakr
b,AsA,f
v < (A-2)
Het veronderstellen van de afwezigheid van externe temperatuurgradiënten correspondeert
met het criterium A-3. De reactiesnelheid corresponderend met de temperatuur aan het
uitwendig oppervlak mag maximum 5% afwijken van de reactiesnelheid bij de
bulktemperatuur om afwezigheid van externe temperatuurgradiënten te verzekeren.
05.0r
rr
b,w
b,ws,w<
− (A-3)
Mits substitutie wordt een criterium opgesteld met enkel waarneembare/berekenbare
grootheden.

Appendix A: Transportlimitaties
70
a
b
b
rvp
ETR
3.0T
Hrd<
α
∆− (A-4)
1.2 Interne gradiënten
Indien de benuttingsgraad voldoende groot is ( 950.>η ) kan de poriediffusielimitering als
verwaarloosbaar beschouwd worden. Deze voorwaarde is voldaan voor kleine waarden van de
Thiele-modulus ( 1<<ϕ ). Echter de Thiele-modulus veronderstelt kennis met betrekking tot
de reactiekinetiek. Aangezien de Weisz-modulus is gebaseerd op de gemeten
productiesnelheid wordt deze modulus vaak in deze context gebruikt. De Weisz-modulus
wordt gedefinieerd als: 2ϕη=Φ (A-5)
De definitie van de Weisz-modulus wordt voor een nde orde kinetiek weergegeven door:
( )s,Aa,eff
2v
obswp
CDa2
r1n ρ+=Φ (A-6)
Het criterium voor de afwezigheid van interne massatransportlimitaties voor een nde orde
kinetiek en bolvormige katalysatordeeltjes resulteert in het volgende criterium:
( ) 1CD36
rd2
1n
s,Aa,eff
v2p <<
+ (A-7)
Het verwaarlozen van interne temperatuurgradiënten voor een bolvormige geometrie is
gerechtvaardigd indien aan het volgende criterium is voldaan:
a
s
sp
rpobsw
2p
ETR
0.3T
Hrd<
λ
∆−ρ (A-8)
2. Resultaten
De intrinsieke kinetiek is bestudeerd in een gasfase Berty-reactor met een Pt/ZSM-22
katalysator met 0,5 gew% Pt. Met betrekking tot de fysische eigenschappen van de
fluïdumfase wordt een gewogen gemiddelde van de warmtegeleidingscoëfficiënt,
warmtecapacitieit, viscositeit, dichtheid en moleculair gewicht berekend uit de waarden voor
de individuele componenten. De binaire diffusiviteit van benzeen met stikstof en waterstof
wordt berekend op basis van de Fuller-Shettler-Gidings vergelijking (Perry et al;, 1997), zoals
voorzien in EUROKIN. De effectieve diffusiviteit wordt op basis van de binaire diffusiviteit,
de Knudsen diffusiviteit en de tortuositeit berekend. De informatie in verband met de
katalysator is reeds aanwezig in het programma.

Appendix A: Transportlimitaties
71
Bij de onderzochte instelcondities is aan alle criteria ruimschoots voldaan om de afwezigheid
van transportlimitaties te garanderen. Instelcondities bij hoge drukken en hoge H2/HC
verhouding zijn het meest kritisch voornamelijk met betrekking tot het optreden van
inwendige concentratiegradiënten. Echter kritische experimentele condities voor dit specifieke
criterium resulteren in waarden met een voldoende marge ten opzichte van de limietwaarde.
De hoogste conversie van benzeen wordt gerealiseerd bij de volgende experimentele
instelcondities: T = 473K, XHC = 0.02, ptot = 30 bar, H2/HC = 10 en W/FHC,0 = 23 kgkat
s/molHC.
uitwendig massatransport: 0.005 < 0.100
uitwendig warmtetransport: 0.242 < 1.550
inwendig massatransport: 0.051 < 0.080
inwendig warmtetransport: 0.020 < 1.550
De geselecteerde experimentele condities voldoen aan de opgestelde criteria zodat het
experimenteel waarnemen van de intrinsieke kinetiek gegarandeerd wordt.

72
Appendix B: Hydrogeneringsexperimenten op benzeen
exp nr. ptot (bar) XHC H2/HC T (K) W/F (kgkat s/mol HC) X r (mol/kgkat s) 1 10 0.020 5.0 448 23.04 18.92 8.41E-03 2 10 0.020 5.0 448 23.04 19.37 8.54E-03 3 10 0.020 5.0 448 23.04 19.29 8.37E-03 4 10 0.020 5.0 448 69.16 53.96 7.50E-03 5 10 0.020 10.0 448 69.16 81.59 1.26E-02 6 10 0.020 5.0 448 34.60 25.18 7.34E-03 7 10 0.020 7.5 448 69.16 80.57 1.08E-02 8 10 0.020 5.0 448 23.04 18.62 8.22E-03 9 10 0.020 5.0 473 23.42 19.15 8.17E-03 10 10 0.020 10.0 473 69.17 81.26 1.22E-02 11 10 0.020 5.0 498 23.31 8.37 3.59E-03 12 10 0.020 5.0 423 23.12 16.30 7.05E-03 13 10 0.010 5.0 448 35.67 11.29 2.90E-03 14 30 0.020 5.0 473 22.96 58.03 2.53E-02 15 20 0.020 5.0 473 23.06 40.70 1.77E-02 16 10 0.020 5.0 473 23.10 19.47 8.43E-03 17 30 0.020 5.0 473 23.08 58.62 2.54E-02 18 30 0.020 5.0 498 23.33 54.25 2.30E-02 19 20 0.020 5.0 448 23.33 38.67 1.68E-02 20 20 0.020 5.0 423 23.33 21.77 9.50E-03 21 20 0.020 5.0 498 23.32 27.33 1.17E-02 22 30 0.020 5.0 423 23.14 29.38 1.27E-02 23 30 0.020 5.0 448 23.14 50.73 2.19E-02 24 30 0.015 5.9 498 23.14 39.19 1.69E-02 25 30 0.020 10.0 423 23.14 47.90 2.07E-02 26 30 0.020 10.0 498 23.14 82.31 3.56E-02 27 30 0.020 10.0 473 23.06 82.33 3.57E-02 28 30 0.020 10.0 448 23.06 72.30 3.14E-02 29 10 0.020 5.0 423 32.28 23.69 7.14E-03 30 10 0.020 5.0 448 32.28 28.93 8.96E-03 31 10 0.020 5.0 423 32.28 23.51 7.29E-03 32 20 0.015 10.0 448 32.28 64.93 2.01E-02 33 20 0.015 10.0 473 32.28 69.30 2.15E-02 34 20 0.015 10.0 498 32.28 62.68 1.94E-02 35 20 0.015 10.7 423 32.28 52.82 1.64E-02 36 30 0.020 5.0 423 32.28 47.53 1.47E-02 37 30 0.020 5.0 448 32.28 63.74 1.97E-02 38 30 0.020 5.0 473 32.28 65.55 2.03E-02 39 30 0.020 5.0 498 32.28 60.21 1.87E-02 40 20 0.020 7.5 498 32.28 57.39 1.78E-02 41 20 0.020 5.0 498 48.41 49.24 1.02E-02 42 30 0.010 10.0 498 48.41 70.72 1.46E-02 43 20 0.020 5.0 498 48.41 48.42 1.00E-02
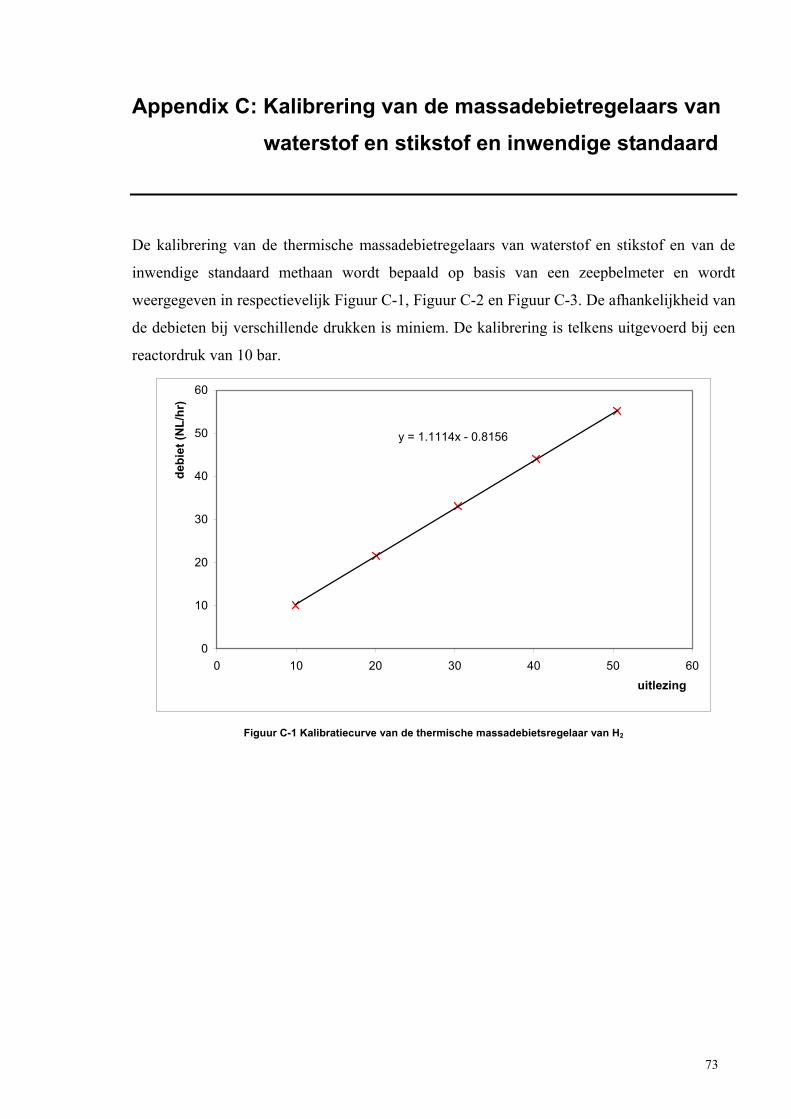
73
Appendix C: Kalibrering van de massadebietregelaars van waterstof en stikstof en inwendige standaard
De kalibrering van de thermische massadebietregelaars van waterstof en stikstof en van de
inwendige standaard methaan wordt bepaald op basis van een zeepbelmeter en wordt
weergegeven in respectievelijk Figuur C-1, Figuur C-2 en Figuur C-3. De afhankelijkheid van
de debieten bij verschillende drukken is miniem. De kalibrering is telkens uitgevoerd bij een
reactordruk van 10 bar.
y = 1.1114x - 0.8156
0
10
20
30
40
50
60
0 10 20 30 40 50 60uitlezing
debi
et (N
L/hr
)
Figuur C-1 Kalibratiecurve van de thermische massadebietsregelaar van H2

Appendix C: Kalibrering van de massadebietregelaars van waterstof en stikstof en inwendige standaard
74
y = 6.549x + 10.704
0
25
50
75
100
125
150
175
200
225
250
275
300
0 5 10 15 20 25 30 35 40 45 50uitlezing
debi
et (N
L/hr
)
Figuur C-2 Kalibratiecurve van de thermische massadebietsregelaar van N2
y = 0.085x - 0.7162
0
1
2
3
4
5
6
7
8
9
10
0 10 20 30 40 50 60 70 80 90 100
uitlezing
debi
et (N
L/hr
)
Figuur C-3 Kalibratiecurve van de thermische massadebietsregelaar van CH4

75
Appendix D: Kalibratiefactoren van de gaschromatograaf
Aan de componenten, die gedetecteerd worden door de GC met FID detector, worden
kalibratiefactoren toegekend. De kalibratie wordt gerealiseerd door het injecteren van een
binair mengsel van benzeen, cyclohexaan en methaan en de standaardcomponent heptaan. De
kalibratiefactor van de standaardcomponent wordt gelijkgesteld aan 1. Uit de volgende
formule worden de kalibratiefactoren van de verschillende componenten berekend.
∑=
= n
1j)j(CF)j(A
)i(CF)i(A)i(mf (D-1)
De kalibratiefactoren (CFi) van de verschillende factoren worden weergegeven in Tabel D-1.
component kalibratiefactor (CFi)
heptaan 1
benzeen 0.92
cyclohexaan 1.00
methaan 1.01
Tabel D-1: Kalibratiefactoren van de verschillende componenten

Referentielijst
N. Dewachtere, PhD Thesis Universiteit Gent (1998)
Single-event kinetische modellering van het katalytisch kraken van gasolie
I. Fovel, Laboratorium voor Petrochemische Techniek Universiteit Gent, 1999-2000
Hydrogenering van tolueen op een hydrokrakingskatalysator
L.H. Hosten, Chem. Eng. Sci., 29, 2247-2252, 1974
Experimental design for precise parameter estimation in single response models
S.D. Lin en M.A. Vannice, J. Catal., 143, 539-572 (1993)
Hydrogenation of aromatic hydrocarbons over supported Pt catalysts
C. Morin, D. Simon, P. Sautet , J. Phys. Chem. B, 107, 2995-3002 (2003)
Density-Functional Study of the adsorption and vibration spectra of benzene molecules on
Pt(111)
C. Morin, D. Simon, P. Sautet, J. Phys. Chem. B, 108, 5653-5665 (2004)
Chemisorption of benzene on Pt(111), Pd(111) and Rh(111) metal surfaces: a structural an
vibrational comparison from first principles
C. Morin, D. Simon, P. Sautet, J. Phys. Chem. B, 108, 12084-12091 (2004)
Trends in the chemisorption of aromatic molecules on a Pt(111) surface: benzene, naphtalene,
and anthracene from first principles calculations
Podkolzin S.G., Watwe R.M., Yan Q., de Pablo J.J. and Dumesic J.A., J. Phys. Chem. B, 105,
8550-8562 (2001).
DFT Calculations and Monte Carlo Simulations of the Co-adsorption of Hydrogen Atoms and
Ethylidyne Species on Pt(111)

M.V. Rahaman en M.A. Vannice, J. Catal., 127, 251-266, 251-266 (1991)
The hydrogenation of toluene and o-, m-, and p-xylene over palladium
J.L. Rousset, L. Stievano, F.J. Cadete Santos Aires, C. Geantet, A.J. Renouprez, M. Pellarin,
J. Catal., 197, 335-343 (2001)
Hydrogenation of toluene over γ -Al2O3 supported Pt, Pd and Pt-Pd model catalysts obtained
by laser vaporization of bulk metals
M. Saeys, M.-F. Reyniers, G.B. Marin, J. Phys. Chem. B, 106, 7489-7498 (2002)
Density Functional Study of benzene adsorption on Pt(111)
M. Saeys, J.W. Thybaut, M. Neurock, G.B. Marin,
Molecular Physics, 102, 267-272 (2004)
Kinetic models for catalytic reactions from first principles: benzene hydrogenation
M. Saeys, M.-F. Reyniers, M. Neurock, G.B. Marin,
J. Phys. Chem. B, 109, 2064-2073 (2005)
Ab initio reaction path analysis of benzene hydrogenation to cyclohexane on Pt(111)
T. Salmi, D. Yu, J.-P Mikkola, J. Wärnå, P. Mäki-Arvela, E. Toukoniitty, S. Toppinen
Ind. Eng. Chem. Res., 43, 4540-4545 (2004)
Advanced kinetic concepts and experimental methods for catalytic three-phase processes
A. Stanislaus en B.H. Cooper, Catal. Rev.-Sci. Eng., 36(1), 75-102 (1994)
Aromatic hydrogenation catalysis: a review
J. Thybaut, PhD Thesis Universiteit Gent (2002)
Production of low aromate fuels: kinetics and industrial apllication of hydrocracking
J. Thybaut, Laboratorium voor Petrochemische Techniek Universiteit Gent, 1997-1998
Gasfase hydrokraken van naftenische en aromatische modelcomponenten in een reactor met
volkomen vermenging

J. Thybaut, M. Saeys en G.B. Marin, Chem. Eng. J., 90, 117-229 (2002)
Hydrogenation kinetics of toluene on Pt/ZSM-22
K. Van Geem, Laboratorium voor Petrochemische Techniek Universiteit Gent, 2000-2001
Effect van de vertakking en het aantal aromatische ringen op de hydrogenering van aromaten
V. Vangenende, Laboratorium voor Petrochemische Techniek Universiteit Gent, 2003-2004
Development of a single-event model for the catalytic hydrogenation of aromatic compounds
J. Verstraete, PhD Thesis Gent University (1997)
Kinetische studie van de katalytische reforming van nafta over een Pt-Sn/Al2O3 katalysator
R.Z.C. Van Meerten en J.W.E. Coenen, J. Catal., 46, 13 (1977)
Gas-phase benzene hydrogenation on nickel-silica catalyst. 4. rate equations and curve fitting
Xu, C., Tsai Y.L. and Koel B.E.J., Phys. Chem., 98, 585-594 (1994), Adsorption of
cyclohexane and benzene on ordered SN/Pt(111) surface alloys.




















