
Leverresectie of levertransplantatie voor leverpolycystose ... · leverziekte bij ADPKD. Bovendien...
59
Academiejaar 2011 - 2012 Leverresectie of levertransplantatie voor leverpolycystose? Een update van de literatuur Pieter Verpoort Promotor: Prof. Dr. X. Rogiers Scriptie voorgedragen in de 2 de Master in het kader van de opleiding MASTER IN DE GENEESKUNDE
Transcript of Leverresectie of levertransplantatie voor leverpolycystose ... · leverziekte bij ADPKD. Bovendien...

Academiejaar 2011 - 2012
Leverresectie of levertransplantatie voor
leverpolycystose? Een update van de literatuur
Pieter Verpoort
Promotor: Prof. Dr. X. Rogiers
Scriptie voorgedragen in de 2de
Master in het kader van de opleiding
MASTER IN DE GENEESKUNDE

“De auteur(s) en de promotor geven de toelating deze scriptie voor consultatie beschikbaar te stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder de beperkingen van het auteursrecht, in het bijzonder met betrekking tot de verplichting uitdrukkelijk de bron te vermelden bij het aanhalen van resultaten uit deze scriptie.” Datum 02/05/2012 Handtekening student Handtekening promotor Naam student Naam promotor

I
Voorwoord
In deze masterproef worden de resultaten gerapporteerd van een literatuurstudie waarbij men een
vergelijking maakt tussen twee invasieve technieken om symptomen bij patiënten met ernstige
polycystische leverziekte te verlichten. Additioneel werd de prevalentie van deze ingrepen in het UZ Gent
opgezocht. De bevindingen worden besproken en gesitueerd in de desbetreffende literatuur.
Deze masterproef kwam tot stand in het kader van de opleiding tot “Master in de Geneeskunde” en bood
mij de mogelijkheid om op een didactische manier in contact te komen met het wetenschappelijk
onderzoek.
In het algemeen wens ik iedereen te bedanken die heeft bijgedragen tot het ontstaan van deze masterproef.
Ik wens in het bijzonder Prof. Dr. Xavier Rogiers te bedanken voor zijn deskundig en opbouwend advies.
Zijn begeleiding in de voor mij aanvankelijk onbekende wereld van het wetenschappelijk onderzoek heeft
me warm gemaakt voor deze tak van de geneeskunde. De ruimte die hij me liet om zelfstandig ideeën aan
te brengen en uit te werken maakte dit tot een erg creatieve en leerrijke ervaring.
Hiernaast wens ik ook het personeel van het Transplantatiecentrum UZ Gent te bedanken voor hun geduld
en begeleiding bij de vele vragen die ik had omtrent transplantatie bij deze groep van patiënten.
Alsook wens ik mijn familie en vrienden te bedanken voor hun feedback en input bij het nalezen van de
masterproef. In het bijzonder wens ik te bedanken: Dr. Guido Leman, Roosmarijn Van Cauwenberge,
Philippe Leune en Tessa De Vriendt voor het nalezen en verbeteren van deze masterproef.
Pieter Verpoort
Gent, april 2012

II
Inhoudstafel
VOORWOORD I
INHOUDSTAFEL II
I. INLEIDING 1
1. Definitie 1
2. Types polycystische leverziekte 2
A. Geassocieerd aan polycystische nierziekte 2
B. Geïsoleerde vorm 3
3. Pathogenese 5
4. Klinisch 6
A. Natuurlijke evolutie 6
B. Symptomen 6
C. Complicaties 7
D. Leverfunctie 7
E. Omvang 8
5. Diagnose 9
A. Differentieel diagnose 9
B. Technische onderzoeken 9
6. Classificatie 11
7. Behandelingsmethodes 12
A. Medische behandeling 12
B. Chirurgische behandelingen 13
II. VRAAGSTELLING 18
III. METHODE 18
1. Literatuuronderzoek 18
2. Recente data 19
A. Registry data 19
B. UZ Gent 19

III
IV. RESULTATEN 20
1. Literatuuronderzoek 20
A. Inleiding 20
B. Resectie 20
C. Transplantatie 25
2. Recente data 35
A. Registry data 35
3. UZ gent 38
A. Conservatieve chirurgie 38
B. Transplantatie 38
V. DISCUSSIE 40
1. Ernstige polycystische leverziekte 40
2. Resectie 40
3. Transplantatie 42
VI. CONCLUSIE 43
V. REFERENTIES 45
1
I. Inleiding
1. Definitie
Polycystische leverziekten (PCLD) zijn genetische stoornissen waarbij er zich progressief multiple cysten
ontwikkelen in het leverparenchym van de patiënt. (1, 2) In tegenstelling tot enkelvoudige levercysten
zijn de levercysten bij PCLD uitgebreider in aantal en grootte. Ze vertonen gelijkenissen met normale
enkelvoudige cysten maar zijn hiervan te onderscheiden door hun aantal, grootte, bilobaire distributie en
aanwezigheid van verscheidene microcysten. (3) Over het kritische aantal cysten om een multicystische
lever te onderscheiden van een lever met verschillende enkelvoudige cysten is er echter nog geen
consensus. (4, 5) Enkele auteurs nemen hier een standpunt in: vanaf vijf cysten (4, 6), zes cysten (7), een
lever met meer dan 50% cystische betrokkenheid van het parenchym (8, 9) of ten minste 20 cysten met
een doorsnede groter dan 1cm in de lever in de afwezigheid van infectieuze, parasitaire of traumatische
oorzaken. (10)
Polycystische leverziekte komt solitair voor en in associatie met polycystische nierziekte. De solitaire
vorm heeft een autosomale overerving. Polycystische nierziekte, gekenmerkt door multiple cysten in de
nier, komt voor als een autosomaal dominante vorm (ADPKD) en zeldzaam als autosomaal recessieve
vorm (ARPKD). (1, 2) De autosomaal dominante vormen komen het frequentst voor. (2, 4)
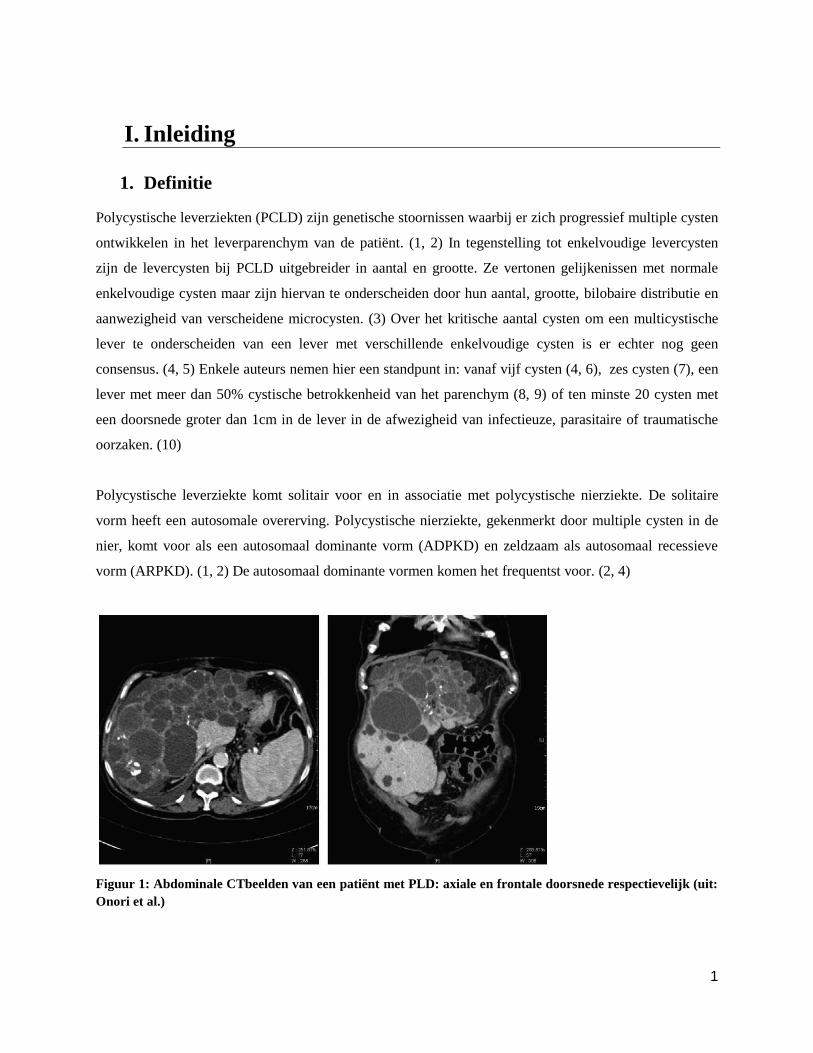
Figuur 1: Abdominale CTbeelden van een patiënt met PLD: axiale en frontale doorsnede respectievelijk (uit:
Onori et al.)

2
2. Types polycystische leverziekte
A. Geassocieerd aan polycystische nierziekte
1. Autosomaal dominant polycystische nierziekte (ADPKD)
Autosomaal dominant polycystische nierziekte is een aandoening waarbij er lever- en niercysten ontstaan.
Deze kunnen vergroten, wat renaal resulteert in een abnormale nierstructuur en nierinsufficiëntie. Vaak
leidt dit tot dialyse en renale transplantatie. (2) End stage renal disease (ESRD) wordt vastgesteld in 50%
van de patiënten met ADPKD in de 5e levensdecade en is daarom de meest voorkomende
levensbedreigende, erfelijke ziekte met een prevalentie van 1/400 tot 1/1000 nieuwgeborenen. (11)
PKD1 en PKD2
Cysten bij ADPLD worden veroorzaakt door mutaties in het PKD1 gen, coderend voor polycystine 1
(PC1) en PKD2, coderend voor polycystine2 (PC2). (2, 12) Meer dan 85% van de ADPKD-cases worden
veroorzaakt door mutaties in het PKD1 gen, bij bijna alle resterende gevallen ligt de oorzaak in een PKD2
gen mutatie. (2, 13) Er zijn gevallen beschreven waarbij patiënten noch PKD1, noch PKD2 mutaties
bezaten welke suggereren dat er ten minste nog één derde onbekend gen verantwoordelijk is voor
ADPKD. (14) Verschillende studies bij volwassenen hebben aangetoond dat patiënten met PKD1
mutaties meer niercysten, grotere cysten, meer voorkomende hypertensie en een snellere progressie tot
end-stage nierfalen vertonen dan patiënten met een PKD2 mutatie. Gelijkaardig worden er bij kinderen
met een PKD1 mutatie meer en grotere niercysten gevonden dan leeftijdsgenoten met een PKD2 mutatie.
(2, 15) Patiënten met mutaties in het PKD2-gen hebben gewoonlijk een later begin van de ziekte en een
levensverwachting die gemiddeld 16 jaar hoger ligt dan patiënten met een PKD1-gen mutatie. (14, 16)
Levercysten
Levercysten zijn de meest voorkomende extrarenale manifestaties van ADPKD en worden gevonden in
60-75% van de ADPKD patiënten aan dialyse. (2, 17, 18) Deze cysten verschijnen op een latere leeftijd
dan de renale maar zijn het uitgebreidst in patiënten met een ernstigste nierdysfunctie en zijn dus
geassocieerd aan het verloop van de cystische nierziekte. (2, 19) Hoewel de evolutie tot leverfalen in
ADPKD ongebruikelijk is, speelt cystische leverziekte een significante rol in de morbiditeit en is het
verantwoordelijk voor 10% van de mortaliteit van ADPKD patiënten aan dialyse. (2, 18)
Vrouwelijke hormoonstimulatie
Ernstige cystische leverziekte tast voornamelijk vrouwen aan en is uitgebreider in vrouwen met multipele
zwangerschappen of het gebruik van exogene vrouwelijke steroïdhormonen. (2, 19)

3
De neiging voor het ontwikkelen van massieve levercysten bij vrouwen wordt ook typisch gezien bij
patiënten met de geïsoleerde vorm van PCLD (zie verder). Eén longitudinale studie van anovulatoire
vrouwen met ADPKD met vervangende hormoontherapie suggereert dat oestrogeen selectief de
hepatische cystziekte verergert. (20)
2. Autosomaal recessieve polycystische nierziekte (ARPKD)
ARPKD is een zeldzame toestand die bij 1/40.000 levend geborenen voorkomt en heeft een hoog
mortaliteitsratio. (2, 21) In overlevende patiënten is de leverziekte de grootste oorzaak van morbiditeit en
mortaliteit en wordt het gekenmerkt door biliaire ontwikkelingsstoornissen met congenitale hepatische
fibrosis (CHF), galwegdilatatie en cyst formatie. (2, 22)
PKHD1
Mutaties in het polycystische nier- en leverziekte 1 gen (PKHD1) veroorzaken deze aandoening. Dit gen
codeert voor het eiwit fibrocystine/polyductine. (2, 23, 24) De ziekte presenteert zich typisch met sterk
vergrootte nieren in utero of in de postnatale periode, met een sterfte in 30% kort na de geboorte door
respiratoire moeilijkheden ten gevolge van de vergrote nieren. (2, 25) De meest voorkomende
complicaties van ARPKD omvatten: hypertensie (60-100%), portale hypertensie veroorzaakt door
ernstige leverfibrose of Caroli’s ziekte (30-75%) en chronische longziekte (ongeveer 11%). (2, 26) Ook
intracraniële aneurysmata en bijnierinsufficiëntie komen vaker voor bij patiënten met ARPKD. (2, 27, 28)
Over het algemeen zijn levercysten zeldzaam in kinderen. Hun voorkomen stijgt echter met toenemende
leeftijd en wordt naar alle waarschijnlijkheid onderschat door de CT en ultrasound studies. (2, 29)
B. Geïsoleerde vorm
In het begin van de 20ste eeuw nam men aan dat polycystische leverziekte steeds samen voorkwam met
polycystische nierziekte. Zeldzame gevallen van geïsoleerde polycystische levers waren varianten met
nieren waarbij de cystvorming was uitgesteld en de niercysten dus nog tot ontwikkeling moesten komen.
(30) In de 2e helft van de 20ste eeuw begon het inzicht te groeien dat geïsoleerde PLD een aparte ziekte-
entiteit kon zijn en ook het autosomaal dominant patroon van overerving kwam aan het licht. (30, 31)
Pas in een studie in 2003 werd deze gedachte ook bevestigd door een linkage analyse van acht Finse
families. De genetische afzonderlijke oorzaak en zijn genetische heterogeniteit zijn hierbij aangetoond.
(30, 32)
PRKCSH en SEC63
Mutaties in de genen PRKCSH of SEC63 zijn verantwoordelijk voor de vorming van levercysten. (30, 33,
34) In tegenstelling tot PCKD zijn cerebrale aneurysmata niet geassocieerd aan geïsoleerde PCLD. (35)

4
Geïsoleerde PCLD is een zeldzame ziekte met een geschatte incidentie van <0.01%. Verschillende
autopsie studies hebben echter aangetoond dat het bestaan ervan niet zoveel zeldzamer voorkomt dan dat
van ADPKD. Deze discrepantie kan te maken hebben met de mildere en minder frequent voorkomende
symptomatologie van geïsoleerde PCLD in vergelijking met ADPKD. We kunnen stellen dat globaal
gezien de incidentie van PCLD onderschat is. (30, 31, 36, 37) Een recente studie door Hoevenaren et al.
(30, 38) suggereert dat geïsoleerde PCLD patiënten minder geassocieerde comorbiditeiten vertonen, een
groter aantal levercysten en een groter cystvolume hebben op het moment van presentatie dan hun
lotgenoten met ADPKD. De verklaring hiervoor is dat ADPKD gewoonlijk in een vroeger ziektestadium
wordt gediagnostiseerd en daarom het volume van hun cysten kleiner is op het moment van introductie
dan patiënten met geïsoleerde PLD, die pas verder in hun ziekteverloop worden opgemerkt. (30)
Lokalisatie
PRKCSH is gelokaliseerd op de korte arm van chromosoom 19 en vormt de codering voor substraat 80K-
H van het proteïne kinase hepatocystine welke een subunit vormt van glucosidase II. (30, 39) SEC63 is
gelocaliseerd op de lange arm van chromosoom 6. Het vormt een deel van een multi-component
translocon. (30, 33, 40) Mutaties in elk van deze gebieden resulteren in proteïne maturatie defecten.
Aangenomen wordt dat 20 tot 30% van de geïsoleerde PLD hierdoor veroorzaakt worden. Dit suggereert
dat er additionele, nog te ontdekken genen zijn, die ook kunnen resulteren in defecten van proteïne-
maturatie. (30, 33) Histopathologisch vertoont ADPLD opvallende gelijkenissen met de cystische
leverziekte bij ADPKD. Bovendien suggereren dierenstudies een pathogenetische common pathway. (30,
41) Waarom ADPLD zich enkel beperkt tot cystvorming in de lever en ADPKD lever en nier aantast
blijft onduidelijk. (30)
Figuur 2: Diagram van de genetica bij polycystische leverziekte (Gemodificeerde figuur van Onori et al. (2))

5
3. Pathogenese
Morfologische studies van individuele levercysten hebben aangetoond dat de levercysten ontstaan uit
biliaire microhamartomas, ook von Meyerburg’s complexen (30, 42) genoemd, die voortkomen door
proliferatie uit biliaire ductules en uit peribiliaire klieren. (30, 43, 44) Kenmerkend is dat, wanneer ze
groter worden, ze loskomen van hun oorsprong. De expansie van de levercysten is het gevolg van een
collectief effect van de proliferatie van het cystaflijnend epitheel, vloeistof secretie in de cyst, remodulatie
van de extracellulaire matrix die de cyst omringt en neovascularisatie. (30, 45) PCLD cysten beginnen
dus als focale dilataties die in grootte toenemen en loskomen van de originele ductule om een autonome
gesloten cyste te vormen. Terwijl de cyste verder toeneemt in grootte, verdringt dit het omliggende
parenchym. (46)
Figuur 3: Onstaan van de levercysten in PLD (uit: Onori et al. (2))
Eens er cystes ontwikkeld zijn, speelt de vergroting van reeds bestaande cysten een grotere bijdrage tot de
vergroting van de lever dan de ontwikkeling van nieuwe cysten. (47) Op 60 jarige leeftijd hebben 80%
van de patiënten met ADPKD levercysten. Bij vrouwen zijn ze dan groter in aantal en afmeting. (3)
Levercysten worden zelden gevonden vóór de puberteit, komen op met de start van de puberteit, zijn meer
prevalent en prominent in vrouwen (zie ook hoger) en stijgen drastisch in grootte tijdens de vruchtbare
levensjaren. (14)

6
Figuur 4: Prevalentie van lever- en niercysten in ADPKD (uit: Everson et al. (1))
4. Klinisch
A. Natuurlijke evolutie
Levercysten bij PCLD vertonen typisch een traag en goedaardig verloop. De meeste patiënten zijn
asymptomatisch waarbij de levercysten toevallig worden ontdekt of opgespoord in associatie met een
PCKD presentatie. Symptomen komen meer voor naarmate de levensduur van de patiënt toeneemt. Ze
zijn het gevolg van de massieve vergroting van de lever, complicaties gerelateerd aan de cysten (wat
eerder ongewoon is) of een combinatie van beide. (2, 35, 48) Vroeger overleden de meeste patiënten met
ADPKD aan nierfalen nog voordat hun lever hen parten speelde. De patiënten vertonen nu dus vaker
symptomen dan vroeger doordat niertransplantatie en dialyse hun levensverwachting hebben verlengd.
(49) Bij sommige patiënten kan er een vooruitstekend abdomen worden bemerkt zonder dat er sprake is
van symptoomklachten. (1)
B. Symptomen
De meest voorkomende symptomen die interventie vereisen zijn abdominale distentie, abdominale pijn en
vroege verzadiging. (47, 50) Andere symptomen zijn: vermoeidheid, nausea, regurgitatie en
kortademigheid. (47) Omdat langdurende symptomen en discomfort tot significante anorexia, malnutritie
en fysieke instabiliteit kunnen leiden, moet er actief ingegrepen worden voor deze lange-termijn gevolgen
optreden. Deze symptomen zijn een typische indicatie voor de massieve vergroting van de lever en komen
voor in de setting van een reeds lang gevestigde ziekte. (4) Een totale cyst op parenchym ratio groter dan
1 geeft een predicatieve waarde voor het ontwikkelen van dergelijke symptomen (zie ook later). (51)

7
Tabel 1: Geregistreerde hepatische en extrahepatische complicaties (uit: Arnold et al. (4))
C. Complicaties
Acute complicaties treden op in minder dan 5% van de patiënten en kunnen acute interventie vereisen.
Deze bestaan uit cyst-hemorragie, -ruptuur, -infectie, uterus prolaps, obstructieve icterus, portale
hypertensie, transsudatieve en exsudatieve ascites en Budd-Chiari syndroom. (35, 47, 50, 52-56). Cyst
infectie vormt een groot probleem voor de patiënt met PLD en toont morbiditeitsratio’s van 3% en
mortaliteitsratio’s van 2% bij ADPKD patiënten met end-stage renaal lijden (ESRD). (4, 18, 57) Klassiek
presenteren deze patiënten zich met koorts, leukocytose en rechter hypochonderpijn. Deze acute
presentatie kan evolueren tot sepsis en dood indien de cystinfectie niet op tijd wordt vastgesteld en
agressief behandeld, in het bijzonder voor patiënten met ESRD met nood aan dialyse. (4, 58) Data stellen
dat de infecties meestal monobacterieel zijn met gram negatieve bacteriën zoals E.coli, dat het
voornaamste verwekkend organisme is. (4, 58-60)
D. Leverfunctie
Ondanks een gemiddeld gewicht meer dan 10kg bij verwijdering van de lever, vertoont het
leverparenchym over het algemeen een goed behoud van zijn functie. (4, 18, 51) De lever kan dus
niettegenstaande de multiple cystinname zijn synthetische functie in bijna alle gevallen bewaren. (61)
8
Patiënten ontwikkelen ook zelden hepatisch-gerelateerde pathologieën zoals leverfalen en portale
hypertensie. (3, 62) Cysten in de nieren lijden daarentegen wel tot progressief nierfalen. In associatie met
niercysten zijn aneurysmata en valvulaire hartziekten aangetoond. (3, 10, 63)
E. Omvang
De uitgebreidheid van de ziekte kan anders zijn dan de klinische presentatie laat uitschijnen. Sommige
patiënten ontwikkelen symptomen door een paar grote cysten die compressie uitoefenen op omliggende
organen. Anderen ondervinden geen symptomen totdat er extensieve hepatomegalie optreedt door
multiple, diffuse cysten. Door de verschillen in anatomie en verschillen in de symptomen van hepatische
cysten bij PCLD-patiënten vereisen deze een specifieke aanpak. (35) Everson et al. delen hun patiënten
arbitrair op in 2 groepen: massieve of minimale inname. Ze baseren zich hiervoor op een definitie voor de
massa-inname waarbij men een ratio van het totaal levercyst/parenchym volume berekend. Als deze ratio
> 1 bedraagt, wordt men ingedeeld tot de groep met massieve inname. (51) Wanneer men deze definitie
hanteert, zijn de meeste symptomatische cases beperkt tot patiënten met massieve hepatische cystinname,
waarbij abdominale pijn, abdominale ongemak en kortademigheid gecorreleerd zijn aan de ernst van de
cystische leverziekte. (20)
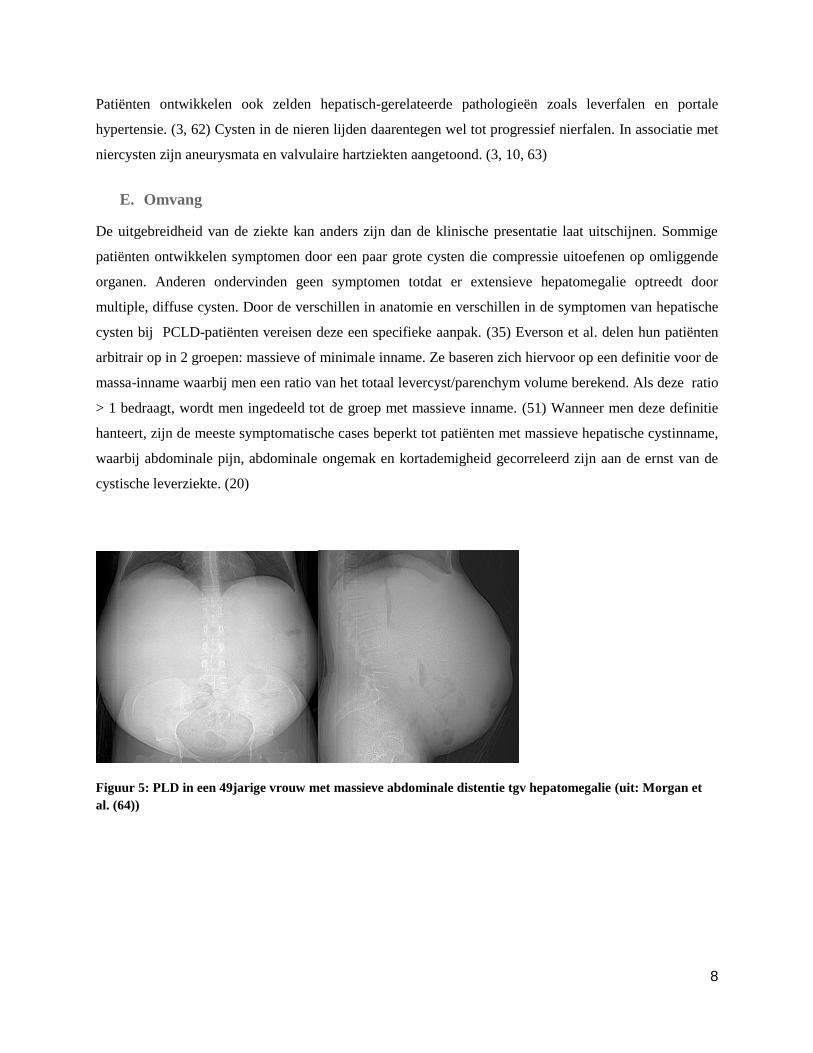
Figuur 5: PLD in een 49jarige vrouw met massieve abdominale distentie tgv hepatomegalie (uit: Morgan et
al. (64))

9
Figuur 6: Symptomen per gender (uit: van Keimpema et al. (65))
5. Diagnose
A. Differentieel diagnose
Management van PCLD vereist kennis van andere ziektes die multiple cysten kunnen veroorzaken omdat
de behandelingen zeer uiteenlopend kunnen zijn. De verschillende cystische ziekten van de lever
omvatten: banale levercysten, echinococcus cysten, neoplastische levercysten en levermetastasen met
centrale necrose (neuro-endocrien, ovarieel, colon, pancreas en nier). De combinatie van klinische
presentatie, lever-echografie en CT leiden meestal tot de diagnose. Soms is een leverbiopsie nodig om de
definitieve diagnose te maken. (35)
B. Technische onderzoeken
1. Echo, CT en MRI
PCLD wordt gediagnostiseerd door gebruik van echografie, magnetische resonantie imaging (MRI) of
computer-assisted tomografie (CT). (30) Abdominale echografie en CT zijn complementaire onderzoeken
en de methodes bij uitstek voor investigatie van PLD. (4, 66) Op echo zijn de cysten anechogeen en
zichtbaar als rond met een glad oppervlak en met distaal een versterking van de echo. (4, 67, 68) Op CT
vertonen deze cysten een typische gladde contour en bevatten een vloeistof met waarden tussen de +5 en
+20 HU. De cyste is duidelijk afgegrensd ten opzichte van het omringende leverparenchym. (4, 68) Bij
veranderingen in de wanddikte of contour, het verschijnen van septa, veranderingen in echo of CT-
attenuatie of een snelle groei, zou men ogenblikkelijk op een hemorragie, infectie of neoplastische laesie
zoals een cystadenoma of cystadenocarcinoma moeten bedacht zijn. (4, 66, 69)
10
Beeldvorming met CT kan ook andere complicaties eigen aan ADPKD aantonen (eg cerebraal
aneusysma, diverticulose, inguinale hernia). (3) Op MRI vertonen deze cysten een hyperintensiteit op T2-
gewogen beelden en een hypointensiteit op T1-gewogen beelden, tenzij verwikkeld met een hemorragie.
(3) Het gebruik van de CT en echo in de evaluatie van abdominale klachten heeft de ontdekking van
levercysten talrijker gemaakt. Het vinden van multiple renale cysten in een patiënt met multiple massieve
levercysten of talrijke levercysten en een familiale geschiedenis samenhangend met ADPKD, maakt de
diagnose tamelijk eenvoudig. Dit in tegenstelling tot de diagnosestelling van PLD bij een patiënt zonder
renale cysten en/of zonder significante cystische betrokkenheid van de lever, wat een stuk moeilijker is.
(4)
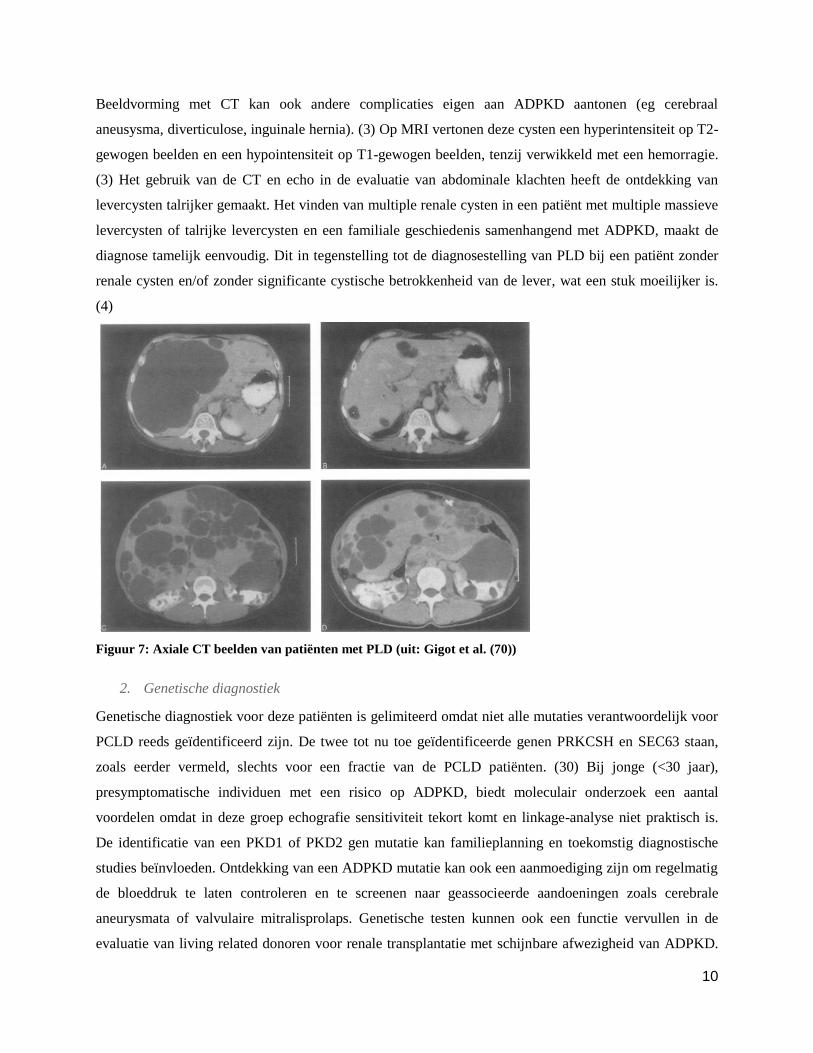
Figuur 7: Axiale CT beelden van patiënten met PLD (uit: Gigot et al. (70))
2. Genetische diagnostiek
Genetische diagnostiek voor deze patiënten is gelimiteerd omdat niet alle mutaties verantwoordelijk voor
PCLD reeds geïdentificeerd zijn. De twee tot nu toe geïdentificeerde genen PRKCSH en SEC63 staan,
zoals eerder vermeld, slechts voor een fractie van de PCLD patiënten. (30) Bij jonge (<30 jaar),
presymptomatische individuen met een risico op ADPKD, biedt moleculair onderzoek een aantal
voordelen omdat in deze groep echografie sensitiviteit tekort komt en linkage-analyse niet praktisch is.
De identificatie van een PKD1 of PKD2 gen mutatie kan familieplanning en toekomstig diagnostische
studies beïnvloeden. Ontdekking van een ADPKD mutatie kan ook een aanmoediging zijn om regelmatig
de bloeddruk te laten controleren en te screenen naar geassocieerde aandoeningen zoals cerebrale
aneurysmata of valvulaire mitralisprolaps. Genetische testen kunnen ook een functie vervullen in de
evaluatie van living related donoren voor renale transplantatie met schijnbare afwezigheid van ADPKD.

11
(1) In Europa zijn deze testen voor PCLD beschikbaar. Genetische sequentie van PRKCSH behoort daar
ook toe. Omdat de sensitiviteit van echografie in PCLD ongedefinieerd is, lijkt het gebruik van PRKCSH
tests om presymptomatische patiënten te identificeren met een risico voor de ziekte misschien zelfs nog
relevanter dan bij ADPKD. (1)
3. Labo
Laboratoriumwaarden met inbegrip van alkalische fosfatase, gamma glutamyl transferase (GGT),
bilirubine en aspartaat aminotransferase (AST) zijn meestal normaal. Bij symptomatische patiënten die
een evaluatie ondergaan voorafgaand aan een chirurgische ingreep, worden vaker abnormale
bloedwaarden gevonden. In deze groep patiënten zijn het alkalische fosfatase en de GGT-waarden
gestegen in 30-47% (1.0-2.0 maal de normale upper limit) en 60-70% (1.5-7.0 maal de upper limit)
respectievelijk. (4, 47, 70-72) De lichte elevatie in GGT correleert dan met de hepatische cyst belasting.
(51) AST kan licht gestegen zijn (1.0- 2.0 maal de normale upper limit) in 27% en bilirubine waarden zijn
in 17% gestegen. (4, 47, 72) Er is een lichte maar significante daling van de antipyrine klaring en de first-
pass eliminatie van cholaat bij patiënten met massieve cystische leverziekte. (73)
6. Classificatie
De symptomen zijn gerelateerd aan de distributie, het aantal cysten en de grootte van de cysten. Daarom
is het van belang deze patiënten ook op deze manier te klasseren. Aan de hand van de classificatie kan
vervolgens een behandeling worden opgesteld.
1. Morino et al.
Morino et al. classificeren PLD als type 1 wanneer er een gelimiteerd aantal grote, voornamelijk
oppervlakkige cysten worden gevonden. Wanneer vele kleine cysten verspreid over de lever aanwezig
zijn, inclusief de achterste segmenten, worden ze tot type 2 gerekend. (4, 74)
2. Gigot et al.
Gigot et al. voorzien een meer gedetailleerd classificatieschema, ook gebaseerd op de grootte en het
aantal cysten maar als bijkomende factor de hoeveelheid resterend leverparenchym. Zo worden de patiënt
in 3 groepen/ types opgesplitst. Type I bestaat uit patiënten met een gelimiteerd aantal (<10) grote cysten
(>10cm). Type II wordt vertegenwoordigd door patiënten met een diffuse inname van het leverparenchym
door multiple medium-sized cysten, maar met nog grote gebieden leverparenchym vrij van cystische
inname op preoperatieve CT-opname.

12
Type III bestaat uit de ernstige vorm van APLD-patiënten met massieve, diffuse inname van het
leverparenchym door kleine en medium-sized levercysten en slechts enkele gebieden van vrij
leverparenchym tussen de cysten. (70)
3. Schnelldorfer et al.
Schnelldorfer et al. gebruiken een vergelijkbaar schema gebaseerd op de graad van symptomen,
distributie van de levercysten, volume van vrij of relatief cystvrij parenchym en vasculaire anatomie van
de niet ingenomen lever. (10) Patiënten met een normale leverfunctie worden hierbij op basis van
klinische en radiologische bevindingen in 4 groepen ingedeeld:
Tabel 2: Classificatie volgens Schnelldorfer et al. (uit: Schnelldorfer et al. (10))
Verder in dit werk zal de classificatie van Gigot gehanteerd worden.
7. Behandelingsmethodes
De aanpak van PLD is gerelateerd aan de klachten van de patiënt. Alleen wanneer de patiënt symptomen
vertoont, is een behandeling nodig. De symptoomvrije patiënten zouden idealiter op jaarbasis moeten
worden geëvalueerd met een echo of CT. Deze procedure is niet aangenaam maar zeer geruststellend voor
de patiënt. Er is voorlopig nog geen bestaande consensus over de optimale behandeling van
symptomatische patiënten. (75)
A. Medische behandeling
Verbindingen zoals cimetidine en somatostatine hadden vroeger reeds in vitro aangetoond de secretine
release te inhiberen. (76) Daarom dat sommige auteurs postoperatief het gebruik van somatostatine-
analogen hadden aangeraden in de hoop hiermee persisterende cystdrainage of vloeistof accumulatie te
reduceren. (77, 78) Pas in 2009 werd aan de hand van een dubbelblinde randomized controlled trial
(RCT) ook daadwerkelijk het effect van lantreotide, een somatostatine-analoog, aangetoond waarbij op 6
maanden tijd een volumeafname van 2.9% werd vastgesteld. (79) Drie andere RCT bevestigen de werking
van somatostatine-analogen met een afname van 4.45% over 6 maand, 4.95% na 1 jaar en 4% na 1 jaar.
(80-82)

13
In de laatste serie werd het grootste effect van lantreotide gezien gedurende de eerste 6 maanden van
behandeling met een volumeafname van 4%. Vervolgens bleef het volume de 6 maanden hierop volgend
behouden. Na het beëindigen van de therapie ondervond men na 6 maanden terug een stijging van 4%.
Lantreotide is dus effectief in het verminderen van het volume de eerste 6 maanden van behandeling maar
moet continu gebruikt worden om zijn effect te behouden. (82) Vaak is 4% echter te weinig om de
symptomen ten gevolge van een hepatomegalie te verlichten. Dit is desalniettemin een behandeling met
veel toekomstperspectief.
B. Chirurgische behandelingen
Verschillende operatietechnieken zijn uitgedacht om het cystvolume te reduceren en zo de complicaties
ten gevolge van de hepatomegalie te behandelen. Operatietechnieken zijn, net als de reeds besproken
indeling van patiënten, afhankelijk van de grootte, het aantal en de uitgebreidheid van de levercysten. (10)
Lange termijn gegevens van geopereerde patiënten, onafhankelijk van de gebruikte techniek, zijn schaars
en er bestaan ook geen gestandaardiseerde selectiecriteria voor operatie. (10, 83)
Alle patiënten moeten grondig geëvalueerd worden op significante symptomen, de performance status
moet worden bekeken, alsook de graad van renale en hepatische dysfunctie, die de morbiditeit en
mortaliteit kunnen beïnvloeden. De risico’s en beperkingen van de ingreep moeten in een informed
consent aan de patiënt worden meegedeeld voor men een chirurgische ingreep kan uitvoeren. (84)
1. Aspiratie
Eenvoudige aspiratie van de cysten werd vroeger als diagnose én behandeling uitgevoerd. (4) Echter, door
de hoge incidentie aan recidieven van zowel de cysten als de symptomen van de patiënt, tot 100% in
sommige series, en het risico om infecties in te brengen, is deze techniek geen eerste keuze meer om de
symptomen definitief te behandelen. (6, 9, 70, 75, 85-89) Bij sommige gevallen van biliaire obstructie of
geïnfecteerde hepatische cysten speelt CT- of Echo-geleide aspiratie van problematische cysten nog
steeds een rol indien de probleemcysten accuraat geïdentificeerd kunnen worden. Voornamelijk in een
setting van geïnfecteerde levercysten lijkt percutane aspiratie in combinatie met antibiotica een optie,
hoewel dit gebaseerd is op slechts een beperkt aantal patiënten-series. (4, 58)
2. Aspiratie en sclerose
Wanneer percutane aspiratie gecombineerd wordt met sclerotherapie, lijkt de verlichting van symptomen
iets langer te duren, wat te danken is aan de destructie van de secretoire capaciteit van het biliaire epitheel
dat de cyste binnenin bekleed. (4, 90) Er zijn verschillende series uitgevoerd bij patiënten met PLD
waarbij de recurrence rate na sclerotherapie varieerde tussen de 20% à 50%. (90, 91)
14
Aangenomen wordt dat factoren zoals type sclerosans, volume van sclerosans ingespoten, het aantal
herhaalde sessies en de tijd dat de drainagecatheter ter plaatse bleef een variabele invloed uitoefenen op
de uitkomst van deze procedure. (90, 91) Op lange termijn geeft deze procedure ook voor solitair grote
cysten bij PLD weinig verlichting omdat cyst inkrimping alleen onvoldoende bleek te zijn. (75, 87). Ook
is deze procedure niet vrij van complicaties: intracystiche bloeding, infectie, hemothorax en pleurale
effusie zijn er enkele van. (4, 90, 92, 93)
3. Transjugulaire intrahepatische portosystemische shunts (TIPS)
Het gebruik van transjugulaire intrahepatische portosystemische shunts (TIPS) voor de behandeling van
PLD patiënten met een portale hypertensie houdt een zeker risico in op intraperitoneale bloeding. Daarom
blijft het gebruik van een TIPS bij patiënten met PLD een algemeen aanvaarde contra-indicatie. (94) Er is
echter nooit een directe evidentie beschreven in de literatuur. Enkele auteurs hebben deze interventie
uitgevoerd in een beperkt aantal cases met een normaal bloedingsrisico als uitkomst. (95) Door het gebrek
aan data is het desondanks niet mogelijk een duidelijke aanbeveling te maken.
4. Fenestratie
Open
Vóór men laparoscopische technieken begon te gebruiken was laparotomie met fenestratie de standaard
therapie voor ongecompliceerde hepatische cysten. De open fenestratie werd voor het eerst beschreven
door Lin et al. in 1968. (96) De techniek berust op een deroofing van zowel oppervlakkig als dieper
gelegen cysten met drainage in de peritoneale holte. De morbiditeit en mortaliteit beschreven bij open
fenestratie variëren tussen 0%-66% en 0%-11% respectievelijk. Recidief van symptomen voor
laparotomie met fenestratie beschreven in de grootste series variëren van 11%-26%. (9, 70, 72, 85)
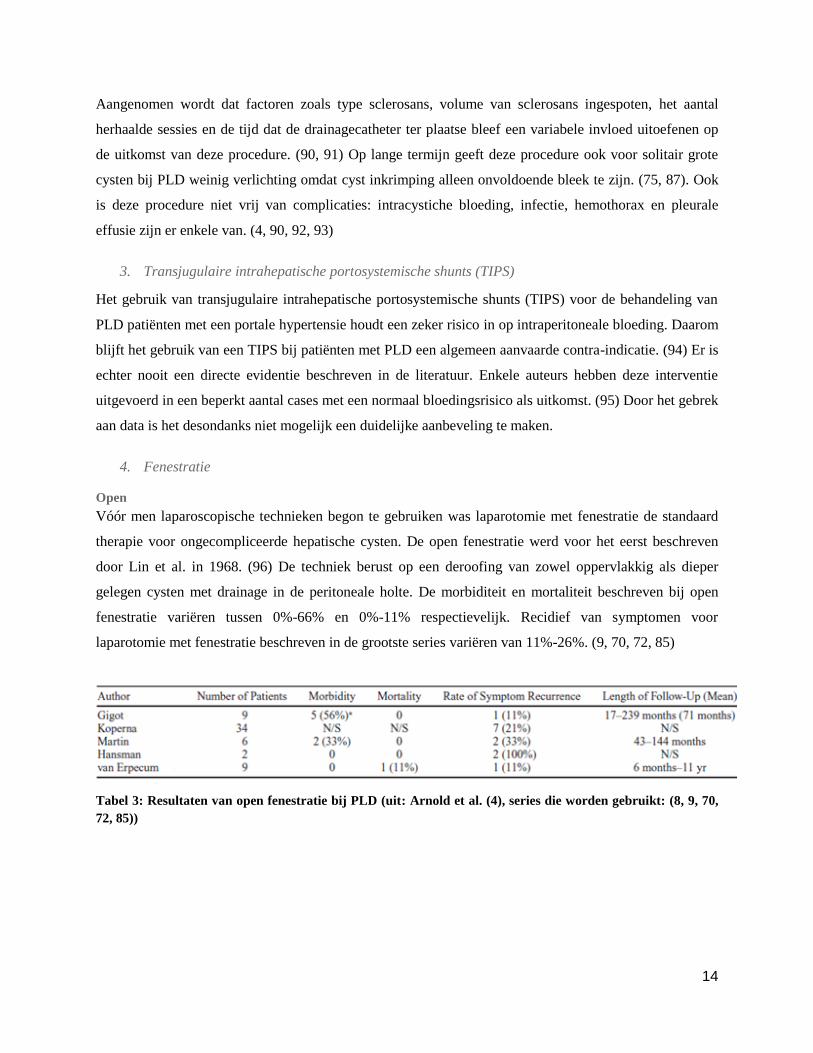
Tabel 3: Resultaten van open fenestratie bij PLD (uit: Arnold et al. (4), series die worden gebruikt: (8, 9, 70,
72, 85))
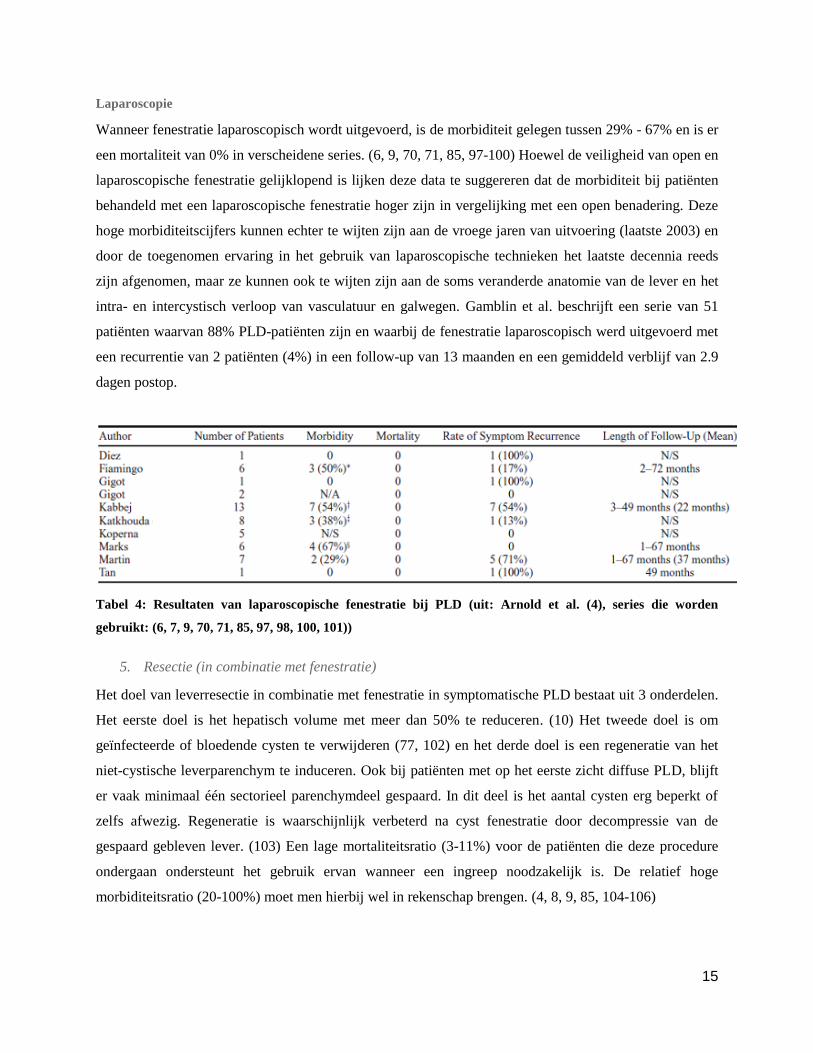
15
Laparoscopie
Wanneer fenestratie laparoscopisch wordt uitgevoerd, is de morbiditeit gelegen tussen 29% - 67% en is er
een mortaliteit van 0% in verscheidene series. (6, 9, 70, 71, 85, 97-100) Hoewel de veiligheid van open en
laparoscopische fenestratie gelijklopend is lijken deze data te suggereren dat de morbiditeit bij patiënten
behandeld met een laparoscopische fenestratie hoger zijn in vergelijking met een open benadering. Deze
hoge morbiditeitscijfers kunnen echter te wijten zijn aan de vroege jaren van uitvoering (laatste 2003) en
door de toegenomen ervaring in het gebruik van laparoscopische technieken het laatste decennia reeds
zijn afgenomen, maar ze kunnen ook te wijten zijn aan de soms veranderde anatomie van de lever en het
intra- en intercystisch verloop van vasculatuur en galwegen. Gamblin et al. beschrijft een serie van 51
patiënten waarvan 88% PLD-patiënten zijn en waarbij de fenestratie laparoscopisch werd uitgevoerd met
een recurrentie van 2 patiënten (4%) in een follow-up van 13 maanden en een gemiddeld verblijf van 2.9
dagen postop.
Tabel 4: Resultaten van laparoscopische fenestratie bij PLD (uit: Arnold et al. (4), series die worden
gebruikt: (6, 7, 9, 70, 71, 85, 97, 98, 100, 101))
5. Resectie (in combinatie met fenestratie)
Het doel van leverresectie in combinatie met fenestratie in symptomatische PLD bestaat uit 3 onderdelen.
Het eerste doel is het hepatisch volume met meer dan 50% te reduceren. (10) Het tweede doel is om
geïnfecteerde of bloedende cysten te verwijderen (77, 102) en het derde doel is een regeneratie van het
niet-cystische leverparenchym te induceren. Ook bij patiënten met op het eerste zicht diffuse PLD, blijft
er vaak minimaal één sectorieel parenchymdeel gespaard. In dit deel is het aantal cysten erg beperkt of
zelfs afwezig. Regeneratie is waarschijnlijk verbeterd na cyst fenestratie door decompressie van de
gespaard gebleven lever. (103) Een lage mortaliteitsratio (3-11%) voor de patiënten die deze procedure
ondergaan ondersteunt het gebruik ervan wanneer een ingreep noodzakelijk is. De relatief hoge
morbiditeitsratio (20-100%) moet men hierbij wel in rekenschap brengen. (4, 8, 9, 85, 104-106)
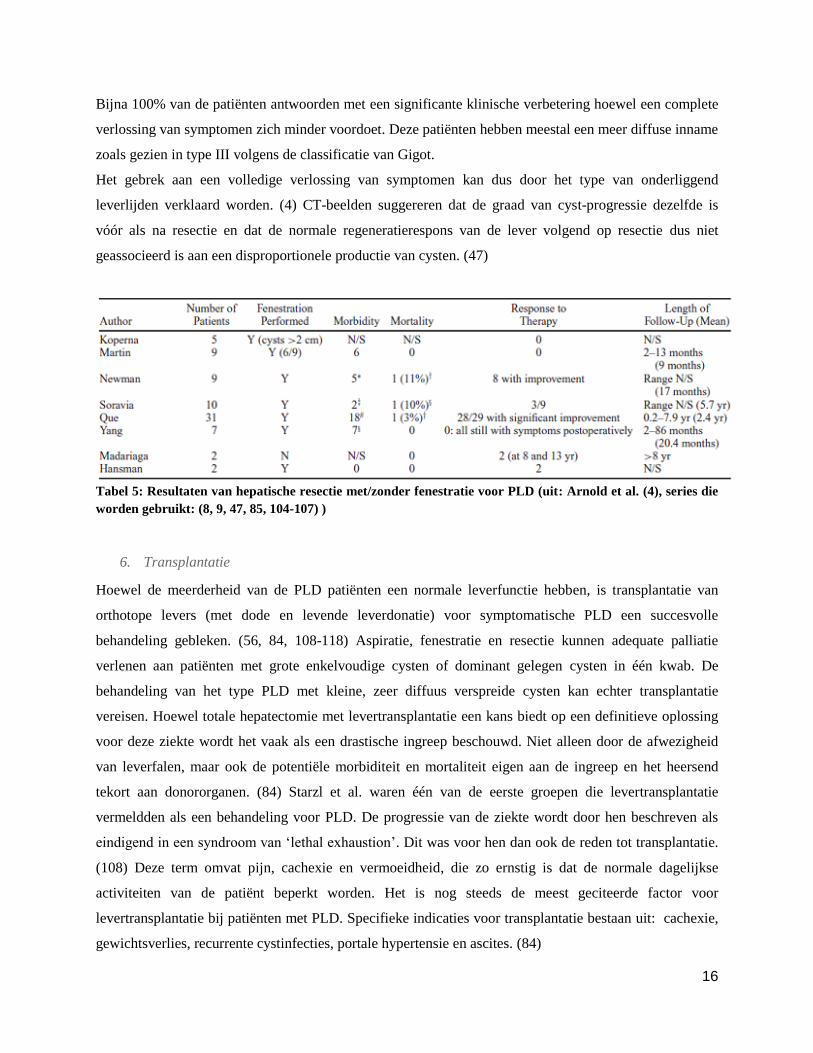
16
Bijna 100% van de patiënten antwoorden met een significante klinische verbetering hoewel een complete
verlossing van symptomen zich minder voordoet. Deze patiënten hebben meestal een meer diffuse inname
zoals gezien in type III volgens de classificatie van Gigot.
Het gebrek aan een volledige verlossing van symptomen kan dus door het type van onderliggend
leverlijden verklaard worden. (4) CT-beelden suggereren dat de graad van cyst-progressie dezelfde is
vóór als na resectie en dat de normale regeneratierespons van de lever volgend op resectie dus niet
geassocieerd is aan een disproportionele productie van cysten. (47)
Tabel 5: Resultaten van hepatische resectie met/zonder fenestratie voor PLD (uit: Arnold et al. (4), series die
worden gebruikt: (8, 9, 47, 85, 104-107) )
6. Transplantatie
Hoewel de meerderheid van de PLD patiënten een normale leverfunctie hebben, is transplantatie van
orthotope levers (met dode en levende leverdonatie) voor symptomatische PLD een succesvolle
behandeling gebleken. (56, 84, 108-118) Aspiratie, fenestratie en resectie kunnen adequate palliatie
verlenen aan patiënten met grote enkelvoudige cysten of dominant gelegen cysten in één kwab. De
behandeling van het type PLD met kleine, zeer diffuus verspreide cysten kan echter transplantatie
vereisen. Hoewel totale hepatectomie met levertransplantatie een kans biedt op een definitieve oplossing
voor deze ziekte wordt het vaak als een drastische ingreep beschouwd. Niet alleen door de afwezigheid
van leverfalen, maar ook de potentiële morbiditeit en mortaliteit eigen aan de ingreep en het heersend
tekort aan donororganen. (84) Starzl et al. waren één van de eerste groepen die levertransplantatie
vermeldden als een behandeling voor PLD. De progressie van de ziekte wordt door hen beschreven als
eindigend in een syndroom van ‘lethal exhaustion’. Dit was voor hen dan ook de reden tot transplantatie.
(108) Deze term omvat pijn, cachexie en vermoeidheid, die zo ernstig is dat de normale dagelijkse
activiteiten van de patiënt beperkt worden. Het is nog steeds de meest geciteerde factor voor
levertransplantatie bij patiënten met PLD. Specifieke indicaties voor transplantatie bestaan uit: cachexie,
gewichtsverlies, recurrente cystinfecties, portale hypertensie en ascites. (84)
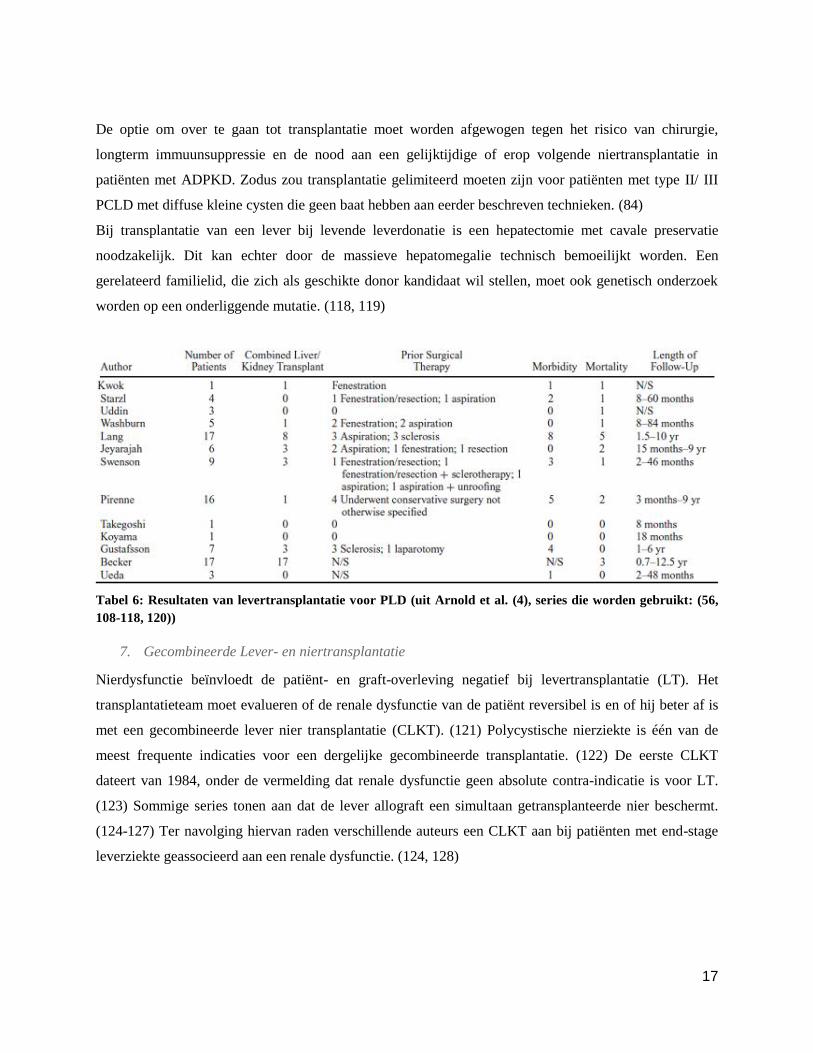
17
De optie om over te gaan tot transplantatie moet worden afgewogen tegen het risico van chirurgie,
longterm immuunsuppressie en de nood aan een gelijktijdige of erop volgende niertransplantatie in
patiënten met ADPKD. Zodus zou transplantatie gelimiteerd moeten zijn voor patiënten met type II/ III
PCLD met diffuse kleine cysten die geen baat hebben aan eerder beschreven technieken. (84)
Bij transplantatie van een lever bij levende leverdonatie is een hepatectomie met cavale preservatie
noodzakelijk. Dit kan echter door de massieve hepatomegalie technisch bemoeilijkt worden. Een
gerelateerd familielid, die zich als geschikte donor kandidaat wil stellen, moet ook genetisch onderzoek
worden op een onderliggende mutatie. (118, 119)
Tabel 6: Resultaten van levertransplantatie voor PLD (uit Arnold et al. (4), series die worden gebruikt: (56,
108-118, 120))
7. Gecombineerde Lever- en niertransplantatie
Nierdysfunctie beïnvloedt de patiënt- en graft-overleving negatief bij levertransplantatie (LT). Het
transplantatieteam moet evalueren of de renale dysfunctie van de patiënt reversibel is en of hij beter af is
met een gecombineerde lever nier transplantatie (CLKT). (121) Polycystische nierziekte is één van de
meest frequente indicaties voor een dergelijke gecombineerde transplantatie. (122) De eerste CLKT
dateert van 1984, onder de vermelding dat renale dysfunctie geen absolute contra-indicatie is voor LT.
(123) Sommige series tonen aan dat de lever allograft een simultaan getransplanteerde nier beschermt.
(124-127) Ter navolging hiervan raden verschillende auteurs een CLKT aan bij patiënten met end-stage
leverziekte geassocieerd aan een renale dysfunctie. (124, 128)

18
II. Vraagstelling
In deze scriptie wordt gepoogd een antwoord te bieden op de vraag welke behandeling de beste oplossing
biedt bij patiënten met ernstige PLD. Hieronder worden patiënten gerekend met een PLD type II of type
III volgens de classificatie van Gigot die ernstige symptomen met een gedaalde levenskwaliteit ten
gevolge van de massieve hepatomegalie vertonen. Theoretisch denkt men hierbij aan resectie of
transplantatie als behandelingsoptie. Verschillende factoren spelen een rol in de behandelingskeuze: de
morbiditeit en mortaliteit eigen aan beide ingrepen, de kans op mogelijk recidiverende symptomen en
specifieke problemen die optreden bij deze ingrepen. Aan de hand van een uitgebreid literatuuronderzoek
en een onderzoek naar de vertegenwoordiging van beide methodes in het UZ Gent wordt dit nagegaan.
III. Methode
1. Literatuuronderzoek
Om tot deze scriptie te komen, werd voornamelijk de databank Pubmed gebruikt. De databank Isi web of
science werd ook geraadpleegd. Dankzij het SFX-systeem verbonden aan de Universiteit van Gent kon
men de volledige versie van de artikels verkrijgen.
Het invoeren van zoektermen als polycystic ,liver, disease, guideline of polycystic, liver, disease met als
filter guideline in de database Pubmed leverde geen bruikbare resultaten op. Ook een gelijkaardige
zoektocht op Isi web of science leverde geen bruikbare resultaten. We stellen hierbij vast dat er
(voorlopig) nog geen guidelines beschikbaar zijn voor polycystsische leverziekte.
Invoeren van de zoektermen: polycystic, liver, disease met als limit randomized controlled trial (RCT) in
Pubmed leverde 4 bruikbare artikels op die handelen over de efficiënte behandeling van polycystische
leverziekte met somatostatine-analogen. (79-82) Opzoeking in Isi web of science leverde dezelfde
bruikbare resultaten op. Voor de chirurgische behandelingen werden er geen resultaten gevonden.

19
Voor reviewartikels:
Invoeren van de zoektermen polycystic liver disease met als limit review leverde 27 relevante titels op.
Onder relevant verstaat men onderwerpen die handelen over leverpolycystose. Invoeren van de
zoektermen polycystic liver disease met als limits: max 10 jaar oud, Humans, English, leverde 92 artikels
met relevante titel op die geen case-reports waren. Invoeren van de termen polycystic liver treatment
review met als limits: max 10 jaar oud, Humans, English, leverde 12 artikels op met een relevante titel.
Voor artikels specifiek handelend over resectie:
Na invoeren van de termen polycystic liver, resection, Sugery limits: English bekomt men 88 artikels.
Voor artikels specifiek handelend over transplantatie:
Na invoeren van de zoektermen polycystic liver disease, transplantation met als limits: human, English
bekomt men 183 artikels.
Ook werd specifiek een artikel van Guglielmi et al. (129) opgezocht om informatie over de minimale
hoeveelheid residueel levervolume in te schatten bij leverresectie.
2. Recente data
A. Registry data
Recente gegevens over transplantatie voor PLD-patiënten in Europa zijn bekomen dankzij Eurotransplant.
Via hun site ( www.eurotransplant.org ) en de site van de European Liver Transplant Registry
(www.ELTR.org) werden wachtlijsten en overlevingsdata verworven. UNOS-data werden bekomen via
hun site ( www.UNOS.org ) waar de meeste informatie gemakkelijk te vinden was over transplantatie in
de USA. De MOHAN foundation wou geen data doorsturen en ook via hun site was geen relevante data
te vinden over transplantaties in India.
B. UZ Gent
Gegevens van het UZ gent omtrent transplantatie bij patiënten met PLD werden bekomen dankzij
raadpleging van de levertransplantatie databank. Resultaten van resectie bij patiënten met PLD werden
gevonden via de leverresectie databank
20
IV. Resultaten
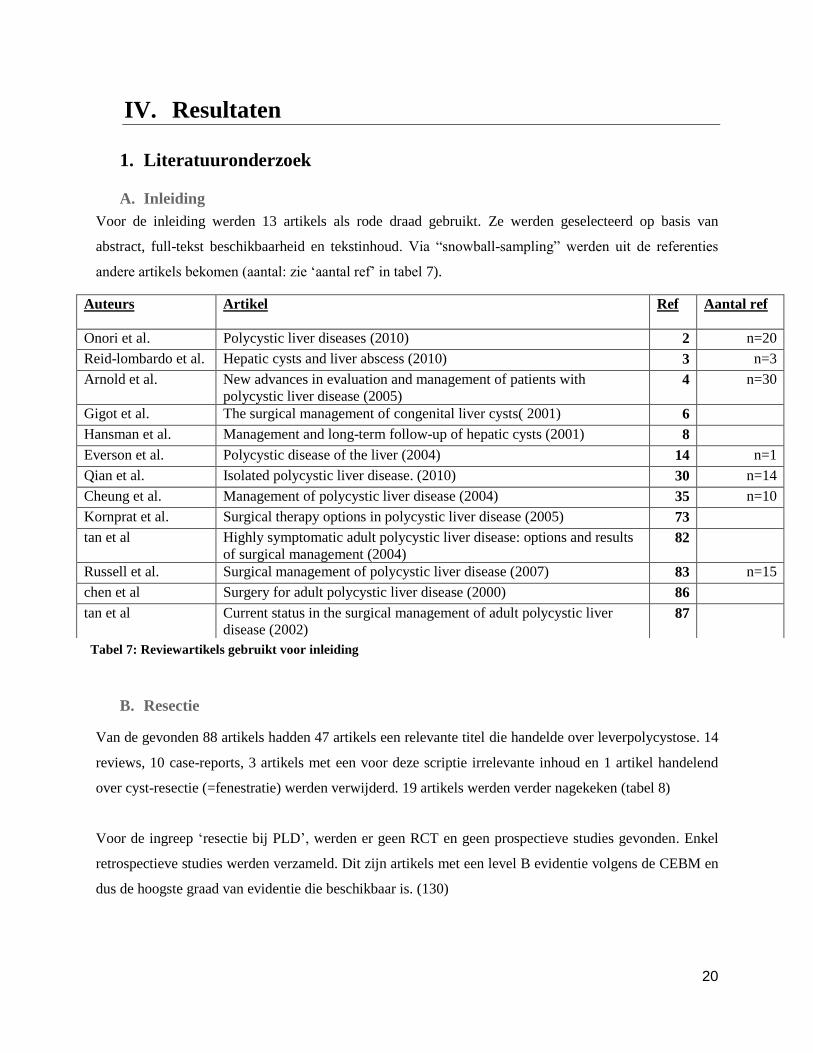
1. Literatuuronderzoek
A. Inleiding
Voor de inleiding werden 13 artikels als rode draad gebruikt. Ze werden geselecteerd op basis van
abstract, full-tekst beschikbaarheid en tekstinhoud. Via “snowball-sampling” werden uit de referenties
andere artikels bekomen (aantal: zie ‘aantal ref’ in tabel 7).
B. Resectie
Van de gevonden 88 artikels hadden 47 artikels een relevante titel die handelde over leverpolycystose. 14
reviews, 10 case-reports, 3 artikels met een voor deze scriptie irrelevante inhoud en 1 artikel handelend
over cyst-resectie (=fenestratie) werden verwijderd. 19 artikels werden verder nagekeken (tabel 8)
Voor de ingreep ‘resectie bij PLD’, werden er geen RCT en geen prospectieve studies gevonden. Enkel
retrospectieve studies werden verzameld. Dit zijn artikels met een level B evidentie volgens de CEBM en
dus de hoogste graad van evidentie die beschikbaar is. (130)
Auteurs
Artikel Ref Aantal ref
Onori et al. Polycystic liver diseases (2010) 2 n=20
Reid-lombardo et al. Hepatic cysts and liver abscess (2010) 3 n=3
Arnold et al. New advances in evaluation and management of patients with
polycystic liver disease (2005) 4 n=30
Gigot et al. The surgical management of congenital liver cysts( 2001) 6
Hansman et al. Management and long-term follow-up of hepatic cysts (2001) 8
Everson et al. Polycystic disease of the liver (2004) 14 n=1
Qian et al. Isolated polycystic liver disease. (2010) 30 n=14
Cheung et al. Management of polycystic liver disease (2004) 35 n=10
Kornprat et al. Surgical therapy options in polycystic liver disease (2005) 73
tan et al Highly symptomatic adult polycystic liver disease: options and results
of surgical management (2004) 82
Russell et al. Surgical management of polycystic liver disease (2007) 83 n=15
chen et al Surgery for adult polycystic liver disease (2000) 86
tan et al Current status in the surgical management of adult polycystic liver
disease (2002) 87
Tabel 7: Reviewartikels gebruikt voor inleiding
21
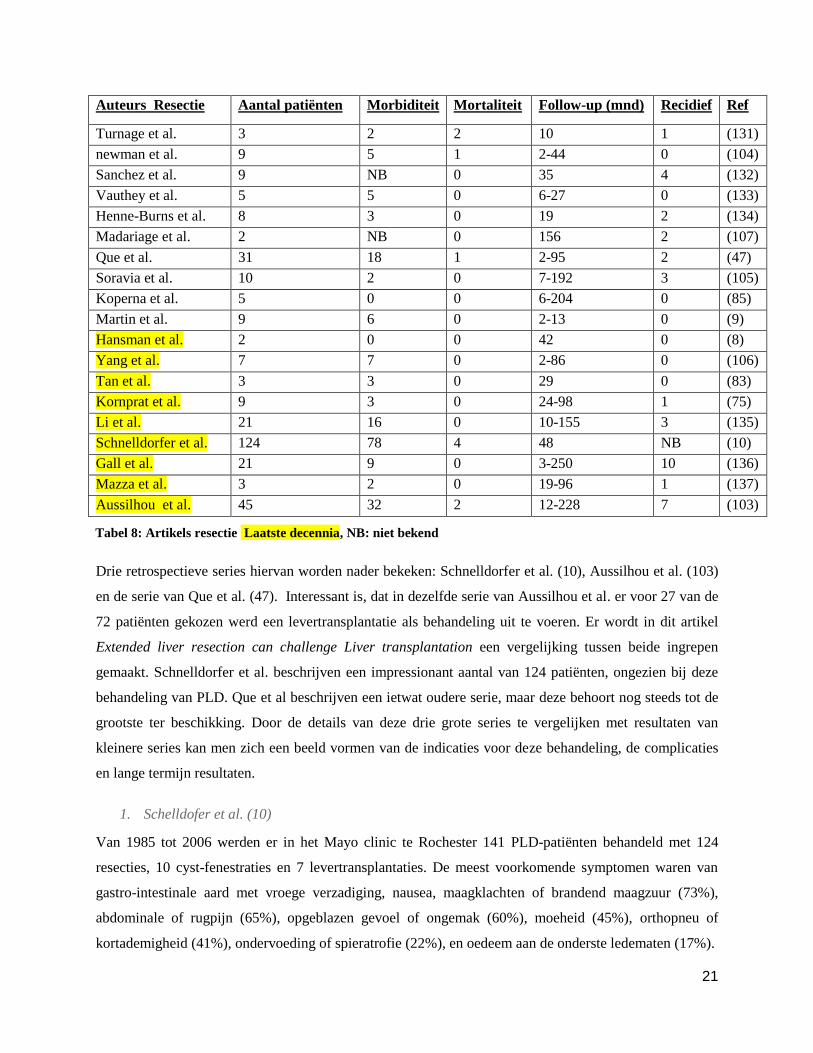
Auteurs Resectie
Aantal patiënten Morbiditeit Mortaliteit Follow-up (mnd) Recidief Ref
Turnage et al. 3 2 2 10 1 (131)
newman et al. 9 5 1 2-44 0 (104)
Sanchez et al. 9 NB 0 35 4 (132)
Vauthey et al. 5 5 0 6-27 0 (133)
Henne-Burns et al. 8 3 0 19 2 (134)
Madariage et al. 2 NB 0 156 2 (107)
Que et al. 31 18 1 2-95 2 (47)
Soravia et al. 10 2 0 7-192 3 (105)
Koperna et al. 5 0 0 6-204 0 (85)
Martin et al. 9 6 0 2-13 0 (9)
Hansman et al. 2 0 0 42 0 (8)
Yang et al. 7 7 0 2-86 0 (106)
Tan et al. 3 3 0 29 0 (83)
Kornprat et al. 9 3 0 24-98 1 (75)
Li et al. 21 16 0 10-155 3 (135)
Schnelldorfer et al. 124 78 4 48 NB (10)
Gall et al. 21 9 0 3-250 10 (136)
Mazza et al. 3 2 0 19-96 1 (137)
Aussilhou et al. 45 32 2 12-228 7 (103)
Drie retrospectieve series hiervan worden nader bekeken: Schnelldorfer et al. (10), Aussilhou et al. (103)
en de serie van Que et al. (47). Interessant is, dat in dezelfde serie van Aussilhou et al. er voor 27 van de
72 patiënten gekozen werd een levertransplantatie als behandeling uit te voeren. Er wordt in dit artikel
Extended liver resection can challenge Liver transplantation een vergelijking tussen beide ingrepen
gemaakt. Schnelldorfer et al. beschrijven een impressionant aantal van 124 patiënten, ongezien bij deze
behandeling van PLD. Que et al beschrijven een ietwat oudere serie, maar deze behoort nog steeds tot de
grootste ter beschikking. Door de details van deze drie grote series te vergelijken met resultaten van
kleinere series kan men zich een beeld vormen van de indicaties voor deze behandeling, de complicaties
en lange termijn resultaten.
1. Schelldofer et al. (10)
Van 1985 tot 2006 werden er in het Mayo clinic te Rochester 141 PLD-patiënten behandeld met 124
resecties, 10 cyst-fenestraties en 7 levertransplantaties. De meest voorkomende symptomen waren van
gastro-intestinale aard met vroege verzadiging, nausea, maagklachten of brandend maagzuur (73%),
abdominale of rugpijn (65%), opgeblazen gevoel of ongemak (60%), moeheid (45%), orthopneu of
kortademigheid (41%), ondervoeding of spieratrofie (22%), en oedeem aan de onderste ledematen (17%).
Tabel 8: Artikels resectie Laatste decennia, NB: niet bekend

22
Symptomen van de galwegen zoals icterus (3%) of cholangitis (1%) waren zeldzaam. De gemiddelde
follow-up bedroeg 7 jaar. De primaire indicaties voor operatie waren: symptomen die de kwaliteit van
leven of de performance status in 131 patiënten verminderen, ernstige veneuze obstructie van de lever met
refractaire ascites bij 6 patiënten, geïnfecteerde levercysten na percutane drainage in 3 en
ongecontroleerde intra cystische bloeding bij één persoon na percutane interventie. 124 patiënten
ondergingen een resectie met cystfenestratie. Dit omvatte lobectomie bij 67 patiënten, uitgebreide
lobectomie bij 30 patiënten en resectie van meerdere leversegmenten bij 27 patiënten. Gemiddeld werden
er 4 leversegmenten gereseceerd. De Leverresectie werd uitgevoerd wanneer er ten minste één sectorieel
gedeelte parenchym relatief gespaard bleef van cystinname.
Complicaties
Bij 63% van de patiënten waren er complicaties. Majeure complicaties (Clavien graad III tot IV) traden
op in 31% van de patiënten. Patiënten met preoperatieve chronische nierinsufficiëntie liepen een groter
risico op perioperatieve complicaties (71%) in vergelijking met patiënten met een normale nierfunctie
(53%; P= 0.041). Vooral ascites en gallekkage vormden de meest voorkomende postoperatieve
complicaties. Re-operatie was nodig om bloedingen te controleren bij 7 patiënten, levertransplantatie voor
leverfalen bij 2 patiënten, gastro-jejunostomie bij één patiënt en abdominale spoeling bij één patiënt.
Perioperatieve sterfte omvatte 4 patiënten (3%), met als oorzaak abdominale sepsis, hart aritmie tijdens
een additionele levertransplantatie, herseninfarct en intracraniële hemorragie.
2. Aussilhou et al. (103)
Vijfenveertig patiënten ondergingen in het Beaujon ziekenhuis in Frankrijk een hepatische resectie met
cyst fenestratie. 29 patiënten (65%) waren Gigot type II en 16 (35%) Gigot type III. De resectie bedroeg
altijd meer dan 2 segmenten en was geassocieerd aan een extensieve cyst fenestratie van de resterende
lever. De gemiddelde reductie van het levervolume bedroeg 75% (range 50-88%) en het gemiddeld
resterend volume non-cystiche lever was 843 ml (range 480-1720). Er was geen operatieve mortaliteit,
maar de algemene morbiditeit postoperatief bedroeg 71%. Resectie van de lever werd uitgevoerd wanneer
het volume van de toekomstige restant-lever minstens 30% van de totale, niet cystische lever zou
bedragen. Dit om de kans op post hepatectomie leverfalen te minimaliseren.

23
Complicaties
De meest frequente complicaties waren biliaire lekkage in 9 (20%) patiënten en ascites in 19 (42%)
patiënten met een gemiddeld verlies van 2160 ml ascites over een gemiddelde periode van 11 dagen en
een maximum van 7 weken. Andere specifieke complicaties: pleurale effusie met nood aan drainage in 9
(20%) patiënten, transiënt leverfalen in 4 (9%) en hemorragie in 9 (20%). Zes (13%) patiënten hadden
nood aan een her-operatie ten gevolge van hun hemorragie en één omwille van een biliair lek. Van de 45
patiënten werden er 43 overlevenden klinisch gevolgd voor een periode van gemiddeld 41 maanden (
range 12-228). De doodsoorzaak van de 2 overleden patiënten was PLD gerelateerd. Eén patiënt overleed
aan een latere levertransplantatie en de tweede aan een thoracale hemorragie na een pleurale punctie.
Persisterende symptomen werden bij 3 (7%) patiënten vastgesteld ten gevolge van insufficiënte
cystfenestratie van de gespaarde lever. Recidief van symptomen werd opgemerkt na een periode van
gemiddeld 3 jaar in 7 (16%) patiënten waarvan 2 ten gevolge van een recidiverende hepatomegalie. Al
deze symptomatische cysten werden met percutaneuze sclerotherapie behandeld. Onder de 43
overlevenden waren 36 patiënten (83%) tevreden met de ingreep.
3. Que et al. (47)
In deze serie ondergingen er 31 patiënten een leverresectie met fenestratie. De indicaties tot chirurgie
bestonden uit abdominale distentie (n=30), abdominale pijn (n=26), vroege verzadiging (n=21),
vermoeidheid (n=17), dyspnoe in ruglig (n=13), geïnfecteerde cyste (n=3), dialyse hypotensie (n=3),
galwegobstructie (n=2), ernstige ascites (n=2) en uterus prolaps. Leverresectie bestond uit 13
lobectomies, 2 uitgebreide leverresecties en 16 non anatomische resecties (non lobaire resecties, dan wel
intrasegmenteel of intersegmenteel). Gemidddeld werden er 4 segmenten gereseceerd per patiënt.
Resectie werd de patiënten aangeboden wanneer deze 2 of meer leversegmenten hadden die relatief vrij
waren van cysten. Uit deze beschrijving valt echter niet op te maken hoeveel Gigot klasse II
respectievelijk klasse III dit beslaat.
Complicaties
Achttien patiënten ondervonden complicaties waaronder transiënte pleurale effusie (n=11), transiënte
ascites (n=7), her-operatie omwille van hemorragie (n=4), trombose van een arterioveneuze fistel (n=3),
infectie (n=2) en een transiënte gallekkage (n=2). Follow-up gebeurde voor 29 van de 30 overlevende
patiënten. Eén patiënt stierf ten gevolge van een intracraniële bloeding 33 dagen postoperatief. Er kon
geen aneurysma aangetoond worden tijdens de autopsie. De gemiddelde follow-up bedroeg 33.6 maanden
(range 2.4 - 94.8). Pre-operatief bedroeg de lever gemiddeld 9357 cm2, post-operatief gemiddeld 3567
cm2.

24
Het levervolume bleef in de meeste gevallen stabiel, behalve bij 2 patiënten: één patiënt had objectieve
progressie en ook de jongste patiënte, 34 jaar, vertoonde een vergroting van de lever. Postoperatief
bevatte haar residuele leversegmenten nog enkele smalle en middelgrote cysten. Het is vooral door de
expansie van deze cysten dat de lever weer was toegenomen in grootte.
Performance status
Van de 30 patiënten met preoperatieve ECOG performance status 0 (n=1), 1 (n=13), 2 (n=15) en 3 (n=2)
waren er 28 met adequate follow-up tevreden met de ingreep en vertoonden een onmiddellijke, en in de
meeste gevallen langdurige, verbetering in hun QOL en ECOG performance status. Met de ECOG kan de
performance status objectief weergegeven worden in 6 categorieën: 0) volledig actief; 1) enkel beperkt
voor zware fysieke activiteiten; 2) ambulant, capabel om voor zichzelf te zorgen, kan licht werk uitvoeren
en is actief >50% van de wakkere uren; 3) enkel capabel in beperkte mate voor zichzelf te zorgen en is
gebonden aan stoel of bed >50% van de wakkere uren; 4) compleet gehandicapt en kan niet voor zichzelf
zorgen en 5) dood. (163) Eén patiënt was ontevreden met de procedure en vond niet dat haar QOL was
verbeterd met de ingreep, ondanks een duidelijke reductie in de levergrootte aangetoond via CT-
onderzoek. Twee patiënten meenden onterecht dat hun lever weer in volume was toegenomen, maar
vonden dat hun QOL nog steeds beter was dan voor de ingreep.
4. Recidiverende pijn
Pathologische analyse in de serie van Aussilhou et al toonde evidentie aan van intracystische bloeding of
infectie in 14 (31%) en 5 (11%) patiënten respectievelijk. (103) Spontane bloeding is dan ook een
frequente oorzaak van acute abdominale pijn. (102) MRI, welke zeer accuraat is om dergelijke
complicaties op te sporen, kan helpen bij de planning deze cysten mee te nemen in de resectie om het
risico op recidiverende pijn te minimaliseren. Intracystische bloeding kon worden gezien in 3 patiënten
van de serie met recidiverende pijn zonder recidiverende hepatomegalie. Dit bedraagt de helft van de
patiënten met recidiverende pijn in deze serie. (103)
5. Technische moeilijkheden
Resectie
Technische moeilijkheden bij resectie van patiënten met PCLD, zoals in deze 3 grote series, worden ook
bevestigd in kleinere series (10, 84, 104, 106, 131, 135, 138). Niet alleen de massieve hepatomegalie
maar ook de rigiditeit van de levers bemoeilijkt de mobiliteit en de toegang tot de bloedvaten. (10)
Ondanks stap-per-stap punctie, fenestratie en cyst-vloeistof aspiratie blijft blootleggen en mobilisatie van
deze zeer grote levers moeilijk.

25
In de 3 series zijn dikwijls operatieve of postoperatieve bloedtransfusie noodzakelijk geweest: in 50% van
de gevallen bij Aussilhou et al. (ondanks Pringle maneuver), gemiddeld 7U ( range 0 - 61U) bloed
perioperatief in de Que et al. serie, en in 80% van de gevallen in de serie van Schelldorfer et al. met een
gemiddelde van 6 +- 1U bloed. (10, 47, 103).
Fenestratie
Bij fenestratie moet men extra aandachtig zijn voor de bloedvaten en galwegen in en rond de cystwand
die moeilijk te identificeren zijn en onder compressie staan, wat resulteert in een hoger operatief risico en
postoperatief risico op gallekkage en hemorragie. (9, 106) Deze complicaties komen in de 3 series ook
sterk naar voor. Ascites is echter de grootste bron van morbiditeit met risico van verlengde drainage,
infectie, lekkage doorheen de abdominale wonde en pleurale effusie. (47, 103) De oorzaak van ascites is
multifactorieel en kan verklaard worden door persisterende cyst secretie, veneuze outflow obstructie,
lymfe lekkage, renale dysfunctie en malnutritie met een laag albuminelevel. (10) De kwaliteit van het
resterende leverparenchym speelt hierbij een niet te onderschatten rol. Histologische veranderingen in het
niet-cystische leverparenchym zijn vaak geassocieerd aan outflow aantasting. Deze omvatten onder
andere vasculaire veranderingen en fibrosevorming die worden toegeschreven aan de secretie van
cytokines en groeifactoren. (46) Bij ¾ van de patiënten in de serie van Aussilhou et al. werden bij analyse
van de chirurgische specimen, outflow aantastingen vastgesteld. Meer zelfs, de analyse toont aan dat
patiënten met ascites veel meer fibrose-vorming vertoonden dan patiënten die geen post-operatieve ascites
hadden. (103) Of deze histologische veranderingen reversibel zijn, dient nog te worden onderzocht.
C. Transplantatie
Na selectie op relevante titels (artikels handelend over polycystische leverziekte) en verwijderen van
case-reports, werden de gevonden 183 artikels gereduceerd tot 28 artikels. Na alsnog verwijderen van 5
case-reports, 1 letter to the editor, 1 reviewartikel en 1 artikel met irrelevante info voor deze scriptie
werden 20 artikels nader bekeken. (zie tabel 9)
Voor de ingreep transplantatie bij PLD werden er geen RCT en geen prospectieve studies gevonden.
Enkel retrospectieve studies werden verzameld. Dit zijn artikels met een level B evidentie volgens de
CEBM en dus de hoogste graad van evidentie die beschikbaar is. (130)
26
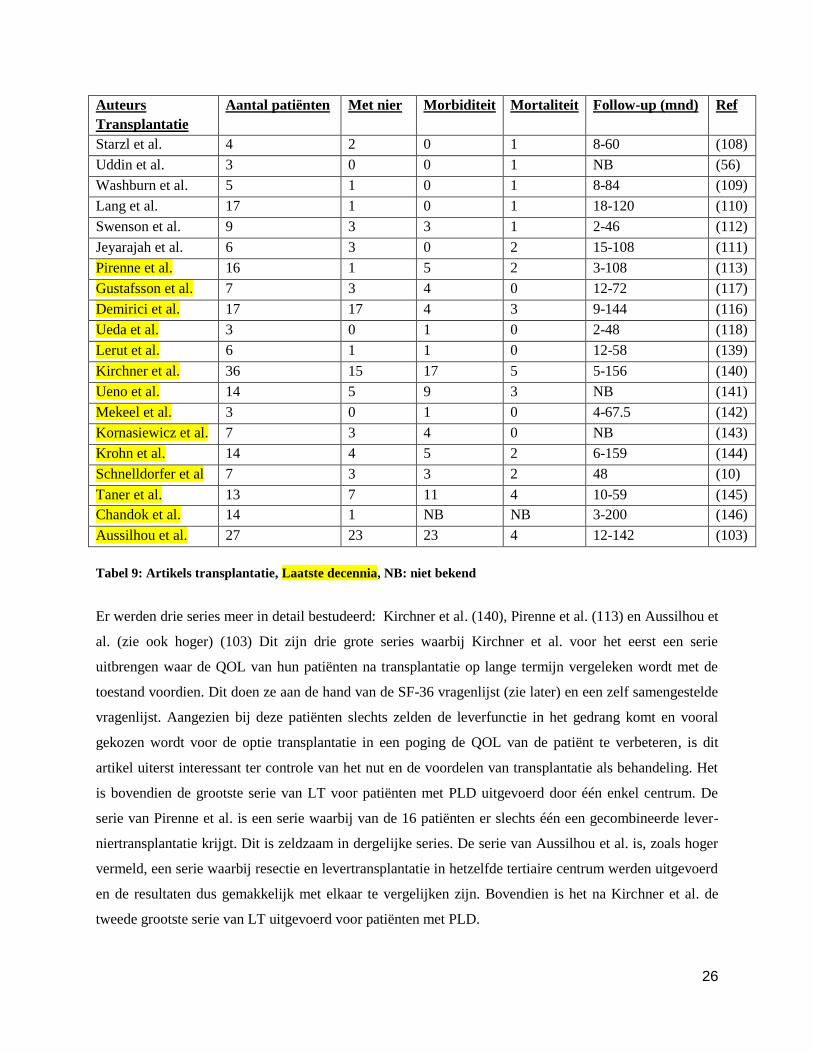
Auteurs
Transplantatie
Aantal patiënten Met nier Morbiditeit Mortaliteit Follow-up (mnd) Ref
Starzl et al. 4 2 0 1 8-60 (108)
Uddin et al. 3 0 0 1 NB (56)
Washburn et al. 5 1 0 1 8-84 (109)
Lang et al. 17 1 0 1 18-120 (110)
Swenson et al. 9 3 3 1 2-46 (112)
Jeyarajah et al. 6 3 0 2 15-108 (111)
Pirenne et al. 16 1 5 2 3-108 (113)
Gustafsson et al. 7 3 4 0 12-72 (117)
Demirici et al. 17 17 4 3 9-144 (116)
Ueda et al. 3 0 1 0 2-48 (118)
Lerut et al. 6 1 1 0 12-58 (139)
Kirchner et al. 36 15 17 5 5-156 (140)
Ueno et al. 14 5 9 3 NB (141)
Mekeel et al. 3 0 1 0 4-67.5 (142)
Kornasiewicz et al. 7 3 4 0 NB (143)
Krohn et al. 14 4 5 2 6-159 (144)
Schnelldorfer et al 7 3 3 2 48 (10)
Taner et al. 13 7 11 4 10-59 (145)
Chandok et al. 14 1 NB NB 3-200 (146)
Aussilhou et al. 27 23 23 4 12-142 (103)
Tabel 9: Artikels transplantatie, Laatste decennia, NB: niet bekend
Er werden drie series meer in detail bestudeerd: Kirchner et al. (140), Pirenne et al. (113) en Aussilhou et
al. (zie ook hoger) (103) Dit zijn drie grote series waarbij Kirchner et al. voor het eerst een serie
uitbrengen waar de QOL van hun patiënten na transplantatie op lange termijn vergeleken wordt met de
toestand voordien. Dit doen ze aan de hand van de SF-36 vragenlijst (zie later) en een zelf samengestelde
vragenlijst. Aangezien bij deze patiënten slechts zelden de leverfunctie in het gedrang komt en vooral
gekozen wordt voor de optie transplantatie in een poging de QOL van de patiënt te verbeteren, is dit
artikel uiterst interessant ter controle van het nut en de voordelen van transplantatie als behandeling. Het
is bovendien de grootste serie van LT voor patiënten met PLD uitgevoerd door één enkel centrum. De
serie van Pirenne et al. is een serie waarbij van de 16 patiënten er slechts één een gecombineerde lever-
niertransplantatie krijgt. Dit is zeldzaam in dergelijke series. De serie van Aussilhou et al. is, zoals hoger
vermeld, een serie waarbij resectie en levertransplantatie in hetzelfde tertiaire centrum werden uitgevoerd
en de resultaten dus gemakkelijk met elkaar te vergelijken zijn. Bovendien is het na Kirchner et al. de
tweede grootste serie van LT uitgevoerd voor patiënten met PLD.

27
Door de details van deze drie grote series te vergelijken met resultaten van kleinere series, kan men zich
een beeld vormen van de indicaties voor deze behandeling, de complicaties en lange termijn resultaten.
1. Kirchner et al (140)
Kirchner et al. beschrijven een serie waarbij 36 patiënten een levertransplantatie (n=21) of lever-nier
transplantatie ondergingen (n=15). Zoals reeds hoger aangehaald nam men hier aan dat lever-nier
transplantatie de beste behandeling is wanneer een patiënt met gecombineerde polycystische nier- en
leverziekte naast ernstig PLD ook nierinsufficiëntie vertoont. Drieëndertig patiënten ontvingen een
kadaver full-size lever, 2 patiënten een kadaver split lever, en 1 patiënt ontving een living donor split
lever. Al de patiënten kloegen over abdominale distentie en ernstig discomfort, maar ook cachexie of
ernstige malnutritie (n=25), recurrente cyst infectie (n=7), dyspnoe (n=8) en portale hypertensie met
oesofageale varices bloedingen (n=2) of ascites (n=10) behoorden tot de symptomen.
Nierfunctie
Nierfunctie (creatinine clearance, serum creatinine of glomerulaire filtratie rate) werd gecontroleerd voor
transplantatie en op het einde van de follow-up. Vijftien patiënten ondergingen gecombineerde lever-nier
transplantatie wegens progressieve nierinsufficiëntie (GFR< 30 ml/min) (n=4) of compleet nierfalen
(n=11) met dialyseafhankelijkheid. Men moet immers ook in rekening brengen dat de patiënten na
transplantatie vaak nefrotoxische immunosuppressiva zoals calcineurine inhibitoren krijgen toegediend,
wat de nierfunctie dikwijls nog doet afnemen. De levertransplantatie werd uitgevoerd volgens een
gestandaardiseerde procedure. Bij een gecombineerde transplantatie werd de nier getransplanteerd
volgend op de lever. Wanneer de patiënt zeer grote polycystische nieren had met klinische problemen
zoals intracystische bloeding of cyst infectie, werden één of beide nieren verwijderd vóór implantatie.
Complicaties
Complicaties: bij 10 patiënten was een heroperatie nodig na transplantatie wegens intra-abdominale
bloeding (n=3), gal lekkage (n=3), thrombectomie van de rechter arteria hepatica (n=1, living split
transplant), lekkage van de uretrocystoneostomie (n=1), maag-ulcer perforatie (n=1) en sigmoid colon
perforatie (n=1). De gemiddelde duur van hospitalisatie voor gecombineerde transplantatie was
significant langer dan die van enkelvoudige levertransplantatie. (62 vs 35 dagen). Twee re-transplantaties
waren nodig binnen de eerste dagen. Op korte tijd overleden 5 van de 36 patiënten. Vier patiënten
stierven aan sepsis na 29, 45, 45 en 47 dagen. Eén patiënt had initieel problemen met de levergraft die niet
naar behoren functioneerde en werd geretransplanteerd na 1 dag. Hij stierf ten gevolge van een
myocardinfarct en pneumonie 61 dagen na de initiële transplantatie.
28
Survival
Er werd geen overlijden meer vastgesteld na de eerste 2 maand in deze groep. Dat brengt de 1- en 5-jaars
overleving op 86%. Van de 5 patiënten die in de serie van Kirchner et al. stierven aan een infectie waren
er 3 patiënten die zowel polycystische lever- als nierziekte hadden, maar een enkelvoudige
levertransplantatie hadden ondergaan. De auteur vermoedt dat het verhoogde risico op sepsis in deze
patiënten kan verklaard worden door bacteriële reservoirs in de nog resterende polycystische nieren. Een
andere verklaring zou de slechte nutritionele toestand van deze patiënten kunnen zijn. (140)
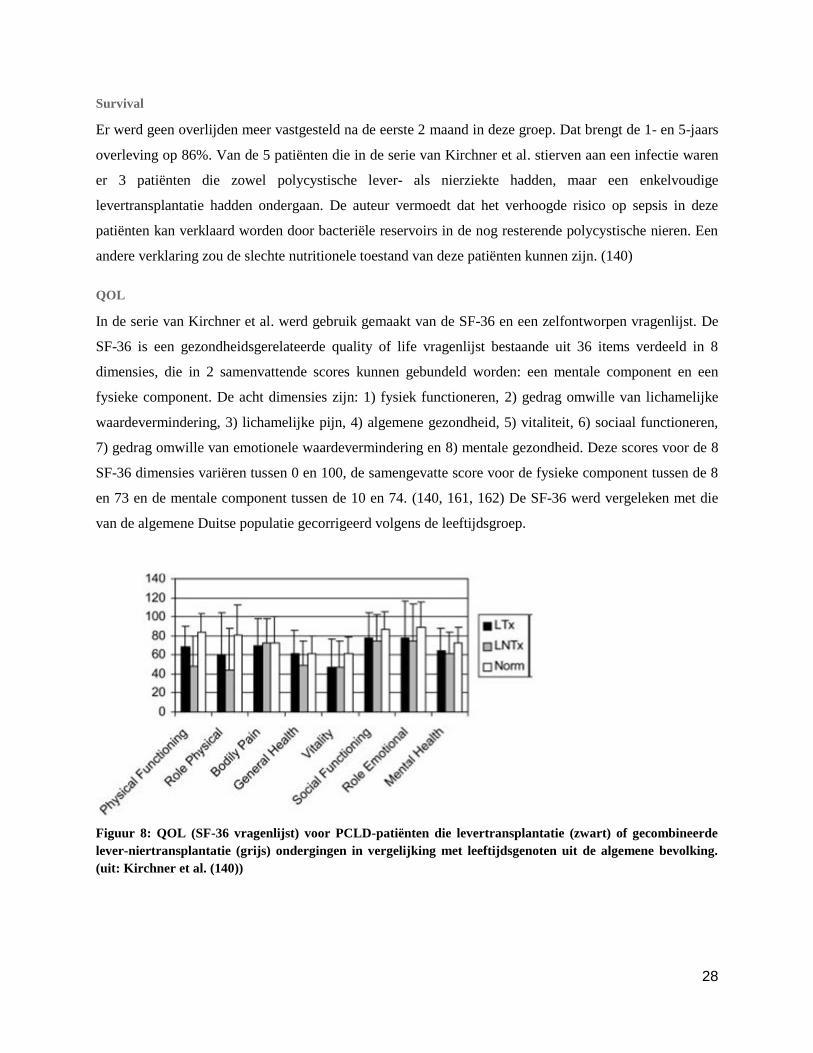
QOL
In de serie van Kirchner et al. werd gebruik gemaakt van de SF-36 en een zelfontworpen vragenlijst. De
SF-36 is een gezondheidsgerelateerde quality of life vragenlijst bestaande uit 36 items verdeeld in 8
dimensies, die in 2 samenvattende scores kunnen gebundeld worden: een mentale component en een
fysieke component. De acht dimensies zijn: 1) fysiek functioneren, 2) gedrag omwille van lichamelijke
waardevermindering, 3) lichamelijke pijn, 4) algemene gezondheid, 5) vitaliteit, 6) sociaal functioneren,
7) gedrag omwille van emotionele waardevermindering en 8) mentale gezondheid. Deze scores voor de 8
SF-36 dimensies variëren tussen 0 en 100, de samengevatte score voor de fysieke component tussen de 8
en 73 en de mentale component tussen de 10 en 74. (140, 161, 162) De SF-36 werd vergeleken met die
van de algemene Duitse populatie gecorrigeerd volgens de leeftijdsgroep.
Figuur 8: QOL (SF-36 vragenlijst) voor PCLD-patiënten die levertransplantatie (zwart) of gecombineerde
lever-niertransplantatie (grijs) ondergingen in vergelijking met leeftijdsgenoten uit de algemene bevolking.
(uit: Kirchner et al. (140))

29
Er bleek geen groot verschil te zijn in de dimensies ‘emotionele waardevermindering’, ‘sociaal
functioneren’ of ‘mentale gezondheid’ wat een succesvolle integratie in de sociale en familiale structuren
weerspiegelt. Er bleek geen verschil te zijn in algemene gezondheid en lichamelijke pijn wat nochtans te
verwachten valt bij immuun gecompromitteerde transplantatiepatiënten. De SF-36 dimensies ‘fysiek
functioneren’, ‘gedrag door fysieke capaciteit’ en ‘vitaliteit’ hadden de neiging lager uit te vallen in de
transplantpatiënten, maar dit bleek statistisch niet significant. Wanneer men rekening houdt met de
ernstige beperkingen gaande van ondervoeding tot cachexie en de reeds lang aanslepende symptomen
vóór operatie lijkt deze licht lagere uitval makkelijk te verklaren. Bij enkelvoudige levertransplantatie zijn
deze verschillen minder uitgesproken dan bij gecombineerde transplantatie. (140)
De zelf samengestelde vragenlijst analyseerde fysieke, emotionele en sociale veranderingen vóór en na
transplantatie in detail. Overeenkomstig de SF-36 voelde de meerderheid van de patiënten zich
uitstekend, zeer goed of goed na transplantatie. Bij de subgroep van enkelvoudige levertransplanten
voelde iedereen zich beter dan voor de ingreep, net als 91% van de groep patiënten die lever- en
niertransplantatie ondergingen. Een gedetailleerde analyse toont een verbetering van alle majeure
symptomen. (zie figuur 9) Vermoeidheid, fysieke fitheid, anorexia, braken, fysieke aantrekkelijkheid en
interesse in seks verbeterden na transplantatie. Algemeen gezien hebben patiënten met PCLD dus een
verbetering van hun levenskwaliteit na lever of gecombineerde lever-nier transplantatie. (140)

30
Figuur 9: Parameters van de QOL vóór en na de transplantatie weergegeven in boxplots. Elke parameter is
ingedeeld in 5 categorieën: 1) geen verslechtering, 2) lichte verslechtering, 3) milde verslechtering, 4) sterke
waardevermindering en 5) extreme waardevermindering. P= statistische significantie. PRE= preop en POST=
postop. N.S.= niet significant (uit: Kirchner et al. (140))
2. Pirenne et al (113)
In deze serie waarbij 16 patiënten een orthotope levertransplantatie (OLT) kregen, onderging slechts één
patiënt terzelfdertijd een niertransplantatie wegens geassocieerde progressieve nierinsufficiëntie. De
indicaties voor levertransplantatie waren ernstige fysieke beperkingen als gevolg van hepatomegalie:
abdominale distensie en pijn, vermoeidheid, anorexia en maagklachten, en verschillende gradaties van
dyspneu. Klinisch gevorderde ondervoeding was aanwezig in 4 patiënten, cholestase in 5 en portale
hypertensie in 3 patiënten.
Complicaties
Biliaire strictuur (n=2), revisie voor bloeding en de novo hepatitis (n=1), pneumothorax en subfrenische
collectie (n=1), pneumonie en tijdelijke nood aan tracheotomie (n=1), en CMV-infectie (n=1) vormden de
complicaties. Twee patiënten stierven in deze studie: één patiënt die reeds herhaalde mislukte pogingen
tot massareductie van de lever had ondergaan en intraoperatief overleed aan een massieve bloeding en
pulmonaire luchtembolen, en één patiënt 6 jaar post-OLT aan longkanker.

31
Met een follow-up tussen de 1.5 tot 10 jaar is de patiënt en graft survival rate 87.5%. Van de 13 die
overleefden en enkel een levertransplantatie doorgemaakt hadden, had één patiënt een bijkomende
niertransplantatie nodig 4 jaar post-OLT.
Effect
Fysieke symptomen gerelateerd aan de abdominale distensie verdwenen onmiddellijk bij alle patiënten
zodat ze terug de draad van hun professionele en sociale leven konden opnemen.
Technische moeilijkheden en oplossingen
Een factor die klassiek wordt aangegeven als limiterend voor het gebruik van deze behandelingsoptie is
de technische moeilijkheid van de procedure, veroorzaakt door de organomegalie (110, 120) Volgens de
auteur kan OLT voor PLD echter getransformeerd worden van een uitdagende operatie tot een
ongecompliceerde ingreep wanneer enkele principes gevolgd worden. Deze bestaan uit: het niet proberen
controleren van de suprahepatische vene in het begin van de ingreep, de dissectie van de leverhilus, het
gebruik van een portale bypass, vena cava en block met de lever verwijderen ipv het behouden van de
natieve cava, en cystaspiratie enkel uitvoeren nadat arteriële en veneuze inflow is onderbroken. Op deze
manier zou laceratie van de vena cava met fatale bloeding en pulmonale luchtembolen, welke het geval
was in één patiënt van deze serie, afgewend kunnen worden. Overvloedig bloeden van een cyste wordt
door de afklemming van veneuze en arteriële aanvoer beperkt. De meeste van deze technieken gelijken op
deze toegepast door Jeyarajah et al (111). Men hoopt op deze manier de operatietijd te verkorten en
bloedtransfusie te beperken.
Eerdere interventies
Gelijklopend met series van Swenson et al. (112), Starzl et al. (108) en Kirchner et al. (140) ondervond
men ook in deze serie dat de chirurgie veel ingewikkelder werd en er een grotere neiging tot bloedingen
plaatsvond, indien de patiënt reeds eerdere operaties voor PLD had ondergaan. In hoog-symptomatische
patiënten met kleine cysten diffuus verspreid over de lever, biedt OLT onmiddellijke verlichting.
Conservatieve therapie bij deze patiënten is niet alleen verbonden aan morbiditeit en mortaliteit eigen aan
de interventie, vaak biedt dit slechts gedeeltelijke verlichting of verlichting beperkt in tijd door het diffuse
karakter van het cystisch proces. Dit resulteert in een hogere kans op recidieven (zie eerder). Daarnaast
bemoeilijkt conservatieve chirurgie aanzienlijk OLT en kan het zo de kans op definitieve behandeling
reduceren. (108, 109, 112)

32
Snellere LT
Men deelt ook dezelfde mening als Kirchner et al. dat transplantatie op een vroeger tijdstip dan
eindstadium leverlijden met ondervoeding en fysieke uitputting zou moeten uitgevoerd worden. Zo kan
men infecties verminderen postop. Ook de bijkomende hoge immuunsupressie kan sneller infectie
uitlokken in deze verzwakte patiënten.
Nier-levertransplantatie
In contrast met de meeste andere series, voert men hier slechts één keer een gecombineerde nier-lever
transplantatie uit. Dit is op de 16 transplantaties uitgevoerd in deze serie slechts 6%. In andere series
loopt dit op tot gemiddeld 53% ( range 0-100%). (108, 109, 111, 112, 114, 116, 117, 120, 140, 147, 148)
In de meeste series blijkt dus meer dan de helft van de transplantaties voor deze groep van patiënten met
ernstige PLD een gecombineerde lever-nier transplantatie te zijn. De gedachte hierachter is dat een
progressieve nierinsufficiëntie blijft toenemen na LT en calcineurine inhibitoren deze progressie nog
versnellen. Bovendien is er een voordeel op het vlak van immuniteit wanneer men bij nier-lever
transplantatie beide organen van dezelfde donor afkomstig zijn ten opzichte van een latere derde-partij
niertransplantatie. (149) In deze serie had maar één patiënt 4 jaar na enkelvoudige OLT nood aan een
niertransplantatie. Dit is in sterk contrast met het verslag van Jeyarajah et al (111) die ondervonden dat tot
33% van de patiënten die een enkelvoudige levertransplantatie ondergingen later behoefte hadden aan een
nier transplantatie. De lage incidentie aan niertransplantaties na OLT in de serie van Pirenne et al. kan te
wijten zijn aan de lage dosering van calcineurine inhibitoren na OLT, ook aangeraden door andere
auteurs. (150, 151)
3. Aussilhou et al (103)
In deze serie, reeds hoger genoemd (zie resultaten, resectie) ondergingen 27 patiënten een
levertransplantatie waarvan 23 (85%) simultaan een niertransplantatie. Drie patiënten (11%) hadden een
Gigot type II en 24 patiënten (89%) een type III PLD. Dit is de op één na grootse single-center studie (na
Kirchner et al.). De nierfunctie werd aanzien als significant insufficiënt wanneer de GFR minder dan 40
ml/min bedroeg. Drierentwintig patiënten (85%) beantwoordden aan deze criteria, waaronder 9
hemodialyse afhankelijk waren, en ondergingen een gecombineerde lever-niertransplantatie.

33
Technische problemen
Bij 8 patiënten (30%) werden er vooral tijdens de hepatectomie van de acceptor technische moeilijkheden
ondervonden met een noodzaak van IVC- clamping bij 5 ervan. Drie van deze 8 patiënten, inclusief 2
patiënten die reeds eerdere resecties als PLD behandeling hadden gekregen, hadden een transfusie van
meer dan 5Units bloed nodig. Al de patiënten hadden een transfusie nodig met een gemiddelde van 7.7
units (range 1-24U).
Complicaties
In totaal ondervonden er 23 patiënten (85%) postoperatieve complicaties. 12 (44%) re-operaties waren
noodzakelijk omwille van een gallekkage (n=4), een hemorragie (n=2) en peritonitis veroorzaakt door
een spontane gastro-intestinale perforatie (n=6). Onder deze twaalf patiënten hadden reeds 4 patiënten een
vroegere laparotomie voor PLD behandeling ondergaan. Veertien patiënten in deze serie vertoonden
ondervoeding, 11 ervan ondervonden ernstige complicaties, waarvan 9 ascites. Post-operatieve sterfte was
te wijten aan multiorgaan falen (MOF) met sepsis in 2 patiënten, hartfalen in één en arterieel graft
thrombose in één, wat de mortaliteit in deze serie op 15% brengt.
Analyse
Analyse postop toonde aan dat in 22 patiënten (81%) het niet cystische parenchym abnormale
eigenschappen vertoonde met fibrosevorming en congestie in 12 en 7 patiënten respectievelijk.
Survival en QOL
De 5-jaarsoverleving van de patiënten was 85% en de gemiddelde follow-up van de overlevende
patiënten duurde gemiddeld 36 maanden (range 12-142). Alle patiënten waren tevreden verlost te zijn van
hun hepatomegalie. Twee patiënten hadden echter persisterende wondsymptomen en 3 kloegen over
intermitterende last, veroorzaakt door grote polycystische nieren.
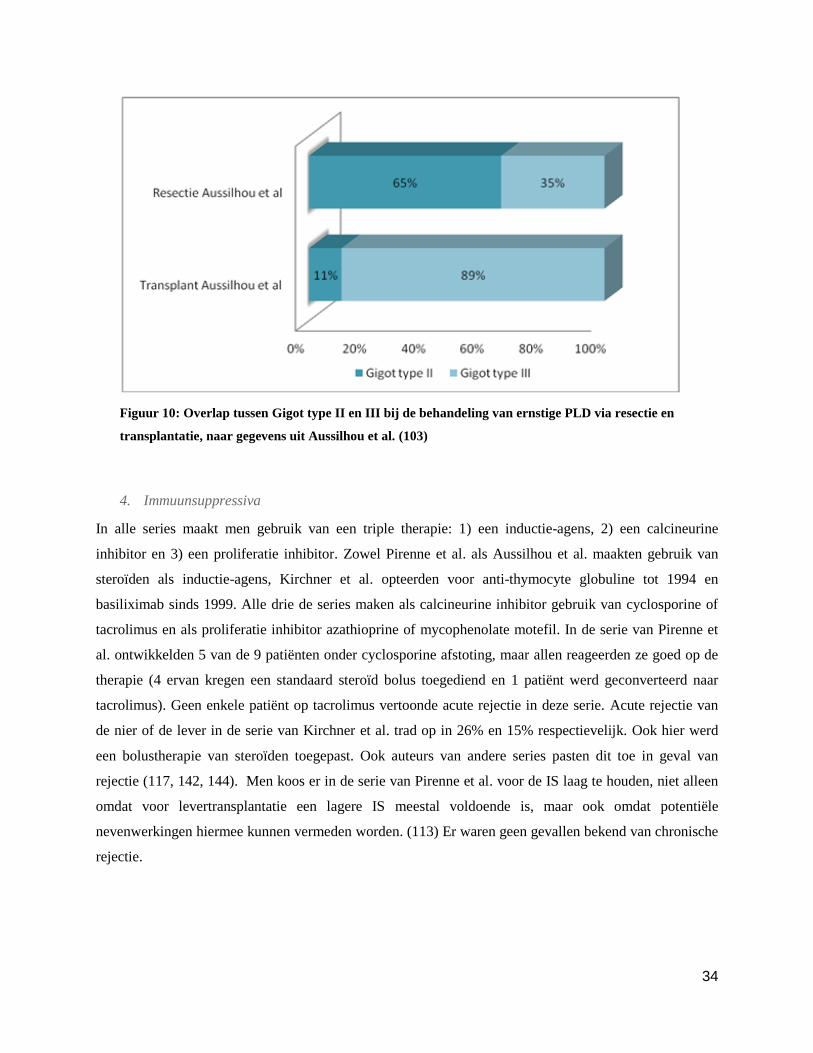
Overlap
Een vergelijking tussen classificaties van de patiënten die een transplantatie of resectie in deze serie
kregen, geeft de mogelijke overlap tussen de behandelingen van PLD Gigot type II en III mooi weer
(figuur 10). Dit benadrukt dat de keuze tussen transplantatie of resectie niet louter op cystgrootte en
verspreiding kan gebaseerd worden, maar ook andere parameters de keuze moeten sturen. De titel van het
artikel: extended liver resection for polycystic liver disease can challenge liver transplantation is
eigenlijk ongelukkig gekozen omdat deze figuur aantoont dat de twee ingrepen werden toegepast op twee
verschillende patiëntengroepen en er dus geen sprake is van een ‘challenge’. Deze serie bevestigt hiermee
dat de twee ingrepen elkaar aanvullen en niet concurreren voor de behandeling van ernstige PLD.
34
Figuur 10: Overlap tussen Gigot type II en III bij de behandeling van ernstige PLD via resectie en
transplantatie, naar gegevens uit Aussilhou et al. (103)
4. Immuunsuppressiva
In alle series maakt men gebruik van een triple therapie: 1) een inductie-agens, 2) een calcineurine
inhibitor en 3) een proliferatie inhibitor. Zowel Pirenne et al. als Aussilhou et al. maakten gebruik van
steroïden als inductie-agens, Kirchner et al. opteerden voor anti-thymocyte globuline tot 1994 en
basiliximab sinds 1999. Alle drie de series maken als calcineurine inhibitor gebruik van cyclosporine of
tacrolimus en als proliferatie inhibitor azathioprine of mycophenolate motefil. In de serie van Pirenne et
al. ontwikkelden 5 van de 9 patiënten onder cyclosporine afstoting, maar allen reageerden ze goed op de
therapie (4 ervan kregen een standaard steroïd bolus toegediend en 1 patiënt werd geconverteerd naar
tacrolimus). Geen enkele patiënt op tacrolimus vertoonde acute rejectie in deze serie. Acute rejectie van
de nier of de lever in de serie van Kirchner et al. trad op in 26% en 15% respectievelijk. Ook hier werd
een bolustherapie van steroïden toegepast. Ook auteurs van andere series pasten dit toe in geval van
rejectie (117, 142, 144). Men koos er in de serie van Pirenne et al. voor de IS laag te houden, niet alleen
omdat voor levertransplantatie een lagere IS meestal voldoende is, maar ook omdat potentiële
nevenwerkingen hiermee kunnen vermeden worden. (113) Er waren geen gevallen bekend van chronische
rejectie.
35
5. Overlevingsgraad
In de series van Kirchner et al., Pirenne et al. en Aussilhou et al., bedroeg de vijfjaarsoverleving
respectievelijk 86%, 87,5% en 85%. Andere series bevestigen deze cijfers: een survival rate van 81%
waarvan de meeste doden de eerste 2 maand niet overleefden. Dit toont een aanzienlijk risico aan van
vroegtijdige complicaties maar uitstekende langetermijn resultaten. Infecties waren ook bij deze series de
hoofdoorzaak van sterfte (63%). 18.5% stierf ten gevolge van complicaties door intra-operatieve
bloedingen en 18.5% door andere oorzaken. (108, 109, 111-114, 116, 117, 120)
2. Recente data
A. Registry data
1. Orgaantekort
Er is een duidelijk tekort aan donororganen voor transplantatie. Door het toenemend succes is er immers
meer nood aan geschikte organen. Dit wordt duidelijk wanneer men kijkt naar de lange wachtlijsten voor
levertransplantatie in Europa en de United states. (zie figuren 11 en 12). De wachtlijst is aan het stagneren
in de US volgens de cijfers van UNOS, maar blijft nog stijgen in de landen verbonden aan Eurotransplant.
(152, 153)
Figuur 11: Wachtlijst registraties in US 200-2010 (UNOS)
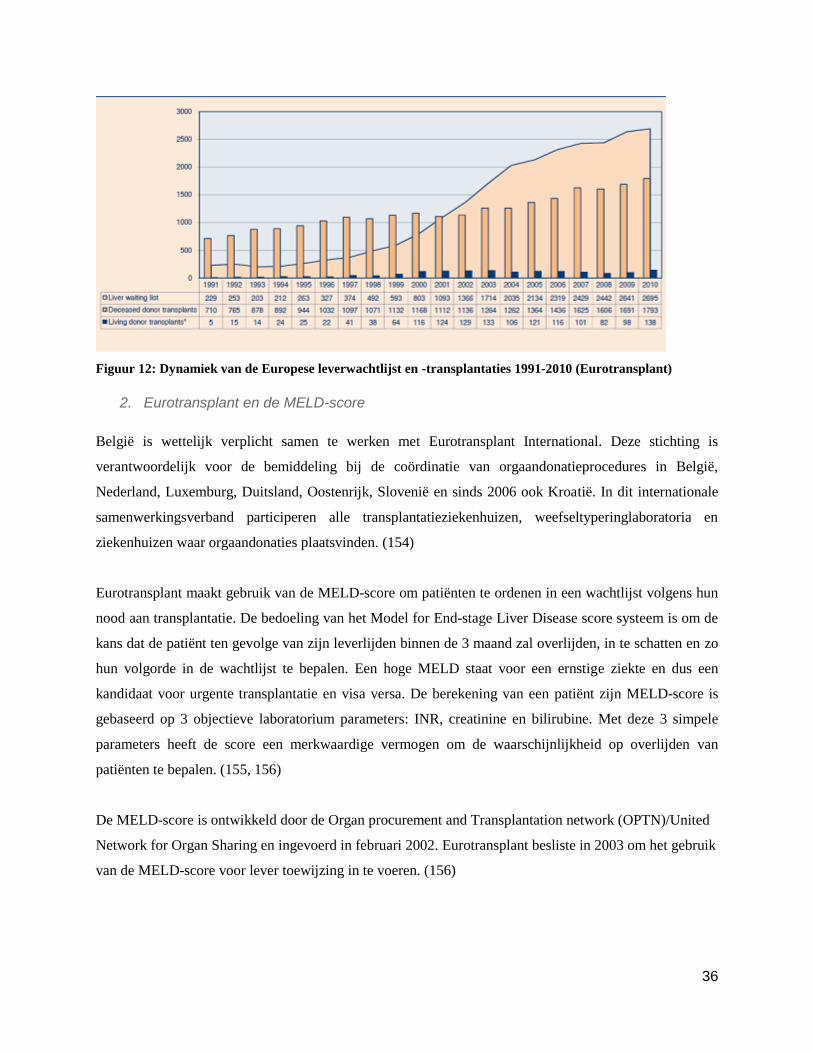
36
Figuur 12: Dynamiek van de Europese leverwachtlijst en -transplantaties 1991-2010 (Eurotransplant)
2. Eurotransplant en de MELD-score
België is wettelijk verplicht samen te werken met Eurotransplant International. Deze stichting is
verantwoordelijk voor de bemiddeling bij de coördinatie van orgaandonatieprocedures in België,
Nederland, Luxemburg, Duitsland, Oostenrijk, Slovenië en sinds 2006 ook Kroatië. In dit internationale
samenwerkingsverband participeren alle transplantatieziekenhuizen, weefseltyperinglaboratoria en
ziekenhuizen waar orgaandonaties plaatsvinden. (154)
Eurotransplant maakt gebruik van de MELD-score om patiënten te ordenen in een wachtlijst volgens hun
nood aan transplantatie. De bedoeling van het Model for End-stage Liver Disease score systeem is om de
kans dat de patiënt ten gevolge van zijn leverlijden binnen de 3 maand zal overlijden, in te schatten en zo
hun volgorde in de wachtlijst te bepalen. Een hoge MELD staat voor een ernstige ziekte en dus een
kandidaat voor urgente transplantatie en visa versa. De berekening van een patiënt zijn MELD-score is
gebaseerd op 3 objectieve laboratorium parameters: INR, creatinine en bilirubine. Met deze 3 simpele
parameters heeft de score een merkwaardige vermogen om de waarschijnlijkheid op overlijden van
patiënten te bepalen. (155, 156)
De MELD-score is ontwikkeld door de Organ procurement and Transplantation network (OPTN)/United
Network for Organ Sharing en ingevoerd in februari 2002. Eurotransplant besliste in 2003 om het gebruik
van de MELD-score voor lever toewijzing in te voeren. (156)

37
Ook UNOS united network for organ sharing, een non-profit privé organisatie in Noord-Amerika en
MOHAN foundation, een non-profit privé organisatie in India (ook kantoren in USA) maken gebruik van
deze MELD score. (152, 157)
Deze score past echter niet volledig bij de PLD-patiënten. Veel patiënten zijn niet competitief voor een
overleden donor transplantatie, omdat ze een relatief normale synthetische functie van hun lever
behouden en dus normale laboratorium parameters hebben. Om deze patiënten een kans te geven om te
concurreren op de wachtlijst voor overleden donor grafts, kunnen hen exceptionele punten worden
toegekend. Ze kunnen een standaard exceptiescore van 20 MELD-punten krijgen in België wanneer ze
voldoen aan 4 criteria: 1) massieve PLD (totaal cyst/ parenchym>1) en complicaties die enkel kunnen
verholpen worden met transplantatie; 2) klinisch merkbare leverziekte als gevolg van massieve PLD
inclusief: gewichtsverlies, ascites en portale hypertensie; 3) falen van non-transplant gerelateerde
interventies of contraindicaties voor verdere non-transplant gerelateerde interventies; en 4) contra-
indicaties voor non-transplant gerelateerde interventies en voldoen aan criteria 1 en 2. Daarboven kunnen
ze extra MELD-punten krijgen afhankelijk van de ernst van sommige symptomen. (158) Zonder deze
uitzonderingspunten kunnen de meeste patiënten enkel in aanmerking voor een Living Donor
Transplantatie (LDLT). (159)
3. Overlevingsgraad
De hoge overlevingscijfers van de levertransplant series worden bevestigd (85% na 5 jaar) wanneer men
kijkt naar gegevens ven de European Liver Transplant Registry. Deze cijfers liggen een pak hoger
wanneer men dit vergelijkt met andere transplantaties (zie figuur 13) met een 5-jaarsoverleving van 59-
76%. (160)
38
Figuur 13: overlevingscijfers bij PLD en andere leverziekten als eerste indicatie. (ELTR)
3. UZ gent
A. Conservatieve chirurgie
In de periode 2000 – (13/04/) 2012 werden er 2 patiënten conservatief chirurgisch behandeld in het UZ
Gent: één met een fenestratie en één met een resectie.
B. Transplantatie
In dezelfde periode zijn er 23 transplantaties uitgevoerd voor ernstige PLD. Hiervan waren er 5
gecombineerde lever-niertransplantaties (22%). Onder de patiënten waren er 18 vrouwen en 5 mannen,
een m/v ratio van 28%. De gemiddelde leeftijd bedroeg 52 jaar met een range tussen de 37- 71. Drie
patiënten overleden aan de operatie. Vijf patiënten kregen gelijktijdig een niertransplantatie. Ook in het
UZ Gent blijkt er dus een hoge overlevingsgraad te zijn: 87% peroperatief.
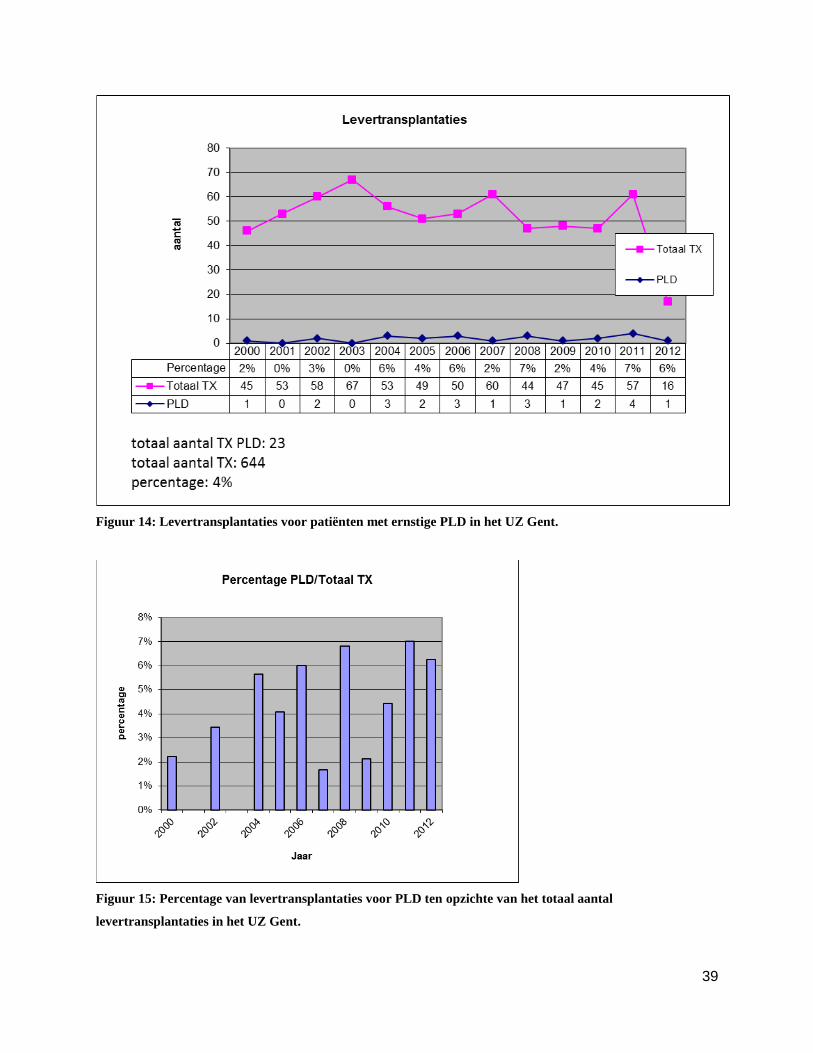
39
Figuur 14: Levertransplantaties voor patiënten met ernstige PLD in het UZ Gent.
Figuur 15: Percentage van levertransplantaties voor PLD ten opzichte van het totaal aantal
levertransplantaties in het UZ Gent.

40
V. Discussie
1. Ernstige polycystische leverziekte
Enkel patiënten met ernstige PCLD hebben baat bij een invasieve ingreep zoals resectie of transplantatie.
Deze ernstige PCLD is een klinische en anatomische (via beeldvorming) vast te stellen situatie. Hieronder
verstaan we patiënten met een Gigot klasse II of klasse III die symptomatische gevolgen van hun
hepatomegalie ondervinden of (zeldzaam) cyst-gebonden symptomen. De symptomen veroorzaakt door
de hepatomegalie bestaan voornamelijk uit abdominale distentie, abdominale pijn, vroege verzadiging,
vermoeidheid, nausea, regurgitatie en kortademigheid. (symptomen, zie tabel 1) Deze hebben een
duidelijke weerslag op de performance status en de gezondheidsstatus van de patiënt. Om de kwaliteit van
leven objectief weer te geven en long-term effecten van de ingreep te controleren kan gebruik gemaakt
worden van de Medical Outcomes Study form 36 (SF-36). Voor de functionele status kan men gebruik
maken van de Eastern Coöperative Oncology Group Performance Status (ECOG-PS).
2. Resectie
Uit de resultaten van de resectieseries kan men opmaken dat deze ingreep vaak een grote kans heeft op
complicaties en een hoge kans op recidief postoperatief. Resectie bij PLD-patiënten heeft niet alleen een
grotere kans op complicaties, maar ook op een langere operatietijd en een groter bloedverlies dan resectie
bij patiënten met een niet-cystische, niet-cirrotische lever. Zo was in de serie van Schnelldorfer et al. het
bloedverlies van de PLD-patiënten 2.3 keer groter, de perioperatieve morbiditeit 2.3 keer groter en de
operatietijd 1.4 keer langer dan in vergelijking met resectie voor andere indicaties. (10, 164) Zoals uit de
resultaten van alle resectieseries blijkt, vormen vooral gallekkages, hemorragie en ascites de grootste
groep complicaties. De grootste reden voor de gallekkage en het bloedverlies is de vervorming van de
normale anatomie met een grotere kans op beschadiging van de bloedvaten en galwegen tot gevolg. De
oorzaak van ascites is multifactorieel en kan verklaard worden door persisterende cyst secretie, veneuze
outflow obstructie, lymfe lekkage, nierdysfunctie en malnutritie met een laag albuminelevel. Eenmaal de
vroegtijdige complicaties verdwenen zijn, neemt de performance status toe. Onder andere >75% van de
patiënten in de serie van Aussilhou et al. vertoonden een langdurige verbetering en 96% van de patiënten
in de serie van Que et al. waren tevreden met de ingreep en ondervonden een directe en behouden
verbetering van hun QOL. Doordat de complicaties van voorbijgaande aard zijn en de sterke verbetering
van de QOL hierna, kan men de hoge morbiditeit postoperatief aanvaarden.

41
Recidief van symptomen ten gevolge van hepatomegalie bij patiënten die een resectie hadden ondergaan,
werd vastgesteld bij 2 patiënten (4%) in de serie van Aussilhou et al. In de serie van Que et al. nam het
levervolume terug toe bij 2 patiënten (6%) ontdekt door sequentiële CT-scans respectievelijk 2 en 3 jaar
postop. In de serie van Schnelldorfer et al. vertoonden minstens 20 personen (16%) een toegenomen
levervolume na resectie met een gemiddelde follow-up van 4 jaar. Wanneer ten minste één sectorieel
gedeelte parenchym relatief gespaard bleef van cyst inname, voerde men in deze serie een resectie uit.
Hoeveel percent van het oorspronkelijk cystvrije leverparenchym de restlever na resectie
vertegenwoordigd, kan hieruit niet worden opgemaakt. Er werden in deze serie slechts 7 personen als
primaire ingreep getransplanteerd voor PLD. Additioneel werden er echter na resectie 7 personen (6%)
getransplanteerd ten gevolge van leverfalen direct postoperatief (n=2) of tijdens follow-up (gemiddeld
7jaar) (n=2) en oncontroleerbare symptomen tijdens de follow-up (n=3). Het kritisch residueel
levervolume voor patiënten zonder onderliggende leverziekte werd onderzocht in de literatuur en de
limiet voor een veilige resectie wordt door verschillende auteurs in het algemeen tussen de 20-30%
gelegd. (129, 165-169) Bij patiënten met een beschadigde lever, veroorzaakt door een onderliggende
leverziekte (cirrose, steatose en cholestase), ligt de grens van het residueel levervolume in de literatuur
tussen de 30-40%. (129, 170-172) Belangrijk hierbij is de berekening van het functioneel residueel
levervolume en een accurate evaluatie van de leverfunctie. Deze twee variabelen zijn immers de
belangrijkste factoren voor een adequate werking van de residuele lever. (129) De grens gesteld door
Aussilhou et al. van 30% van het totale cystvrij leverparenchym als restlever in deze groep van patiënten
lijkt een veilige waarde te zijn. PLD-patiënten vallen immers onder de categorie van onderliggende
leverziekte. Het gespaard parenchym heeft vaak een histologische verandering ondergaan met
fibrosevorming maar de leverfunctie blijft meestal uitstekend bewaard. Aan de hand van programma’s
zoals QReads version 4.1.8. clinical image software (Mayo Clinic, Rochester, MN) of Myrian (Aussilhou
et al.) kan het volume van de lever berekend worden met onderverdeling van parenchymvolume en
cystvolume. Naar alle waarschijnlijkheid was de restlever in de serie van Schnelldorfer et al. vaak < 30%
van het relatief cystvrije leverparenchym. In deze serie hadden ze dan vaker een transplantatie moeten
uitvoeren als primaire ingreep met als resultaat minder recidieven en reddings-transplantaties. In de serie
van Aussilhou et al. werd er ook één additionele transplantatie uitgevoerd in de follow-up na resectie,
maar er wordt niet meegedeeld voor welke indicatie. In de serie van Que et al. werd er in de follow-up
(gemiddeld 2.4 jaar) geen additionele transplantatie uitgevoerd. In deze serie moesten er minstens 2
aangrenzende leversegmenten relatief vrij zijn van cysten om tot resectie over te gaan. Deze voorwaarde
lijkt ook een veilig levervolume te garanderen maar de grens van 30% van het relatief cystvrije
leverparenchym als restlever is gemakkelijker objectiveerbaar via computersoftware en minder
persoonsafhankelijk. Verder onderzoek rond deze minimumwaarde is aangewezen. Het totaal

42
recidiefcijfer voor symptomen na resectie in de serie van Aussilhou et al. bedroeg 16%, veroorzaakt door
hepatomegalie in 4%, cystbloeding in 8%, cholangitis in 2% en niervergroting in 2%. Dit komt ongeveer
overeen met het recidiefcijfer van de andere series: 18% ( zie tabel 8: aantal patiënten met recidief
(n=36)/ totaal aantal patiënten (n=202) exclusief de serie van Schnelldofer et al.). Preoperatieve MRI kan
helpen bij de planning bloedende cysten mee te nemen in de resectie. Zo kan het recidiefcijfer aan
cystbloedingen tot een minimum herleid worden. Recidiverende hepatomegalie kan in geselecteerde
gevallen verhinderd worden door gebruik van medicatie. Vier RCT hebben immers de werking van
somatostatine-analogen aangetoond met reductie van het levervolume. (79-82) (zie vroeger). Deze
vaststelling biedt veel perspectieven: toediening van deze medicatie kan een operatie uitstellen of
faciliteren en kan postoperatief in geselecteerde gevallen worden toegediend om recidief van
hepatomegalie te voorkomen. Eén jaar is voorlopig de langste termijn dat lantreotide getest geweest is. Of
het gunstige effect verschillende jaren behouden blijft, moet nog worden bevestigd.
3. Transplantatie
Transplantatie zou moeten worden aangeboden als primaire ingreep voor patiënten met ernstige PLD, best
nog voor zijn eindstadium-complicaties manifest worden. Transplantatie in patiënten met eindstadium
PCLD, zichtbaar door ernstige beperking, zwakte en malnutritie, vertoont immers een hogere infectie-
gerelateerde mortaliteit in de levertransplantatie series. Ook de postoperatieve immuunsuppressiva
verzwakken de immuniteit. Een transplantatie in geselecteerde kandidaten lijkt een grote kans op
verbeterde uitkomst, een zinvol herstel en terugkeer naar hun vroeger functioneren en kwaliteit van leven
te bieden. Het tekort aan donororganen zou geen argument mogen zijn een zorgvuldig geselecteerde
patiënt een transplantatie te ontzeggen. De opmerkelijke verbetering van de QOL en van de sociale status
samen met de meerderheid van de patiënten die opnieuw zouden opteren voor een transplantatie,
bewijzen immers de effectiviteit van deze ingreep. Wanneer patiënten met ernstige PLD bijkomende
nierinsufficiëntie vertonen is een gecombineerde lever-niertransplantatie de meest aangewezen
behandeling. Men kan immers verwachten dat de progressieve nierinsufficiëntie blijft toenemen na LT en
dat calcineurine inhibitoren deze progressie nog kunnen versnellen. Los van het voordeel van één enkele
operatie biedt een gecombineerde lever-niertransplantatie van eenzelfde donor een immunologische
bescherming voor de nier. Door het tekort aan nieren als donororganen moet men het aantal
gecombineerde lever-niertransplantaties wel beperken tot de patiënten voor wie deze duidelijk
geïndiceerd zijn. Dit zijn patiënten met gevorderd nierfalen (pre-dialyse of dialysestadium) of met een
gedaalde nierfunctie waarvan men niet verwacht dat het veroorzaakt wordt door een abdominaal
compartiment syndroom (cfr vena cava compressie, renale of renaal vasculaire compressie veroorzaakt
door de hepatomegalie).

43
In de serie van Kirchner et al. werd er een gecombineerde lever-niertransplantatie uitgevoerd wanneer de
patiënt een GFR<30% had. Zes patiënten van de 21 die enkel een OLT hadden gekregen vertoonden
echter reeds een GFR <30% op het einde van de follow-up. In de serie van Pirenne et al. kreeg slechts één
patiënt (16%) een gecombineerde transplantatie. Dit is uitzonderlijk bij series van PLD-patiënten.
Additioneel was er bij één patiënt een niertransplantatie nodig na OLT. De GFR van deze patiënten kan
echter niet correct uit de data worden afgeleid en dus is de correcte nierfunctie van deze patiënten niet
gekend. Verdere data om de GFR preop en na lange follow-up te vergelijken bij OLT voor PLD blijft
noodzakelijk maar men kan stellen dat een grens getrokken op 40ml/min (zoals in de serie van Aussilhou
et al.) om een gecombineerde lever-niertransplantatie uit te voeren, een aannemelijke waarde is.
VI. Conclusie
Figuur 16: Algoritme behandelingskeuze (Gunstige anatomie= vasculaire structuren en galwegen)
Al het voorgaande in beschouwing genomen lijkt transplantatie voor deze groep van patiënten de meest
aangewezen optie: een definitieve oplossing met een sterk verbeterde QOL en performance status. De
mortaliteit is aanvaardbaar en bij de laagste onder de levertransplantaties. Het grote donortekort dwingt
ons echter tot creativiteit.

44
Resectie biedt hier een alternatieve oplossing wanneer er minstens 30% van het relatief cystvrije
leverparenchym als restvolume overblijft na resectie en de anatomische structuren adequate aan- en
afvoer van stoffen kunnen garanderen. Omdat de QOL sterk verbetert en bijna alle patiënten tevreden zijn
met het resultaat van de ingreep kan het hoge morbiditeitscijfer, dat van voorbijgaande aard is, aanvaard
worden. Het recidiefcijfer van gemiddeld 18% is hoog, maar kan nog voor een stuk verminderd worden.
De patiënten die niet voldoende cystvrij parenchym als restlever zouden overhouden na resectie hebben
weinig voordeel bij deze ingreep. Resectie bij deze groep van patiënten uitvoeren geeft immers geen
garantie op een succesvolle uitkomst: door de vaak histologische veranderingen is niet het volledige
restvolume van de lever actief waardoor er sneller leverfalen kan optreden. Daarnaast biedt dit dan vaak
slechts gedeelte verlichting of verlichting beperkt in tijd door het diffuse karakter van het cystisch proces.
Het stelt hen dan onnodig bloot aan potentiële morbiditeit en mortaliteit en brengt ook hun kans op
definitieve behandeling door transplantatie in gevaar. Wanneer de patiënt wel voldoet aan de voorwaarde,
moet er preoperatief een CT-angiografie worden afgenomen om adequate bloedvoorziening en veneuze
afvoer te garanderen in de cystvrije segmenten die de toekomstige lever gaan uitmaken. Hierbij moet men
vooral aandacht hebben voor de vena porta en de sushepatische venen. Ook een adequate galafvoer moet
verzekerd worden. Wanneer men ernstige twijfels heeft bij de postoperatieve functionaliteit van de
vasculaire structuren of ernstige problemen met de galafvoer verwacht, gaat de voorkeur uit naar
transplantatie. Beschadiging van de vasculatuur en galwegen met bloedingen en gallekkage tot gevolg,
vormen immers de meest voorkomende complicaties bij resectie en bemoeilijken de procedure. Enkel
wanneer er adequate bloedvoorziening, veneuze afvoer en minimale beschadiging van de galwegen kan
gegarandeerd worden, kan men tot resectie overgaan. Wanneer, in zeldzame gevallen, leverfalen duidelijk
blijkt uit de labowaarden is transplantatie uiteraard een noodzaak. In de meeste gevallen zijn de
labowaarden echter normaal tot licht gestegen en kan vanuit deze parameters geen onderscheid gemaakt
worden tussen de 2 behandelingsopties. Door de relatief hoge incidentie aan niercysten en de
mogelijkheid om een gecombineerde lever-niertransplantatie uit te voeren moet men ook steeds de
nierfunctie nakijken. Wanneer de GFR minder dan 40ml/min bedraagt, moet men overwegen een
gecombineerde lever-niertransplantatie uit te uitvoeren.
De denkpiste in deze scriptie dient in de praktijk nog verder te worden getoetst. Idealiter zou onderzoek in
dit vakgebied met nog meer langetermijn gegevens worden aangevuld. Tot slot dient benadrukt te worden
dat patiënten steeds individueel moeten worden bekeken door een multidisciplinair team om de beste
keuze van behandeling te kunnen stellen. Een dergelijk team zou minstens moeten bestaan uit een
hepatobiliair chirurg, nefroloog, radioloog en gastro-entroloog, bij voorkeur in een tertiair centrum.

45
V. Referenties
1. Everson GT, Helmke SM, Doctor B. Advances in management of polycystic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2(4):563-76. Epub 2008/12/17. 2. Onori P, Franchitto A, Mancinelli R, Carpino G, Alvaro D, Francis H, et al. Polycystic liver diseases. Dig Liver Dis. 2010;42(4):261-71. Epub 2010/02/09. 3. Reid-Lombardo KM, Khan S, Sclabas G. Hepatic cysts and liver abscess. Surg Clin North Am. 2010;90(4):679-97. Epub 2010/07/20. 4. Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol. 2005;100(11):2569-82. Epub 2005/11/11. 5. Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343(8901):824-7. Epub 1994/04/02. 6. Gigot JF, Metairie S, Etienne J, Horsmans Y, van Beers BE, Sempoux C, et al. The surgical management of congenital liver cysts. Surg Endosc. 2001;15(4):357-63. Epub 2001/06/08. 7. Katkhouda N, Hurwitz M, Gugenheim J, Mavor E, Mason RJ, Waldrep DJ, et al. Laparoscopic management of benign solid and cystic lesions of the liver. Ann Surg. 1999;229(4):460-6. Epub 1999/04/15. 8. Hansman MF, Ryan JA, Holmes JH, Hogan S, Lee FT, Kramer D, et al. Management and long-term follow-up of hepatic cysts. American Journal of Surgery. 2001;181(5):404-10. 9. Martin IJ, McKinley AJ, Currie EJ, Holmes P, Garden OJ. Tailoring the management of nonparasitic liver cysts. Ann Surg. 1998;228(2):167-72. Epub 1998/08/26. 10. Schnelldorfer T, Torres VE, Zakaria S, Rosen CB, Nagorney DM. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg. 2009;250(1):112-8. Epub 2009/06/30. 11. Harris PC, Torres VE. Polycystic Kidney Disease, Autosomal Dominant. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews. Seattle WA: University of Washington, Seattle; 1993. 12. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-68. Epub 2009/05/21. 13. Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. American journal of physiology Renal physiology. 2007;292(3):F930-45. Epub 2006/11/09. 14. Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40(4):774-82. Epub 2004/09/24. 15. Fencl F, Janda J, Blahova K, Hribal Z, Stekrova J, Puchmajerova A, et al. Genotype-phenotype correlation in children with autosomal dominant polycystic kidney disease. Pediatric nephrology. 2009;24(5):983-9. Epub 2009/02/06. 16. Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353(9147):103-7. Epub 1999/02/19. 17. Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. American journal of kidney diseases : the official journal of the National Kidney Foundation. 1993;22(4):520-5. Epub 1993/10/01. 18. Grunfeld JP, Albouze G, Jungers P, Landais P, Dana A, Droz D, et al. Liver changes and complications in adult polycystic kidney disease. Advances in nephrology from the Necker Hospital. 1985;14:1-20. Epub 1985/01/01.

46
19. Gabow PA, Johnson AM, Kaehny WD, Manco-Johnson ML, Duley IT, Everson GT. Risk factors for the development of hepatic cysts in autosomal dominant polycystic kidney disease. Hepatology. 1990;11(6):1033-7. Epub 1990/06/01. 20. Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, et al. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26(5):1282-6. Epub 1997/11/15. 21. Sessa A, Meroni M, Righetti M, Battini G, Maglio A, Puricelli SL. Autosomal recessive polycystic kidney disease. Contributions to nephrology. 2001(136):50-6. Epub 2001/11/02. 22. Schneider BL MM. Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transpl. 2005;9:634-9. 23. Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. American journal of human genetics. 2002;70(5):1305-17. Epub 2002/03/19. 24. Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nature genetics. 2002;30(3):259-69. Epub 2002/03/29. 25. Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatric nephrology. 1997;11(3):302-6. Epub 1997/06/01. 26. Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111(5 Pt 1):1072-80. Epub 2003/05/03. 27. Lilova MI, Petkov DL. Intracranial aneurysms in a child with autosomal recessive polycystic kidney disease. Pediatric nephrology. 2001;16(12):1030-2. Epub 2002/01/17. 28. Yonemura K, Yasuda H, Fujigaki Y, Oki Y, Hishida A. Adrenal insufficiency due to isolated adrenocorticotropin deficiency complicated by autosomal recessive polycystic kidney disease. Renal failure. 2003;25(3):485-92. Epub 2003/06/14. 29. Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1(1):64-9. Epub 2007/08/21. 30. Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis. 2010;17(2):181-9. Epub 2010/03/12. 31. Feldman M. Polycystic disease of the liver. Am J Gastroenterol. 1958;29(1):83-6. Epub 1958/01/01. 32. Tahvanainen P, Tahvanainen E, Reijonen H, Halme L, Kaariainen H, Hockerstedt K. Polycystic liver disease is genetically heterogeneous: clinical and linkage studies in eight Finnish families. J Hepatol. 2003;38(1):39-43. Epub 2002/12/14. 33. Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nature genetics. 2004;36(6):575-7. Epub 2004/05/11. 34. Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. American journal of human genetics. 2003;72(3):691-703. Epub 2003/01/17. 35. Cheung J, Scudamore CH, Yoshida EM. Management of polycystic liver disease. Can J Gastroenterol. 2004;18(11):666-70. Epub 2004/11/27. 36. Melnick PJ. Polycystic liver; analysis of seventy cases. AMA archives of pathology. 1955;59(2):162-72. Epub 1955/02/01. 37. Karhunen PJ, Tenhu M. Adult polycystic liver and kidney diseases are separate entities. Clinical genetics. 1986;30(1):29-37. Epub 1986/07/01.

47
38. Hoevenaren IA, Wester R, Schrier RW, McFann K, Doctor RB, Drenth JP, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver international : official journal of the International Association for the Study of the Liver. 2008;28(2):264-70. Epub 2007/10/12. 39. Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nature genetics. 2003;33(3):345-7. Epub 2003/02/11. 40. Skowronek MH, Rotter M, Haas IG. Molecular characterization of a novel mammalian DnaJ-like Sec63p homolog. Biological chemistry. 1999;380(9):1133-8. Epub 1999/10/30. 41. Gao H, Wang Y, Wegierski T, Skouloudaki K, Putz M, Fu X, et al. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Human molecular genetics. 2010;19(1):16-24. Epub 2009/10/06. 42. H. vM. Uber die cystenleber. Beitr Path Anat. 1918;64:477A/532. 43. Bistritz L, Tamboli C, Bigam D, Bain VG. Polycystic liver disease: experience at a teaching hospital. Am J Gastroenterol. 2005;100(10):2212-7. Epub 2005/09/27. 44. Perrone RD, Grubman SA, Rogers LC, Lee DW, Moy E, Murray SL, et al. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. The American journal of physiology. 1995;269(3 Pt 1):G335-45. Epub 1995/09/01. 45. Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Current opinion in gastroenterology. 2009;25(3):265-71. Epub 2009/04/08. 46. Nichols MT, Gidey E, Matzakos T, Dahl R, Stiegmann G, Shah RJ, et al. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40(4):836-46. Epub 2004/09/24. 47. Que F, Nagorney DM, Gross JB, Jr., Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108(2):487-94. Epub 1995/02/01. 48. Bleeker-Rovers CP, Vos FJ, Corstens FH, Oyen WJ. Imaging of infectious diseases using [18F] fluorodeoxyglucose PET. The quarterly journal of nuclear medicine and molecular imaging : official publication of the Italian Association of Nuclear Medicine. 2008;52(1):17-29. Epub 2007/07/28. 49. VE T. - Treatment of polycystic liver disease: one size does not fit all. D - 8110075. (- 1523-6838 (Electronic)):T - ppublish. 50. Hansman MF, Ryan JA, Jr., Holmes JHt, Hogan S, Lee FT, Kramer D, et al. Management and long-term follow-up of hepatic cysts. Am J Surg. 2001;181(5):404-10. Epub 2001/07/13. 51. Everson GT, Scherzinger A, Berger-Leff N, Reichen J, Lezotte D, Manco-Johnson M, et al. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988;8(6):1627-34. Epub 1988/11/01. 52. Rosenfeld L, Abergel A, Bonny C, Poincloux L, Gayard P, Garcier JM, et al. [Complicated polycystic liver disease with intracystic hemorrhage and obstructive jaundice]. Gastroenterol Clin Biol. 2001;25(8-9):818-22. Epub 2001/10/13. Polykystose hepatique compliquee d'une hemorragie intra-kystique et d'un ictere compressif. 53. Ergun H, Wolf BH, Hissong SL. Obstructive jaundice caused by polycystic liver disease. Radiology. 1980;136(2):435-6. Epub 1980/08/01. 54. Ratcliffe PJ, Reeders S, Theaker JM. Bleeding oesophageal varices and hepatic dysfunction in adult polycystic kidney disease. British medical journal. 1984;288(6427):1330-1. Epub 1984/05/05.

48
55. Tang SJ, Jensen DM, Gralnek IM, Roth BE. Portal hypertensive enteropathy in a patient with polycystic liver disease: a unique endoscopic finding. Gastrointestinal endoscopy. 2002;56(6):924-6. Epub 2002/11/26. 56. Uddin W, Ramage JK, Portmann B, Wilson P, Benjamin I, Tan KC, et al. Hepatic venous outflow obstruction in patients with polycystic liver disease: pathogenesis and treatment. Gut. 1995;36(1):142-5. Epub 1995/01/01. 57. Abascal J, Moya M, Martin F. Infection of hepatic cysts in polycystic disease. World J Surg. 1984;8(3):424-5. Epub 1984/06/01. 58. Telenti A, Torres VE, Gross JB, Jr., Van Scoy RE, Brown ML, Hattery RR. Hepatic cyst infection in autosomal dominant polycystic kidney disease. Mayo Clinic proceedings Mayo Clinic. 1990;65(7):933-42. Epub 1990/07/01. 59. Kwok CG, McDougall IR. Persistent fever in a patient with polycystic kidney and liver diseases and bilateral hip prostheses. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 1996;37(12):2062-5. Epub 1996/12/01. 60. Bourgeois N, Kinnaert P, Vereerstraeten P, Schoutens A, Toussaint C. Infection of hepatic cysts following kidney transplantation in polycystic disease. World J Surg. 1983;7(5):629-31. Epub 1983/09/01. 61. Qian Q, Li A, King BF, Kamath PS, Lager DJ, Huston J, 3rd, et al. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37(1):164-71. Epub 2002/12/25. 62. Sallee M, Rafat C, Zahar JR, Paulmier B, Grunfeld JP, Knebelmann B, et al. Cyst infections in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4(7):1183-9. Epub 2009/05/28. 63. Rossetti S, Chauveau D, Kubly V, Slezak JM, Saggar-Malik AK, Pei Y, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361(9376):2196-201. Epub 2003/07/05. 64. Morgan DE, Lockhart ME, Canon CL, Holcombe MP, Bynon JS. Polycystic liver disease: multimodality imaging for complications and transplant evaluation. Radiographics. 2006;26(6):1655-68; quiz Epub 2006/11/15. 65. van Keimpema L, de Koning DB, van Hoek B, van den Berg AP, van Oijen MGH, de Man RA, et al. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 2011;31(1):92-8. 66. Wan SK, Cochlin DL. Sonographic and computed tomographic features of polycystic disease of the liver. Gastrointestinal radiology. 1990;15(4):310-2. Epub 1990/01/01. 67. Spiegel RM, King DL, Green WM. Ultrasonography of primary cysts of the liver. AJR Am J Roentgenol. 1978;131(2):235-8. Epub 1978/08/01. 68. Levine E, Cook LT, Grantham JJ. Liver cysts in autosomal-dominant polycystic kidney disease: clinical and computed tomographic study. AJR Am J Roentgenol. 1985;145(2):229-33. Epub 1985/08/01. 69. Korobkin M, Stephens DH, Lee JK, Stanley RJ, Fishman EK, Francis IR, et al. Biliary cystadenoma and cystadenocarcinoma: CT and sonographic findings. AJR Am J Roentgenol. 1989;153(3):507-11. Epub 1989/09/01. 70. Gigot JF, Jadoul P, Que F, Van Beers BE, Etienne J, Horsmans Y, et al. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management? Ann Surg. 1997;225(3):286-94. Epub 1997/03/01. 71. Kabbej M, Sauvanet A, Chauveau D, Farges O, Belghiti J. Laparoscopic fenestration in polycystic liver disease. Br J Surg. 1996;83(12):1697-701. Epub 1996/12/01. 72. van Erpecum KJ, Janssens AR, Terpstra JL, Tjon ATRT. Highly symptomatic adult polycystic disease of the liver. A report of fifteen cases. J Hepatol. 1987;5(1):109-17. Epub 1987/08/01. 73. Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Mayo Clinic proceedings Mayo Clinic. 1990;65(7):1020-5. Epub 1990/07/01.

49
74. Morino M, De Giuli M, Festa V, Garrone C. Laparoscopic management of symptomatic nonparasitic cysts of the liver. Indications and results. Ann Surg. 1994;219(2):157-64. Epub 1994/02/01. 75. Kornprat P, Cerwenka H, Bacher H, El-Shabrawi A, Tillich M, Langner C, et al. Surgical therapy options in polycystic liver disease. Wien Klin Wochenschr. 2005;117(5-6):215-8. Epub 2005/05/07. 76. Oektedalen O, Opstad PK, Demuckadell OBS, Fausa O, Flaten O. Basal Hyperchlorhydria and Its Relation to the Plasma-Concentrations of Secretin, Vasoactive Intestinal Polypeptide (Vip) and Gastrin during Prolonged Strain. Regul Peptides. 1983;5(3):235-44. 77. Vauthey JN, Maddern GJ, Blumgart LH. Adult Polycystic Disease of the Liver. Brit J Surg. 1991;78(5):524-7. 78. Paliard P, Partensky C. [Polycystic liver disease with abdominal pain and jaundice treated by two successive "fenestration" procedure (author's transl)]. Gastroenterol Clin Biol. 1980;4(12):854-7. Epub 1980/12/01. Traitement par fenestration iterative d'une forme douloureuse, puis cholestatique de polykystose hepatique. Interet possible de la cimetidine dans la reduction de la secretion kystique. 79. van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, et al. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137(5):1661-8 e1-2. Epub 2009/08/04. 80. Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, et al. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5(5):783-9. Epub 2010/02/27. 81. Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. Journal of the American Society of Nephrology : JASN. 2010;21(6):1052-61. Epub 2010/05/01. 82. Chrispijn M, Nevens F, Gevers TJ, Vanslembrouck R, van Oijen MG, Coudyzer W, et al. The long-term outcome of patients with polycystic liver disease treated with lanreotide. Alimentary pharmacology & therapeutics. 2012;35(2):266-74. Epub 2011/11/25. 83. Tan YM, Ooi LL. Highly symptomatic adult polycystic liver disease: options and results of surgical management. ANZ J Surg. 2004;74(8):653-7. Epub 2004/08/19. 84. Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13(38):5052-9. Epub 2007/09/19. 85. Koperna T, Vogl S, Satzinger U, Schulz F. Nonparasitic cysts of the liver: results and options of surgical treatment. World J Surg. 1997;21(8):850-4; discussion 4-5. Epub 1997/11/05. 86. Ammori BJ, Jenkins BL, Lim PC, Prasad KR, Pollard SG, Lodge JP. Surgical strategy for cystic diseases of the liver in a western hepatobiliary center. World J Surg. 2002;26(4):462-9. Epub 2002/03/23. 87. Chen MF. Surgery for adult polycystic liver disease. J Gastroenterol Hepatol. 2000;15(11):1239-42. Epub 2000/12/29. 88. Tan YM, Ooi L, Mack POP. Current status in the surgical management of adult polycystic liver disease. Annals Academy of Medicine Singapore. 2002;31(2):217-22. 89. Saini S, Mueller PR, Ferrucci JT, Jr., Simeone JF, Wittenberg J, Butch RJ. Percutaneous aspiration of hepatic cysts does not provide definitive therapy. AJR Am J Roentgenol. 1983;141(3):559-60. Epub 1983/09/01. 90. Bean WJ, Rodan BA. Hepatic cysts: treatment with alcohol. AJR Am J Roentgenol. 1985;144(2):237-41. Epub 1985/02/01. 91. Tikkakoski T, Makela JT, Leinonen S, Paivansalo M, Merikanto J, Karttunen A, et al. Treatment of symptomatic congenital hepatic cysts with single-session percutaneous drainage

50
and ethanol sclerosis: technique and outcome. Journal of vascular and interventional radiology : JVIR. 1996;7(2):235-9. Epub 1996/03/01. 92. Dumot JA, Fields MS, Meyer RA, Shay SS, Conwell DL, Brzezinski A. Alcohol sclerosis for polycystic liver disease and obstructive jaundice: use of a nasobiliary catheter. Am J Gastroenterol. 1994;89(9):1555-7. Epub 1994/09/01. 93. Andersson R, Jeppsson B, Lunderquist A, Bengmark S. Alcohol sclerotherapy of non-parasitic cysts of the liver. Br J Surg. 1989;76(3):254-5. Epub 1989/03/01. 94. AJ S. The use and misuse of tranjugular intrahepatic portasystemic shunts. Curr Gastroenterol Rep. 2000;2:61-71. 95. Shin ES, Darcy MD. Transjugular intrahepatic portosystemic shunt placement in the setting of polycystic liver disease: questioning the contraindication. Journal of vascular and interventional radiology : JVIR. 2001;12(9):1099-102. Epub 2001/09/06. 96. Lin TY, Chen CC, Wang SM. Treatment of non-parasitic cystic disease of the liver: a new approach to therapy with polycystic liver. Ann Surg. 1968;168(5):921-7. Epub 1968/11/01. 97. Diez J, Decoud J, Gutierrez L, Suhl A, Merello J. Laparoscopic treatment of symptomatic cysts of the liver. Br J Surg. 1998;85(1):25-7. Epub 1998/02/14. 98. Fiamingo P, Tedeschi U, Veroux M, Cillo U, Brolese A, Da Rold A, et al. Laparoscopic treatment of simple hepatic cysts and polycystic liver disease. Surg Endosc. 2003;17(4):623-6. Epub 2003/02/08. 99. Katkhouda N, Mavor E, Gugenheim J, Mouiel J. Laparoscopic management of benign cystic lesions of the liver. J Hepatobiliary Pancreat Surg. 2000;7(2):212-7. Epub 2000/09/12. 100. Tan YM, Ooi LL, Soo KC, Mack PO. Does laparoscopic fenestration provide long-term alleviation for symptomatic cystic disease of the liver? ANZ J Surg. 2002;72(10):743-5. Epub 2003/01/22. 101. Marks J, Mouiel J, Katkhouda N, Gugenheim J, Fabiani P. Laparoscopic liver surgery. A report on 28 patients. Surg Endosc. 1998;12(4):331-4. Epub 1998/04/16. 102. Chauveau D, Fakhouri F, Grunfeld JP. Liver involvement in autosomal-dominant polycystic kidney disease: therapeutic dilemma. Journal of the American Society of Nephrology : JASN. 2000;11(9):1767-75. Epub 2000/08/31. 103. Aussilhou B, Doufle G, Hubert C, Francoz C, Paugam C, Paradis V, et al. Extended Liver Resection for Polycystic Liver Disease Can Challenge Liver Transplantation. Annals of Surgery. 2010;252(5):735-41. 104. Newman KD, Torres VE, Rakela J, Nagorney DM. Treatment of highly symptomatic polycystic liver disease. Preliminary experience with a combined hepatic resection-fenestration procedure. Ann Surg. 1990;212(1):30-7. Epub 1990/07/01. 105. Soravia C, Mentha G, Giostra E, Morel P, Rohner A. Surgery for adult polycystic liver disease. Surgery. 1995;117(3):272-5. Epub 1995/03/01. 106. Yang GS, Li QG, Lu JH, Yang N, Zhang HB, Zhou XP. Combined hepatic resection with fenestration for highly symptomatic polycystic liver disease: A report on seven patients. World J Gastroenterol. 2004;10(17):2598-601. Epub 2004/08/10. 107. Madariaga JR, Iwatsuki S, Starzl TE, Todo S, Selby R, Zetti G. Hepatic resection for cystic lesions of the liver. Ann Surg. 1993;218(5):610-4. Epub 1993/11/01. 108. Starzl TE, Reyes J, Tzakis A, Mieles L, Todo S, Gordon R. Liver-Transplantation for Polycystic Liver-Disease. Arch Surg-Chicago. 1990;125(5):575-7. 109. Washburn WK, Johnson LB, Lewis WD, Jenkins RL. Liver transplantation for adult polycystic liver disease. Liver transplantation and surgery : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 1996;2(1):17-22. Epub 1996/01/01. 110. Lang H, von Woellwarth J, Oldhafer KJ, Behrend M, Schlitt HJ, Nashan B, et al. Liver transplantation in patients with polycystic liver disease. Transplant Proc. 1997;29(7):2832-3. Epub 1997/11/20.

51
111. Jeyarajah DR, Gonwa TA, Testa G, Abbasoglu O, Goldstein R, Husberg BS, et al. Liver and kidney transplantation for polycystic disease. Transplantation. 1998;66(4):529-32. Epub 1998/09/12. 112. Swenson K, Seu P, Kinkhabwala M, Maggard M, Martin P, Goss J, et al. Liver transplantation for adult polycystic liver disease. Hepatology. 1998;28(2):412-5. Epub 1998/08/08. 113. Pirenne J, Aerts R, Yoong K, Gunson B, Koshiba T, Fourneau I, et al. Liver transplantation for polycystic liver disease. Liver Transpl. 2001;7(3):238-45. Epub 2001/03/13. 114. Takegoshi K, Tanaka K, Nomura H, Miyagi K, Taira S, Takayanagi N. Successful living donor liver transplantation for polycystic liver in a patient with auto somal-dominant polycystic kidney disease. Journal of Clinical Gastroenterology. 2001;33(3):229-31. 115. Koyama I, Fuchinoue S, Urashima Y, Kato Y, Tsuji K, Kawase T, et al. Living related liver transplantation for polycystic liver disease. Transplant International. 2002;15(11):578-80. 116. Demirci G, Becker T, Nyibata M, Lueck R, Bektas H, Lehner F, et al. Results of combined and sequential liver-kidney transplantation. Liver Transpl. 2003;9(10):1067-78. Epub 2003/10/04. 117. Gustafsson BI, Friman S, Mjornstedt L, Olausson M, Backman L. Liver transplantation for polycystic liver disease - Indications and outcome. Transplantation Proceedings. 2003;35(2):813-4. 118. Ueda M, Egawa H, Oike F, Taira K, Uryuhara K, Fujimoto Y, et al. Living-donor liver transplantation for polycystic liver disease. Transplantation. 2004;77(3):480-1. Epub 2004/02/18. 119. Everson GT, Taylor MR. Management of polycystic liver disease. Curr Gastroenterol Rep. 2005;7(1):19-25. Epub 2005/02/11. 120. Kwok MK, Lewin KJ. Massive hepatomegaly in adult polycystic liver disease. The American journal of surgical pathology. 1988;12(4):321-4. Epub 1988/04/01. 121. Mai ML GT. Renal dysfunction and orthotopic liver transplantation: management strategies and indications for combined liver and kidney transplantation Liver Transpl. 2004;9:166. 122. Grewal HP, Brady L, Cronin DC, Loss GE, Siegel CT, Oswald K, et al. Combined liver and kidney transplantation in children. Transplantation. 2000;70(1):100-5. 123. Margreiter R, Kramar R, Huber C, Steiner E, Niederwieser D, Judmaier G, et al. Combined liver and kidney transplantation. Lancet. 1984;1(8385):1077-8. Epub 1984/05/12. 124. Gonwa TA, Nery JR, Husberg BS, Klintmalm GB. Simultaneous liver and renal transplantation in man. Transplantation. 1988;46(5):690-3. Epub 1988/11/01. 125. Rasmussen A, Davies HF, Jamieson NV, Evans DB, Calne RY. Combined transplantation of liver and kidney from the same donor protects the kidney from rejection and improves kidney graft survival. Transplantation. 1995;59(6):919-21. Epub 1995/03/27. 126. Hiesse C, Samuel D, Chraibi A, Bensadoun H, Blanchet P, Bellamy J, et al. Combined liver and kidney transplantation in nephropathy associated with liver disease. Transplant Proc. 1995;27(1):1262-3. Epub 1995/02/01. 127. Eid A, Moore SB, Wiesner RH, DeGoey SR, Nielson A, Krom RA. Evidence that the liver does not always protect the kidney from hyperacute rejection in combined liver-kidney transplantation across a positive lymphocyte crossmatch. Transplantation. 1990;50(2):331-4. Epub 1990/08/01. 128. Buckel E, Morales J, Brahm J, Fierro MFA, Silva G, Segovia R, et al. Combined liver and kidney transplantation in a multicenter transplantation program in Chile. Transplantation Proceedings. 2005;37(8):3380-1. 129. Guglielmi A, Ruzzenente A, Conci S, Valdegamberi A, Iacono C. How much remnant is enough in liver resection? Dig Surg. 2012;29(1):6-17. Epub 2012/03/24. 130. http://www.cebm.net/index.aspx?o=1025. online 2011.

52
131. Turnage RH, Eckhauser FE, Knol JA, Thompson NW. Therapeutic dilemmas in patients with symptomatic polycystic liver disease. The American surgeon. 1988;54(6):365-72. Epub 1988/06/01. 132. Sanchez H, Gagner M, Rossi RL, Jenkins RL, Lewis WD, Munson JL, et al. Surgical management of nonparasitic cystic liver disease. Am J Surg. 1991;161(1):113-8; discussion 8-9. Epub 1991/01/01. 133. Vauthey JN, Maddern GJ, Kolbinger P, Baer HU, Blumgart LH. Clinical experience with adult polycystic liver disease. Br J Surg. 1992;79(6):562-5. Epub 1992/06/01. 134. Henne-Bruns D, Klomp HJ, Kremer B. Non-parasitic liver cysts and polycystic liver disease: results of surgical treatment. Hepatogastroenterology. 1993;40(1):1-5. Epub 1993/02/01. 135. Li TJ, Zhang HB, Lu JH, Zhao J, Yang N, Yang GS. Treatment of polycystic liver disease with resection-fenestration and a new classification. World J Gastroenterol. 2008;14(32):5066-72. 136. Gall TM, Oniscu GC, Madhavan K, Parks RW, Garden OJ. Surgical management and longterm follow-up of non-parasitic hepatic cysts. HPB (Oxford). 2009;11(3):235-41. Epub 2009/07/11. 137. Mazza OM, Fernandez DL, Pekolj J, Pfaffen G, Sanchez Claria R, Molmenti EP, et al. Management of nonparasitic hepatic cysts. J Am Coll Surg. 2009;209(6):733-9. Epub 2009/12/05. 138. Vons C, Chauveau D, Martinod E, Smadja C, Capron F, Grunfeld JP, et al. Liver resection in patients with polycystic liver disease. Gastroenterologie Clinique Et Biologique. 1998;22(1):50-4. 139. Lerut J, Ciccarelli O, Rutgers M, Orlando G, Mathijs J, Danse E, et al. Liver transplantation with preservation of the inferior vena cava in case of symptomatic adult polycystic disease. Transplant international : official journal of the European Society for Organ Transplantation. 2005;18(5):513-8. Epub 2005/04/12. 140. Kirchner GI, Rifai K, Cantz T, Nashan B, Terkamp C, Becker T, et al. Outcome and quality of life in patients with polycystic liver disease after liver or combined liver-kidney transplantation. Liver Transpl. 2006;12(8):1268-77. Epub 2006/06/03. 141. Ueno T, Barri YM, Netto GJ, Martin A, Onaca N, Sanchez EQ, et al. Liver and kidney transplantation for polycystic liver and kidney-renal function and outcome. Transplantation. 2006;82(4):501-7. Epub 2006/08/24. 142. Mekeel KL, Moss AA, Reddy KS, Douglas DD, Vargas HE, Carey EJ, et al. Living donor liver transplantation in polycystic liver disease. Liver Transpl. 2008;14(5):680-3. Epub 2008/04/25. 143. Kornasiewicz O, Dudek K, Bugajski M, Najnigier B, Krawczyk M. Choice of transplantation techniques and indications for liver transplantation in polycystic liver disease in patients with no signs of end-stage liver disease. Transplant Proc. 2008;40(5):1536-8. Epub 2008/07/01. 144. Krohn PS, Hillingso JG, Kirkegaard P. Liver transplantation in polycystic liver disease: a relevant treatment modality for adults? Scand J Gastroenterol. 2008;43(1):89-94. Epub 2008/10/22. 145. Taner B, Willingham DL, Hewitt WR, Grewal HP, Nguyen JH, Hughes CB. Polycystic liver disease and liver transplantation: single-institution experience. Transplant Proc. 2009;41(9):3769-71. Epub 2009/11/18. 146. Chandok N, Uhanova J, Marotta P. Clinical outcomes of liver transplantation for polycystic liver disease: a single center experience. Annals of hepatology. 2010;9(3):278-81. Epub 2010/08/20. 147. Klupp J, Bechstein WO, Lobeck H, Neuhaus P. [Orthotopic liver transplantation in therapy of advanced polycystic liver disease]. Der Chirurg; Zeitschrift fur alle Gebiete der

53
operativen Medizen. 1996;67(5):515-21. Epub 1996/05/01. Orthotope Lebertransplantation zur Therapie der fortgeschrittenen polycystischen Lebererkrankung. 148. Cavallari G, Nardo B, Montalti R, Beltempo P, Bertelli R, Puviani L, et al. Combined liver-kidney transplantation: experience at Bologna's transplant center. Transplant Proc. 2004;36(3):541-2. Epub 2004/04/28. 149. Benedetti E, Pirenne J, Troppmann C, Hakim N, Moon C, Gruessner RW, et al. Combined liver and kidney transplantation. Transplant International. 1996;9(5):486-91. 150. Harrison JD, Mirza DF, Gunson BK, Mayer AD, Buckels JA, McMaster P. Development of indications and results during 10 years of orthotopic liver transplantation. Clinical transplants. 1993:153-60. Epub 1993/01/01. 151. Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation. 1998;66(1):59-66. Epub 1998/07/29. 152. http://www.UNOS.org. online 2011. 153. http://www.eurotransplant.org/cms/mediaobject.php?file=ar_2010.pdf. online 2011. 154. http://www.uzgenttransplant.be/transplantatie/eurotransplant.asp. online 2011. 155. Roberts J. Clinical liver transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2001;1(1):18-20. Epub 2002/07/04. 156. http://www.eurotransplant.org/cms/index.php?page=public_meld. online 2011. 157. http://www.mohanfoundation.org. online 2011. 158. Eurotransplant Manual. August 2010;27:65-6; 86. 159. Merion RM, Schaubel DE, Dykstra DM, Freeman RB, Port FK, Wolfe RA. The survival benefit of liver transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2005;5(2):307-13. Epub 2005/01/13. 160. www.ELTR.org. online 2011. 161. McHorney CA, Ware JE, Jr., Raczek AE. The MOS 36-Item Short-Form Health Survey (SF-36): II. Psychometric and clinical tests of validity in measuring physical and mental health constructs. Medical care. 1993;31(3):247-63. Epub 1993/03/01. 162. Younossi ZM, Boparai N, Price LL, Kiwi ML, McCormick M, Guyatt G. Health-related quality of life in chronic liver disease: the impact of type and severity of disease. Am J Gastroenterol. 2001;96(7):2199-205. Epub 2001/07/27. 163. http://www.ecog.org/general/perf_stat.html. online 2011. 164. Gali B, Findlay JY, Plevak DJ, Rosen CB, Dierkhising R, Nagorney DM. Right hepatectomy for living liver donation vs right hepatectomy for disease: intraoperative and immediate postoperative comparison. Arch Surg. 2007;142(5):467-71; discussion 71-2. Epub 2007/05/23. 165. Abdalla EK, Barnett CC, Doherty D, Curley SA, Vauthey JN. Extended hepatectomy in patients with hepatobiliary malignancies with and without preoperative portal vein embolization. Arch Surg. 2002;137(6):675-80; discussion 80-1. Epub 2002/06/07. 166. Shoup M, Gonen M, D'Angelica M, Jarnagin WR, DeMatteo RP, Schwartz LH, et al. Volumetric analysis predicts hepatic dysfunction in patients undergoing major liver resection. Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract. 2003;7(3):325-30. Epub 2003/03/26. 167. Vauthey JN, Chaoui A, Do KA, Bilimoria MM, Fenstermacher MJ, Charnsangavej C, et al. Standardized measurement of the future liver remnant prior to extended liver resection: methodology and clinical associations. Surgery. 2000;127(5):512-9. Epub 2000/05/20. 168. Abdalla EK, Adam R, Bilchik AJ, Jaeck D, Vauthey JN, Mahvi D. Improving resectability of hepatic colorectal metastases: expert consensus statement. Annals of surgical oncology. 2006;13(10):1271-80. Epub 2006/09/07.

54
169. Kishi Y, Abdalla EK, Chun YS, Zorzi D, Madoff DC, Wallace MJ, et al. Three hundred and one consecutive extended right hepatectomies: evaluation of outcome based on systematic liver volumetry. Ann Surg. 2009;250(4):540-8. Epub 2009/09/05. 170. de Meijer VE, Kalish BT, Puder M, Ijzermans JN. Systematic review and meta-analysis of steatosis as a risk factor in major hepatic resection. Br J Surg. 2010;97(9):1331-9. Epub 2010/07/20. 171. Ferrero A, Vigano L, Polastri R, Muratore A, Eminefendic H, Regge D, et al. Postoperative liver dysfunction and future remnant liver: where is the limit? Results of a prospective study. World J Surg. 2007;31(8):1643-51. Epub 2007/06/07. 172. Suda K, Ohtsuka M, Ambiru S, Kimura F, Shimizu H, Yoshidome H, et al. Risk factors of liver dysfunction after extended hepatic resection in biliary tract malignancies. Am J Surg. 2009;197(6):752-8. Epub 2008/09/10.


















