UJET DE LA THESE - univ-oran1.dz · 2019-01-21 · dans le processus de cancérisation autres que...
99
1 R R RE E EP P PU U UB B BL L LI I IQ Q QU U UE E E A A AL L LG G GE E ER R RI I IE E EN N NN N NE E E D D DE E EM M MO O OC C CR R RA A AT T TI I IQ Q QU U UE E E E E ET T T P P PO O OP P PU U UL L LA A AI I IR R RE E E MINISTERE DE L'ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE SCIENTIFIQUE UNIVERSITE D'ORAN ES-SENIA FACULTE DES SCIENCES DEPARTEMENT DE BIOLOGIE LABORATOIRE DE BIOLOGIE DU DEVELOPPEMENT Thèse présentée en vue de l'obtention du diplôme de Doctorat Option: Embryogenèse & Oncogenèse Présentée par ABERKANE Meriem Samia SUJET DE LA THESE: Etude de l’implication des gènes du métabolisme des xénobiotiques : GST, MDR1, CYP3A5, UGT1A1 et TS dans la survenue du cancer colorectal dans la population de l’Ouest Algérien Devant le jury composé de : - Président : Pr Saidi D. Faculté des Sciences, Université d’Oran. - Directrice de thèse : Pr El Kebir F.Z. Faculté des Sciences, Université d’Oran. - Examinateur : Pr Colonna P. Hôpital Européen Georges Pompidou, Paris. - Examinateur : Pr Mehtar N. Université des Sciences et Technologies d’Oran. - Examinateur : Pr Djellali L. Faculté de Médecine. Université d’Oran. - Examinateur : Pr Benlatreche C. Faculté de Médecine. Université de Constantine. - Invité : Dr Boudjema A. Université des Sciences et Technologies d’Oran. Année 2009
Transcript of UJET DE LA THESE - univ-oran1.dz · 2019-01-21 · dans le processus de cancérisation autres que...

1
RRREEEPPPUUUBBBLLLIIIQQQUUUEEE AAALLLGGGEEERRRIIIEEENNNNNNEEE DDDEEEMMMOOOCCCRRRAAATTTIIIQQQUUUEEE EEETTT PPPOOOPPPUUULLLAAAIIIRRREEE
MINISTERE DE L'ENSEIGNEMENT SUPERIEUR ET DE LA RECHERCHE SCIENTIFIQUE
UNIVERSITE D'ORAN ES-SENIA
FACULTE DES SCIENCES DEPARTEMENT DE BIOLOGIE
LABORATOIRE DE BIOLOGIE DU DEVELOPPEMENT
Thèse présentée en vue de l'obtention du diplôme de Doctorat
Option: Embryogenèse & Oncogenèse
Présentée par
ABERKANE Meriem Samia
SUJET DE LA THESE:
Etude de l’implication des gènes du métabolisme des xénobiotiques :
GST, MDR1, CYP3A5, UGT1A1 et TS dans la survenue du cancer
colorectal dans la population de l’Ouest Algérien
Devant le jury composé de :
- Président : Pr Saidi D. Faculté des Sciences, Université d’Oran.
- Directrice de thèse : Pr El Kebir F.Z. Faculté des Sciences, Université d’Oran.
- Examinateur : Pr Colonna P. Hôpital Européen Georges Pompidou, Paris.
- Examinateur : Pr Mehtar N. Université des Sciences et Technologies d’Oran.
- Examinateur : Pr Djellali L. Faculté de Médecine. Université d’Oran.
- Examinateur : Pr Benlatreche C. Faculté de Médecine. Université de Constantine.
- Invité : Dr Boudjema A. Université des Sciences et Technologies d’Oran.
Année 2009

2
A mes parents,
A mes enfants Mokhtar et Karim
A mon mari Abdelkader
A ma sœur Kenza
A mon frère Abou Khalil et son épouse Lamia
A mes neveux
A Mme Zahia Chentouf-Mentouri
A Melle Martine Muffat-Joly
A ma belle famille
A tous mes amis

3
REMERCIEMENTS
Madame le professeur FZ El Kebir, vous qui m’avez accueillie dans votre
laboratoire, encouragée et conseillée, tout au long de ce travail, soyez assurée
de ma sincère gratitude.
Monsieur le Professeur D SAIDI, votre compétence reconnue de tous n’a d’égal
que votre disponibilité et votre gentillesse. Vous avez accepté de présider ce
jury, croyez à ma profonde reconnaissance.
Madame le Professeur N MEHTAR, vous m’avez permis d’accéder à votre
laboratoire pour la réalisation d’une partie de ce travail et me faites aujourd’hui
l’honneur de l’évaluer. Je vous en remercie et tiens à vous exprimer ma gratitude.
Madame le Professeur C BENLATRECHE, vous avez accepté de juger ce travail,
j’en suis honorée et vous prie de croire en ma sincère reconnaissance.
Monsieur le Professeur P COLONNA, après m’avoir encadrée pour la réalisation
de mon premier mémoire aujourd’hui vous me faites l’honneur d’examiner mon
travail avec la disponibilité et la modestie qui vous caractérisent. Je vous en
remercie du fond du cœur.

4
Monsieur le professeur L DJELLALI, je vous suis infiniment reconnaissante de
m’avoir ouvert votre service clinique et vous remercie d’accepter de juger mon
travail.
Je tiens à remercier tout particulièrement le Dr A BOUDJEMA pour son amitié,
sa disponibilité, ses encouragements et sa précieuse aide.
Je remercie Madame le Professeur MA LORIOT ainsi que Madame AM
HOULLIET du laboratoire de toxicologie moléculaire de la Faculté de Médecine
du Centre Universitaire Paris-Descartes (INSERM U-775) pour leur
collaboration scientifique et leur amitié.
Je remercie également mes collègues et amis pour leur contribution à la
réalisation de ce travail, en particulier le Dr B LARBAOUI, Le Dr F
CHENNTOUF, le Dr T SAHRAOUI, Mr M FODIL, et Melle F ZEMMANI.
Enfin, je remercie les organismes et institutions qui ont permis de mener à
terme ce travail :
- Le Laboratoire de toxicologie moléculaire de la Faculté de Médecine du
Centre Universitaire Paris-Descartes (INSERM U-775)
- Le Laboratoire de Biologie du Développement de l’Université d’Oran.
- Le service d’Oncologie du CHU d’Oran.
- L’Agence Nationale pour le Développement de la Recherche en Santé
(ANDRS).
- La Faculté de Médecine de l’Université d’Oran.
- Le Laboratoire de Génétique Moléculaire et Cellulaire de l’USTO.

5
Résumé
En Algérie, les cancers colorectaux (CCR) sont en troisième position après ceux du poumon et de la vessie chez
l’homme et ceux du sein et du col utérin chez la femme. Les gènes considérés comme ayant une implication
significative dans la mise en place du processus de cancérisation sont les oncogènes, les anti-oncogènes, les
gènes de réparation de l’ADN et les gènes du métabolisme des xénobiotiques. Les xénobiotiques sont des
molécules étrangères à l’organisme dont font partie les médicaments, les polluants de l’eau ou de
l’atmosphère, les additifs alimentaires et certains composés naturels des aliments. De nombreux
xénobiotiques sont potentiellement carcinogènes et font l’objet d’une métabolisation dans l’organisme au
cours de laquelle interviennent de nombreuses enzymes.
L’objectif de ce travail est de rechercher d’éventuelles associations entre les polymorphismes des gènes du
métabolisme et du transport des xénobiotiques : GSTM1, GSTT1, GSTP1 (313A>G et 341C>T), CYP3A5
6986A>G, MDR1, UGT1A1((TA)6>(TA)7 et -3156 G>A), TS (2R>3R et délétion/insertion de 6pb)
et la survenue du CCR sur une population constituée de 99 malades et 101 contrôles, tous originaires de
l’Ouest Algérien.
Aucune relation d’association entre les polymorphismes des gènes GST, CYP3A5, MDR1, UGT1A1 et la
survenue du CCR n’a été retrouvée dans notre population suggérant ainsi l’intervention d’autres mécanismes
dans le processus de cancérisation autres que ces gènes. En revanche, une association entre le polymorphisme
délétion/insertion de 6pb du gène de la thymidylate synthétase (TS) et la survenue du CCR a été retrouvée
dans notre population. En effet, les individus porteurs de l’insertion 6pb (allèle 6pb) dans le gène de la TS
avaient plus de risque de développer un CCR que les individus présentant la délétion de 6pb (allèle 0pb)
(OR=1.92, p=0.018). De plus, une différence statistiquement significative a aussi été retrouvée entre les cas et
les contrôles concernant les génotypes, suggérant que les deux génotypes 0pb/6pb et 6pb/6pb sont associés à
un plus haut risque de CCR.
La TS est une enzyme capitale dans la biosynthèse de la thymidine requise pour la synthèse et la réparation
de l’ADN. Elle intervient aussi dans le métabolisme des folates, connus pour leur rôle protecteur vis-à-vis du
CCR. Ce polymorphisme délétion/insertion de 6pb, bien que siégeant dans la région 3’UTR non codante du
gène de la TS pourrait affecter la stabilité ou la structure secondaire de l’ARNm de la TS régulant de ce fait le
taux d’expression de cette enzyme. Nos résultats suggèrent que l’insertion de 6pb serait corrélée à une plus
faible activité de la TS augmentant de ce fait la susceptibilité à développer le CCR via une déficience dans le
métabolisme des folates. De plus, un manque de reserve en thymidine du à la diminution du taux de TS
entrainerait probablement la cellule sur une voie de cancérisation à cause de l’incorporation d’uracile au lieu
de la thymine dans la chaine d’ADN.
Par ailleurs, les enzymes codées par ces gènes peuvent moduler la réponse et/ou la toxicité à des molécules utilisées dans la chimiothérapie du CCR comme le 5Fluorouracile, l’Irinotécan ou l’Oxaliplatine.
Ce premier travail nous ouvre donc des perspectives intéressantes dans la recherche de corrélations entre certains polymorphismes et la survenue d’autres pathologies mais aussi la réponse individuelle à la chimiothérapie du CCR.
Mots clés : Cancer colorectal, CCR, Xénobiotiques, GST, CYP3A5, MDR1, UGT1A1, TS.

6
SOMMAIRE
I/ Introduction ............................................................................................................................................8
II/ Aspect clinique................................................................................................................................... 10
1 Description ....................................................................................................................................................... 10
2 Traitement ....................................................................................................................................................... 10
2.1 Chirurgie ............................................................................................................................................................................................. 10 2.2 Radiothérapie................................................................................................................................................................................... 11 2.3 Chimiothérapie ................................................................................................................................................................................ 11
III Aspect génétique ................................................................................................................................ 13
1 Cancer du colon héréditaire ..................................................................................................................... 13
1.1 La polypose adénomateuse familiale (PAF) ..................................................................................................................... 13 1.2 Le syndrome de Lynch (HNPCC) ............................................................................................................................................. 14
2 Mécanismes moléculaires dans la cancérogenèse colorectale ................................................... 14
2.1 L’instabilité des microsatellites............................................................................................................................................... 15 2.2 L’instabilité chromosomique .................................................................................................................................................... 15
IV Pharmacogénétique des agents anticancéreux utilisés dans la chimiothérapie du CCR
...................................................................................................................................................................... 28
1 Mécanismes d’action des agents anticancéreux cytotoxiques .................................................... 29
2 Pharmacogénétique du 5-Fluorouracile ............................................................................................. 29
3 Pharmacogénétique de l’Irinotécan ...................................................................................................... 33
4 Pharmacogénétique de l’Oxaliplatine .................................................................................................. 36
But de l’étude ........................................................................................................................................... 37
V Population et méthodes..................................................................................................................... 40
1 Population d’étude........................................................................................................................................ 40
2 Gènes explorés ................................................................................................................................................ 40
2.1 Gène CYP3A5: SNP 6986A>G .................................................................................................................................................... 40 2.2 Gènes GST : délétion de GSTM1, délétion de GSTT1 et SNPs : GSTP1 313A>G et GSTP1 341C>T ......... 41 2.3 Gène UGT1A1: Le VNTR UGT1A1*28(TA)6>(TA)7 et le SNP -3156 G>A ............................................................ 41 2.4 Gène MDR : SNP 3435C>T .......................................................................................................................................................... 42 2.5 Gène TS: Répétition 2R>3R dans la région 5’UTR du gène TS et délétion/insertion de 6pb dans sa
région 3’UTR ............................................................................................................................................................................................. 42
3 Méthodes d’exploration génétique ......................................................................................................... 43
3.1 Extraction et dosage de l’ADN ................................................................................................................................................. 43 3.2 Exploration de la délétion des gènes GSTT1, GSTM1 et du SNP GSTP1 313A>G par PCR multiplex,
digestion enzymatique et analyse des RFLP. .......................................................................................................................... 44 3.3 Analyse des SNPs : GSTP1 341C>T, MDR1 3435C>T, CYP3A5 6986A>G et UGT1A1 -3156 G>A par
PCR quantitative en temps réel....................................................................................................................................................... 45 3.4 Exploration des polymorphismes UGT1A1*28 (TA)6>(TA)7, TS5’ « 2R>3R » et TS3’
« délétion/insertion de 6pb » par analyse de fragments ................................................................................................... 49

7
4 Analyse statistique ....................................................................................................................................... 52
VI Résultats et discussion ........................................................................................................................ 53
1 Résultats et discussion du génotypage des sujets pour les gènes GSTM1, GSTT1, et GSTP1
exons 5 et 6 ............................................................................................................................................................ 53
2 Résultats et discussion du génotypage des sujets pour le gène MDR1 .............................................. 64
3 Résultats et discussion du génotypage des sujets pour le gène CYP3A5........................................... 67
4 Résultats et discussion du génotypage des sujets pour le gène UGT1A1 ............................... 71
5 Résultats et discussion du génotypage des sujets pour le gène TS ........................................... 77
Conclusion et perspectives....................................................................................................................... 86
Réferences Bibliographiques .............................................................................................................. 88
ANNEXES .................................................................................................................................................... 97

Revue bibliographique
8
I/ INTRODUCTION
Le cancer colorectal est un cancer siégeant dans la région du côlon et du rectum (partie
terminale du colon). Il est caractérisé par une prolifération anormale de cellules dans le gros
intestin et par la formation de carcinomes glandulaires ou adénocarcinomes. Les cancers du
côlon et du rectum étant assez semblables, ils sont regroupés sous le terme de cancer
colorectal (CCR).
Le cancer colorectal constitue un principal problème de santé dans le monde. En effet, il
représente la deuxième cause de mortalité par cancer dans le monde, avec 400.000 décès
environ annuellement (European Journal of Cancer, 2005).
Il est 10 fois plus fréquent aux états unis d’Amérique qu’en Afrique. En 2006, on lui
incombait plus de 55 000 décès aux états unis (Jemal et al, 2006). Ces différences sont la
conséquence du rôle essentiel de l’alimentation dans sa survenue (Irigaray et al, 2007). En
Europe, il représente le second cancer, en termes de fréquence, chez la femme, après le cancer
du sein et le troisième chez l'homme après le cancer du poumon et celui de la prostate (Boyle
et al, 2005).
En Algérie et particulièrement à Oran les cancers colorectaux sont en troisième position après
ceux du poumon et de la vessie chez l’homme et ceux du sein et du col utérin chez la femme
avec une augmentation de plus de 50% des cas entre 1986 et 2000 (Registre du cancer d’Oran,
2006).
A l’échelon moléculaire, le développement d’un adénome puis d’un cancer colorectal est le
résultat d’une accumulation d’évènements génétiques complexes altérant le fonctionnement
normal d’un certain nombre de gènes impliqués dans le contrôle de la prolifération et de la
division cellulaire. Les gènes considérés comme ayant une implication significative dans la
mise en place du processus tumoral sont les oncogènes, les anti-oncogènes, les gènes de
réparation de l’ADN et les gènes du métabolisme des xénobiotiques (Vogelstein et al, 1988).
L’organisme vivant est constamment exposé aux composés exogènes carcinogènes tels que la
dioxine, les nitrosoamines et les hydrocarbures aromatiques polycycliques (HAP). Pour

Revue bibliographique
9
neutraliser leurs effets toxiques, l'organisme possède un système multienzymatique complexe
permettant l'élimination de ces substances hydrophobes par les urines ou la bile. Toute
variation dans l'activité de ces enzymes pourra potentiellement avoir des répercussions
significatives sur le devenir des composés médicamenteux et carcinogènes et sur les quantités
des métabolites produits. Actuellement, il est clairement établi que les polymorphismes
génétiques des enzymes du métabolisme des médicaments (EMX) influencent la susceptibilité
individuelle vis-à-vis de certaines pathologies comme les cancers. Ces analyses se regroupent
sous le terme de pharmacogénétique (Cross et al, 2004). Un autre volet de cette discipline
consiste à œuvrer pour une meilleure connaissance des cibles moléculaires des
chimiothérapies afin de permettre l’identification de facteurs génétiques prédictifs de la
réponse et/ou de la toxicité aux médicaments utilisés dans la chimiothérapie de différents
cancers, dont les cancers colorectaux (Loriot et al, 2004).
L’objectif de la présente étude est la recherche d’éventuelles associations entre les
polymorphismes des gènes impliqués dans le métabolisme des xénobiotiques et la survenue
du cancer colorectal sur une population de l’Ouest Algérien. Ce premier travail nous ouvre
des perspectives intéressantes dans la recherche de corrélations entre certains polymorphismes
et la réponse individuelle à la chimiothérapie du CCR.

Revue bibliographique
10
II/ ASPECT CLINIQUE
1 DESCRIPTION
Le gros intestin appelé aussi colon, se situe dans la cavité abdominale où il prend la forme
d’un « U » renversé. Il fait suite à l’intestin grêle dans la fosse iliaque droite puis remonte le
long de l’abdomen, qu’il traverse dans sa partie supérieure, pour redescendre ensuite jusque
dans la fosse iliaque gauche où il forme une boucle en S (colon sigmoïde). Il se continue
ensuite avec le rectum puis, 15 cm plus bas, avec le canal anal pour se terminer par l’anus.
Les tumeurs malignes du colon se développent dans 70 pour cent des cas dans le sigmoïde ou
le rectum, et sont issues de la muqueuse (couche interne) ou plus souvent d'un polype (tumeur
bénigne qui s'est cancérisée).
Les polypes adénomateux naissent de l'épithélium glandulaire, les cellules se développant
sans restriction et sans se différencier. Ils peuvent être sessiles ou pédiculés, composés
d'abord de cellules de type tubulaire ou de type villeux. Au niveau des cryptes, là où se
renouvelle l'épithélium, on peut observer des zones de cancer colique bien différencié. Il
existe en outre des polypes hyperplasiques, mais dont l'évolution vers le cancer n'est pas
fréquente. Le polype adénomateux semble être la maladie pré cancéreuse à partir de laquelle
se développe le cancer. Les polypes sont au moins 10 fois plus fréquents que les cancers.
2 TRAITEMENT
2.1 CHIRURGIE
La chirurgie est le traitement de référence des cancers colorectaux. Pour traiter un cancer
colorectal potentiellement curable, la première démarche est le traitement de la tumeur
primitive par exérèse chirurgicale. L’évaluation préopératoire et l’examen

Revue bibliographique
11
anatomopathologique de la tumeur primitive permettent de poser l’indication d’un traitement
complémentaire.
L’examen anatomopathologique précise le grade de la tumeur (extension en profondeur de la
tumeur, présence de ganglions envahis, présence de métastases à distance).
2.2 RADIOTHERAPIE
Pour les tumeurs colorectales localement avancées, la radiothérapie est un traitement
complémentaire à la chirurgie qui permet non seulement de diminuer les risques de récidive
locorégionale, mais aussi d’augmenter la survie globale des patients. La radiothérapie n’a en
aucun cas une visée curative dans le traitement des cancers colorectaux.
2.3 CHIMIOTHERAPIE
Les résultats souvent décevants de la chirurgie exclusive dans les formes évoluées conduisent
à lui associer une chimiothérapie adjuvante. Les molécules communément utilisées dans la
chimiothérapie du cancer colorectal sont les suivantes :
2.3.1 Le 5-Fluorouracile
De tous les médicaments anticancéreux, le 5-fluorouracile figure parmi les plus anciens et les
moins onéreux (Aparicio et al, 2002).
Synthétisé en 1957 par Heidelberger à l’Université du Wisconsin (USA), le 5-fluorouracile est
l’anticancéreux le plus utilisé en chimiothérapie du CCR. Il a été développé à partir de
l’uracile, base azotée qui entre dans la composition de l’ARN. Un atome de fluor remplace un
atome d’hydrogène en position 5, d’où le nom de 5-fluorouracile ou encore 5FU. Le
fluorouracile est un antimétabolite qui « mime » l’uracile, précurseur de la thymine, base

Revue bibliographique
12
nécessaire à la synthèse d’ADN (De Chaisemartin et al, 2005). Il inhibe la thymidylate
synthétase empêchant la méthylation de l’uracile en thymine et ses métabolites sont
incorporés dans l’ADN et l’ARN bloquant ainsi leur synthèse. Comme la plupart des
antimétabolites, le fluorouracile agit sur les cellules en phase S du cycle cellulaire (phase de
synthèse d’ADN). Le fluorouracile se fixe aux protéines du sang et diffuse rapidement dans
l’organisme. Il franchit la barrière hémato-encéphalique où sa concentration est faible mais
persistante. Il est métabolisé dans le foie. Seule une petite fraction est éliminée par le rein. La
prévention des complications toxiques du fluorouracile passe par une étude pharmacologique.
Ainsi une toxicité sévère est à redouter pour les patients présentant un déficit congénital en
une enzyme, la dihydropyrimidine déshydrogénase (DPD) qui intervient à la première étape
du catabolisme du fluorouracile et dégrade plus de 80% de cette molécule.
2.3.2 Oxaliplatine
L'oxaliplatine associée au 5-fluorouracile a démontré son efficacité dans les cancers du côlon
de stade III et IV. Cette association est régulièrement prescrite dans ces deux indications. Ce
composé bloque la réplication de l'ADN et les enzymes responsables de son métabolisme,
comme les glutathion S-transférases (GST), jouent un rôle dans l'efficacité de cette thérapie
(Stoehlmacher et al, 2002).
2.3.3 Irinotécan ou CPT-11
L'Irinotécan (Campto) utilisé dans le traitement du cancer du côlon est une pro-drogue dont le
métabolite actif, le SN-38, inhibiteur de la topo-isomérase I, est 100 à 1000 fois plus actif que
la molécule mère. Ce métabolite est produit par des estérases notamment la carboxylestérase
puis normalement glucurono-conjugué (SN-38G) par une uridinediphosphate-
glucuronosyltransférase de type 1 (UGT1) qui le rend inactif et atoxique avant d'être éliminé
par la bile et les urines. Il peut subir un cycle entérohépatique en cas de déconjugaison par les
glucuronidases bactériennes intestinales et redevenir actif et toxique au niveau intestinal sur
les cellules à renouvellement rapide comme les cellules du colon (De Jong et al, 2006).

Revue bibliographique
13
III ASPECT GENETIQUE
1 CANCER DU COLON HEREDITAIRE
Bien que nécessairement réducteurs et schématiques, au regard de nos connaissances encore
fragmentaires, quelques aspects prédominants émergent dans la compréhension des
mécanismes de la cancérogenèse. S’agissant du cancer colorectal, ces mécanismes
moléculaires ont été tout d’abord mis en évidence sur deux variantes héréditaires principales :
la polypose adénomateuse familiale (PAF) et le syndrome de Lynch ou HNPCC (Hereditary
Non Polyposis Colorectal Cancer) qui constituent environ 5% de tous les CCR. Les
recherches sur ces deux syndromes ont permis de mieux comprendre les bases moléculaires
du cancer colorectal intervenant dans un cadre sporadique c'est-à-dire en dehors d'un contexte
héréditaire prédisposant au cancer du côlon (Aaltonen et al, 1993).
1.1 LA POLYPOSE ADENOMATEUSE FAMILIALE (PAF)
La polypose adénomateuse familiale (PAF) est un syndrome autosomique dominant en
rapport avec des altérations du gène APC (Adenomatous Polyposis Coli). Cette maladie
représente 1 % des cancers colorectaux et affecte environ un individu sur 10 000 (Bjork et al,
1999). Des centaines voire des milliers de polypes sont observés tout le long du colon vers
l'âge de 30-40 ans. Vers 40 ans, un ou plusieurs de ces polypes dégénèrent, entraînant un
cancer colique évolutif.
Le gène APC est situé sur le bras long du chromosome 5 et code pour une protéine de 2843
acides aminés. Cette dernière contient une variété de domaines fonctionnels impliqués dans la
transcription, la régulation du cycle cellulaire, la différentiation, et l'apoptose. Des mutations
dans le gène APC ont été démontrées en 1991 (Kinzler et al 1991) et à ce jour, plus de 800
mutations ont été identifiées. Les mutations de phase prédominent et des mutations non-sens
sont retrouvées dans un tiers des cas, tandis que les grandes délétions et mutations faux sens
représentent des causes plus rares de PAF (Nilbert et al 2008).

Revue bibliographique
14
1.2 LE SYNDROME DE LYNCH (HNPCC)
Le syndrome HNPCC (hereditary non polyposis colon cancer) représente environ 5 % des
CCR. Il constitue une prédisposition héréditaire au cancer, dont les critères de reconnaissance
reposent sur des informations individuelles et généalogiques. En effet, des critères dits
d’Amsterdam ont été définis pour ce syndrome : 3 sujets atteints de cancer appartenant au
spectre (cancer colorectal, endomètre, intestin grêle, voies urinaires), l’un étant lié au premier
degré aux 2 autres, 2 générations étant atteintes, et un des cancers au moins s’étant révélé
avant l’âge de 50 ans (Lynch et al, 1996). Le syndrome HNPCC est lié, dans environ 70 %
des cas, à une altération constitutionnelle d'un gène de réparation des mésappariements de
l’ADN, les gènes MMR (MisMatch Repair). Les cellules tumorales présentent alors un
phénotype particulier appelé MSI (Microsatellite Instability). L'instabilité des microsatellites
(MSI) se caractérise par la variation anormale du nombre de séquences répétées dans l'ADN
tumoral comparé à l'ADN du même patient provenant de tissu sain. Celle-ci est présente dans
95 % des cas de HNPCC mais également dans environ 15 % des cancers du côlon non
héréditaires (ou sporadiques) (Lamoril et al 2006).
2 MECANISMES MOLECULAIRES DANS LA CANCEROGENESE COLORECTALE
Il existe deux principales voies de cancérogenèse colorectale et toutes deux résultent d’une
instabilité génétique, l’une, la plus fréquente, à l’échelon chromosomique (instabilité
chromosomique nucléaire « CIN »), l’autre à l’échelon des nucléotides (instabilité des
microsatellites « MSI »). Ces deux voies différentes à l’échelle moléculaire donnent des
lésions semblables au plan morphologique (les adénomes), mais dont le génie évolutif vers le
cancer est différent et plus important dans la voie de l’instabilité des microsatellites (Hamilton
et al, 2006).

Revue bibliographique
15
2.1 L’INSTABILITE DES MICROSATELLITES
Ce mécanisme concerne 15 % des cancers colorectaux sporadiques et est observé de façon
plus claire dans le cadre du syndrome de Lynch. Ces cancers sont appelés MSI ou RER+
(Replication Error). Les cellules cancéreuses ont un contenu en ADN normal (diploïdie),
n’ont pas de pertes chromosomiques et ont des anomalies des gènes MMR (Mitchell et al,
2002). Ces gènes codent pour des protéines dont le rôle est de détecter et de réparer les erreurs
de réplication de l’ADN survenues au cours de la mitose. La mutation ou la méthylation de la
région promotrice des gènes MMR induit une déficience de ce système de réparation et les
mutations vont s’accumuler, préférentiellement au niveau des microsatellites, régions du
génome particulièrement sujettes aux erreurs de réplication (Won-Suk Lee et al, 2008).
2.2 L’INSTABILITE CHROMOSOMIQUE
Celle-ci représente le mécanisme moléculaire de cancérogenèse le plus fréquent dans le
cancer colorectal et concerne 80 à 85 % des cancers colorectaux sporadiques. Ces cancers
sont appelés LOH+ (Loss of Heterozygoty) et sont caractérisés par la survenue de pertes
allèliques (pertes de fragments chromosomiques ou pertes d’hétérozygotie). Les cellules
cancéreuses ont un contenu anormalement élevé en ADN (hyperploïdie), des pertes
chromosomiques fréquentes et des mutations fréquentes (Lièvre et al, 2005). Ces altérations
touchent trois principales catégories de gènes : les oncogènes, les anti-oncogènes et les gènes
du métabolisme des xénobiotiques.
2.2.1 Oncogènes
Physiologiquement, les proto-oncogènes ont une action stimulatrice sur la division cellulaire
mais leur expression est soumise à une régulation fine durant le cycle cellulaire. Ils sont
susceptibles d'être activés en oncogènes lorsqu'ils subissent des altérations somatiques

Revue bibliographique
16
(mutation ponctuelle, translocation ou amplification) ou plus rarement constitutionnelles.
Dans cette catégorie, les plus documentés sont les suivants:
Gène ras
En analysant les tumeurs du côlon d'un point de vue moléculaire, plusieurs gènes impliqués
dans la cancérogenèse ont déjà pu être mis en évidence. Parmi ceux qui sont le plus souvent
incriminés, l’oncogène « ras », en particulier K-ras et N-ras (Gosse-Brun et al, 1998).
Au nombre de trois (K-ras, H-ras et N-ras), les protéines « ras » sont au cœur de nombreuses
voies de signalisation. Elles intègrent et interprètent divers signaux venus de l'extérieur pour
les véhiculer à l'intérieur de la cellule. Pour cela, elles oscillent entre deux états : actif et
inactif. Pour être actives, les protéines « ras » doivent être ancrées à la face interne de la
membrane plasmique. Sous cette forme, elles déclenchent notamment la prolifération des
cellules. Lorsqu'une mutation intervient dans l’un des gènes ras, les protéines sont alors
produites sous une forme continuellement active et les cellules ne cessent de se diviser en
multipliant les erreurs. Le gène ras (K-ras et N-ras) est l'objet de mutations somatiques dans
plus de 50% des tumeurs coliques, et dans environ 50% des adénomes de plus de 1 cm de
diamètre. En revanche, il est peu muté dans les petits adénomes (moins de 10%). Les
mutations retrouvées dans le CCR ne portent pas sur le gène H-ras, mais sur les codons 12 et
13 du K-ras et 12, 13, 61 du N-ras (Soh et al, 1993). Le rôle exact de ces mutations n'est pas
connu. Elles pourraient transformer un petit adénome en grand adénome dysplasique, ou bien
être présentes d'emblée dans les cellules très proliférantes.
Gène c-src
Le src cellulaire « c-src » est un homologue humain de l'oncogène viral du sarcome de Rous,
« v-src ». Le gène c-src a été impliqué dans le développement et la progression de nombreux
cancers humains (Warmuth et al, 2003), y compris le cancer colorectal, et semble nettement
activé dans ce dernier (Iravani et al, 1998). La première étude ayant étayé cette hypothèse est

Revue bibliographique
17
celle qui a rapporté une mutation ponctuelle du gène src au codon 531 à l’origine d’une
activation de c-src (Irby et al 1999).
Gène c-myc
A l’origine, la première implication de ce gène dans les cancers humains a été mise en
évidence dans les lymphomes de Burkitt chez lesquels la translocation t (8-14) (q24;q32)
conduit à la juxtaposition du gène c-myc devant l'élément enhancer du gène codant pour les
chaînes lourdes des immunoglobulines. Cette hyper expression tissu-spécifique de c-myc est
l'élément clé dans l'établissement du processus néoplasique de ces lymphomes (Soussi, 2001).
Depuis, c-myc a été incriminé dans différents cancers dont le cancer colorectal. En effet,
l’amplification de la région 8q24 a été récemment retrouvée associée à un risque augmenté de
CCR (Gruber et al, 2007).
2.2.2 Gènes suppresseurs de tumeurs ou anti-oncogènes
Les gènes suppresseurs de tumeurs ou anti-oncogènes, identifiés grâce aux formes
héréditaires de cancer (ex : gène APC) se comportent dans leur état normal comme des
inhibiteurs de la division cellulaire. Leur mode de fonctionnement est récessif au niveau
cellulaire, c'est-à-dire que, pour que le cancer apparaisse, les deux allèles d'un même anti-
oncogène doivent être inactivés (par mutations ponctuelles, délétions ou une combinaison des
deux), selon la théorie du double événement mutationnel décrit par Knudson dans le cas du
rétinoblastome (Knudson et al, 1971).
Gène APC
Des mutations qui inactivent l’anti-oncogène APC sur le chromosome 5q sont responsables
des tous premiers dérèglements observés lors de la constitution des adénomes de petite taille.
Au niveau du chromosome 5q, on observe une perte de l'hétérozygotie (LOH), c'est-à-dire que
seul un des deux chromosomes parait affecté. Cependant, en général, l'autre copie est

Revue bibliographique
18
inactivée (d'après la théorie de Knudson) ; on évoque alors la présence d'une nouvelle protéine
dominante synthétisée par l'allèle muté. La mutation sur le gène APC est très précoce dans le
développement tumoral et il semblerait qu’il soit muté dans 80 % des cancers du côlon
(Powell et al 1992).
Gène MCC
Le gène MCC (Mutated in Colorectal Cancer) se situe très près du gène APC sur le
chromosome 5q21, et a été retrouvé muté dans environ 10 à 15% des cancers colorectaux. Il
semble avoir un rôle suppresseur probable en association avec le gène APC (Kohonen-Corish
et al, 2007).
Gène DCC
Une perte de l'hétérozygotie est retrouvée sur le chromosome 18q, dans 70% des cancers
colorectaux évolués, mais dans seulement 10% des adénomes débutants. A ce niveau a été
identifié un gène appelé DCC (Deleted in Colon Carcinoma) contenant plus de 29 exons, et
codant pour une protéine transmembranaire. Le gène DCC est exprimé dans toutes les cellules
muqueuses normales et semble être responsable des interactions cellulaires de l'épithélium
normal. Son inactivation serait responsable des troubles de l'adhésion cellulaire, de l'invasion
et des métastases dans le CCR (Beggs et al, 2008)
Gène p53
Le gène p53 code pour un facteur transcriptionnel capable de réguler le cycle cellulaire,
l'apoptose et la réparation de l'ADN (Olivier et al 2003). Ce gène localisé sur le chromosome
17p13 est le plus communément muté dans tous les types de cancers (Levine et al, 1994).
Concernant le CCR, les mutations du domaine central du gène p53 sont les plus souvent
incriminées et les polymorphismes qui lui sont le plus souvent associés sont situés dans le
codon 72 de l’exon 4 (Schneider-Stock et al, 2004; Mammano et al, 2008).

Revue bibliographique
19
2.2.3 Gènes du métabolisme et du transport des carcinogènes
Les xénobiotiques, dont les carcinogènes, sont des molécules de faible masse moléculaire
étrangères à l’organisme. Il s’agit par exemple des médicaments, des polluants de l’eau ou de
l’atmosphère, des additifs alimentaires mais également de certains composés naturels des
aliments. Les substances les plus souvent impliqués dans les processus cancérigènes (cancers
respiratoires, de la vessie, de la peau, des voies aérodigestives supérieures, des systèmes
lymphatique et hématopoïétique et des voies digestives) sont les hydrocarbures aromatiques
polycycliques (HAP) et les amines hétérocycliques (AH). Parmi les expositions
environnementales aux HAP les plus fréquentes, des produits naturels, des médicaments, et
des polluants de l’environnement, toxines végétales et animales, dérivés des combustibles
domestiques et industriels, solvants, colorants, additifs alimentaires, pesticides, herbicides,
etc….
Généralement hydrophobes, les xénobiotiques ont pour tendance naturelle de s’accumuler
dans les phases lipidiques des membranes cellulaires. Elles entraînent ainsi une mort
inéluctable des organismes si ceux-ci ne s’étaient dotés, au cours de l’évolution, de systèmes
enzymatiques permettant leur détoxication et leur élimination.
Le processus de détoxication se déroule selon trois phases regroupant différentes enzymes de
biotransformation (phase I et II), et des transporteurs (phase III) (Figure 1).
Les réactions de phase I, dites de fonctionnalisation, permettent l’introduction d’une fonction
chimique nouvelle (-OH, NH2, COOH) rendant la molécule plus polaire. Les xénobiotiques
pénètrent facilement dans la cellule s’ils sont hydrophobes. Ils sont alors pris en charge par
ces enzymes, dont les plus importantes sont les cytochromes P450 (CYP450) qui catalysent
une réaction de mono oxygénation et peuvent transformer ces carcinogènes en métabolites
très électrophiles, voire génotoxiques.
Les réactions de phases II, dites de conjugaisons, permettent l’ajout d’un radical hydrophile et
sont réalisées soit directement sur le xénobiotique inchangé, soit sur les métabolites
« fonctionnalisés » générés par la phase I. Ces réactions de conjugaison avec un groupement
(glutathion, acide glucuronique, méthyl, acétyl…), augmentent l’hydrophilie de la molécule,
facilitent son transport et activent son élimination par voies rénale et biliaire. Les enzymes de

Revue bibliographique
20
la phase II sont, entre autres, les glutathion S-transférases (GST), uridine diphosphate
glucorono-transférase (UGT1A1), la glutathion peroxydase, la sulfotransférase etc… (Allorge
et al, 2004).
Les enzymes de phase III permettent l’élimination des conjugués hydrophiles. Ce sont
essentiellement des glycoprotéines membranaires comme la P-glycoporotéine P-gp, produit
du gène MDR1 « Multi Drug Resistance » (Schinkel et al, 1997) permettant le transport actif
des xénobiotiques et des conjugués de la phase II hors de la cellule (Silverman JA, 1999).
Elles participent à l’excrétion hors de ces cellules par la voie biliaire (Stieger B et al 1998) et
sont à l’origine de résistances multiples car elles diminuent la quantité de médicament
présente dans la cellule (Silverman et al 1999). Toutefois, le métabolisme des xénobiotiques
ne conduit pas toujours à la détoxication de ces composés. Dans certains cas, le produit oxydé
ou réduit peut manifester une très forte réactivité par attaque électrophile ou nucléophile des
macromolécules environnantes (protéines, acides nucléiques). Ainsi, la formation d’adduits
sur l’ADN peut entraîner l’apparition de mutations ponctuelles et initier des processus de
cancérogenèse, tandis que la formation d’adduits aux protéines peut entraîner une nécrose
tissulaire et des phénomènes d’allergie (Dansette et al, 1998). Toutes ces enzymes sont donc
considérées aujourd’hui comme étant potentiellement impliquées dans la survenue de certains
cancers via leur rôle de détoxication et d’élimination des substances carcinogènes auxquelles
est souvent exposé l’organisme.

Revue bibliographique
21
Figure 1: Représentation schématique du métabolisme des xénobiotiques.
Les xénobiotiques (X) sont généralement des molécules hydrophobes, qui pénètrent facilement dans la cellule.
Ils peuvent en être expulsés par des protéines comme la P-gp ou être métabolisés par fonctionnalisation (Phase
I, exemple : les CYP 450) puis/ou par conjugaison (Phase II, exemple : les GST), en produits plus hydrophiles
(XOH et XOR), ce qui facilite leur élimination hors de la cellule. L’élimination est directe ou effectuée par
l’intermédiaire de protéines dites de Phase III, comme les MRP « Multidrug Resistance Proteins » dont la P-gp
(Beaune et al, 2000).
Gène CYP450
Les cytochromes P450 (CYP450) forment une super famille multi génique codant pour des
enzymes de la phase I, impliquées dans le métabolisme oxydatif de diverses molécules aussi
bien des xénobiotiques (médicaments, pesticides, polluants, cancérigènes…) que des
substances endogènes (hormones stéroïdiennes, vitamines, acides gras…) (Perera et al, 1997).
Chez l’homme, les formes prépondérantes de cytochromes P450 sont représentées par
plusieurs familles et sous familles présentant de fortes homologies dans leurs séquences en
acides aminés (Nebert et al, 1987).
Actuellement, plus de 57 cytochromes P450s sont connus, la plupart d’entre eux possèdent
une expression type cellulaire et tissu-spécifique (Ding et al, 2003). Les réactions de

Revue bibliographique
22
transformations catalysées par ces enzymes s’inscrivent dans un processus de détoxication
évitant l’accumulation de molécules potentiellement toxiques dans l’organisme.
Les cytochromes P450 ont un rôle majeur dans le développement tumoral de par leur fonction
dans le métabolisme de nombreuses substances carcinogènes (Murray et al, 2000). En effet,
ces enzymes peuvent parfois catalyser l’activation chimique de certains composés et produire
des métabolites toxiques voire cancérigènes (Bethke et al, 2007).
Les enzymes CYP450 participent également au métabolisme de nombreuses hormones
stéroïdes et d’acides gras connus pour leur implication dans le développement de certains
cancers (Li et al, 2005; Rodriguez-Antona et al, 2006).
Parmi les composés impliqués dans l’étiologie des cancers colorectaux, les hydrocarbures
aromatiques polycycliques (HAP). Ces derniers sont générés essentiellement au cours de la
cuisson de la viande à haute température et requièrent une activation métabolique par les
CYP450 par hydroxylation avant d’exercer leur effet génotoxique (Windmill et al, 1997).
Les enzymes CYP3A comme les 3A4, 3A5, 3A7 et 3A43, représentent la sous famille la plus
importante des CYP450 dans le métabolisme des xénobiotiques parce qu’ils constituent plus
de 30% de la totalité des protéines CYP450 dans le foie et métabolisent plus de 50% des
médicaments (Shimada et al 1994; Westlind et al, 2001). La plupart de ces protéines sont très
polymorphes et des mutations dans leurs gènes respectifs abolissent, réduisent ou altèrent
leurs fonctions enzymatiques respectives. A ce jour, plusieurs études se sont focalisées sur le
rôle des polymorphismes des gènes CYP1A1 (Ye et al, 2002) de CYP1A2 (Chen et al, 2005)
de CYP1B1 (Gibson et al, 2003) et de CYP2E1 (Park et al, 2003) dans les cancers du colon
ou de l’estomac.
Concernant le gène CYP3A5, localisé sur le chromosome 7q22.1, il existe plus de 34 variants
allèliques (Human Cytochrome P450 (CYP) Allele Nomenclature Committee :
http://www.cypalleles.ki.se, mise à jour du 09/09/2008). Le plus connu est le SNP (single
nucleotide polymorphism) CYP3A5*3C qui consiste en une transistion 6986A>G dans
l’intron 3 qui crée un site d’épissage alternatif et code pour un ARNm tronqué avec un codon
stop prématuré aboutissant à une absence d’activité de la protéine (Kuehl et al, 2001). Ce
SNP est retrouvé chez les afro-américains, les caucasiens et les japonais à une fréquence de

Revue bibliographique
23
50 à 80% (Kuehl et al, 2001; Garsa et al, 2005 ; Quaranta et al, 2006) et une étude récente a
retrouvé une association de ce SNP avec la diminution du risque de cancer de l’œsophage
(Dandara et al, 2005). Cependant, à ce jour l’impact des polymorphismes génétiques de
CYP3A5 sur le cancer colorectal n’a pas été clairement établi.
Gènes GST
Parmi les enzymes du métabolisme de phase II, les glutathion S-transférases (GST) ; Les GST
sont des enzymes polymorphes impliquées dans la conjugaison du glutathion réduit à des
composés électrophiles nocifs. Ces enzymes occupent une place très importante dans le
système de défense cellulaire (Hayes et al, 1999). Ce sont des enzymes solubles avec une
masse moléculaire d'environ 25 kDa, dimériques et largement répandues dans la faune et la
flore. Chaque sous-unité GST porte deux sites de fixation : le premier est spécifique du
glutathion réduit (GSH), site « G », alors que le second l'est pour le substrat « S »
médicamenteux ou un autre xénobiotique endogène ou exogène, site « H » (Alexandrie et al,
2002).
Les fonctions biologiques remplies par les GST sont diverses. Les plus importantes sont la
détoxication des xénobiotiques et l’activation de certains substrats. En effet, les GST
catalysent la conjugaison du GSH à différents substrats de la phase I nocifs pour la cellule.
Par conséquent, elles constituent une importante ligne de défense protégeant les composants
cellulaires (ADN, lipides et protéines) des effets délétères induits par ces composés (Giachelia
et al, 2007). Les GST jouent aussi un rôle important dans le transport de composés endogènes
comme les hormones, les stéroïdes, l’acide urique et la bilirubine (Hayes et al, 2000 ;
Cavalieri et al, 2000).
Bien que la grande majorité des xénobiotiques soit neutralisée par les GST, il existe plusieurs
cas pour lesquels la catalyse impliquant ces enzymes ne conduit pas à une détoxication
complète. Ainsi, certains conjugués néoformés sont instables et peuvent être clivés donnant
des métabolites souvent très toxiques ce qui expose la cellule à la menace chimique de ces
derniers (Lo et al, 2007).

Revue bibliographique
24
Les GST représentent une importante famille d'isoenzymes réparties en huit classes : mu
(GSTM), alpha (GSTA), pi (GSTP), thêta (GSTT), zêta (GSTZ), sigma (GST), kappa (GSTK)
et oméga (GSTO) (Hayes et al, 1999).
Les cinq classes de GST, GSTA, GSTM, GSTP, GSTT et GSTZ, présentent des
polymorphismes génétiques et les plus étudiés et documentés d’entre eux sont ceux relatifs
aux sous-classes GSTM1, T1 et P1 car elles sont à l’origine de changements au niveau de
l’activité enzymatique de ces protéines (Hayes et al, 2005) (tableau 1).
Les différentes enzymes de la classe GSTM sont codées par les gènes localisés sur le
chromosome 1p13 (Ross et al, 1993). Les GSTP sont codées par un unique gène situé sur le
chromosome 11q13 (Board et al, 1989) et enfin, la classe des GSTT qui est composée de deux
sous unités : GSTT1, et GSTT2 toutes les deux codées par des gènes localisés sur le
chromosome 22q11 (Sherratt et al, 1997).
Tableau 1 : Principaux polymorphismes caractérisés pour les classes GST les plus étudiées (Habdous et al,
2004).
Classe GST Gène Variants allèliques Conséquences
Theta hGSTT1 hGSTT1*A Thr104
hGSTT1*B Thr104Pro
hGSTT1*0 Absence de l’enzyme
Mu hGSTM1 hGSTM1*A Lys173
hGSTM1*B Lys173Asn
hGSTM1*0 Absence de l’enzyme
Pi hGSTP1 hGSTP1*A Ile105/Ala114
hGSTP1*B Ile105Val/Ala114
hGSTP1*C Ile105Val/Ala114Val
hGSTP1*D Ile105/Ala114Val

Revue bibliographique
25
Trois allèles sont décrits pour GSTT1: GSTT1*A, GSTT1*B et GSTT1*0 correspondant
respectivement à l’allèle sauvage, à une variation nucléotidique au niveau du codon 310
(310A>C) qui transforme le résidu thréonine 104 en proline et à une délétion du gène
GSTT1. Pour GSTM1, la variation nucléotidique (2619G>C) et une délétion complète du
gène GSTM1 sont responsables de la présence de trois allèles nommés : GSTM1*A,
GSTM1*B et GSTM1*0. GSTP1 présente quatre variants allèliques nommés GSTP1*A,
GSTP1*B, GSTP1*C et GSTP1*D. Ces variants résultent de la présence de deux variations
nucléotidiques 313A>G et 341C>T au niveau de la séquence codante. Ces transitions
nucléotidiques transforment le codon ATC (isoleucine) en position 105 dans les GSTP1*A et
GSTP1*D en GTC (valine) dans les GSTP1*B et GSTP1*C. Quant au codon GCG (alanine)
en position 114, initialement présent dans les GSTP1*A et 1*B, il est transformé en GTG
(valine) dans les GSTP1*C et GSTP1*D (Habdous et al, 2004).
La délétion à l’état homozygote des gènes GSTT1 (allèle hGSTT1*0) ou GSTM1 (allèle
hGSTM1*0) a pour conséquence une totale absence d’activité de ces deux enzymes. Tandis
que les substitutions sur le gène GSTP1 aboutissent à des changements d’acides aminés qui
s’opèrent dans le site actif de GSTP1 et conduisent à une plus faible activité de cette enzyme
due à une diminution de son affinité pour le substrat (Zimniak et al, 1994 ; Ali-Osman et al,
1997;Van der Logt et al 2004).
Etant donné le rôle que jouent les GST dans la détoxication des composés carcinogènes,
plusieurs études épidémiologiques ont examiné l'incidence de leurs polymorphismes
génétiques sur le risque de survenue et/ou d'aggravation de nombreuses pathologies comme
les cancers et les maladies cardiovasculaires.
Dans une méta-analyse, Benhamou et al concluent à une augmentation du risque de cancer du
poumon chez les sujets ayant l'allèle nul GSTM1*0 (Benhamou et al, 2002) ; tandis que
d’autres travaux ont associé ce génotype à une augmentation du risque du cancer de la
vessie (Kempkes et al, 1996 ; Engel et al, 2002) mais également à celui du cancer du larynx
(D’Errico et al, 1999). GSTM1 a également été largement étudié dans la relation avec le CCR
à cause de son taux d’expression important dans le tractus gastro-intestinal et son rôle dans la
détoxification des dérivés carcinogènes dans l’alimentation (Lin et al, 1995). La plupart de ces

Revue bibliographique
26
études semblent aboutir à une association entre l’allèle nul GSTM1 et le risque accru au CCR
(Zhong et al, 1993 ; Moore et al, 2005).
Concernant le gène GSTT1, un risque relativement élevé de survenue d'astrocytomes et de
méningiomes chez les sujets porteurs de l'allèle nul GSTT1*0 a été rapporté (Elexpuru-
Camiruaga et al, 1995). De plus, une augmentation du risque de survenue du cancer de la
vessie a également été associée à cet allèle dans différentes études cas/témoins (Ma et al,
2002 ; Lee et al, 2002) mais à l’inverse du génotype GSTM1 nul, la délétion homozygote de
GSTT1 semble n’avoir aucune incidence sur le risque de survenue du cancer du poumon
(D’Errico et al, 1999).
Par ailleurs, la corrélation entre le gène GSTT1 et la survenue du CCR a fait l’objet de
nombreuses études aussi (Chenevix-Trench et al 1995 ; Deakin et al, 1996) dont quelques
unes qui ont retrouvé une association significative entre le génotype nul GSTT1 et une
augmentation du risque de survenue de ce cancer (Deakin et al, 1996) ; cependant cette
association est contestée dans d’autres travaux (De Jong et al, 2002 ; Van der Logt et al,
2004).
Concernant le gène GSTP1, de nombreuses études ont incriminé le polymorphisme dans le
codon 105 du gène GSTP1 dans le risque de CCR (Harris et al, 1998 ; Sachse et al, 2002)
mais les données des différentes méta-analyses ne vont pas toutes dans le même sens et le
risque probable d’une association de ce SNP avec la survenue du CCR reste encore à
déterminer (Houlston et al, 2001).
Ces différentes associations demeurent difficiles à établir vu les différences ethniques
importantes qui existent pour les polymorphismes de ces gènes (Longuemaux et al, 1999;
Park et al, 1999 ; Wormhoudt et al, 1999). En effet, certains polymorphismes SNPs qui ont
été associés avec le CCR dans une population pourrait être sans effet sur le risque de la
survenue de ce cancer dans d’autres populations.

Revue bibliographique
27
Gène MDR1
La P-glycoprotéine (P-gp) est une protéine de la membrane cellulaire de 170 kDa codée chez
l’homme par le gène MDR1 « Multidrug Resistance » localisé en 7q21.1. Ce gène s’étend sur
200kb et comprend 28 exons (Gottesman et al ; 1995).
La P-gp est exprimée de manière constitutive au niveau de certains organes notamment au
niveau de l’intestin et des cellules endothéliales des capillaires sanguins de la barrière hémato-
encéphalique. C’est un transporteur membranaire qui participe à la protection du milieu
intracellulaire en rejetant à l’extérieur de la cellule des molécules présentes dans le milieu
intracellulaire entre autres des substances potentiellement carcinogènes, des métabolites
cellulaires toxiques mais aussi des molécules médicamenteuses. Dans ce dernier cas, ceci peut
constituer une barrière importante dans la réponse thérapeutique (Gottesman et al, 1996).
Différentes études ont montré que ce gène était très polymorphe avec plus de 50 SNPs
différents (Cascorbi et al, 2001). Etant donné le rôle physiologique de la P-gp , de nombreuses
études ont pu mettre en évidence une association entre des SNPs altérant sa fonction et
certains cancers comme la leucémie myéloide chronique (Urayama et al, 2007), le cancer du
sein (Turgut et al, 2007) et le cancer rénal (Siegsmund et al, 2002).Une attention particulière
s’est portée sur un SNP particulier, MDR 3435C>T localisé dans l’exon 26. Bien que ce SNP
soit une mutation silencieuse codant pour l'isoleucine, ce polymorphisme semble affecter
l'expression et la fonction de la P-gp de façon importante (Hoffmeyer et al, 2000).
Concernant le cancer colorectal certaines études ont retrouvé une augmentation du risque de
sa survenue avec l’allèle 3435T de ce SNP mais également l’allèle 2677G du SNP du SNP
MDR1 2677G > T/A situé dans l’exon 21 (Osswald et al, 2007). Cependant cette corrélation
reste controversée (Petrova et al, 2008).
Par ailleurs, il existe d’importantes différences ethniques concernant les polymorphismes de
ce gène, ce qui contribue à rendre plus difficile la recherche d’associations entre la plupart de
ces SNPs et la survenue de cancers (Ozawa et al, 2004).

Revue bibliographique
28
IV PHARMACOGENETIQUE DES AGENTS ANTICANCEREUX UTILISES
DANS LA CHIMIOTHERAPIE DU CCR
Les médicaments sont souvent une cause importante de morbidité et de mortalité même
lorsque leur prescription est faite correctement. Une étude française a montré que ces
accidents étaient à l’origine d’environ 10 à 20% des admissions dans un service d’urgence
(Queneau et al, 2003). Une autre étude attribue aux médicaments 44.000 à 98.000 décès par
an aux Etats-Unis (Kohn et al, 2000).
A l’inverse, des médicaments très utilisés sont inactifs chez certains patients. Une telle
variabilité de la réponse aux médicaments dépend de facteurs environnementaux
(alimentation, interaction médicamenteuse, tabagisme), de l’état du malade (sévérité de la
maladie, pathologies associées, âge), d’erreurs thérapeutiques, mais aussi de déterminants
génétiques.
La pharmacogénétique est l’étude des relations entre la variabilité du génome et la réponse
thérapeutique. Elle étudie les mécanismes d’origine génétique intervenant dans la réponse
aux médicaments dans le but d’optimiser les traitements médicamenteux, tant en termes
d’efficacité que de sécurité d’emploi. Cette branche de la pharmacologie s’est imposée il y a
plus de 50 ans lorsqu’on a constaté que les hémolyses aiguës après la prise d’un anti-
paludéen, la primaquine, survenaient chez les malades présentant un déficit héréditaire en
glucose-6-phosphate déshydrogénase (Beutler et al, 1993).
En cancérologie, la prédiction de la toxicité des agents anticancéreux devient un problème
crucial compte tenu de la fenêtre thérapeutique étroite de ces médicaments et des alternatives
possibles entre différents protocoles de chimiothérapie. L’évaluation du risque de toxicité
grave avant la mise en route du traitement et la sélection du protocole thérapeutique
(molécule, posologie, schéma d’administration) en fonction de critères spécifiques au malade,
sont des objectifs majeurs pour l’amélioration de la réponse à la chimiothérapie.
Le médicament est aussi un xénobiotique, son introduction dans l’organisme est suivie de
deux temps, un de transformation, souvent hépatique, et un deuxième d’effet sur la cible, dans

Revue bibliographique
29
un ordre variable. Ces deux temps sont précédés par une phase d’absorption intestinale pour
les médicaments administrés par voie orale. Toutes ces étapes sont assurées par des
transporteurs et des enzymes dont l’expression peut varier en fonction du polymorphisme du
gène concerné.
1 MECANISMES D’ACTION DES AGENTS ANTICANCEREUX CYTOTOXIQUES
L’objectif de la chimiothérapie cytotoxique anticancéreuse est de détruire la totalité des
cellules cancéreuses. La plupart des agents cytotoxiques utilisés en chimiothérapie
anticancéreuse interagissent avec l’ADN ou ses précurseurs : ils inhibent la synthèse de
l’ADN ou induisent des lésions irréparables de l’ADN. Certains agents agissent après la phase
de transcription : ils interagissent avec des protéines et des enzymes impliquées dans la
prolifération/division cellulaire : par exemple, les taxanes se fixent sur la tubuline et
empêchent la division cellulaire. Les drogues cytotoxiques ne sont pas spécifiques des cellules
cancéreuses, elles agissent aussi sur les cellules normales à prolifération rapide telles que les
cellules de la moelle osseuse, les cellules de la muqueuse digestive, les gonades, la peau, les
phanères. C’est de cette non-spécificité que découle leur toxicité.
Après avoir passé en revue les différentes molécules administrées en chimiothérapie du CCR
dans le chapitre précèdent, nous allons examiner dans celui-ci les principales enzymes
intervenant dans le métabolisme de ces molécules, ensuite les incidences des mutations
somatiques sur leurs gènes respectifs en essayant de prévoir quelles sont les perspectives de
prescription personnalisée qu’ouvrent ces travaux récents.
2 PHARMACOGENETIQUE DU 5-FLUOROURACILE
Une méta-analyse réalisée sur des malades atteints d’un cancer colorectal et recevant du 5-FU
a montré que des toxicités sévères surviennent dans environ 1/3 des cas et une toxicité létale
pour 0,5 % des patients (Meta-analysis Group In Cancer, 1998).
Revue bibliographique
30
Les polymorphismes des gènes codants pour des enzymes intervenant dans le métabolisme du
5-FU sont en partie responsables de la survenue de ces effets indésirables (Ulrich et al, 2003).
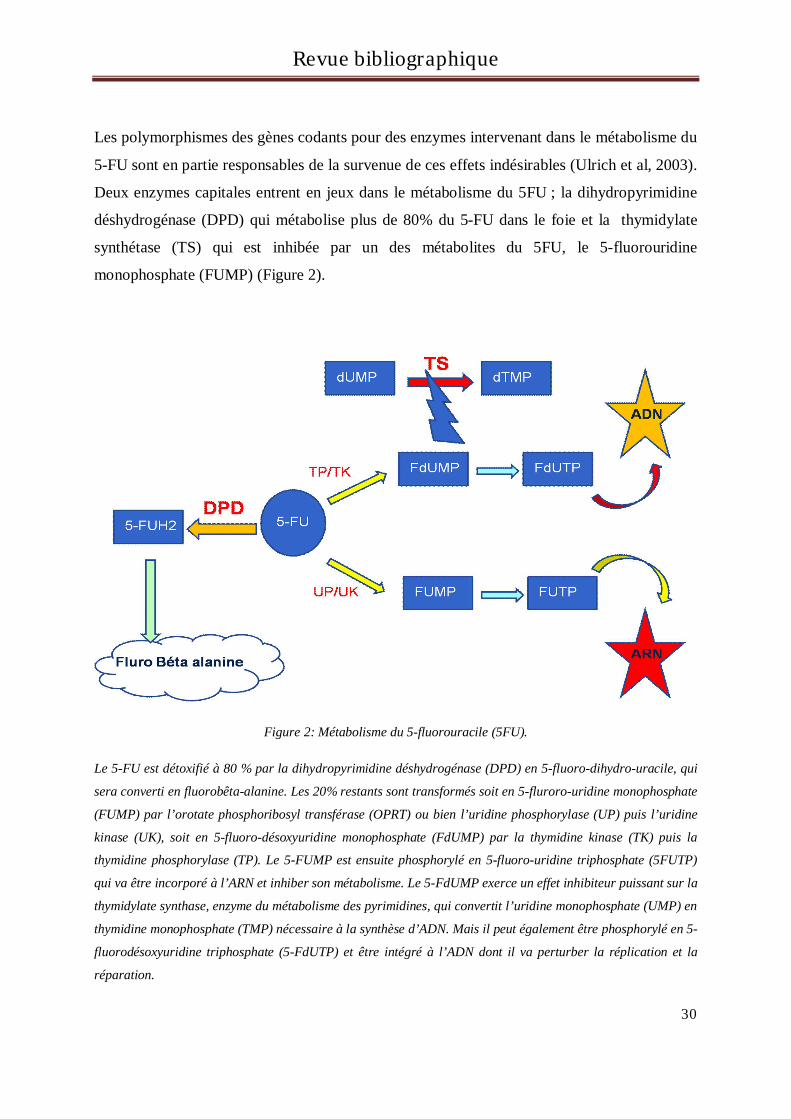
Deux enzymes capitales entrent en jeux dans le métabolisme du 5FU ; la dihydropyrimidine
déshydrogénase (DPD) qui métabolise plus de 80% du 5-FU dans le foie et la thymidylate
synthétase (TS) qui est inhibée par un des métabolites du 5FU, le 5-fluorouridine
monophosphate (FUMP) (Figure 2).
Figure 2: Métabolisme du 5-fluorouracile (5FU).
Le 5-FU est détoxifié à 80 % par la dihydropyrimidine déshydrogénase (DPD) en 5-fluoro-dihydro-uracile, qui
sera converti en fluorobêta-alanine. Les 20% restants sont transformés soit en 5-fluroro-uridine monophosphate
(FUMP) par l’orotate phosphoribosyl transférase (OPRT) ou bien l’uridine phosphorylase (UP) puis l’uridine
kinase (UK), soit en 5-fluoro-désoxyuridine monophosphate (FdUMP) par la thymidine kinase (TK) puis la
thymidine phosphorylase (TP). Le 5-FUMP est ensuite phosphorylé en 5-fluoro-uridine triphosphate (5FUTP)
qui va être incorporé à l’ARN et inhiber son métabolisme. Le 5-FdUMP exerce un effet inhibiteur puissant sur la
thymidylate synthase, enzyme du métabolisme des pyrimidines, qui convertit l’uridine monophosphate (UMP) en
thymidine monophosphate (TMP) nécessaire à la synthèse d’ADN. Mais il peut également être phosphorylé en 5-
fluorodésoxyuridine triphosphate (5-FdUTP) et être intégré à l’ADN dont il va perturber la réplication et la
réparation.

Revue bibliographique
31
Gène DYPD
La DPD (Dihydropyrimidine dehydrogenase) produit du gène DPYD localisé sur le
chromosome 1p21.3 est l’intervenant enzymatique principal dans l’inactivation de plus de
80% du 5FU au niveau hépatique. Cette enzyme semble avoir une activité exclusivement
cytosolique et une expression ubiquitaire (Van Kuilenbourg et al, 2002 (a)).
L’activité de la DPD est très variable selon les individus, et les malades à faible activité ne
peuvent pas métaboliser le 5-FU, d’où la survenue de phénomènes toxiques graves atteignant
le système nerveux, la moelle osseuse et le tractus gastro-intestinal, et pouvant conduire au
décès (Milano et al, 1999).
Le gène codant pour la DPD comprend 23 exons et plus de 39 mutations sur ce gène,
inactivant l’enzyme, ont été répertoriées (Van Kuilenbourg 2004). La plus fréquente est une
substitution intronique au niveau d’un site d’épissage, qui aboutit à l’excision totale de l’exon
14 (allèle DPYD*2A) et correspondant environ à 40–50 % de l’ensemble des variants (Van
Kuilenbourg et al, 2002 (b); De Chaisemartin et al 2005).
Plus de la moitié de ces SNPs ont un effet délétère connu sur l’activité enzymatique de la
DPD (Mc Leod et al, 1998; Van Kuilenbourg et al, 2000) mais l’impact clinique de
nombreux autres SNPs modifiant la séquence protéique de la DPD restent à explorer.
Gène TS
La thymidine synthétase (TS) codée par le gène TS situé sur le chromosome 18p11.32, est une
enzyme capitale dans la biosynthèse des nucléotides. En effet, la TS est une enzyme à deux
substrats qui forme avec le déoxyuridine monophosphate (dUMP) et le coenzyme méthylène
tétrahydrofolate un complexe ternaire où le méthylène tétrahydrofolate cède un groupement
méthyl au dUMP pour le convertir en désoxythymidine monophosphate (dTMP). Ce dernier
est ensuite phosphorylé en désoxythymidine triphosphate (dTTP). Cette conversion est
essentielle pour la disponibilité de la thymidine requise pour la synthèse et la réparation de
l’ADN. La TS est aussi un intervenant essentiel dans le métabolisme des folates étant donné
que la forme circulante des folates dans le sang est surtout le méthyl tétrahydrofolate

Revue bibliographique
32
(Carreras et al, 1995). Mais le rôle le plus étudié de la TS est celui qu’elle joue dans la
réponse à certaines molécules utilisées en chimiothérapie dont le 5FU. En effet, dans la
cascade métabolique du 5-FU, un des métabolites actifs, le 5-FdUMP se fixe à la TS et inhibe
son activité ce qui diminue voire abolit la synthèse de l’ADN des cellules tumorales entre
autres et freine ainsi la progression du cancer (Santi et al, 1974) (Figure 2). Plusieurs études
ont montré une relation inverse entre le taux d’ARNm ou protéique de la TS et la réponse
anti-tumorale au 5-FU (Johnston et al, 1995). Ces travaux ont mis en évidence plusieurs
polymorphismes du gène TS aboutissant à une variation du taux d’expression de cette
enzyme. Parmi ces polymorphismes, le plus documenté est celui se trouvant dans la région
enhancer 5’UTR (untranslated region) du gène TS. En effet, le promoteur de TS comprend
une séquence de 28pb (paires de bases) qui peut être répétée plusieurs fois, les nombres de
copies les plus fréquents dans la population normale étant 2 et 3 répétitions (allèles 2R ou
3R). La présence de trois répétitions se traduit par une augmentation de la transcription et de
l’expression de la thymidylate synthétase (Horie et al, 1995) ; cette augmentation a été
corrélée à une plus faible inhibition de la thymidylate synthase par le 5-FU et une réponse
inefficace voire toxique à cet anticancéreux (Pullarkat et al, 2001).
Par ailleurs, un nouveau polymorphisme G > C touchant le 12ème nucléotide de la 2ème
répétition de 28pb du promoteur de TS entraîne une altération du site de fixation du facteur de
transcription USF-1 (upstream stimulating factor) et modifie l'expression de la thymidylate
synthétase ce qui interfère avec l’expression du gène (Marcuello et al, 2004).
Une autre variation importante a été également mise en évidence. Il s'agit d'une délétion ou
insertion de 6pb dans la région 3'UTR du gène TS (Ulrich et al, 2000). L’effet de ce
polymorphisme sur l’expression de la TS est encore sujet à controverse (Mandola et al, 2004 ;
Dotor et al, 2006). Néanmoins ce dernier polymorphisme a récemment été corrélé à un risque
accru de survenue de certains cancers comme le cancer du poumon ou celui de l’œsophage
(Zhang et al, 2004; Shi et al, 2005).
Globalement, une expression élevée de la thymidylate synthétase est désormais largement
reconnue comme étant une des causes déterminantes de la résistance au 5-FU. Cette donnée
valable pour le cancer colorectal devrait être prise en compte dans le traitement et le
pronostic. L’ensemble de ces données suggèrent que l’étude des polymorphismes du gène de

Revue bibliographique
33
la TS pourrait être utile dans la sélection des patients pour lesquels le 5-FU est efficace et bien
toléré.
3 PHARMACOGENETIQUE DE L’IRINOTECAN
L’Irinotécan ou le CPT-11 (carbonyloxycamptothécine) est une autre molécule utilisée dans la
chimiothérapie du CCR qui abolit la division anarchique des cellules cancéreuses en inhibant
la topo-isomérase I. Les topo-isomérases sont des enzymes assurant la spiralisation et
déspiralisation de l’ADN après avoir crée des coupures transitoires de l’un (topo-isomérase I)
ou des deux (topo-isomérase II) brins d’ADN puis elles assurent la réparation de ses coupures.
Elles permettent une relaxation des forces de torsion générées au moment de la réplication.
Les inhibiteurs des topo-isomérase I ou II stabilisent le complexe de clivage et empêchent
l’étape de réparation et provoquent une coupure définitive des brins d’ADN ce qui induit
l’apoptose des cellules.
L’action de l’Irinotécan sur la topo-isomérase I passe par sa conversion en son métabolite
actif, le SN 38 qui est 100 à 1000 fois plus actif sur la topo-isomérase I que la molécule mère.
Ce métabolite est produit par des estérases notamment la carboxylestérase (CES). Le SN-38
est alors détoxifié par glucorono-conjugaison (SN-38 G) par une glucuronyl-transférase de
type 1 (UGT1) qui le rend inactif et atoxique avant d'être éliminé par la bile et les urines (De
Chaisemartin et al, 2005).
L’Irinotécan peut engendrer une toxicité importante aussi bien hématologique (leucopénies)
que digestive (diarrhées aiguës). Par ailleurs, La réponse au traitement à l’Irinotécan pour un
cancer du côlon de même grade varie énormément d’un patient à l’autre (Yu et al. 2005).
Outre les facteurs environnementaux, les facteurs génétiques comme les insertions, les
délétions, les réarrangements chromosomiques et les polymorphismes peuvent altérer la
composition en acides aminés, l’épissage et l’expression de gènes et ainsi modifier la
pharmacocinétique et la pharmacodynamique de ce médicament (Figure 3).

Revue bibliographique
34
Gène CES
Les gènes codant pour les CES (carboxylestérases) sont localisés en 16q13-q22 et présentent
une forte homologie, faisant évoquer un gène ancestral commun. Il existe trois
isoenzymes humaines connues: la CES 1, CES 2 et CES 3. CES2 a une action catalytique sur
l’Irinotécan 100 et 2000 fois plus importante que CES 1 et CES 3 respectivement (Sanghani et
al, 2004). Il a été mis en évidence plus de onze polymorphismes sur la CES 2 avec des
distributions variables en fonction de l'ethnie (Charasson, 2005; Ceppa, 2005). Cependant, si
des variations dans l'expression de l'ARNm de cette enzyme ont été corrélées avec certains
polymorphismes, les études pharmacogénétiques n’ont pas encore aboutit à un consensus sur
l’association des SNPs de ce gène avec le taux de métabolisme de l'Irinotécan (Wu et al,
2003; Kubo et al, 2005). En revanche, un épissage alternatif affectant l’exon 10 de ce gène
aboutissant à une protéine tronquée de 16 acides aminés dans le site actif de CES 2 a
récemment été corrélé à une baisse d’activité importante de plus de 80% de cette enzyme
(Schiel et al, 2007).
Figure 3 : Métabolisme de l’Irinotécan
L’Irinotécan est converti par la carboxylestérase (CES) en son métabolite actif, le SN 38.
Ce métabolite est détoxifié au niveau hépatique par l’uridine diphosphate glucuronosyl-
transférase 1A1 (UGT1A1) en glucurono-SN38 « SN38G» (De Chaisemartin et al, 2005).

Revue bibliographique
35
Gène UGT1A1
L’UGT1A1 (Uridine diphosphate glucuronosyltransferase 1 family, polypeptide A1) codée
par le gène UGT1A1 sur le chromosome 2q37.1 est un membre de la famille des UGT,
enzymes du métabolisme de phase II, responsables de la détoxification de nombreux
xénobiotiques, mais également de certaines substances endogènes. Le processus de
glucurono-conjugaison consiste en l’ajout d’acide glucuronique à des composés liposolubles
(Kaderlik et al, 1994). Cela permet d’augmenter leur solubilité et leur poids moléculaire, ce
qui empêche leur réabsorption dans les tissus et favorise l’élimination de ces composés dans
la bile ou l’urine. Les UGT sont situées dans la membrane cellulaire au niveau du réticulum
endoplasmique. Ces protéines sont exprimées dans à peu près tous les tissus, mais le sont
majoritairement dans le système hépatique, rénal, gastro-intestinal et cérébral.
L’UGT1A1 est physiologiquement responsable de la glucurono-conjugaison de la bilirubine
mais aussi de nombreux autres xénobiotiques dont l’Irinotécan. Son intervention dans le
métabolisme de ce dernier consiste à catalyser la réaction de glucurono-conjugaison du SN-38
le rendant ainsi inactif et atoxique avant d'être éliminé par la bile et les urines.
Il existe au niveau de la séquence TATA box du gène UGT1A1 un VNTR (variable number
of tandem repeat) UGT1A1*28(TA) 6>(TA)7 à la position -53, consistant en une répétition
d’un dinucléotide (TA) qui influence l’activité transcriptionnelle du promoteur, l’allèle
sauvage UGT1A1*1 correspondant à 6 répétitions. L’allèle comprenant sept répétitions du
dinucléotide suite à une micro-insertion (allèle TA7 ou UGT1A1*28) est associé à plus de
70% de réduction de son expression. Le génotype homozygote UGT1A1*28/*28 est d’ailleurs
une des causes de la maladie de Gilbert, hyper bilirubinémie modérée chronique assez
fréquente (10 à 15 % de la population européenne) (Bosma et al, 1995). Plusieurs études ont
démontré que le génotype UGT1A1*28/*28 est associé à une toxicité accrue de l’Irinotécan,
aussi bien pour sa toxicité hématologique que digestive (Kweekel et al, 2008). En effet, les
patients UGT1A1*28/*28 auraient un risque 9,3 fois plus important de développer une
neutropénie de grade IV que les autres génotypes (Innocenti et al, 2004). Il existe d’autres
polymorphismes candidats présents au niveau du promoteur de ce gène; il s’agit de
UGT1A1-3279G>T et UGT1A1-3156G>A (Innocenti et al, 2002; Zhou Q et al, 2005) et il a
été suggéré par l’équipe d’Innocenti que le SNP UGT1A1-3156G>A serait plus prédictif du

Revue bibliographique
36
statut fonctionnel de l’enzyme UGT1A1 que le VNTR UGT1A1*28(TA) 6>(TA)7 (Innocenti
et al, 2004).
4 PHARMACOGENETIQUE DE L’OXALIPLATINE
L'oxaliplatine est un médicament anti-cancéreux de synthèse du groupe des agents dérivés de
platine. Son action passe par l’inhibition de la synthèse et de la réplication de l’ADN par
formation de ponts intrabrins entre 2 guanines adjacentes par leur site N7 (Kenji Inagaki et al,
1984). La première étape du métabolisme de l'oxaliplatine consiste en la libération d'oxalate
qui est remplacé par 2 ions chlore aboutissant ainsi à la formation du dichlorodachplatine
(dichloro-diaminocyclohexane platinum) (Hagler et al. 1973) et une intoxication aiguë par de
l'oxalate se traduit par des paresthésies des extrémités, des dysesthésies exacerbées par
l'exposition au froid et des myoclonies, pouvant aller jusqu'à des convulsions en cas
d'intoxication massive (Argyriou et al, 2008).
Gène GSTP1
En plus de son rôle potentiel évoqué précédemment dans la survenue du CCR (Sachse et al,
2002; Harris et al, 1998), l’isoenzyme GSTP1 participe à la détoxication des dérivés platine
de l’oxaliplatine dans le système nerveux périphérique et les tumeurs résistantes à
l'oxaliplatine présenteraient une augmentation de l'expression de cette enzyme (Stoehlmacher,
2002). Le polymorphisme de GSTP1 correspondant à la transition 313A>G (Ileu105Val) sur
l'exon 5 (voir tableau 1) provoque une baisse de l'activité de l'enzyme et par conséquent
devrait permettre une meilleure efficacité de la thérapie. Cette hypothèse est confirmée par
l'étude de l’équipe de Stoehlmacher chez 107 patients atteints de cancer colorectal métastasé
traités par oxaliplatine et 5-FU (Stoehlmacher et al, 2002). Paradoxalement, une étude récente
a retrouvé une association entre le variant 105Ile et le risque de neuropathie chez des patients
atteints de cancer gastro-intestinal suggérant de ce fait un effet protecteur de l’allèle variant
105Val par rapport à la toxicité de l’oxaliplatine (Lecomte et al, 2006). Le rôle des
polymorphismes du gène GSTP1 sur le métabolisme de l’oxaliplatine reste donc à déterminer.

Population et méthodes
37
BUT DE L’ETUDE
Les xénobiotiques sont des molécules à faible poids moléculaire, étrangères à l’organisme
dont font partie les médicaments, les polluants de l’eau ou de l’atmosphère, les additifs
alimentaires mais également certains composés naturels des aliments. Beaucoup de ces
molécules sont potentiellement carcinogènes et font l’objet d’une métabolisation lorsqu’elles
arrivent dans l’organisme. Ces processus de biotransformation surviennent essentiellement au
niveau du foie, mais d’autres tissus comme le colon ou le tube digestif sont également
susceptibles de métaboliser ces xénobiotiques. Les processus mis en route sont généralement
multi étapes et font appel à des réactions préliminaires d’oxydation pour rendre les molécules
plus polaires (réaction de phase I), à des réactions de conjugaison avec un ligand comme les
sulfates, l’acide glucuronique, l’acétate ou le glutathion (réactions de phase II) et enfin au
transport de ces molécules (phase III) qui permet l’élimination de ces conjugués hydrophiles
grâce à des glycoprotéines membranaires.
Lors de la phase I, une fonctionnalisation des xénobiotiques est catalysée par certaines
enzymes dont font partie les cytochromes P450 qui catalysent une réaction de mono
oxygénation et peuvent transformer ces xénobiotiques en métabolites très électrophiles, voire
génotoxiques. Parmi ces enzymes, CYP3A5 est exprimée en grande quantité dans le tractus
digestif et son activité enzymatique est impliquée dans l’activation de plusieurs
procarcinogènes comme les nitrosamines, les hydrocarbures aromatiques polycycliques, et les
amines hétérocycliques. Le SNP CYP3A5 6986A>G aboutit à des changements fonctionnels
de cette enzyme et pourrait avoir un impact sur la survenue du CCR. Nous avons donc choisit
de l’explorer afin de rechercher une éventuelle association avec le risque du développement
du CCR d’une part et d’autre part pour déterminer la distribution de sa fréquence dans notre
population.
Pour les enzymes de la phase II du métabolisme des xénobiotiques, notre choix s’est porté sur
les gènes codant pour la famille des enzymes GST. Ces enzymes occupent une place très
importante dans le système de défense cellulaire de part leur rôle de conjugaison du glutathion
réduit à des composés électrophiles nocifs permettant la détoxication des xénobiotiques et

Population et méthodes
38
l’activation de certains substrats. Celles ci pourraient être également impliquées dans la
toxicité à certains agents anticancéreux comme l’oxaliplatine Etant donné ce rôle capital,
plusieurs études épidémiologiques ont examiné l'incidence de leurs polymorphismes
génétiques sur le risque de survenue et/ou d'aggravation de nombreuses pathologies comme le
CCR et les conclusions de ces études ne convergent pas toujours. Nous avons voulu examiner
cette relation d’association avec le risque d’émergence du CCR dans notre population. Pour
cela, nous avons choisit d’analyser 4 polymorphismes génétiques différents et de comparer la
distribution de leurs fréquences respectives entre les cas et les contrôles à la recherche d’une
éventuelle association avec un risque majoré de CCR ; il s’agit d’une part, de la délétion totale
des gènes GSTM1 et GSTT1 qui, à l'état homozygote, est associée à une absence complète
d'activité enzymatique et d’autre part, des deux SNPs 313A>G et 341C>T sur GSTP1
impliqués dans la diminution de l’activité enzymatique de GSTP1. De nombreux autres
cancers ont été associés à des polymorphismes des GST, l’évaluation des fréquences de ces
derniers dans la population Algérienne pourrait donc être utilisée dans d’autres études
cas/témoins concernant ces pathologies.
Un autre exemple de polymorphisme portant sur une transférase de la phase II est celui de
UGT1A1 (UDP-glucuronosyltransférase) dont le rôle est de catalyser une réaction de
glucorono-conjugaison des xénobiotiques afin de les rendre plus hydrophiles et faciliter leur
élimination. Concernant ce gène, nous avons recherché une éventuelle association entre les
polymorphismes UGT1A1-3156G >A et le VNTR UGT1A1*28 (TA)6>(TA)7 à la position
-53 et la survenue du CCR sur notre population car ces SNPs induisent une baisse de
transcription de cette enzyme et de ce fait une plus faible activité enzymatique. Par
conséquent, elles peuvent modifier le métabolisme de certains composés carcinogènes. De
plus, plusieurs études ont retrouvé une association de ces polymorphismes avec la toxicité à
l’Irinotécan. Cet anticancéreux étant largement utilisé dans la chimiothérapie du CCR en
Algérie, la connaissance des fréquences de ces SNPs dans notre population serait intéressante
pour évaluer d’une manière globale l’éventuel effet toxique de cette molécule dans notre
contexte. La phase III d’excrétion des xénobiotiques hors de la cellule est assurée par la
protéine P-gp codée par le gène MDR1 « Multidrug Resistance ». Cette protéine, en modulant
les concentrations plasmatiques et intracellulaires des xénobiotiques, dont certaines molécules
carcinogènes, pourrait influencer la susceptibilité au CCR mais aussi la réponse au traitement.

Population et méthodes
39
Dans cette étude nous avons exploré le SNP MDR1 3435C>T de ce gène car il est
responsable d’une baisse significative de l’expression de la protéine P-gp et serait également
un facteur de risque important dans la survenue de certains cancers dont le CCR.
L’exploration du polymorphisme du gène MDR1 a été entreprise également à cause du rôle
primordial de cette protéine dans le phénomène de résistance thérapeutique.
Pour finir, nous nous sommes intéressés à deux polymorphismes du gène codant pour la
thymidylate synthétase à cause du rôle incontournable de cette enzyme dans le métabolisme
des pyrimidines. En effet celle-ci catalyse la réaction de conversion du désoxyuridine
monophosphate en désoxythymidine monophosphate et les deux polymorphismes explorés
dans notre étude ont été incriminés dans la modulation du taux d’expression de cette enzyme.
Il s’agit du polymorphisme de répétition 2R>3R dans la région 5’UTR d’une séquence de
28pb et la délétion/insertion de 6bp en position 1494 dans sa région 3'UTR.
Notre choix de mener une étude cas/témoins sur ces deux polymorphismes de la TS a été
guidé par l’hypothèse qu’une plus faible expression de la TS due à un de ces deux
polymorphismes réduisant le taux de thymidine, pourrait augmenter le risque d’incorporation
de l’uracile au lieu de la thymine dans l’ADN, ce qui pourrait accroitre le risque de cassure de
l’ADN double brin dans les cellules à prolifération rapide comme les cellules du colon.
Par ailleurs, la principale cible du 5-FU, un anticancéreux largement utilisé dans la
chimiothérapie du CCR étant la thymidylate synthétase, de nombreuses études ont été
réalisées pour évaluer l’utilité de l’expression de cette enzyme en tant que marqueur
biologique de réponse et/ou de toxicité au traitement à base de 5-FU. L’exploration de ces
deux polymorphismes sur notre population est de ce fait d’une grande importance pour
retrouver les patients capables d’une réponse optimale à cet anticancéreux en Algérie.
Au delà du but initial de cette première étude en Algérie consistant à rechercher une
éventuelle association entre les polymorphismes des gènes codant pour les enzymes du
métabolisme des xénobiotiques et la survenue du CCR, l’évaluation de la distribution des
fréquences de ces variants dans notre population pourrait servir à envisager d’autres études
cas/contrôles concernant d’autres pathologies dans lesquelles les fonctions de ces enzymes
sont impliquées.

Population et méthodes
40
V POPULATION ET METHODES
1 POPULATION D’ETUDE
Notre travail a été réalisé sur une population de 200 individus dont 101 sujets sains non
apparentés et ne présentant aucune maladie chronique et 99 malades atteints de cancer
colorectal admis au service d’oncologie du CHU d’Oran et âgés entre 20 et 75ans. Tous les
individus recrutés dans cette étude sont originaires de l’Ouest Algérien.
2 GENES EXPLORES
2.1 GENE CYP3A5: SNP 6986A>G
L’hypothèse qui justifie la présente étude est basée sur le fait que l’enzyme CYP3A5 est
exprimée en grande quantité dans le tractus digestif et que son activité enzymatique est
impliquée dans l’activation de plusieurs procarcinogènes dont certaines mycotoxines comme
l’aflatoxine B1 mais aussi les nitrosamines, les hydrocarbures aromatiques polycycliques, et
les amines hétérocycliques produits au cours de la cuisson des aliments (Parke, 1994;
Windmill et al, 1997).
Le SNP CYP3A5 6986A>G (dbSNP Id : rs776746) fait partie des polymorphismes
récemment découverts qui aboutissent à des changements fonctionnels de cette enzyme (Van
Schaik et al, 2002). Ce SNP donne naissance à une protéine tronquée, et nous l’avons exploré
afin de définir la distribution de sa fréquence dans notre population d’une part et d’autre part
pour rechercher une éventuelle association avec le risque du développement du CCR.

Population et méthodes
41
2.2 GENES GST : DELETION DE GSTM1, DELETION DE GSTT1 ET SNPS : GSTP1 313A>G
ET GSTP1 341C>T
Pour cette classe de gène nous avons choisis d’analyser 4 polymorphismes génétiques
différents : d’une part, les délétions totales des gènes GSTM1 et GSTT1 qui, à l'état
homozygote, sont corrélées à une absence complète d'activité enzymatique et d’autre part,
deux SNPs sur GSTP1 : une substitution 313A>G (Ileu105Val) sur l’exon 5 pour le premier
(dbSNP Id : rs1695) et une substitution 341C>T (Ala114Val) sur l’exon 6 pour le second
(dbSNP Id : rs1138272) aboutissant toutes deux à une diminution de l’activité enzymatique
de GSTP1.
Les polymorphismes de ces trois gènes sont largement étudiés par rapport à leur fonction dans
la détoxification des xénobiotiques dont certaines molécules carcinogènes présentes dans
l’alimentation ou dans la fumée de tabac et leurs implications possibles dans la susceptibilité
génétique à développer plusieurs cancers dont le CCR (Chenevix-Trench et 1995; Deakin et
al, 1996; Harris et al, 1998; Sachse et al, 2002). Enfin, ces gènes sont explorés pour leur rôle
dans la détermination génétique individuelle de la réponse aux molécules anticancéreuses
comme l’oxaliplatine utilisée dans la chimiothérapie du CCR (Stoehlmacher et al, 2002;
Lecomte et al, 2006).
2.3 GENE UGT1A1: LE VNTR UGT1A1*28(TA)6>(TA)7 ET LE SNP -3156 G>A
Dans cette étude nous avons recherché une éventuelle association entre les polymorphismes
UGT1A1-3156G >A (dbSNP Id : rs10929302) et le VNTR UGT1A1*28 (TA)6>(TA)7
(dbSNP Id : rs8175347) à la position -53 et la survenue du CCR sur notre population car ces
SNPs ont pour conséquence une baisse de transcription de cette enzyme et de ce fait une plus
faible activité enzymatique. Par conséquent, elles peuvent modifier le métabolisme de certains
composés carcinogènes présents dans l’alimentation. De plus, plusieurs études ont retrouvé
une association des polymorphismes UGT1A1-3156G >A et UGT1A1*28 (TA)6>(TA)7
avec la toxicité à l’Irinotécan (Innocenti et al, 2004; Kweekel et al, 2008). Cet anticancéreux
étant largement utilisé dans la chimiothérapie du CCR en Algérie, la connaissance des

Population et méthodes
42
fréquences de ces SNPs dans notre population serait intéressante pour évaluer d’une manière
globale l’éventuel effet toxique de cette molécule dans notre contexte.
2.4 GENE MDR : SNP 3435C>T
Un seul SNP de ce gène, le SNP MDR1 3435C>T (dbSNP Id : rs1045642) a été exploré dans
cette étude car il est responsable d’une baisse significative de l’expression de la protéine P-gp.
Cette dernière, de part son rôle dans le transport des xénobiotiques dont les médicaments
anticancéreux mais aussi les substances carcinogènes, est en première ligne dans le
phénomène de résistance thérapeutique (Sekine et al, 2001; Nagasubriamanian et al, 2003).
Ce SNP serait également un facteur de risque important dans la survenue de certains cancers
dont le CCR (Osswald et al, 2007). De plus la fréquence de ce SNP diffère selon les ethnies,
et l’évaluation de sa fréquence sur la population Algérienne est encore inconnue.
2.5 GENE TS: REPETITION 2R>3R DANS LA REGION 5’UTR DU GENE TS ET
DELETION/INSERTION DE 6PB DANS SA REGION 3’UTR
Pour ce gène nous avons exploré deux polymorphismes : Le polymorphisme de répétition
2R>3R dans la région 5’UTR d’une séquence de 28pb (dbSNP Id : rs34743033) et la
délétion/insertion de 6bp dans la région 3'UTR (dbSNP Id : rs34489327).
Concernant le polymorphisme de répétition 2R>3R en 5’UTR, le génotype 3R/3R a été
associé à une augmentation du taux d’expression de la TS et à une activité enzymatique plus
importante (Kawakami et al, 1999). De plus, l’allèle 3R confère une efficacité de traduction
de la TS plus élevée que l’allèle 2R (Horie et al, 1995) et une étude a montré que le taux
d’ARNm de TS dans les tissus tumoraux était 3 fois plus bas chez les sujets homozygotes
2R/2R que chez les sujets homozygotes 3R/3R (Pullarkat et al, 2001). Par ailleurs, la délétion
de 6pb dans la région 3’UTR de la TS a été corrélée également à un taux d’expression plus
faible d’ARNm dans les tumeurs colorectales due une à augmentation du taux d’instabilité de
celui-ci (Mandola et al, 2004).

Population et méthodes
43
Notre choix de mener une étude cas/témoins sur ces deux polymorphismes de la TS a été
guidé par l’hypothèse qu’une plus faible expression de la TS due à un de ces deux
polymorphismes réduisant la conversion du dUMP en dTMP pourrait augmenter le risque
d’incorporation de l’uracile au lieu de la thymine dans l’ADN, ce qui pourrait accroitre le
risque de cassure de l’ADN double brin dans les cellules à prolifération rapide comme les
cellules du colon.
Par ailleurs, la principale cible du 5-FU étant la thymidylate synthétase, de nombreuses études
ont été réalisées pour évaluer l’utilité de l’expression de la TS en tant que marqueur
biologique de réponse au traitement à base de 5-FU. L’exploration de ces deux
polymorphismes sur notre population est de ce fait d’une grande importance pour retrouver
les patients capable d’une réponse optimale à cet anticancéreux largement utilisé dans la
chimiothérapie du CCR en Algérie.
3 METHODES D’EXPLORATION GENETIQUE
3.1 EXTRACTION ET DOSAGE DE L’ADN
L’ADN a été extrait à partir de 10 à 20 ml de sang total par la technique aux sels (salting out).
Elle est basée sur le traitement des lysats de cellules blanches par une solution saline associée
à la protéinase K (enzyme protéolytique) (Miller et al, 1988).
L’estimation de la concentration de l’ADN se fait par la mesure de l’absorbance à 260nm.
Sachant qu’une unité de DO à 260nm est équivalente à 50 µg /ml d’ADN, il est possible
d’évaluer la quantité d’ADN d’un échantillon par la formule: facteur de dilution x 50 x DO260.
Un ADN de bonne qualité (sans contamination par les protéines) présente un rapport
DO260/DO280 compris entre 1,5 et 2.

Population et méthodes
44
3.2 EXPLORATION DE LA DELETION DES GENES GSTT1, GSTM1 ET DU SNP GSTP1
313A>G PAR PCR MULTIPLEX, DIGESTION ENZYMATIQUE ET ANALYSE DES RFLP.
Nous avons procédé à un génotypage simultané des sujets pour les trois gènes GST dans un
même tube en réalisant une PCR multiplex (polymerase chain reaction) (Lecomte et al, 2006)
avec des paires d’amorces spécifiques à chacun des gènes (Nedelcheva Kristensen et al, 1998)
(tableau 2).
La PCR a été réalisée dans un volume final de 25µl avec un programme PCR de 30 cycles en
tout avec un « Touch Down » de 68° à 48° consistant à diminuer la température de 1° par
cycle sur 20cycles. Programme de la PCR : 1 cycle de 5min à 94°C suivi de 30sec à 94°C,
30sec de 68°C à 48°C et 30sec à 72°C pendant 20 cycles; 30sec à 94°C, 30sec à 51°C et
30sec à 72°C pendant 10 cycles et enfin 10 min à 72°C.
20µl des produits PCR ont été soumis à une digestion enzymatique par BsmA1 (New England
Biolabs, Saint Quentin en Yvelines, France) suivie d’une analyse des RFLP (restriction
fragment lenght polymorphism) (Lecomte et al, 2006).
Les résultats étaient validés en utilisant le fragment PCR de GSTP1 comme témoin
d’amplification.
Tableau 2 : Séquences des amorces utilisées pour la PCR multiplex des gènes GSTT1, GSTM1, et GSTP1
313A>G (Nedelcheva Kristensen et al, 1998).
Gènes GST Séquences des amorces
GSTT1 Amorce F : TTCCTTACTGGTCCTCACATCTC
Amorce R : TCACCGGATCATGGCCAGCA
GSTM1 Amorce F : GGACTCCCTGAAAAGCTAAAGC
Amorce R : GTTGGGCTCAAATATACGGTGG
GSTP1 313A>G
rs1695
Amorce F : ACCCCAGGGCTCTATGGGAA
Amorce R : TGAGGGCACAAGAAGCCCCT
Population et méthodes
45
Seul le fragment de 176pb de GSTP1 est digéré en deux fragments de 91pb et 85pb lorsqu’on
est en présence de GSTP1 105Val et non digéré en présence de l’allèle 105Ile. La taille des
fragments générés des produits PCR étaient de 450pb pour GSTT1, 250pb pour GSTM1 et
176pb pour GSTP1. La présence du gène GSTM1 ou du gène GSTT1 donne un signal PCR
positif tandis que la délétion ne donne aucun signal. Les produits de digestion sont alors
révélés par électrophorèse sur gel de polyacrylamide à 8%.
3.3 ANALYSE DES SNPS : GSTP1 341C>T, MDR1 3435C>T, CYP3A5 6986A>G ET
UGT1A1 -3156 G>A PAR PCR QUANTITATIVE EN TEMPS REEL.
Les SNPs GSTP1 341C>T, MDR1 3435C>T, UGT1A1-3156 G>A et CYP3A5 6986A>G ont
été détectés au moyen d’une analyse par discrimination allèlique par PCR quantitative en
temps réel (Taqman, ABI PRISM® 7900 Sequence Detection System Applied Biosystems,
Foster City, CA) (Livak et al, 1995) en adaptant la chimie et les conditions recommandées par
la biotechnologie Applied Biosystems (http://snp500cancer.nci.nih.gov/taqman_assays, mise
à jour du 03/12/2008) pour le génotypage de chacun de ces SNPs (Tableau3).
Tableau 3 : Séquences des amorces et des sondes Taqman utilisées pour l’analyse des SNPs : GSTP1 341C>T,
MDR1 3435C>T, CYP3A5 6986A>G et UGT1A1 -3156 G>A
SNP Séquences des amorces Sonde VIC Sonde FAM
GSTP1 341C>T
rs1138272
Amorce F: AGTAGGATGATACATGGTGGTGTCT
Amorce R: GGCAGTGCCTTCACATAGTCAT
CTTGCCCGCCTCCT CTTGCCCACCTCCT
MDR1 3435C>T
rs1045642
Amorce F: CTGTTTGACTGCAGCATTGCT
Amorce R: ATGTATGTTGGCCTCCTTTGCT
CCCTCACGATCTCTT CCCTCACAATCTCTT
UGT1A1-3156G>A
rs10929302
Amorce F: TCCAGAATGGCTAGAGGGTAAGAG
Amorce R: CTTGCTCTCAAAACTCTGGGATAGA
CCTGTCCAAGCTCA CCTGTCTAAGCTCA
CYP3A5 6986A>G
rs776746
Amorce F: CGAATGCTCTACTGTCATTTCTAACCA
Amorce R: TGAAGGGTAATGTGGTCCAAACAG
AGAGAGACTGAAAG AGAGAGATTGAAAG

Population et méthodes
46
La PCR quantitative en temps réel est basée sur une réaction enzymologique, la PCR, et sur la
mesure en continu de son produit. A chaque cycle d’amplification, la quantité d’ADN total ou
d’amplicon est mesurée grâce à un marqueur fluorescent. L’obtention de la cinétique
complète de la réaction de polymérisation permet d’obtenir une quantification absolue de la
quantité initiale d’ADN cible (Tse et al, 2003).
Les sondes d’hydrolyse « Taqman » sont les sondes les plus fréquement utilisées pour la
détection de mutations ponctuelles. Elles sont marquées par deux fluorophores : un à leur
extrêmité 5’ (fluorophore donneur ou émetteur) et l’autre à leur extrêmité 3’ (fluorophore
extincteur ou « Quencher »). Le Quencher à proximité du donneur inhibe l’émission de
fluorescence par ce dernier. Ces sondes sont dessinées de manière à s’hybrider spécifiquement
à leur cible (Figure 4).
Figure 4: Principe de la technique Taqman
P1 et P2 sont les amorces utilisées pour la PCR. La sonde est marquée à son extrêmité 5’ par un fluorophore (F)
par exemple le fluorophore FAM et à son exrêmité 3’ par le Quencher (Q), par exemple le Quencher TAMRA.
Pendant la réaction de PCR, la sonde se fixe à la cible. La Taq polymérase, par son activité nucléasique 5’clive
la sonde et libère la molécule de fluorescence FAM. Schéma tiré du livre : Principes de biologie moléculaire en
biologie clinique (Ameziane et al 2006).

Population et méthodes
47
Pour notre étude, les sondes spécifiques utilisées ont été marquées à leur extrémité 5’ par les
fluorophores FAM et VIC (Absolute Blue QPCR ROX Mix – Abgene ref AB- 4138/B)
respectivement pour l’allèle 1 et l’allèle 2 de chacun des SNPs explorés.
Pour chacun des SNPs explorés, l’analyse a été réalisée en utilisant 1,5µl d’ADN à 25ng dans
un volume réactionnel Taqman final de 5µl (5’nuclease Assay), composé de 2.5 ul du tampon
« Taqman Genotyping Master Mix » (Applied Biosystems, Foster City, CA) à 2X et du
mélange « sonde assay-specific » contenant les amorces spécifiques à chaque SNP et les
sondes VIC et FAM à des concentrations définies par le biosystème d’analyse « Applied
Biosystems » . Ces réactifs sont commandés en fonction de la référence NCBI « rs » (National
Center for Biotechnology Information Reference) de chacun des SNPs et les conditions
« Taqman » appliquées sont celles recommandées par le fabricant (Applied Biosystems)
(http://snp500cancer.nci.nih.gov/taqman_assays, mise à jour du 03/12/2008)
Les réactions ont été faites sur une plaque de 384 puits. Et des contrôles homozygotes
sauvages, homozygotes mutés et hétérozygotes correspondant à chacun des SNPs ont été
ajoutés à la plaque. On obtient donc deux fluorescences pour un hétérozygote et une seule
(VIC ou FAM) pour un homozygote. La figure 5 montre un exemple de SNP où l’allèle
sauvage est C et l’allèle muté est T.
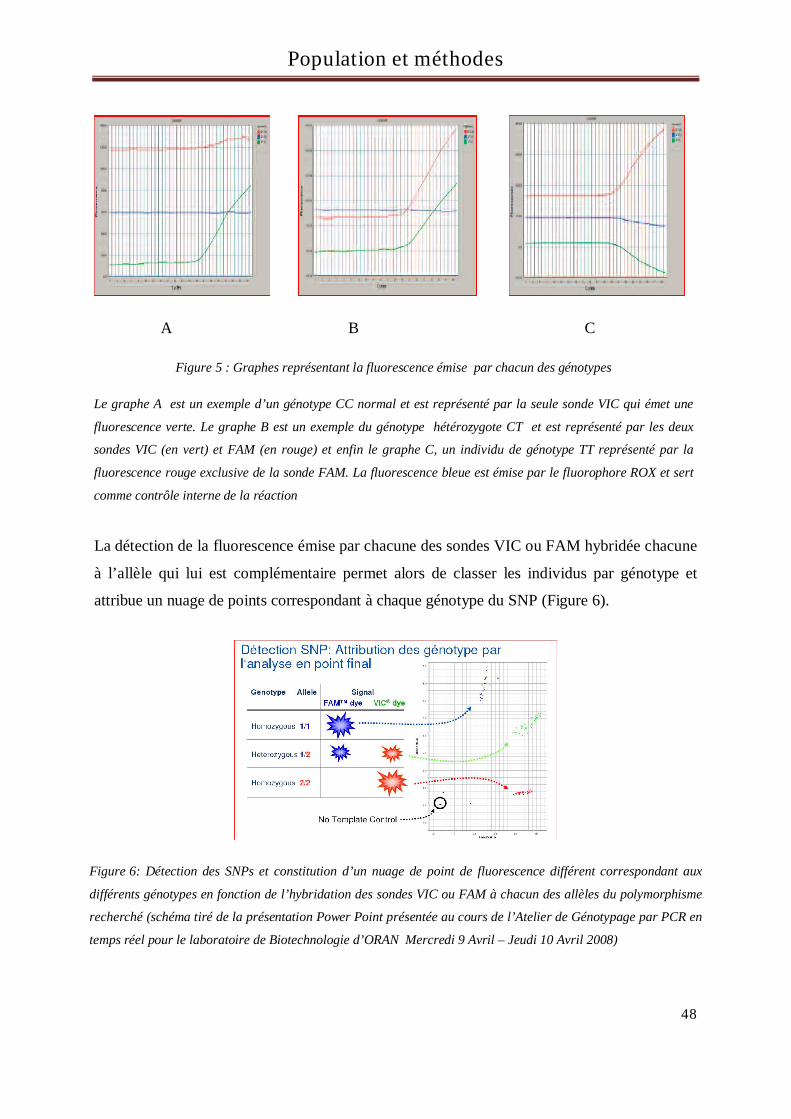
Population et méthodes
48
La détection de la fluorescence émise par chacune des sondes VIC ou FAM hybridée chacune
à l’allèle qui lui est complémentaire permet alors de classer les individus par génotype et
attribue un nuage de points correspondant à chaque génotype du SNP (Figure 6).
A B C
Figure 5 : Graphes représentant la fluorescence émise par chacun des génotypes
Le graphe A est un exemple d’un génotype CC normal et est représenté par la seule sonde VIC qui émet une
fluorescence verte. Le graphe B est un exemple du génotype hétérozygote CT et est représenté par les deux
sondes VIC (en vert) et FAM (en rouge) et enfin le graphe C, un individu de génotype TT représenté par la
fluorescence rouge exclusive de la sonde FAM. La fluorescence bleue est émise par le fluorophore ROX et sert
comme contrôle interne de la réaction
Figure 6: Détection des SNPs et constitution d’un nuage de point de fluorescence différent correspondant aux
différents génotypes en fonction de l’hybridation des sondes VIC ou FAM à chacun des allèles du polymorphisme
recherché (schéma tiré de la présentation Power Point présentée au cours de l’Atelier de Génotypage par PCR en
temps réel pour le laboratoire de Biotechnologie d’ORAN Mercredi 9 Avril – Jeudi 10 Avril 2008)
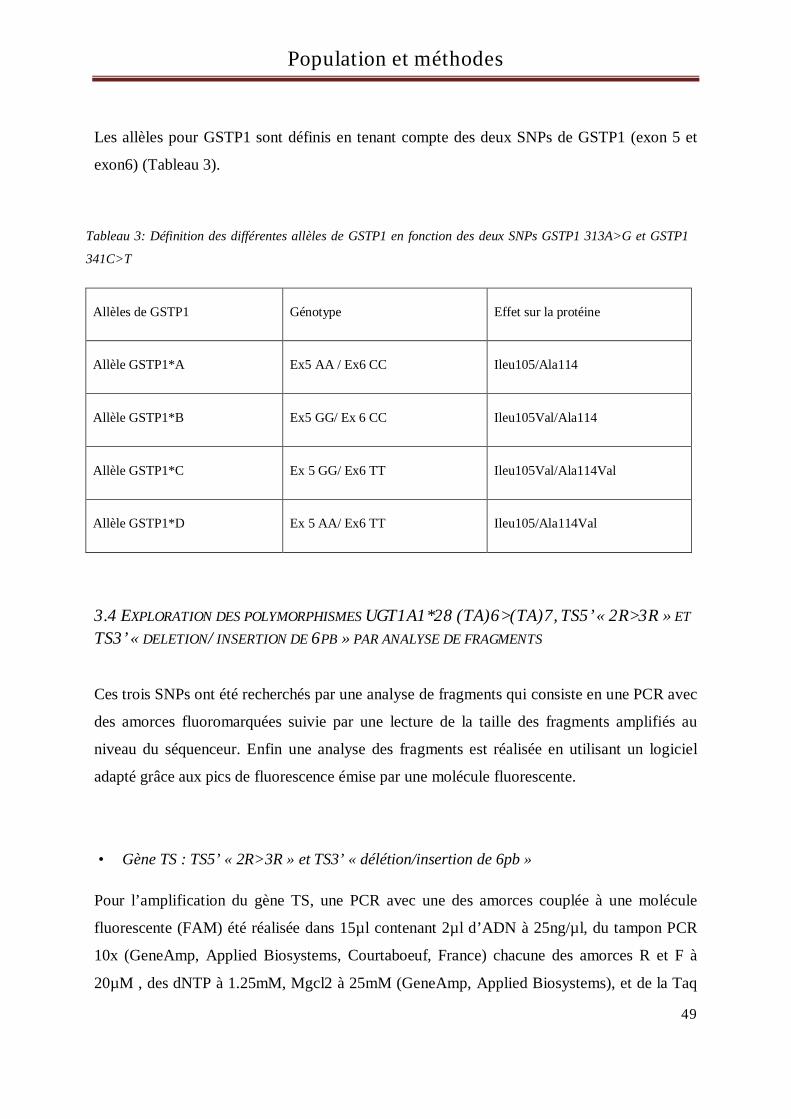
Population et méthodes
49
Les allèles pour GSTP1 sont définis en tenant compte des deux SNPs de GSTP1 (exon 5 et
exon6) (Tableau 3).
3.4 EXPLORATION DES POLYMORPHISMES UGT1A1*28 (TA)6>(TA)7, TS5’ « 2R>3R » ET
TS3’ « DELETION/INSERTION DE 6PB » PAR ANALYSE DE FRAGMENTS
Ces trois SNPs ont été recherchés par une analyse de fragments qui consiste en une PCR avec
des amorces fluoromarquées suivie par une lecture de la taille des fragments amplifiés au
niveau du séquenceur. Enfin une analyse des fragments est réalisée en utilisant un logiciel
adapté grâce aux pics de fluorescence émise par une molécule fluorescente.
• Gène TS : TS5’ « 2R>3R » et TS3’ « délétion/insertion de 6pb »
Pour l’amplification du gène TS, une PCR avec une des amorces couplée à une molécule
fluorescente (FAM) été réalisée dans 15µl contenant 2µl d’ADN à 25ng/µl, du tampon PCR
10x (GeneAmp, Applied Biosystems, Courtaboeuf, France) chacune des amorces R et F à
20µM , des dNTP à 1.25mM, Mgcl2 à 25mM (GeneAmp, Applied Biosystems), et de la Taq
Tableau 3: Définition des différentes allèles de GSTP1 en fonction des deux SNPs GSTP1 313A>G et GSTP1
341C>T
Allèles de GSTP1 Génotype Effet sur la protéine
Allèle GSTP1*A Ex5 AA / Ex6 CC Ileu105/Ala114
Allèle GSTP1*B Ex5 GG/ Ex 6 CC Ileu105Val/Ala114
Allèle GSTP1*C Ex 5 GG/ Ex6 TT Ileu105Val/Ala114Val
Allèle GSTP1*D Ex 5 AA/ Ex6 TT Ileu105/Ala114Val

Population et méthodes
50
Quiagen à U/µl en suivant un programme constitué de 15min à 95°, 30 cycles de 30sec à
94°C, 30sec à 55°C ou 62°C pour TS 5’ et TS 3’ respectivement 30sec à 72°C et enfin 6min
à 72°C.
• UGT1A1*28 (TA)6>(TA)7
Pour l’amplification de UGT1A1, une PCR avec une des amorces couplée à une molécule
fluorescente « FAM » a été réalisée dans 19µl contenant 2µl d’ADN à 25ng/µl, du tampon
PCR 10x (GeneAmp, Applied Biosystems, Courtaboeuf, France), chacune des amorces R et F
à 20µM, des dNTPs à 1.25mM, Mgcl2 à 25mM (GeneAmp, Applied Biosystems), et de la
Taq Gold polymérase à 5U/µl (Applied - N808-0243 ); le programme de la PCR était
constitué de 10min à 94°, 10 cycles de 30sec à 94°C, 45sec à 60°C et 1min à 72° ; 24 cycles
de 30sec à 94°, 45sec à 58°, 1min à 72° et enfin 7min à 72°C.
Un marqueur de taille « Genescan 400HD ROX » (Applied 402985) et un agent dénaturant :
Hi-Di Formamide (Applied 4311320) sont ajoutés aux produits PCR obtenus (tableau 4).
Tableau 4 : Séquences des amorces utilisées pour l’analyse des polymorphismes UGT1A1*28
(TA)6>(TA)7, TS5’ « 2R>3R » et TS3’ « délétion/insertion de 6pb ».
Gène Amorces
TS5’ « 2R>3R »
rs34743033
Amorce F : 5’(FAM) GTGGCTCCTGCGTTTCCCCC 3’
Amorce R: 5’ GCTCCGAGCCGGCCACAGGCAT 3’
TS3’ « délétion/insertion de 6pb »
rs34489327
Amorce F : 5’GACGAATGCAGAACACTTCT 3’
Amorce R : 5’(FAM) AATCTGAGGGAGCTGAGTAAC 3’
UGT1A1*28 (TA)6 >(TA)7
rs8175347
Amorce F : 5’ GCTCCACCTTCTTTATCTCTG 3’
Amorce R : 5’(FAM) CAGCATGGGACACCACTG 3’

Population et méthodes
51
Les produits PCR fluorescents ont été distribués en colonne sur une plaque « MicroAmp
Optical » (Applied N801-0560) pour être séparés par électrophorèse capillaire sur séquenceur
(ABI PRISM 3700 DNA Analyser - Applied Biosystems) et analysés grâce aux logiciel
« GENEMAPPER ».
Pour TS 5’, on attend une bande à 214 pb pour les homozygotes sauvages « 2R/2R » et une
autre à 248 s’il ya répétition de la séquence de 28pb pour les hétérozygotes « 2R/ 3R ». Les
homozygotes « 3R/3R » ont une seule bande à 248pb.
Pour TS 3’, on attend un fragment de 107pb pour les homozygotes sauvages pour la délétion
de 6pb et un autre à 107+ 6 bases s’il ya insertion de 6pb.
Pour le SNP UGT1A1*28 (TA)6>(TA)7 à la position -53, on attend un fragment à 249pb
pour les homozygotes sauvages « UGT1A1*1/*1 », un autre à 251pb pour les homozygotes
mutés « UGT1A1*28/*28 » et deux fragments à 249pb et 251pb pour les hétérozygotes
« UGT1A1*1/*28 ».

Population et méthodes
52
4 ANALYSE STATISTIQUE
Les analyses des résultats obtenus dans cette étude ont été réalisées en utilisant les outils
statistiques classiques de recherche d’association marqueur génétique/maladie : Khi2 Odds
ratio et la signification statistique (p).
La signification statistique d’une association entre un marqueur et une maladie, est testée par
le calcul du Khi2. Le Khi2 de conformité permet d’apprécier la signification ou non d’une
différence de distribution, pour un marqueur donné, entre les populations de malades et de
témoins.
(p) représente le degré de signification qui correspond à la probabilité que l’écart global soit
imputable seulement aux fluctuations du hasard. Lorsque la probabilité ‘p’ est égale ou
inférieure à 0.05 (5%), il y a moins de 5 chances sur 100 que la distribution résulte du hasard.
Ainsi, la différence de distribution entre les populations de malades et de témoins, pour un
marqueur donné, est donc statistiquement significative et le marqueur peut être considéré
comme associé à la maladie. L’Odds Ratio ‘OR’ est utilisé dans les enquêtes de cohorte et de
type cas/témoins, il permet d’apprécier l’intensité d’une association entre un marqueur et une
maladie. L’évaluation du Khi2, de ‘p’ et de OR dans cette étude a été réalisée par l’utilisation
du logiciel Epi info 6.

Résultats et discussion
53
VI RESULTATS ET DISCUSSION
1 RESULTATS ET DISCUSSION DU GENOTYPAGE DES SUJETS POUR LES GENES
GSTM1, GSTT1, ET GSTP1 EXONS 5 ET 6
Les résultats des génotypage des sujets pour les gènes GSTM1 et GSTT1 ont concerné 71
patients atteints de CCR et 49 contrôles tandis que ceux révélant les polymorphismes de
GSTP1 exon 5 ont été obtenus pour 97 CCR et 99 contrôles et ceux de GSTP1 exon 6 ont été
analysés pour 93 CCR et 99 contrôles.
La figure 7 montre le profil d’amplification des gènes GSTM1, GSTT1 mais aussi le profil de
digestion enzymatique par BsmA1 du gène GSTP1 exon 5. Au cours de cette réaction sont
amplifiés simultanément les gènes GSTM1 dont la taille est de 250pb et le gène GSTT1 dont
la taille est de 450pb. Les délétions homozygotes GSTM1 et GSTT1 sont caractérisées par
une absence totale de bande correspondant à l’amplification. Les délétions hétérozygotes ne
sont pas mises en évidence. La distribution des fréquences de ces deux délétions dans notre
population est reportée sur le tableau 5.
Figure 7: Profils d’amplification des gènes GSTM1, GSTT1 et
de digestion enzymatique par BsmA1 du gène GSTP1 exon 5
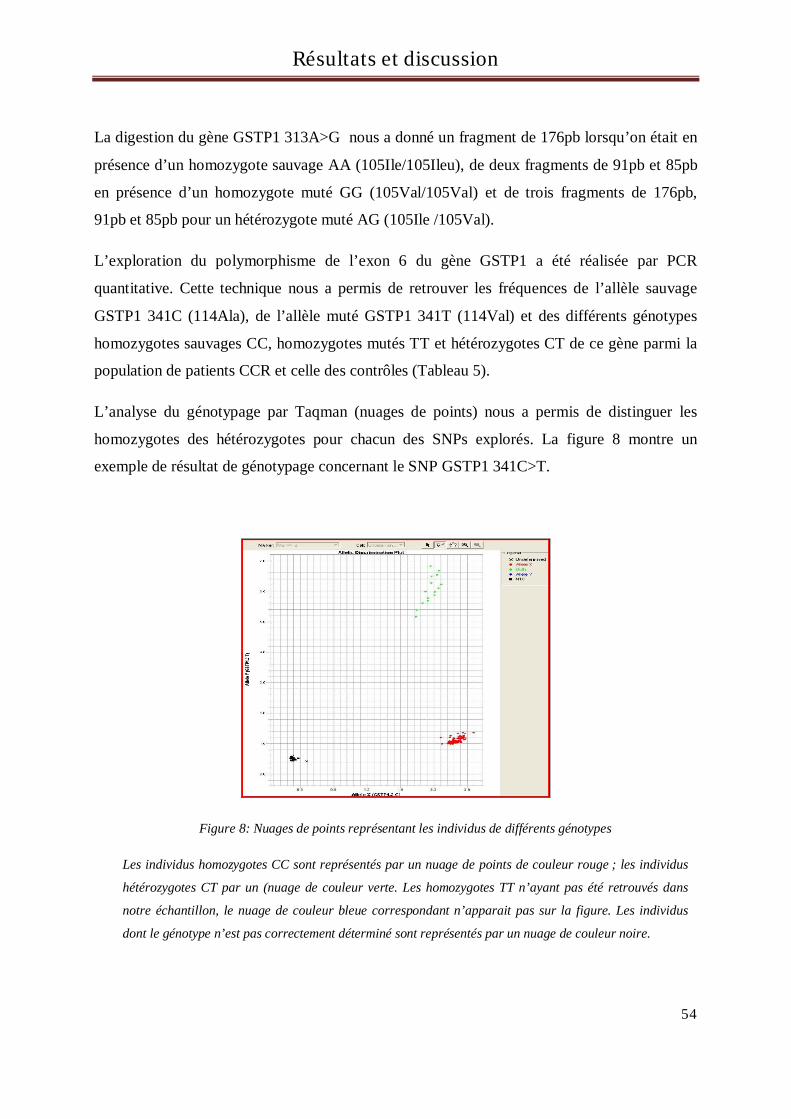
Résultats et discussion
54
La digestion du gène GSTP1 313A>G nous a donné un fragment de 176pb lorsqu’on était en
présence d’un homozygote sauvage AA (105Ile/105Ileu), de deux fragments de 91pb et 85pb
en présence d’un homozygote muté GG (105Val/105Val) et de trois fragments de 176pb,
91pb et 85pb pour un hétérozygote muté AG (105Ile /105Val).
L’exploration du polymorphisme de l’exon 6 du gène GSTP1 a été réalisée par PCR
quantitative. Cette technique nous a permis de retrouver les fréquences de l’allèle sauvage
GSTP1 341C (114Ala), de l’allèle muté GSTP1 341T (114Val) et des différents génotypes
homozygotes sauvages CC, homozygotes mutés TT et hétérozygotes CT de ce gène parmi la
population de patients CCR et celle des contrôles (Tableau 5).
L’analyse du génotypage par Taqman (nuages de points) nous a permis de distinguer les
homozygotes des hétérozygotes pour chacun des SNPs explorés. La figure 8 montre un
exemple de résultat de génotypage concernant le SNP GSTP1 341C>T.
Figure 8: Nuages de points représentant les individus de différents génotypes
Les individus homozygotes CC sont représentés par un nuage de points de couleur rouge ; les individus
hétérozygotes CT par un (nuage de couleur verte. Les homozygotes TT n’ayant pas été retrouvés dans
notre échantillon, le nuage de couleur bleue correspondant n’apparait pas sur la figure. Les individus
dont le génotype n’est pas correctement déterminé sont représentés par un nuage de couleur noire.
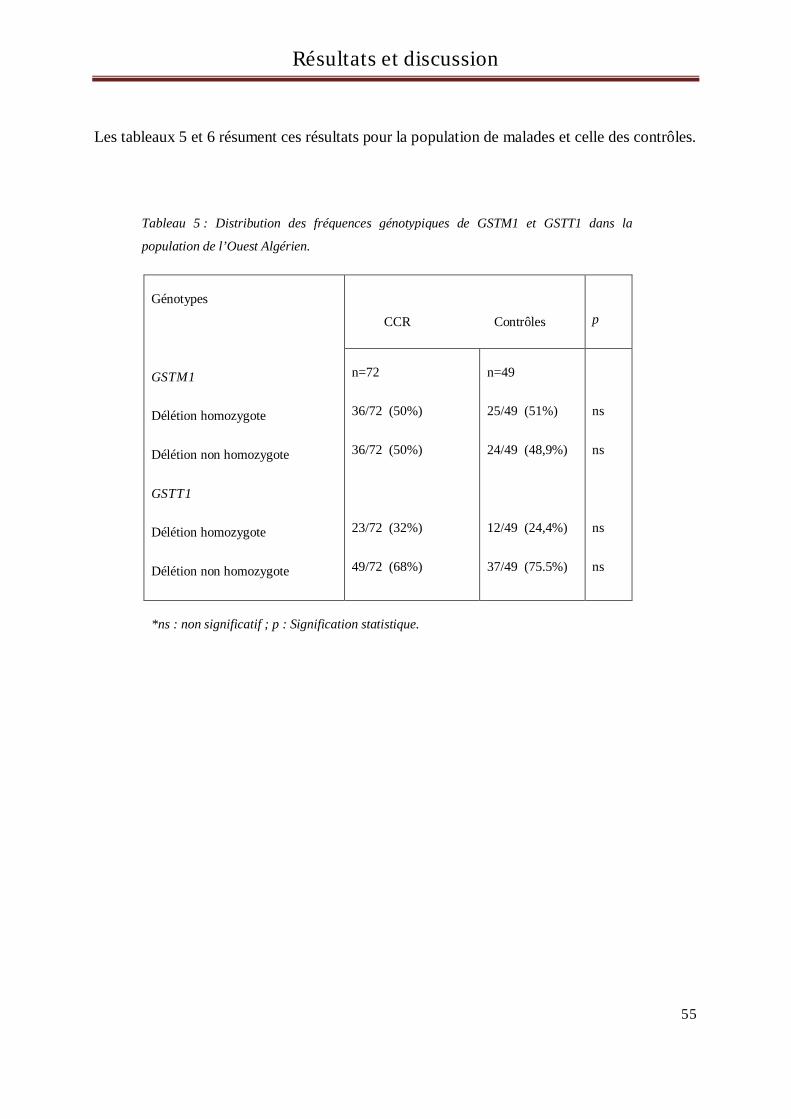
Résultats et discussion
55
Les tableaux 5 et 6 résument ces résultats pour la population de malades et celle des contrôles.
Tableau 5 : Distribution des fréquences génotypiques de GSTM1 et GSTT1 dans la
population de l’Ouest Algérien.
Génotypes
GSTM1
Délétion homozygote
Délétion non homozygote
GSTT1
Délétion homozygote
Délétion non homozygote
CCR Contrôles
p
n=72
36/72 (50%)
36/72 (50%)
23/72 (32%)
49/72 (68%)
n=49
25/49 (51%)
24/49 (48,9%)
12/49 (24,4%)
37/49 (75.5%)
ns
ns
ns
ns
*ns : non significatif ; p : Signification statistique.
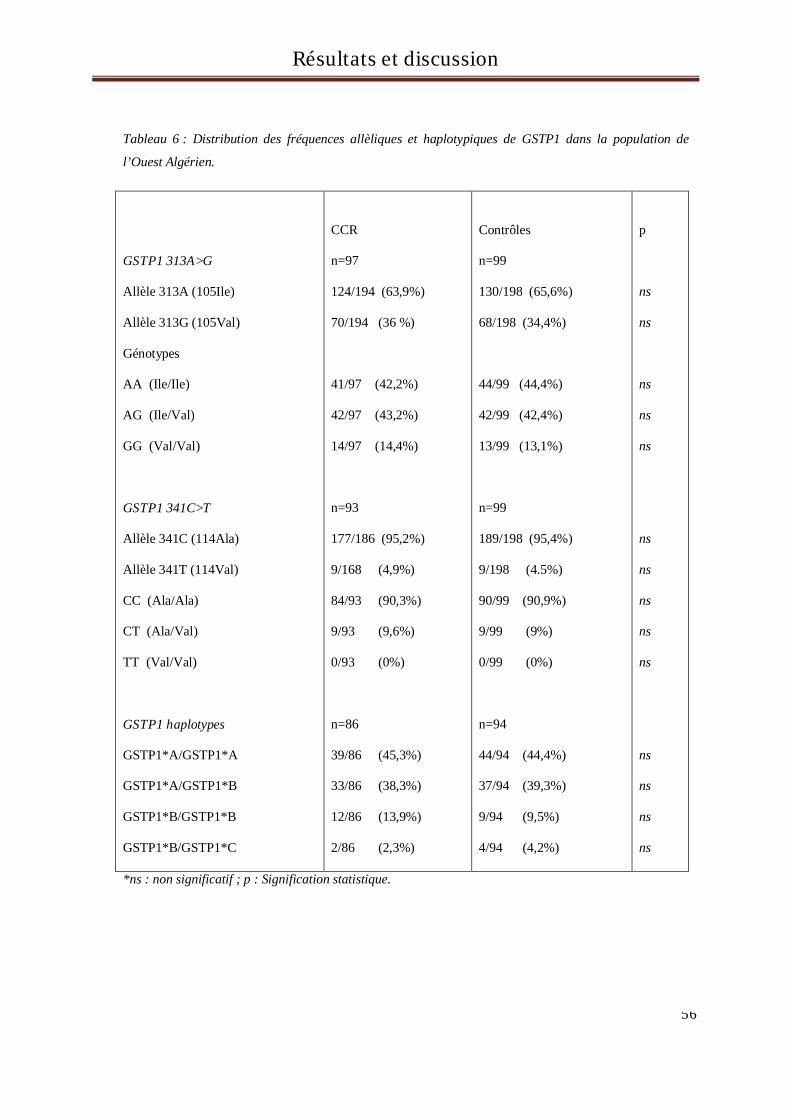
Résultats et discussion
56
Tableau 6 : Distribution des fréquences allèliques et haplotypiques de GSTP1 dans la population de
l’Ouest Algérien.
GSTP1 313A>G
Allèle 313A (105Ile)
Allèle 313G (105Val)
Génotypes
AA (Ile/Ile)
AG (Ile/Val)
GG (Val/Val)
GSTP1 341C>T
Allèle 341C (114Ala)
Allèle 341T (114Val)
CC (Ala/Ala)
CT (Ala/Val)
TT (Val/Val)
GSTP1 haplotypes
GSTP1*A/GSTP1*A
GSTP1*A/GSTP1*B
GSTP1*B/GSTP1*B
GSTP1*B/GSTP1*C
CCR
n=97
124/194 (63,9%)
70/194 (36 %)
41/97 (42,2%)
42/97 (43,2%)
14/97 (14,4%)
n=93
177/186 (95,2%)
9/168 (4,9%)
84/93 (90,3%)
9/93 (9,6%)
0/93 (0%)
n=86
39/86 (45,3%)
33/86 (38,3%)
12/86 (13,9%)
2/86 (2,3%)
Contrôles
n=99
130/198 (65,6%)
68/198 (34,4%)
44/99 (44,4%)
42/99 (42,4%)
13/99 (13,1%)
n=99
189/198 (95,4%)
9/198 (4.5%)
90/99 (90,9%)
9/99 (9%)
0/99 (0%)
n=94
44/94 (44,4%)
37/94 (39,3%)
9/94 (9,5%)
4/94 (4,2%)
p
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
*ns : non significatif ; p : Signification statistique.
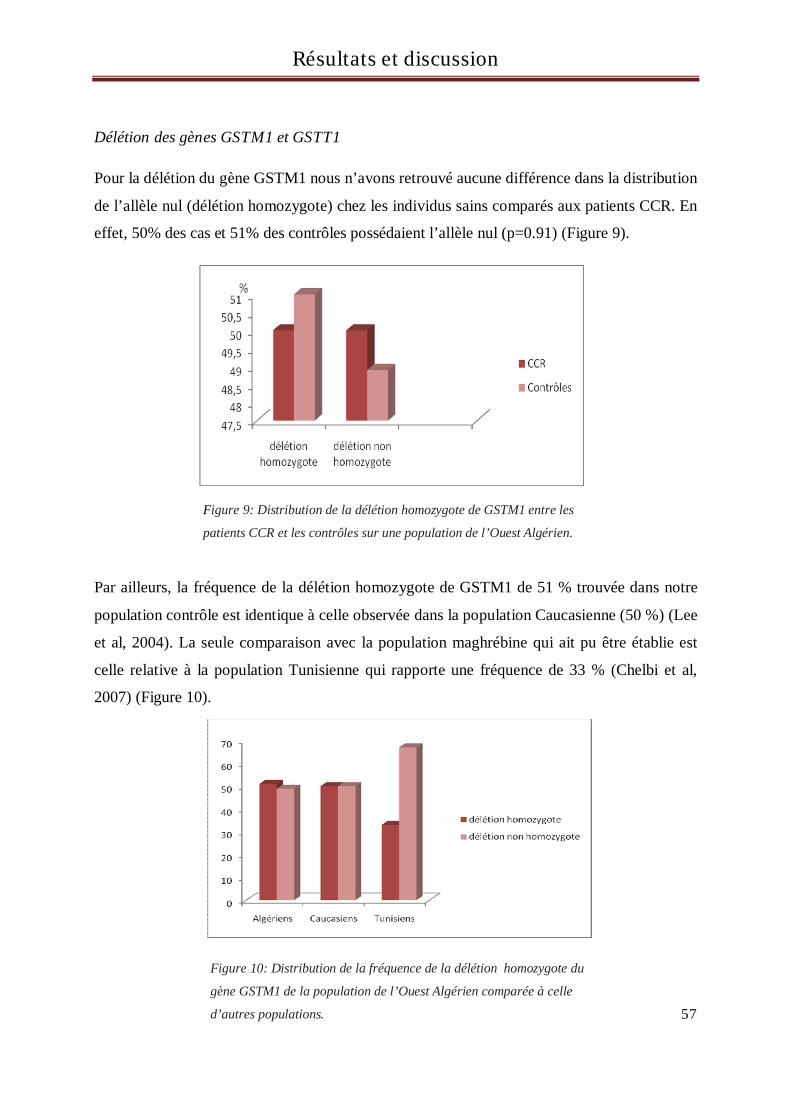
Résultats et discussion
57
Délétion des gènes GSTM1 et GSTT1
Pour la délétion du gène GSTM1 nous n’avons retrouvé aucune différence dans la distribution
de l’allèle nul (délétion homozygote) chez les individus sains comparés aux patients CCR. En
effet, 50% des cas et 51% des contrôles possédaient l’allèle nul (p=0.91) (Figure 9).
Par ailleurs, la fréquence de la délétion homozygote de GSTM1 de 51 % trouvée dans notre
population contrôle est identique à celle observée dans la population Caucasienne (50 %) (Lee
et al, 2004). La seule comparaison avec la population maghrébine qui ait pu être établie est
celle relative à la population Tunisienne qui rapporte une fréquence de 33 % (Chelbi et al,
2007) (Figure 10).
Figure 9: Distribution de la délétion homozygote de GSTM1 entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
Figure 10: Distribution de la fréquence de la délétion homozygote du
gène GSTM1 de la population de l’Ouest Algérien comparée à celle
d’autres populations.
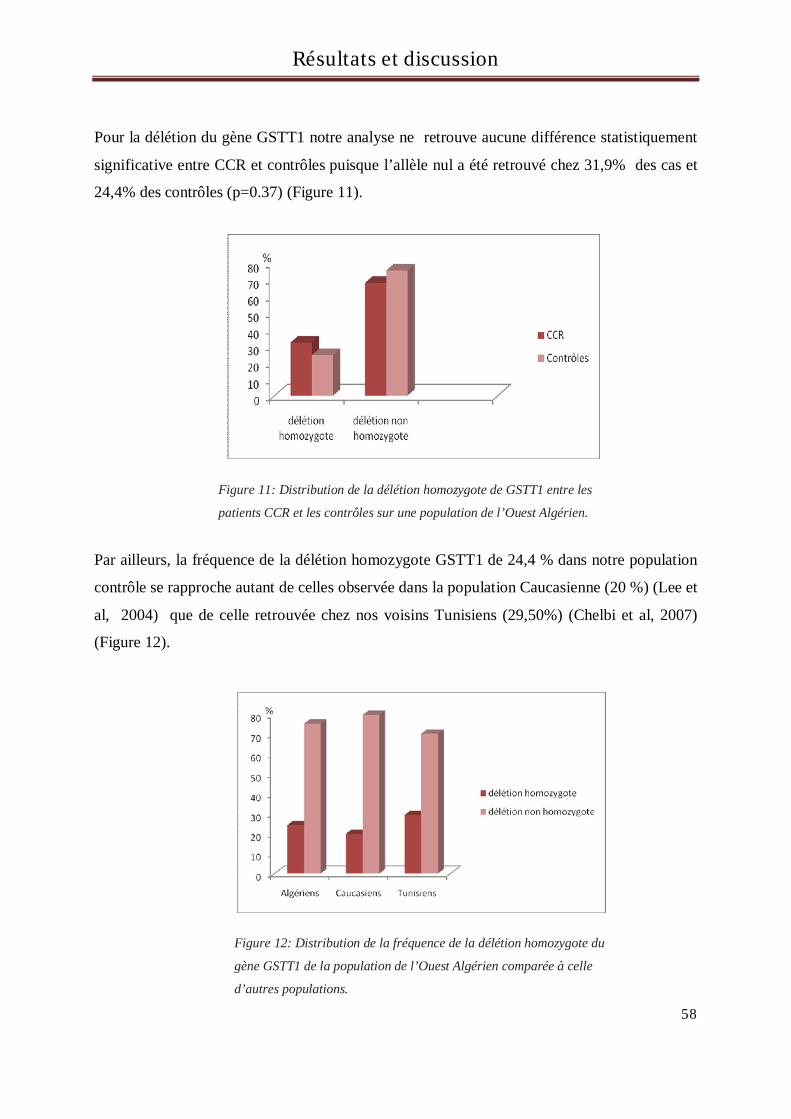
Résultats et discussion
58
Pour la délétion du gène GSTT1 notre analyse ne retrouve aucune différence statistiquement
significative entre CCR et contrôles puisque l’allèle nul a été retrouvé chez 31,9% des cas et
24,4% des contrôles (p=0.37) (Figure 11).
Par ailleurs, la fréquence de la délétion homozygote GSTT1 de 24,4 % dans notre population
contrôle se rapproche autant de celles observée dans la population Caucasienne (20 %) (Lee et
al, 2004) que de celle retrouvée chez nos voisins Tunisiens (29,50%) (Chelbi et al, 2007)
(Figure 12).
Figure 11: Distribution de la délétion homozygote de GSTT1 entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
Figure 12: Distribution de la fréquence de la délétion homozygote du
gène GSTT1 de la population de l’Ouest Algérien comparée à celle
d’autres populations.

Résultats et discussion
59
L’absence d’association entre la délétion homozygote de GSTM1 et la délétion homozygote
de GSTT1 et la survenue du CCR que nous avons retrouvée dans cette étude suggère que les
individus porteurs de ces délétions à l’état homozygote qui abolissent complètement l’activité
de ces deux enzymes, n’ont pas plus de risque de développer un CCR dans notre population
que les sujets non homozygotes.
La relation entre le risque de CCR et le polymorphisme GSTM1 a été largement analysée et
des études portant sur une large cohorte (Houlston et al, 2001 ; Little et al, 2006) n’ont
conclut à aucune association du polymorphisme GSTM1 avec le CCR. Nos résultats sont
conformes à ces observations. En revanche, l’équipe de Sachse et celle de Martínez décrivent
un risque accru au CCR pour les porteurs nuls de la délétion de GSTM1 (Sachse et al, 2002 ;
Martínez et al, 2006).
Concernant, GSTT1, nos résultats sont en accord avec la méta-analyse de Houlston mais en
contradiction avec celle de De Jong qui rapporte des résultats opposés (Houlston et al, 2001)
(De Jong et al, 2002).
Les délétions des gènes GSTM1 et GSTT1 annihilent complètement l’activité enzymatique
des enzymes correspondantes or il a été démontré que ces enzymes sont responsables de la
détoxication de nombreuses molécules électrophiles mutagènes présentes dans la fumée de
tabac ou encore dans l’alimentation. En effet la fumée de tabac est une source riche en
molécules carcinogènes comme les hydrocarbures aromatiques polycycliques, et les amines
aromatiques, et d'autres carcinogènes et ces molécules font parties des nombreux substrats des
GST (Giovannucci et al, 1996 ; Potter et al, 1999). Le colon est également exposé aux HAP et
aux amines hétérocycliques par la consommation d’aliments cuits à températures élevées, ce
qui représente un autre facteur de risque potentiel pour le CCR (Sinha et al, 1999 ; Sinha et al,
2001). Les HAP activés sont des substrats pour les enzymes GSTM1 et GSTT1 qui,
lorsqu’elles sont actives, les conjuguent au glutathion leur faisant perdre ainsi leur potentiel
cancérigène (Prochaska et al, 1998). Cette conjugaison est également induite par la
consommation diététique des isothiocyanates trouvés dans les légumes crucifères (Whitty et
al, 1987). De plus, la formation des adduits d’ADN et le taux de mutations somatiques ont été
rapportés comme étant augmentés chez les individus porteurs de délétions homozygotes pour
GSTM1 et GSTT1 (Smith et al, 1995). Il n’est donc pas surprenant que certains auteurs aient
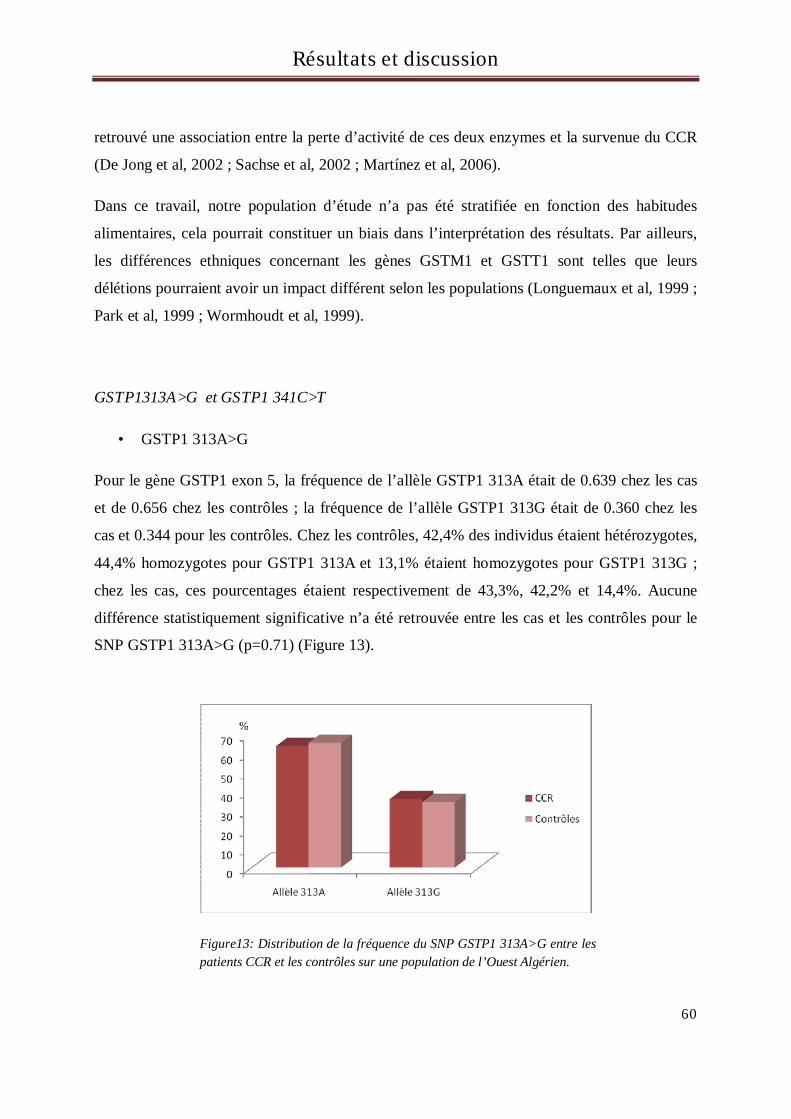
Résultats et discussion
60
retrouvé une association entre la perte d’activité de ces deux enzymes et la survenue du CCR
(De Jong et al, 2002 ; Sachse et al, 2002 ; Martínez et al, 2006).
Dans ce travail, notre population d’étude n’a pas été stratifiée en fonction des habitudes
alimentaires, cela pourrait constituer un biais dans l’interprétation des résultats. Par ailleurs,
les différences ethniques concernant les gènes GSTM1 et GSTT1 sont telles que leurs
délétions pourraient avoir un impact différent selon les populations (Longuemaux et al, 1999 ;
Park et al, 1999 ; Wormhoudt et al, 1999).
GSTP1313A>G et GSTP1 341C>T
• GSTP1 313A>G
Pour le gène GSTP1 exon 5, la fréquence de l’allèle GSTP1 313A était de 0.639 chez les cas
et de 0.656 chez les contrôles ; la fréquence de l’allèle GSTP1 313G était de 0.360 chez les
cas et 0.344 pour les contrôles. Chez les contrôles, 42,4% des individus étaient hétérozygotes,
44,4% homozygotes pour GSTP1 313A et 13,1% étaient homozygotes pour GSTP1 313G ;
chez les cas, ces pourcentages étaient respectivement de 43,3%, 42,2% et 14,4%. Aucune
différence statistiquement significative n’a été retrouvée entre les cas et les contrôles pour le
SNP GSTP1 313A>G (p=0.71) (Figure 13).
Figure13: Distribution de la fréquence du SNP GSTP1 313A>G entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
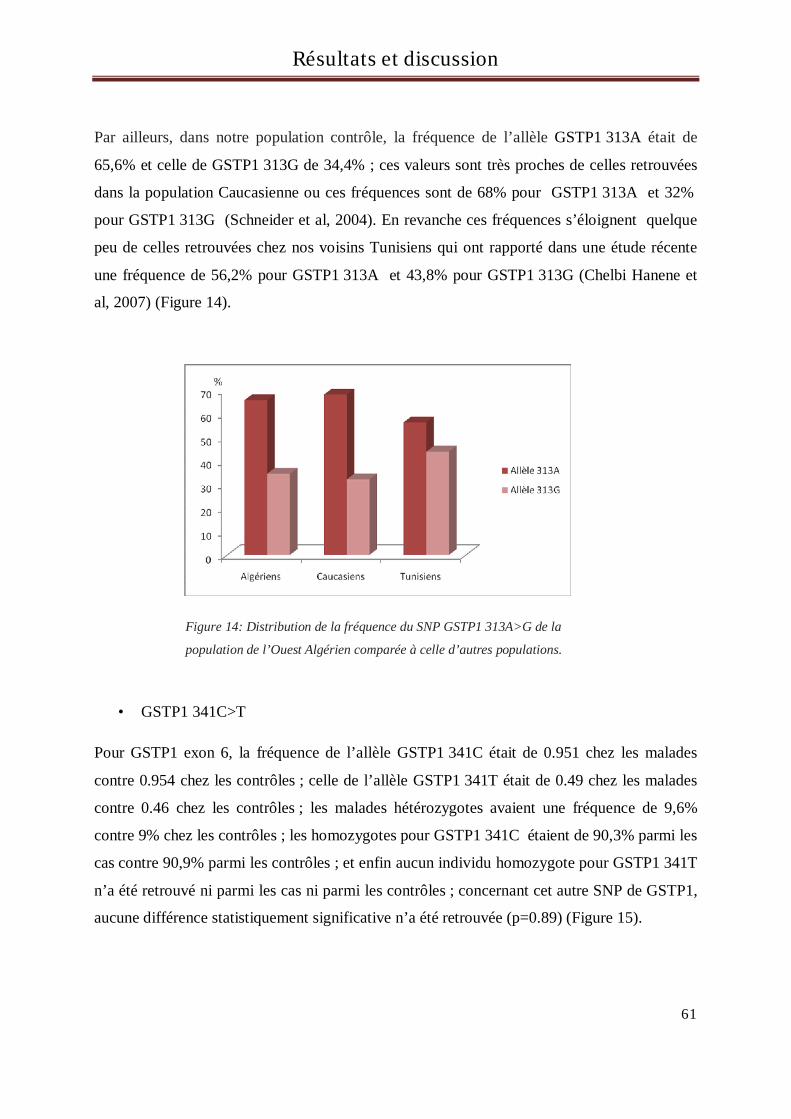
Résultats et discussion
61
Par ailleurs, dans notre population contrôle, la fréquence de l’allèle GSTP1 313A était de
65,6% et celle de GSTP1 313G de 34,4% ; ces valeurs sont très proches de celles retrouvées
dans la population Caucasienne ou ces fréquences sont de 68% pour GSTP1 313A et 32%
pour GSTP1 313G (Schneider et al, 2004). En revanche ces fréquences s’éloignent quelque
peu de celles retrouvées chez nos voisins Tunisiens qui ont rapporté dans une étude récente
une fréquence de 56,2% pour GSTP1 313A et 43,8% pour GSTP1 313G (Chelbi Hanene et
al, 2007) (Figure 14).
• GSTP1 341C>T
Pour GSTP1 exon 6, la fréquence de l’allèle GSTP1 341C était de 0.951 chez les malades
contre 0.954 chez les contrôles ; celle de l’allèle GSTP1 341T était de 0.49 chez les malades
contre 0.46 chez les contrôles ; les malades hétérozygotes avaient une fréquence de 9,6%
contre 9% chez les contrôles ; les homozygotes pour GSTP1 341C étaient de 90,3% parmi les
cas contre 90,9% parmi les contrôles ; et enfin aucun individu homozygote pour GSTP1 341T
n’a été retrouvé ni parmi les cas ni parmi les contrôles ; concernant cet autre SNP de GSTP1,
aucune différence statistiquement significative n’a été retrouvée (p=0.89) (Figure 15).
Figure 14: Distribution de la fréquence du SNP GSTP1 313A>G de la
population de l’Ouest Algérien comparée à celle d’autres populations.
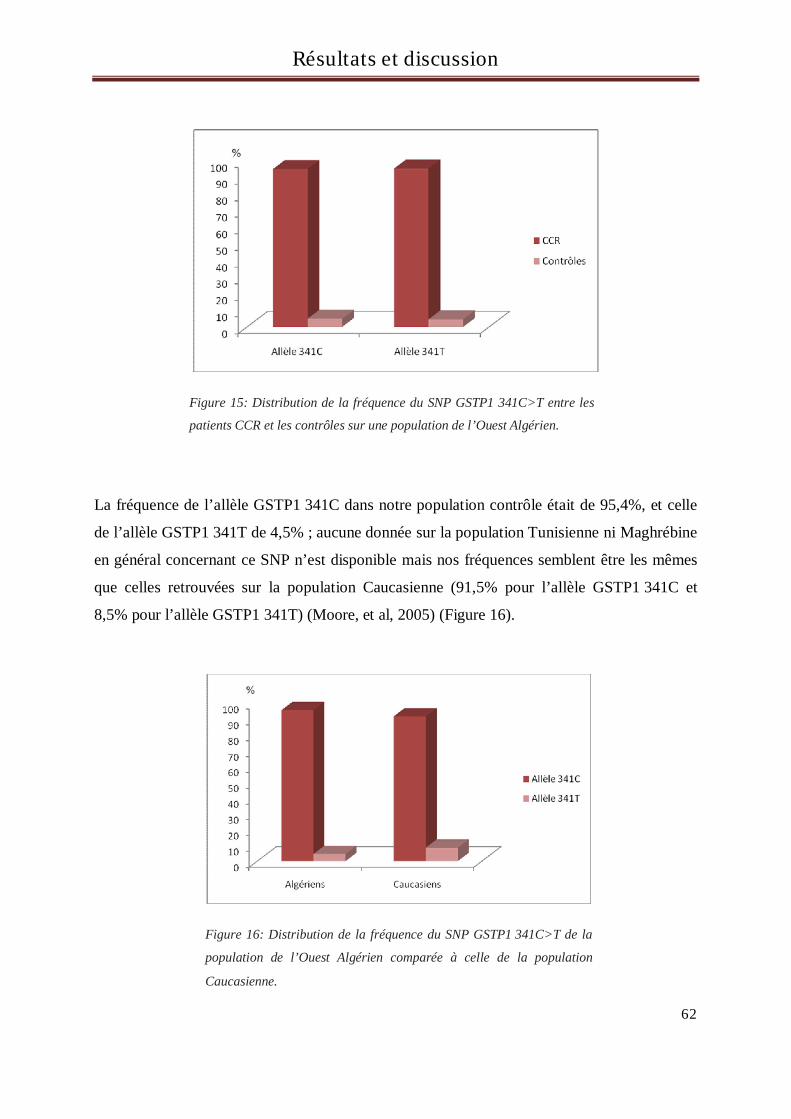
Résultats et discussion
62
La fréquence de l’allèle GSTP1 341C dans notre population contrôle était de 95,4%, et celle
de l’allèle GSTP1 341T de 4,5% ; aucune donnée sur la population Tunisienne ni Maghrébine
en général concernant ce SNP n’est disponible mais nos fréquences semblent être les mêmes
que celles retrouvées sur la population Caucasienne (91,5% pour l’allèle GSTP1 341C et
8,5% pour l’allèle GSTP1 341T) (Moore, et al, 2005) (Figure 16).
Figure 15: Distribution de la fréquence du SNP GSTP1 341C>T entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
Figure 16: Distribution de la fréquence du SNP GSTP1 341C>T de la
population de l’Ouest Algérien comparée à celle de la population
Caucasienne.

Résultats et discussion
63
Nous avons également analysé la fréquence des haplotypes sur ce gène et aucune différence
statistiquement significative n’a été retrouvée entre la population de patients CCR et la
population contrôle.
De par son taux d’expression plus important au niveau du colon et de par son intervention
dans la désactivation des aminés hétérocycliques mutagènes formés dans les aliments cuits,
GSTP1 semble être parmi les GST, le meilleur gène candidat pour la susceptibilité au CCR
(Lin et al, 1994 ; Nijhoff et al, 1995).
Les deux SNPs sur GSTP1, GSTP1 313A>G sur l’exon 5 et GSTP1 341C>T dans l’exon 6
ont une incidence sur la structure tridimensionnelle de l’enzyme et sur la stéréospécificité du
site catalytique (Ali-Osman et al, 1997). En effet, en altérant le site actif de GSTP1, ils
diminuent son affinité pour le substrat aboutissant de ce fait à une plus faible activité de cette
enzyme (Zimniak P et al, 1994 ; Van der Logt et al, 2004). In vitro, l’activité enzymatique de
la GSTP1 est quatre fois plus faible pour les variants allèliques GSTP1*B et GSTP1*C
comparés à l’allèle de référence GSTP1*A et l’allèle GSTP1*D semble très rare (Alexandrie
et al, 2002).
Seules de rares études ont conforté l’hypothèse selon laquelle ces polymorphismes sur le gène
GSTP1 sont associés à la survenue du CCR (Katoh et al, 1996 ; Harris et al, 1998). En
revanche, de nombreux travaux ne retrouvent pas cette association (Welfare et al, 1999 ;
Moore, et al, 2005 ; Ateş et al, 2005). Nous avons recherché cette relation d’association et les
résultats de notre étude sont en accord avec ces derniers puisqu’aucune association
statistiquement significative na été retrouvée entre les variants 313G ou 341T de GSTP1 et
l’augmentation du risque de CCR dans notre population. De même que pour GSTM1 et
GSTT1, cette non association suggère que d’autres facteurs comme l’alimentation pourraient
intervenir dans ce processus de cancérisation et que la modification de l’activité de GSTP1
seule n’aurait aucun impact sur la survenue du CCR.
Les gènes GST ont été largement explorés et les résultats restent à ce jour contradictoire quant
à leur association avec la survenue du CCR mais il reste important de connaitre les
distributions de leurs différents allèles dans notre population notamment dans le domaine de
la pharmacogénétique. En effet, le polymorphisme 313A>G du gène codant pour GSTP1 a pu
être corrélé avec une augmentation significative de la survie dans une série de patients atteints

Résultats et discussion
64
d’un cancer colorectal métastasé traité par une chimiothérapie à base d’oxaliplatine (médiane
de survie à 25 mois pour le génotype Val/Val, 13 mois pour Ile/Val, et 8 mois pour Ile/Ile)
(Stoelmacher et al, 2002). De plus, une étude récente a associé ce SNP à un risque diminué
de neuropathie périphérique cumulative chez des patients atteints de cancer gastro-intestinal
(Lecomte et al, 2006).
Cependant, les conclusions relatives à l’implication de ce polymorphisme dans la survenue
de la toxicité à l’oxaliplatine restent contradictoires et de plus amples études sont nécessaires
afin de déterminer le rôle exact de ce SNP dans la réponse à l’oxaliplatine. Cet anticancéreux
est également prescrit dans la chimiothérapie du CCR en Algérie et une recherche de la
corrélation entre la réponse à cette molécule et les génotypes de GSTP1 sur notre population
de patients est en cours d’analyse. En attendant que ce travail soit réalisé, la distribution des
fréquences génotypiques de ces gènes dans notre population pourrait servir à rechercher des
associations avec d’autres pathologies.
2 RESULTATS ET DISCUSSION DU GENOTYPAGE DES SUJETS POUR LE GENE MDR1
Les résultats génotypiques pour le SNP MDR1 3435 C>T ont été retrouvé pour 94 patients
atteints de CCR et 98 contrôles.
Les fréquences allèliques et génotypiques du SNP MDR1 3435C>T ont été comparées entre
les cas et les contrôles. La fréquence de l’allèle C était de 0,643 chez les cas contre 0,658 chez
les contrôles. Celle de l’allèle T était de 0,356 chez les malades contre 0,341 chez les sujets
sains. Le génotype CC a été retrouvé à une fréquence de 0,414, le génotype CT à 0,457 et TT
à 0,127 dans le groupe des malades. Dans le groupe des sujets sains, la fréquence du génotype
CC était de 0,448 celle de CT de 0,418 et celle de TT de 0,132 (Tableau 7).
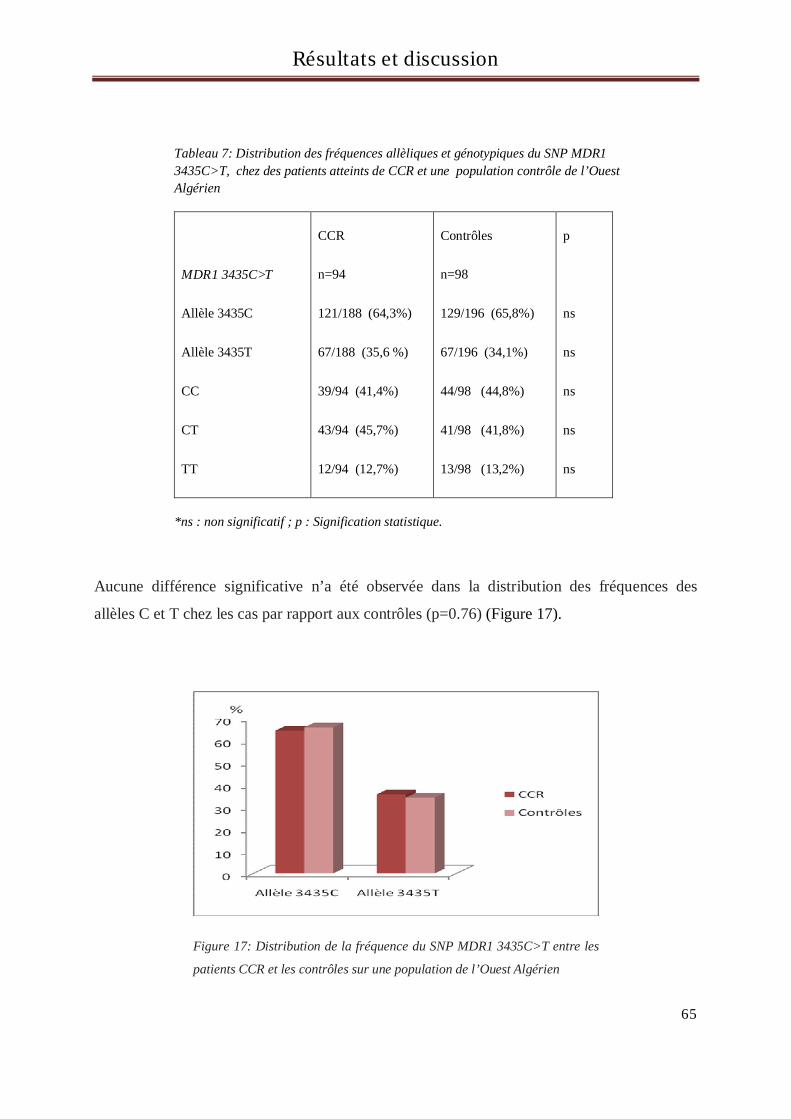
Résultats et discussion
65
Aucune différence significative n’a été observée dans la distribution des fréquences des
allèles C et T chez les cas par rapport aux contrôles (p=0.76) (Figure 17).
Tableau 7: Distribution des fréquences allèliques et génotypiques du SNP MDR1
3435C>T, chez des patients atteints de CCR et une population contrôle de l’Ouest
Algérien
MDR1 3435C>T
Allèle 3435C
Allèle 3435T
CC
CT
TT
CCR
n=94
121/188 (64,3%)
67/188 (35,6 %)
39/94 (41,4%)
43/94 (45,7%)
12/94 (12,7%)
Contrôles
n=98
129/196 (65,8%)
67/196 (34,1%)
44/98 (44,8%)
41/98 (41,8%)
13/98 (13,2%)
p
ns
ns
ns
ns
ns
*ns : non significatif ; p : Signification statistique.
Figure 17: Distribution de la fréquence du SNP MDR1 3435C>T entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien
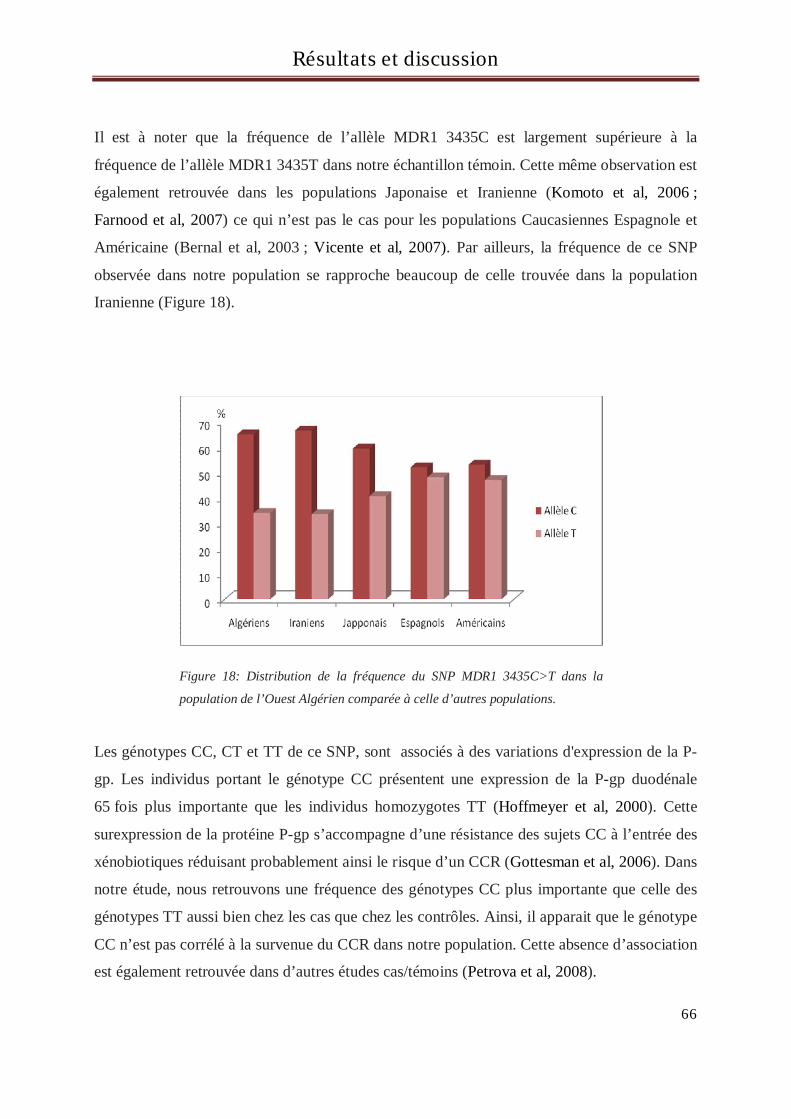
Résultats et discussion
66
Il est à noter que la fréquence de l’allèle MDR1 3435C est largement supérieure à la
fréquence de l’allèle MDR1 3435T dans notre échantillon témoin. Cette même observation est
également retrouvée dans les populations Japonaise et Iranienne (Komoto et al, 2006 ;
Farnood et al, 2007) ce qui n’est pas le cas pour les populations Caucasiennes Espagnole et
Américaine (Bernal et al, 2003 ; Vicente et al, 2007). Par ailleurs, la fréquence de ce SNP
observée dans notre population se rapproche beaucoup de celle trouvée dans la population
Iranienne (Figure 18).
Les génotypes CC, CT et TT de ce SNP, sont associés à des variations d'expression de la P-
gp. Les individus portant le génotype CC présentent une expression de la P-gp duodénale
65 fois plus importante que les individus homozygotes TT (Hoffmeyer et al, 2000). Cette
surexpression de la protéine P-gp s’accompagne d’une résistance des sujets CC à l’entrée des
xénobiotiques réduisant probablement ainsi le risque d’un CCR (Gottesman et al, 2006). Dans
notre étude, nous retrouvons une fréquence des génotypes CC plus importante que celle des
génotypes TT aussi bien chez les cas que chez les contrôles. Ainsi, il apparait que le génotype
CC n’est pas corrélé à la survenue du CCR dans notre population. Cette absence d’association
est également retrouvée dans d’autres études cas/témoins (Petrova et al, 2008).
Figure 18: Distribution de la fréquence du SNP MDR1 3435C>T dans la
population de l’Ouest Algérien comparée à celle d’autres populations.

Résultats et discussion
67
L’analyse de la distribution des fréquences allèliques du SNP 3435C>T du gène MDR1
semble montrer que notre population est très proche de la population Iranienne. De plus,
contrairement à certaines populations, l’allèle MDR1 3435C est prédominant dans la
population Algérienne mais ne semble conférer aucune protection ni susceptibilité particulière
vis-à-vis de la survenue du CCR. Néanmoins, il serait intéressant d’explorer d’autres SNPs du
gène MDR1 en particulier les SNPs se trouvant dans la région promotrice de MDR1 comme
2410T>C, 1910T>C et 692T>C dont l’effet sur l’expression de la P-gp dans la muqueuse
colorectale a été clairement mis en évidence (Taniguchi et al, 2003).
L’importance de l’influence du polymorphisme du gène MDR1 sur la réponse aux
médicaments utilisés dans la chimiothérapie du CCR est telle, qu’il est très intéressant de
compléter cette étude par la recherche des corrélations entre les génotypes de ce SNP vis-à-vis
des molécules anti-cancers et ceci pour une meilleure prise en charge des malades ; ce travail
nécessite du temps pour une bonne évaluation à long terme de cette réponse thérapeutique et
est en cours d’analyse sur la population de l’Ouest Algérien.
3 RESULTATS ET DISCUSSION DU GENOTYPAGE DES SUJETS POUR LE GENE
CYP3A5
Les résultats génotypiques pour le SNP 6986A>G dans l’intron 3 du gène CYP3A5 ont été
retrouvés pour 96 patients atteints de CCR et 99 contrôles.
L’allèle sauvage 6986A de ce polymorphisme est noté CYP3A5*1 tandis que l’allèle muté
6986G s’écrit CYP3A5*3.
La fréquence de l’allèle CYP3A5*1 était de 0,161 chez les cas contre 0,204 chez les
contrôles. Celle de l’allèle CYP3A5*3 était de 0,838 chez les malades contre 0,806 chez les
sujets sains. Concernant la distribution des génotypes, dans notre population d’étude, le
génotype CYP3A5*1/*1 est très rare autant chez les cas (1%) que chez les contrôles (6.1%).
Par ailleurs, le génotype CYP3A5*3/*3 a été retrouvé à une fréquence de 68.7% chez les
CCR et 66.3% chez les contrôles (Tableau 8).
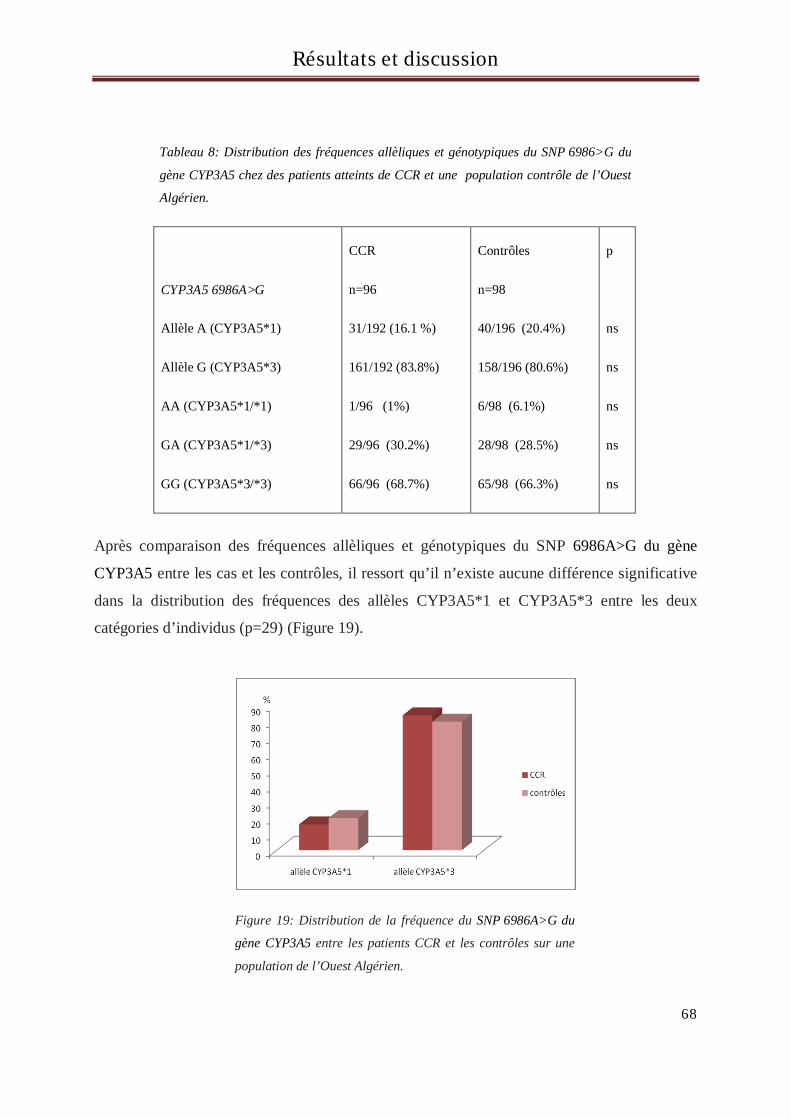
Résultats et discussion
68
Après comparaison des fréquences allèliques et génotypiques du SNP 6986A>G du gène
CYP3A5 entre les cas et les contrôles, il ressort qu’il n’existe aucune différence significative
dans la distribution des fréquences des allèles CYP3A5*1 et CYP3A5*3 entre les deux
catégories d’individus (p=29) (Figure 19).
Tableau 8: Distribution des fréquences allèliques et génotypiques du SNP 6986>G du
gène CYP3A5 chez des patients atteints de CCR et une population contrôle de l’Ouest
Algérien.
CYP3A5 6986A>G
Allèle A (CYP3A5*1)
Allèle G (CYP3A5*3)
AA (CYP3A5*1/*1)
GA (CYP3A5*1/*3)
GG (CYP3A5*3/*3)
CCR
n=96
31/192 (16.1 %)
161/192 (83.8%)
1/96 (1%)
29/96 (30.2%)
66/96 (68.7%)
Contrôles
n=98
40/196 (20.4%)
158/196 (80.6%)
6/98 (6.1%)
28/98 (28.5%)
65/98 (66.3%)
p
ns
ns
ns
ns
ns
Figure 19: Distribution de la fréquence du SNP 6986A>G du
gène CYP3A5 entre les patients CCR et les contrôles sur une
population de l’Ouest Algérien.
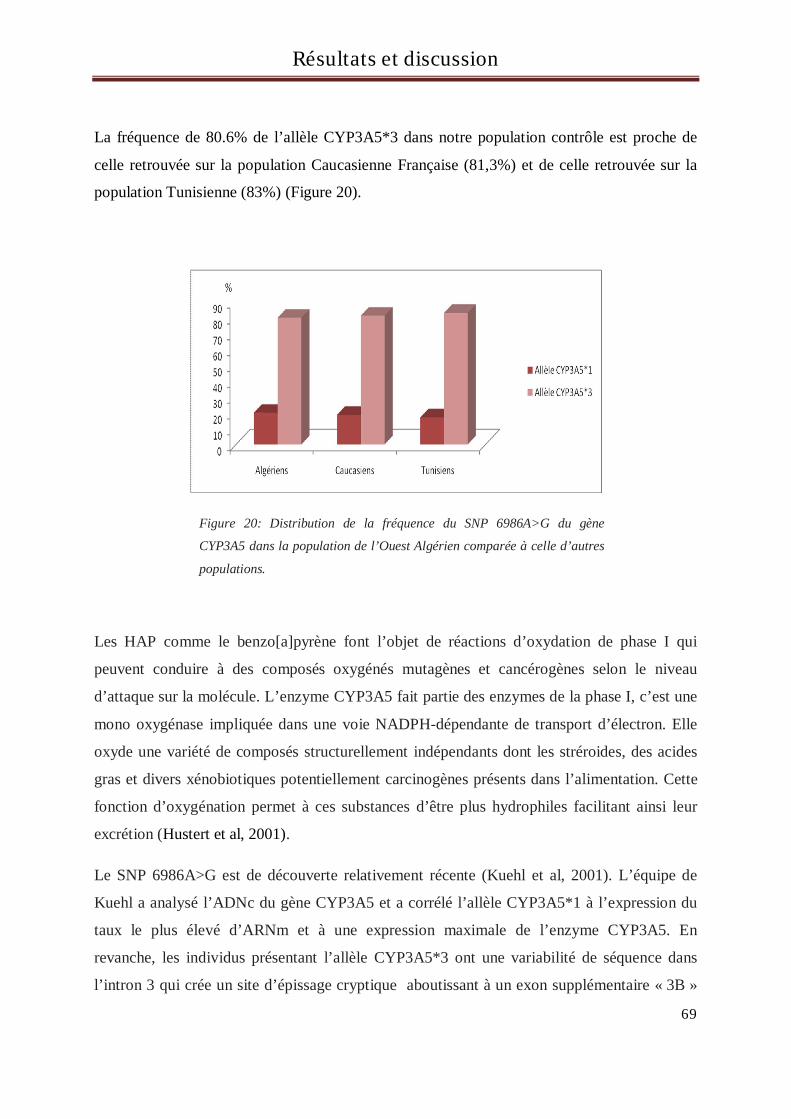
Résultats et discussion
69
La fréquence de 80.6% de l’allèle CYP3A5*3 dans notre population contrôle est proche de
celle retrouvée sur la population Caucasienne Française (81,3%) et de celle retrouvée sur la
population Tunisienne (83%) (Figure 20).
Les HAP comme le benzo[a]pyrène font l’objet de réactions d’oxydation de phase I qui
peuvent conduire à des composés oxygénés mutagènes et cancérogènes selon le niveau
d’attaque sur la molécule. L’enzyme CYP3A5 fait partie des enzymes de la phase I, c’est une
mono oxygénase impliquée dans une voie NADPH-dépendante de transport d’électron. Elle
oxyde une variété de composés structurellement indépendants dont les stréroides, des acides
gras et divers xénobiotiques potentiellement carcinogènes présents dans l’alimentation. Cette
fonction d’oxygénation permet à ces substances d’être plus hydrophiles facilitant ainsi leur
excrétion (Hustert et al, 2001).
Le SNP 6986A>G est de découverte relativement récente (Kuehl et al, 2001). L’équipe de
Kuehl a analysé l’ADNc du gène CYP3A5 et a corrélé l’allèle CYP3A5*1 à l’expression du
taux le plus élevé d’ARNm et à une expression maximale de l’enzyme CYP3A5. En
revanche, les individus présentant l’allèle CYP3A5*3 ont une variabilité de séquence dans
l’intron 3 qui crée un site d’épissage cryptique aboutissant à un exon supplémentaire « 3B »
Figure 20: Distribution de la fréquence du SNP 6986A>G du gène
CYP3A5 dans la population de l’Ouest Algérien comparée à celle d’autres
populations.

Résultats et discussion
70
dérivé de séquences de l’intron 3. Cet allèle code pour un ARNm avec un codon stop
prématuré et la traduction de ce transcrit anormal génère une protéine prématurément
tronquée au niveau de l’acide aminé 102. Cet épissage alternatif serait responsable d’une
importante réduction voire absence de l’enzyme CYP3A5 (Kuehl et al, 2001).
Sachant que l’allèle CYP3A5*3 est synonyme d’une expression plus faible voire nulle de
l’enzyme CYP3A5, il semblerait que dans notre population, l’expression de CYP3A5 serait
plus faible et donc que la perte de l’expression de cette enzyme due au SNP 6986A>G n’ait
pas de lien avec une susceptibilité accrue au CCR. Ceci est en accord avec les conclusions
d’autres études récentes (Petrova et al, 2007 ; Gervasini et al, 2007). Cette absence
d’association, malgré la fréquence élevée de ce SNP dans notre population, pourrait
s’expliquer par le mode d’alimentation différent et donc une exposition à des substances
cancérigènes potentielles différentes dans nos régions qui ne nécessiteraient pas la fonction
enzymatique de CYP3A5 pour être désactivées. De plus, il se pourrait que l’expression de
CYP3A5 ne soit pas exclusivement régulée au niveau transcriptionnel, et que les mécanismes
de régulation de celle-ci pourraient être différents entre les sujets souffrant de CCR et les
individus sains (Bergheim et al, 2005). Cette hypothèse signifierait que le polymorphisme
modifiant la transcription du gène CYP3A5 pourrait être contrebalancé par un autre
mécanisme de régulation qui aboutirait à une expression normale de cette enzyme chez les
sujets malades. Par ailleurs, il se pourrait que ce SNP ait des conséquences différentes en
fonction de l’alimentation et donc de la localisation géographique. Cette hypothèse est étayée
par d’autres travaux ou l’effet de l’allèle CYP3A5*3 semble plutôt protecteur vis-à-vis du
cancer dans d’autres populations. En effet dans ces études ce SNP est associé à une
diminution du risque de cancer de l’œsophage mais aussi du cancer de la prostate (Dandara et
al, 2005 ; Plummer et al, 2003).
Néanmoins, il reste important de connaitre la distribution des fréquences allèliques et
génotypiques de ce SNP dans notre population car cela pourrait servir à établir d’autres
comparaisons dans le cadre d’études cas/témoins à la recherche d’une éventuelle association
avec d’autres pathologies cancéreuses en Algérie.

Résultats et discussion
71
4 RESULTATS ET DISCUSSION DU GENOTYPAGE DES SUJETS POUR LE GENE UGT1A1
• VNTR UGT1A1*28(TA) 6>(TA)7
Concernant le VNTR UGT1A1*28(TA) 6>(TA)7, l’étude a porté sur 72 CCR et 45 individus
sains.
La fréquence de l’allèle UGT1A1*1 correspondant à 6 répétitions de la séquence TA était de
0,68 chez les CCR contre 0,60 chez les contrôles ; la fréquence de l’allèle UGT1A1*28
correspondant à 7 répétitions de TA était de 0,32 chez les CCR contre 0,40 chez les contrôles.
Concernant les génotypes, 47% des CCR et 40% des contrôles étaient homozygotes pour
l’allèle UGT1A1*1, tandis que 11% des CCR et 20% des contrôles étaient homozygotes pour
l’allèle UGT1A1*28. Les hétérozygotes UGT1A1*1/*28 étaient de 42% chez les CCR contre
40% chez les contrôles (Tableau 9).
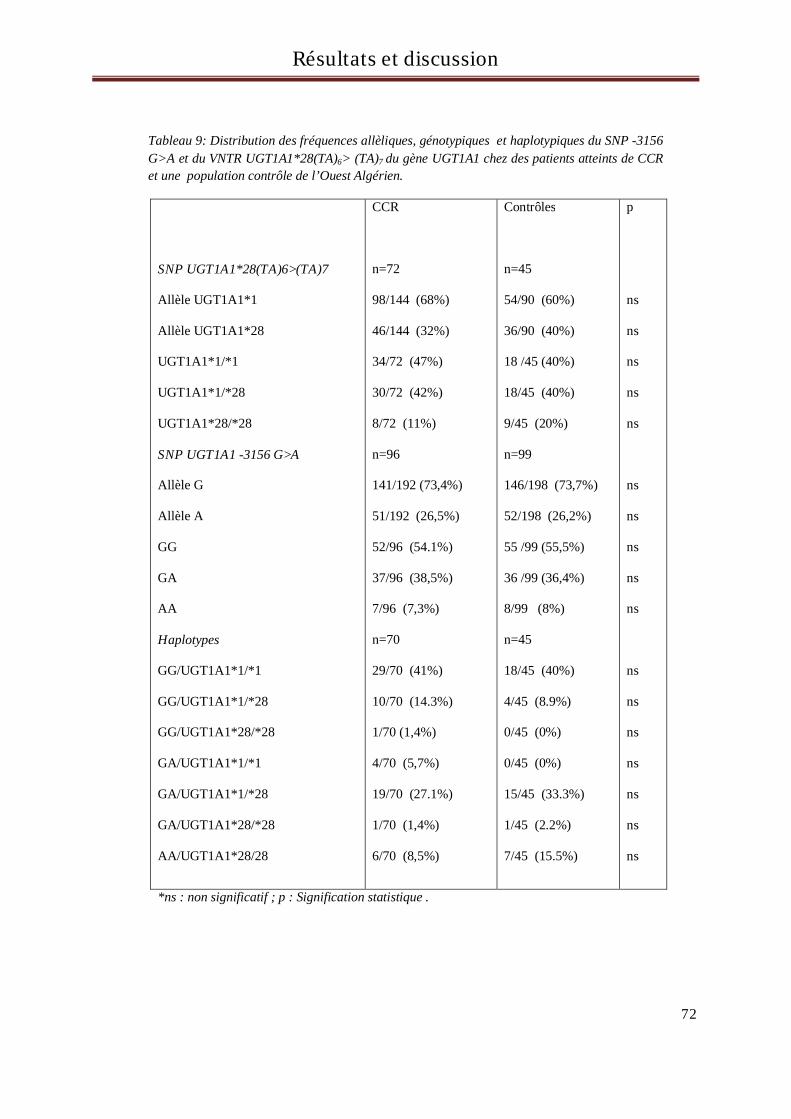
Résultats et discussion
72
Tableau 9: Distribution des fréquences allèliques, génotypiques et haplotypiques du SNP -3156
G>A et du VNTR UGT1A1*28(TA)6> (TA)7 du gène UGT1A1 chez des patients atteints de CCR
et une population contrôle de l’Ouest Algérien.
SNP UGT1A1*28(TA)6>(TA)7
Allèle UGT1A1*1
Allèle UGT1A1*28
UGT1A1*1/*1
UGT1A1*1/*28
UGT1A1*28/*28
SNP UGT1A1 -3156 G>A
Allèle G
Allèle A
GG
GA
AA
Haplotypes
GG/UGT1A1*1/*1
GG/UGT1A1*1/*28
GG/UGT1A1*28/*28
GA/UGT1A1*1/*1
GA/UGT1A1*1/*28
GA/UGT1A1*28/*28
AA/UGT1A1*28/28
CCR
n=72
98/144 (68%)
46/144 (32%)
34/72 (47%)
30/72 (42%)
8/72 (11%)
n=96
141/192 (73,4%)
51/192 (26,5%)
52/96 (54.1%)
37/96 (38,5%)
7/96 (7,3%)
n=70
29/70 (41%)
10/70 (14.3%)
1/70 (1,4%)
4/70 (5,7%)
19/70 (27.1%)
1/70 (1,4%)
6/70 (8,5%)
Contrôles
n=45
54/90 (60%)
36/90 (40%)
18 /45 (40%)
18/45 (40%)
9/45 (20%)
n=99
146/198 (73,7%)
52/198 (26,2%)
55 /99 (55,5%)
36 /99 (36,4%)
8/99 (8%)
n=45
18/45 (40%)
4/45 (8.9%)
0/45 (0%)
0/45 (0%)
15/45 (33.3%)
1/45 (2.2%)
7/45 (15.5%)
p
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
ns
*ns : non significatif ; p : Signification statistique .
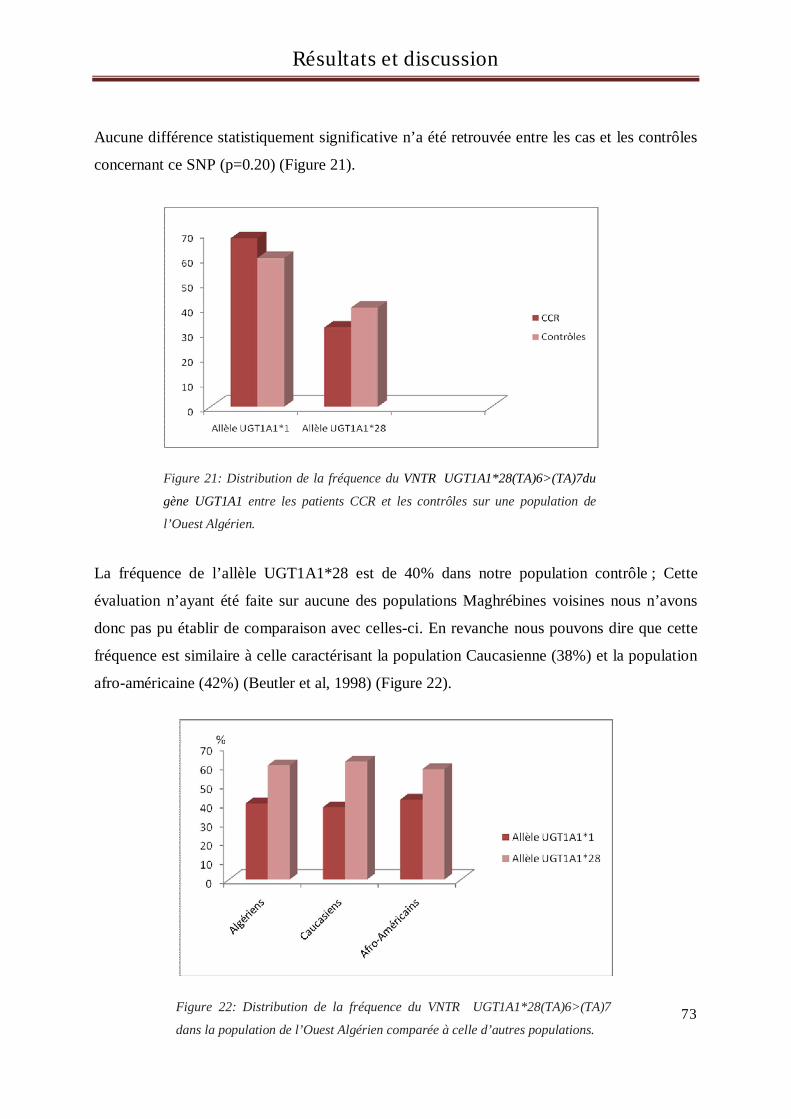
Résultats et discussion
73
Aucune différence statistiquement significative n’a été retrouvée entre les cas et les contrôles
concernant ce SNP (p=0.20) (Figure 21).
La fréquence de l’allèle UGT1A1*28 est de 40% dans notre population contrôle ; Cette
évaluation n’ayant été faite sur aucune des populations Maghrébines voisines nous n’avons
donc pas pu établir de comparaison avec celles-ci. En revanche nous pouvons dire que cette
fréquence est similaire à celle caractérisant la population Caucasienne (38%) et la population
afro-américaine (42%) (Beutler et al, 1998) (Figure 22).
Figure 21: Distribution de la fréquence du VNTR UGT1A1*28(TA)6>(TA)7du
gène UGT1A1 entre les patients CCR et les contrôles sur une population de
l’Ouest Algérien.
Figure 22: Distribution de la fréquence du VNTR UGT1A1*28(TA)6>(TA)7
dans la population de l’Ouest Algérien comparée à celle d’autres populations.
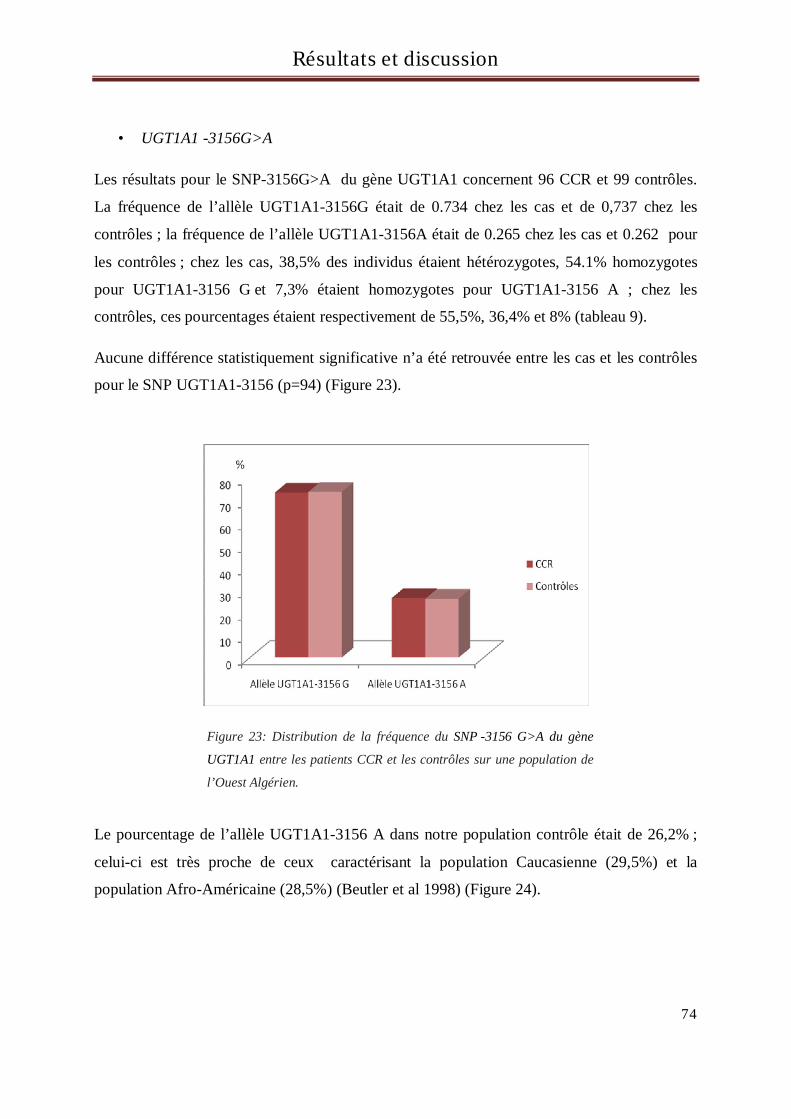
Résultats et discussion
74
• UGT1A1 -3156G>A
Les résultats pour le SNP-3156G>A du gène UGT1A1 concernent 96 CCR et 99 contrôles.
La fréquence de l’allèle UGT1A1-3156G était de 0.734 chez les cas et de 0,737 chez les
contrôles ; la fréquence de l’allèle UGT1A1-3156A était de 0.265 chez les cas et 0.262 pour
les contrôles ; chez les cas, 38,5% des individus étaient hétérozygotes, 54.1% homozygotes
pour UGT1A1-3156 G et 7,3% étaient homozygotes pour UGT1A1-3156 A ; chez les
contrôles, ces pourcentages étaient respectivement de 55,5%, 36,4% et 8% (tableau 9).
Aucune différence statistiquement significative n’a été retrouvée entre les cas et les contrôles
pour le SNP UGT1A1-3156 (p=94) (Figure 23).
Le pourcentage de l’allèle UGT1A1-3156 A dans notre population contrôle était de 26,2% ;
celui-ci est très proche de ceux caractérisant la population Caucasienne (29,5%) et la
population Afro-Américaine (28,5%) (Beutler et al 1998) (Figure 24).
Figure 23: Distribution de la fréquence du SNP -3156 G>A du gène
UGT1A1 entre les patients CCR et les contrôles sur une population de
l’Ouest Algérien.

Résultats et discussion
75
Après avoir comparé les fréquences de chaque SNP seul entre les cas et les malades nous
nous sommes intéressé à établir les différents haplotypes constitués par ces deux SNPs. Nous
n’avons retrouvé, la non plus, aucune différence statistiquement significative entre les cas et
les contrôles (Tableau 9).
Le CCR est en grande partie associé au mode de vie, mais il peut être aussi modulé par des
facteurs génétiques à faible pénétrance. De nombreuses études ont clairement démontré le rôle
du gène UGT1A1 dans le métabolisme des molécules potentiellement carcinogènes produites
par la cuisson des aliments à hautes températures dont les amines hétérocycliques et les
hydrocarbures aromatiques polycycliques issus de la cuisson de la viande (Kaderlik et al,
1994). En effet, la réaction de glucuronidation assurée par cette enzyme est une des premières
voies de détoxification de ces molécules dont les métabolites toxiques se fixent au niveau de
l’ADN cellulaire conduisant à des mutations. (Turesky, 2002). Le tractus gastro-intestinal
étant exposé directement à ces substances toxiques ou pré-cancérigènes, la muqueuse
intestinale représente donc la première barrière à risque. Par ailleurs, certaines études ont
retrouvé une association entre des SNPs d’autres enzymes de la même famille que UGT1A1,
l’enzyme UGT1A7 et la survenue de CCR (Van der Logt et al, 2004 ;Tang et al, 2005 ; Chen
Figure 24: Distribution de la fréquence du SNP UGT1A1-3156G>A dans la
population de l’Ouest Algérien comparée à celle d’autres populations.

Résultats et discussion
76
et al, 2006). Il est donc concevable de supposer que des polymorphismes du gène UGT1A1
réduisant la transcription de cette enzyme comme les SNPs UGT1A1*28(TA)6>(TA)7 sur le
promoteur ou UGT1A1-3156 en amont du promoteur puissent avoir une conséquence sur
l’augmentation du risque de CCR.
Dans notre étude nous n’avons retrouvé aucune différence de distribution de l’allèle
UGT1A1*28 entre les cas et les contrôles. Cela signifie que les individus présentant une plus
faible expression de l’enzyme UGT1A1 due à l’allèle UGT1A1*28 ne présentent pas plus de
risque de développer le CCR par rapport aux patients ne présentant pas cet allèle. Ces
résultats sont en accord avec d’autres études qui aboutissent à la même conclusion (Van der
Logt et al, 2004 ; Tang, et al, 2005).
De même que le VNTR UGT1A1*28(TA)6>(TA)7, le variant UGT1A1 -3156G>A situé dans
une région enhancer en amont du promoteur de ce gène a été corrélé avec une baisse du taux
de transcription du gène UGT1A1 (Innocenti et al, 2002). En se basant sur la possibilité qu’il
puisse de ce fait avoir une influence sur le risque de survenue du CCR, nous l’avons analysé
sur notre population et n’avons retrouvé aucune différence statistiquement significative entre
les cas et les contrôles.
L’importance du génotypage des sujets pour ces deux polymorphismes du gène UGT1A1
réside surtout dans le fait qu’ils soient impliqués dans la toxicité à l’Irinotécan, une molécule
couramment utilisée dans la chimiothérapie du CCR. En effet, plusieurs études ont montré
une association entre ces deux SNPs et une sévère neutropénie après administration
d’Irinotécan (Innocenti et al, 2004).
La réponse au médicament implique deux aspects importants, la quantité de médicament actif
qui atteint la tumeur dans le tissu cible, c’est-à-dire le côlon et également la quantité de
médicament actif qui agit dans la cellule tumorale. Des polymorphismes de UGT1A1 dans
l’ADN constitutif qui affectent toutes les cellules du corps vont moduler la quantité de
médicament actif (SN-38) qui atteint le tissu cible. Le foie est l’organe de prédilection pour le
métabolisme de l’Irinotécan. Les polymorphismes comme UGT1A1*28 affectent la quantité
de SN-38 actif circulant donc par conséquent, la quantité qui se rend au tissu cible. Ceci
pourrait avoir un impact sur la réponse à l’Irinotécan mais aucune étude à ce jour n’a permis

Résultats et discussion
77
de démontrer de façon précise l’impact d’UGT1A1 sur la réponse à la thérapie à base
d’Irinotécan.
Le travail que nous avons réalisé sur ce gène constitue la première étape dans la recherche en
cours de cette corrélation sur notre échantillon d’individus malades soumis a une
chimiothérapie à base d’Irinotécan afin d’optimiser le dosage de cette molécule.
Les fréquences de ces polymorphismes SNPs sur notre population contrôle pourraient aussi
servir dans d’autres études cas /témoins en particulier pour l’hyper bilirubinémie du syndrome
de Gilbert (Bosma et al, 1995). En effet cette pathologie a pour principal facteur causal une
baisse de transcription du gène UGT1A1 due en particulier au polymorphisme
UGT1A1*28(TA)6>(TA)7 et a pour conséquence une diminution de la réaction de
glucoronidation du SN-38 de la bilirubine d’où une accumulation toxique de celle-ci dans le
foie. Aujourd’hui, dans les pays développé, un dépistage systématique pré thérapeutique est
réalisé afin d'adapter les doses d’Irinotécan aux capacités de glucuronidation de l'enzyme
UGT1A1 avant la mise en place d'un traitement à base de cette molécule.
5 RESULTATS ET DISCUSSION DU GENOTYPAGE DES SUJETS POUR LE GENE TS
Pour ce gène, deux polymorphismes ont été explorés : la répétition de la séquence de 28pb
« 2R >3R » en 5’ au niveau du promoteur, et la délétion/insertion de 6pb dans la région 3’ en
aval du codon stop à la position 1494. Pour le premier polymorphisme, les résultats rapportés
dans notre étude concernent 70 CCR et 48 contrôles tandis que pour le second, les résultats
concernent 71 CCR et 48 patients.
• TS répétition 2R>3R
La fréquence de l’allèle 2R correspondant à 2 répétitions de la séquence de 28pb était de 0,52
chez les CCR contre 0,53 chez les contrôles ; celle de l’allèle 3R correspondant à 3 répétitions
de la séquence de 28pb était de 0,48 chez les CCR contre 0,47 chez les contrôles. 31,4% des
CCR et 29% des contrôles étaient homozygotes 2R/2R ; 27,1% des CCR et 23% des contrôles
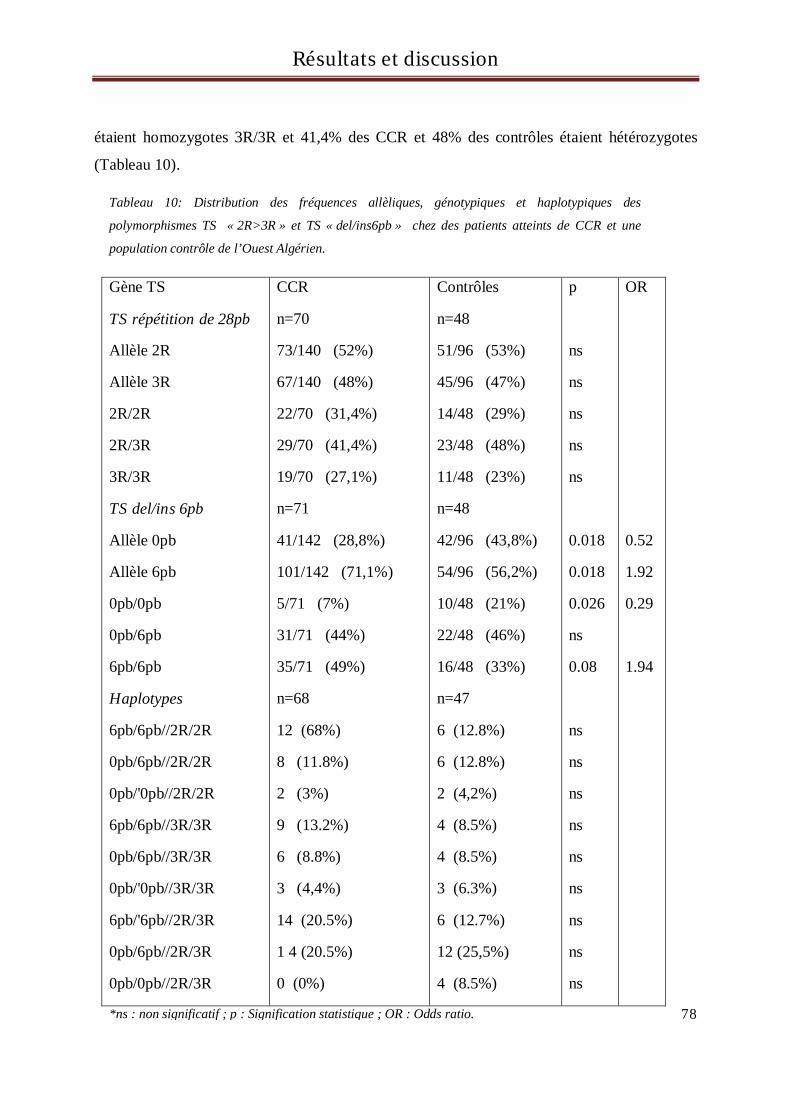
Résultats et discussion
78
étaient homozygotes 3R/3R et 41,4% des CCR et 48% des contrôles étaient hétérozygotes
(Tableau 10).
Tableau 10: Distribution des fréquences allèliques, génotypiques et haplotypiques des
polymorphismes TS « 2R>3R » et TS « del/ins6pb » chez des patients atteints de CCR et une
population contrôle de l’Ouest Algérien.
Gène TS
TS répétition de 28pb
Allèle 2R
Allèle 3R
2R/2R
2R/3R
3R/3R
TS del/ins 6pb
Allèle 0pb
Allèle 6pb
0pb/0pb
0pb/6pb
6pb/6pb
Haplotypes
6pb/6pb//2R/2R
0pb/6pb//2R/2R
0pb/'0pb//2R/2R
6pb/6pb//3R/3R
0pb/6pb//3R/3R
0pb/'0pb//3R/3R
6pb/'6pb//2R/3R
0pb/6pb//2R/3R
0pb/0pb//2R/3R
CCR
n=70
73/140 (52%)
67/140 (48%)
22/70 (31,4%)
29/70 (41,4%)
19/70 (27,1%)
n=71
41/142 (28,8%)
101/142 (71,1%)
5/71 (7%)
31/71 (44%)
35/71 (49%)
n=68
12 (68%)
8 (11.8%)
2 (3%)
9 (13.2%)
6 (8.8%)
3 (4,4%)
14 (20.5%)
1 4 (20.5%)
0 (0%)
Contrôles
n=48
51/96 (53%)
45/96 (47%)
14/48 (29%)
23/48 (48%)
11/48 (23%)
n=48
42/96 (43,8%)
54/96 (56,2%)
10/48 (21%)
22/48 (46%)
16/48 (33%)
n=47
6 (12.8%)
6 (12.8%)
2 (4,2%)
4 (8.5%)
4 (8.5%)
3 (6.3%)
6 (12.7%)
12 (25,5%)
4 (8.5%)
p
ns
ns
ns
ns
ns
0.018
0.018
0.026
ns
0.08
ns
ns
ns
ns
ns
ns
ns
ns
ns
OR
0.52
1.92
0.29
1.94
*ns : non significatif ; p : Signification statistique ; OR : Odds ratio.
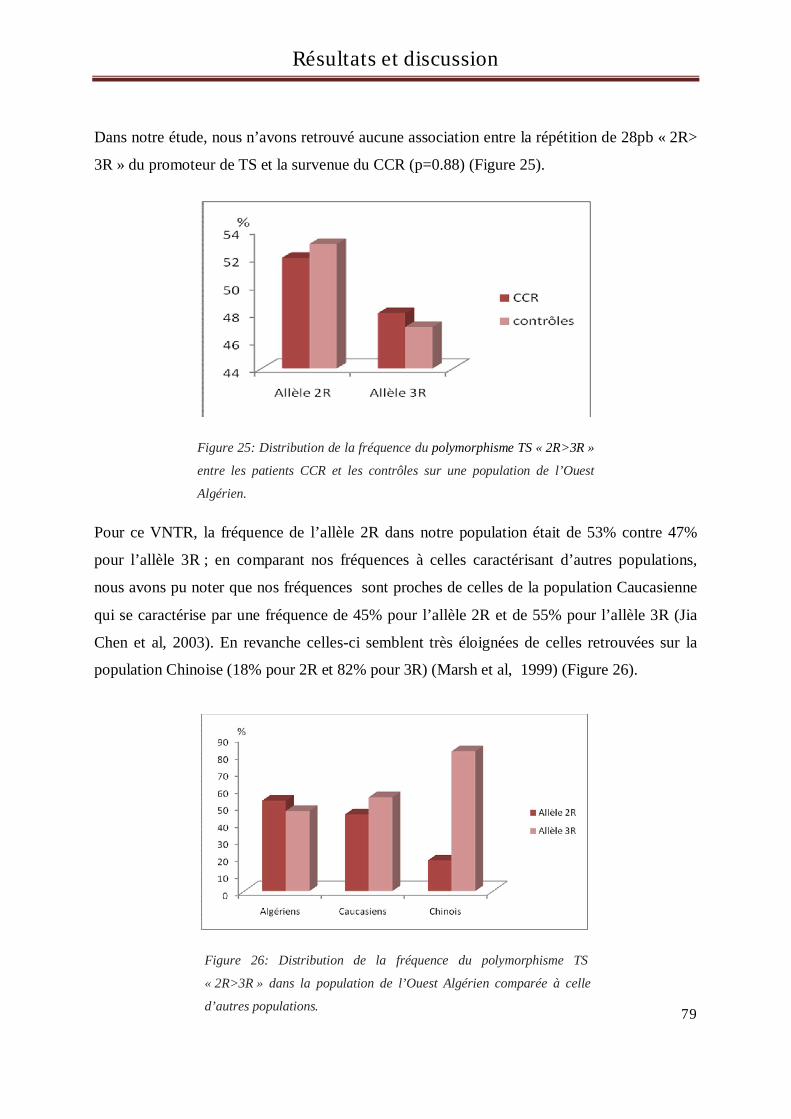
Résultats et discussion
79
Dans notre étude, nous n’avons retrouvé aucune association entre la répétition de 28pb « 2R>
3R » du promoteur de TS et la survenue du CCR (p=0.88) (Figure 25).
Pour ce VNTR, la fréquence de l’allèle 2R dans notre population était de 53% contre 47%
pour l’allèle 3R ; en comparant nos fréquences à celles caractérisant d’autres populations,
nous avons pu noter que nos fréquences sont proches de celles de la population Caucasienne
qui se caractérise par une fréquence de 45% pour l’allèle 2R et de 55% pour l’allèle 3R (Jia
Chen et al, 2003). En revanche celles-ci semblent très éloignées de celles retrouvées sur la
population Chinoise (18% pour 2R et 82% pour 3R) (Marsh et al, 1999) (Figure 26).
Figure 26: Distribution de la fréquence du polymorphisme TS
« 2R>3R » dans la population de l’Ouest Algérien comparée à celle
d’autres populations.
Figure 25: Distribution de la fréquence du polymorphisme TS « 2R>3R »
entre les patients CCR et les contrôles sur une population de l’Ouest
Algérien.
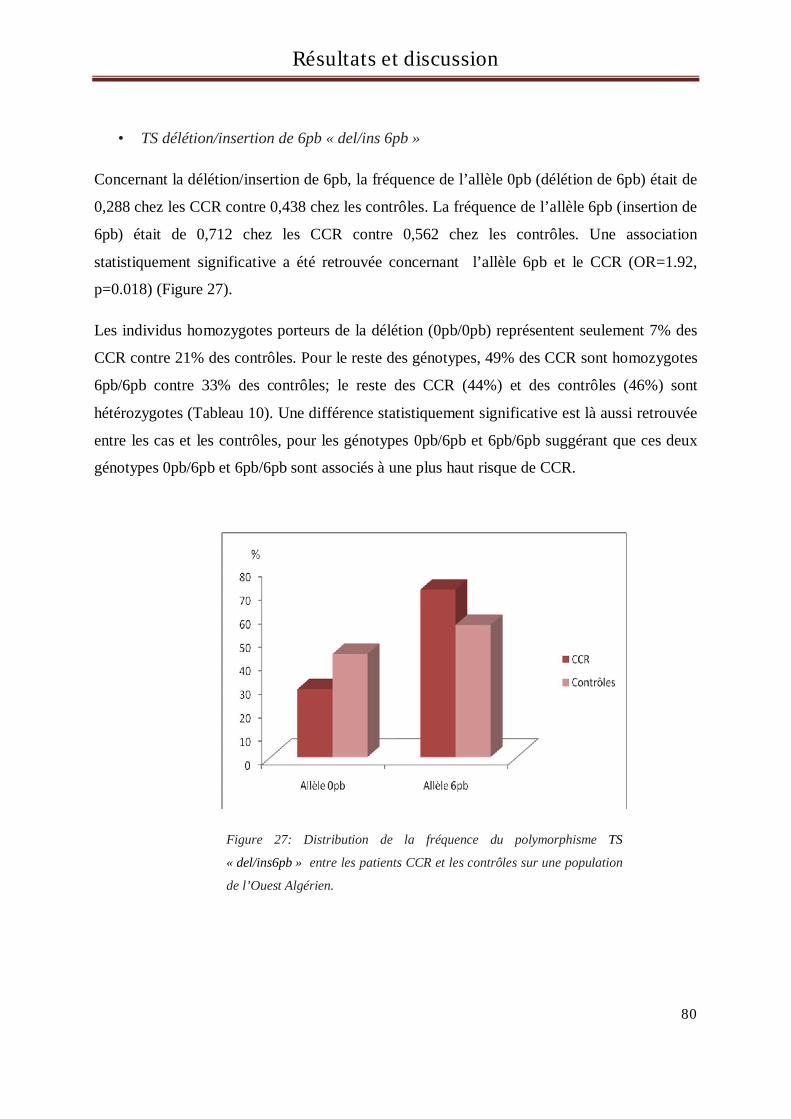
Résultats et discussion
80
• TS délétion/insertion de 6pb « del/ins 6pb »
Concernant la délétion/insertion de 6pb, la fréquence de l’allèle 0pb (délétion de 6pb) était de
0,288 chez les CCR contre 0,438 chez les contrôles. La fréquence de l’allèle 6pb (insertion de
6pb) était de 0,712 chez les CCR contre 0,562 chez les contrôles. Une association
statistiquement significative a été retrouvée concernant l’allèle 6pb et le CCR (OR=1.92,
p=0.018) (Figure 27).
Les individus homozygotes porteurs de la délétion (0pb/0pb) représentent seulement 7% des
CCR contre 21% des contrôles. Pour le reste des génotypes, 49% des CCR sont homozygotes
6pb/6pb contre 33% des contrôles; le reste des CCR (44%) et des contrôles (46%) sont
hétérozygotes (Tableau 10). Une différence statistiquement significative est là aussi retrouvée
entre les cas et les contrôles, pour les génotypes 0pb/6pb et 6pb/6pb suggérant que ces deux
génotypes 0pb/6pb et 6pb/6pb sont associés à une plus haut risque de CCR.
Figure 27: Distribution de la fréquence du polymorphisme TS
« del/ins6pb » entre les patients CCR et les contrôles sur une population
de l’Ouest Algérien.
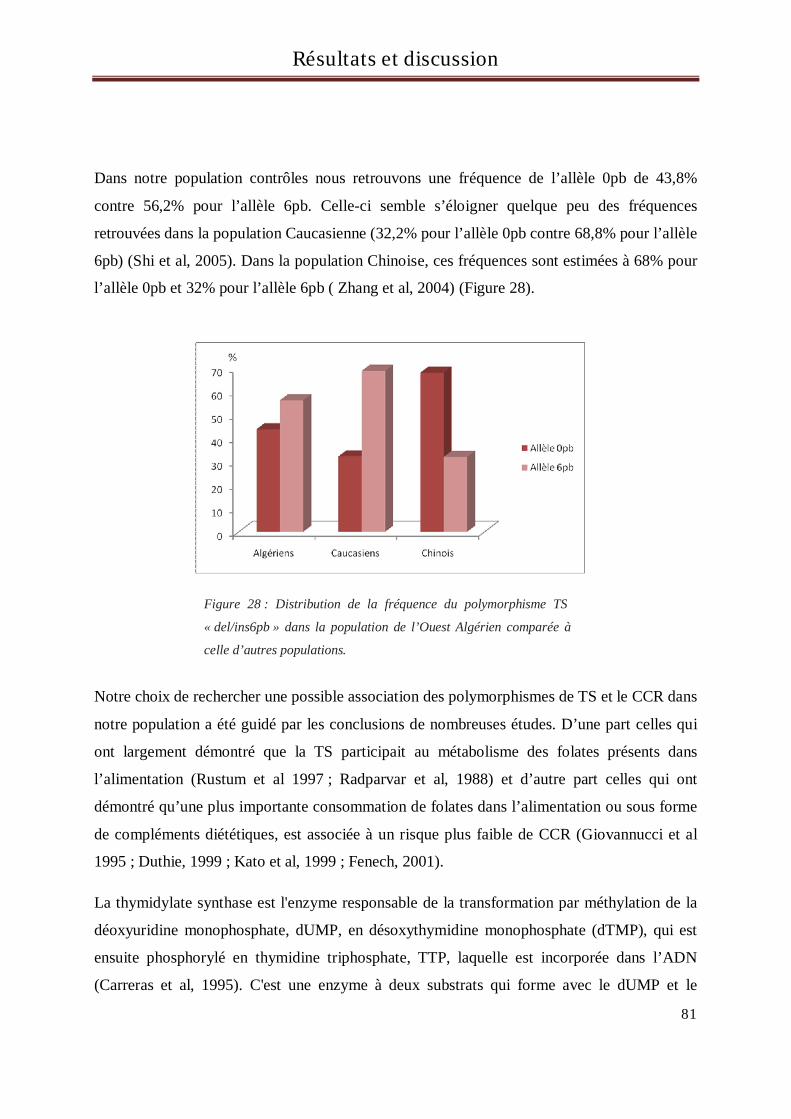
Résultats et discussion
81
Dans notre population contrôles nous retrouvons une fréquence de l’allèle 0pb de 43,8%
contre 56,2% pour l’allèle 6pb. Celle-ci semble s’éloigner quelque peu des fréquences
retrouvées dans la population Caucasienne (32,2% pour l’allèle 0pb contre 68,8% pour l’allèle
6pb) (Shi et al, 2005). Dans la population Chinoise, ces fréquences sont estimées à 68% pour
l’allèle 0pb et 32% pour l’allèle 6pb ( Zhang et al, 2004) (Figure 28).
Notre choix de rechercher une possible association des polymorphismes de TS et le CCR dans
notre population a été guidé par les conclusions de nombreuses études. D’une part celles qui
ont largement démontré que la TS participait au métabolisme des folates présents dans
l’alimentation (Rustum et al 1997 ; Radparvar et al, 1988) et d’autre part celles qui ont
démontré qu’une plus importante consommation de folates dans l’alimentation ou sous forme
de compléments diététiques, est associée à un risque plus faible de CCR (Giovannucci et al
1995 ; Duthie, 1999 ; Kato et al, 1999 ; Fenech, 2001).
La thymidylate synthase est l'enzyme responsable de la transformation par méthylation de la
déoxyuridine monophosphate, dUMP, en désoxythymidine monophosphate (dTMP), qui est
ensuite phosphorylé en thymidine triphosphate, TTP, laquelle est incorporée dans l’ADN
(Carreras et al, 1995). C'est une enzyme à deux substrats qui forme avec le dUMP et le
Figure 28 : Distribution de la fréquence du polymorphisme TS
« del/ins6pb » dans la population de l’Ouest Algérien comparée à
celle d’autres populations.

Résultats et discussion
82
méthylène tétrahydrofolate un complexe où le méthylène tétrahydrofolate cède un groupe
méthyl au dUMP. De plus, la TS est une enzyme clé dans le métabolisme des folates en
catalysant la conversion du 5,10-methylenetetrahydrofolate en dihydrofolate (Radparvar et al,
1988). Il n’est donc pas surprenant qu’une diminution de l’activité de la TS puisse avoir un
impact sur le métabolisme des folates et de ce fait augmenter le risque de cancer colorectal.
Par ailleurs, les cellules du colon ont une division cellulaire rapide et sont ainsi le siège d’une
synthèse d'ADN importante, la réparation de l'ADN est de ce fait essentielle dans ces cellules,
et des mécanismes de réparation d'ADN peuvent être irréversiblement affectés par un manque
de thymidine (Mattano et al, 1990 ; James et al, 1994). En effet, il a été prouvé que la baisse
de dTMP pourrrait aboutir à une incorporation de l’uracile au lieu de la thymine dans l’ADN,
erreur qui ne pourrait être réparée par l’uracile-ADN-glycosylase par manque de thymine et
qui provoquerait alors des cassures de la double chaine d’ADN (Duthi, 1999 ; Fenech ,2001).
C’est d’ailleurs ce mécanisme, entre autres, qui est mis à profit en chimiothérapie à base de
5FU pour agir sur les cellules tumorales. Rappelons que le 5FU est un analogue de l’uracile
qui agit selon plusieurs mécanismes. Il peut être intégré soit à l’ARN sous forme de FUTP,
altérant profondément son métabolisme et sa stabilité, soit à l’ADN sous forme de FdUTP,
provoquant une inhibition de la réplication et une fragmentation de l’ADN. Toutefois, son
activité antitumorale s’exerce principalement sous la forme de FdUMP, métabolite capable
d’inhiber la thymidylate synthétase. Cette inhibition est suivie d’un déficit en thymidine et
d’une incorporation de dUTP dans l’ADN, ce qui provoque l’apoptose cellulaire (Longley et
al, 2003).
Par ailleurs, sachant que de nombreuses analyses fonctionnelles ont depuis longtemps prouvé
que la répétition 3R dans le promoteur de TS contribuait à une plus grande efficacité de
l’expression du gène de la TS (Horie et al, 1995 ; Kai-Huan Yu et al, 2008) et partant de
l’hypothèse qu’une plus grande expression de la TS aboutissait à un plus important
métabolisme des folates, nous avons supposé que l’allèle 3R pouvait par conséquent avoir un
effet protecteur par rapport à la survenue du CCR. Nous n’avons retrouvé aucun résultat
allant dans ce sens. Pourtant l’équipe de Ulrich a envisagé cette possibilité et a aboutit à la
conclusion que les polymorphismes du gène TS et la consommation de folates, sous forme de
compléments diététiques avaient un effet interactif sur le risque de cancer. En effet dans cette
étude cas/contrôles de 510 patients souffrant d’un CCR et 604 individus sains, les auteurs de

Résultats et discussion
83
cette étude ont montré que les individus de génotype 3R/3R et ayant une consommation
importante de folates étaient associés à un risque de CCR plus faible ; tandis que les individus
de génotype 2R/2R ayant une consommation aussi importante de folates étaient associés à un
risque plus élevé de CCR (Ulrich et al, 2002).
Cependant d’autres études qui ont recherché l’impact de ce polymorphisme sur la survenue du
CCR ont trouvé des résultats contradictoires. Entre autres, cette étude prospective cas/témoins
portant sur 270 cas de CCR et 454 contrôles dans laquelle les individus de génotype 2R/2R
présentaient un risque plus faible de CCR (Chen et al, 2003). Dans cette étude, les individus
de génotype 2R/2R avaient un taux de folates plasmatiques inférieur à ceux avec le génotype
3R/3R et ces résultats suggeraient donc que ce risque diminué n’était pas influencé par ce
polymorphisme.
Du point de vue pharmacogénétique, étant donné que la TS est une des cibles du 5FU utilisé
dans la chimiothérapie du CCR, plusieurs équipes se sont penchées sur l’analyse de l’impact
du polymorphisme du promoteur de TS sur la réponse à cette molécule (Lacopetta et al,
2001). Plusieurs études ont alors prouvé qu’il existait une relation inverse entre le taux
d’ARNm ou protéique de TS et la réponse anti-tumorale au 5-FU (Lenz et al, 1998 ; Etienne
et al, 2002). Ces travaux ont montré que les malades homozygotes 2R/2R avaient un taux de
réponse au 5-FU supérieur à celui des malades homozygotes 3R/3R et que la présence de
l’allèle 3R est donc associée à une augmentation de l’activité transcriptionnelle (Horie et al,
1995) et par conséquent une diminution de la réponse au 5-FU, mais aussi à une diminution
de la survie (Pullarkat et al, 2001).
Parallèlement à la réponse au 5-FU, une étude récente a montré que ce polymorphisme de TS
était également prédictif de la toxicité au 5-FU avec des taux de toxicité de 43 %, 18 % et 3 %
respectivement pour les génotypes 2R/2R, 2R/3R et 3R/3R (Lecomte et al, 2004). Les sujets
porteurs d au moins un allèle comportant 2 répétitions sont exposés à un risque plus élevé de
toxicité. Ceci s’expliquant par le fait qu’une diminution du taux d’ARNm de la TS dans les
tissus sains des patients CCR de génotype 2R/2R recevant du 5FU ne confère aucune
protection des cellules normales contre les dommages provoqués par cet antimétabolite à
cause d’une trop grande efficacité dans l’inhibition de la TS.

Résultats et discussion
84
Le plus grand nombre de malades de notre population d’étude étaient de génotype 2R/2R ;
ceux-ci sont plus importants en nombre que les autres génotypes et d’après toutes ces
données, cela signifie que ces patients seraient de meilleurs candidats pour une réponse
optimale au 5FU, mais pourraient aussi être sujets à une toxicité plus importante que les
individus présentant les autres génotypes ; il serait donc intéressant de poursuivre cette étude
par l’analyse de la réponse à la chimiothérapie à base de 5FU de ces malades afin d’une part
de confirmer les résultats de ces différentes conclusions sur notre population, et d’autre part
afin de prévoir la réponse à cet anticancéreux et de prévenir les réactions de toxicité à cette
molécule.
Le second polymorphisme du gène TS exploré dans ce travail est celui relatif à la
délétion/insertion de 6pb dans la région 3’UTR de ce gène. Malgré le faible nombre
d’individus enrôlés dans cette étude pour l’exploration de ce polymorphisme, nos résultats
suggèrent qu’il existe un risque plus grand de CCR associé à l’insertion de 6pb « allèle 6pb »
(p=0.018). La même analyse a montré que les deux échantillons cas et témoins sont en
équilibre de Hardy-Weinberg.
Ce polymorphisme a été découvert lors d’une étude faite par (Ulrich et al, 2000). Dans cette
étude, il est suggéré que ce polymorphisme bien que siégeant dans la région 3’UTR non
codante du gène pourrait affecter la stabilité ou la structure secondaire de l’ARNm de la TS
régulant de ce fait le taux d’expression de cette enzyme. Dans notre étude, nous avons trouvé
que les individus porteurs de l’insertion 6pb avaient plus de risque de développer un CCR.
Etant donné les différentes preuves confortant l’hypothèse que la diminution de l’activité de la
TS aurait un impact sur le métabolismes des folates et par conséquent sur la réparation de
l’ADN, ce résultat suggère que l’insertion de 6pb serait corrélée à une plus faible expression
d’ARNm de la TS et que cette plus faible activité de la TS pourrait avoir un effet augmenté
sur la susceptibilité à développer le CCR via une déficience dans le métabolisme des folates.
Une autre explication serait qu’un manque de reserve en thymidine dû à la diminution du taux
de TS entrainerait la cellule sur une voie de cancérisation à cause de l’incorporation d’uracile
au lieu de la thymine dans la chaine d’ADN. Notre résultat est en accord avec d’autres études
qui ont retrouvé une association entre les génotypes 0pb/6pb et 6pb/6pb et le cancer de
l’œsophage d’une part mais aussi du poumon (Zhang et al, 2004 ; Shi et al, 2005).

Résultats et discussion
85
Cependant , les conclusions de ces travaux et les notres sont en contradiction avec une analyse
de fonction sur la signification biologique de la délétion de 6pb dans la région 3’UTR de TS ;
en effet cette étude a montré que la délétion de 6pb se traduisait par une diminution d’environ
50% du taux d’ARNm de la TS par rapport à l’insertion de 6pb et que donc l’activité
enzymatique était tout aussi diminuée (Mandola et al, 2004).
D’autres études ont exploré cette éventuelle association avec le CCR mais n’ont aboutit à
aucun résultat probant (Ulrich et al, 2000 ; Ulrich et al, 2002 ; Chen et al, 2003). Ceci
signifirait que d’autres mécanismes sont impliqués dans cette voie oncogénique et que ceux-ci
restent encore à élucider.
Concernant ce polymorphisme délétion/insertion de 6pb, l’équipe de Lecomte retrouve aussi
une association entre un taux de survie plus important des individus CCR recevant du 5F en
chimiothérapie et l’insertion de 6pb, ce qui suggère selon les conclusions tirées de l’étude du
polymorphisme 2R>3R que cette insertion de 6pb diminuerait le taux d’expression de la
thymidine synthétase. Ceci serait en accord avec nos résultats concernant l’association de ce
polymorphisme et le CCR et contredit les conclusions de l’équipe de Lenz (Lenz et al, 1998).
Paradoxalement, un travail récent (Dotor et al, 2006) a démontré que la délétion de 6pb était
un facteur prédictif d’une meilleure réponse au 5FU et d’un taux de survie plus important
dans le CCR. Les individus porteurs de l’allèle 6pb dans notre échantillon de malades sont en
revanche plus importants que ceux porteurs de l’allèle insertion6pb ; en se basant sur ce
polymorphisme seul et selon cette étude nos patients auraient un taux de réponse au 5FU
moins élevé.
Ce travail que nous avons réalisé sur ce gène nous a permis d’évaluer les fréquences des deux
polymorphismes de la TS, les plus documentés, sur la population CCR Algérienne d’une part,
ce qui pourrait servir à définir les patients susceptibles de bénéficier correctement d’une
chimiothérapie à base de 5FU ; d’autre part sur la population contrôle ce qui nous a permis de
rapporter les fréquences de ces deux variants de la TS caractérisant la population Algérienne
et de les comparer à d’autres populations. Ces données pourraient servir pour d’autres études
cas/témoins sur d’autres pathologies cancéreuses.

Conclusion et perspectives
86
CONCLUSION ET PERSPECTIVES
La recherche de corrélations entre les polymorphismes des gènes du métabolisme et du
transport des xénobiotiques : GSTM1, GSTT1, GSTP1 « 313A>G » et « 341C>T », CYP3A5
6986A>G, MDR1 3435C>T, UGT1A1 « (TA)6>(TA)7 » et « -3156 G>A », TS « 2R>3R »
et TS « délétion/insertion de 6pb » et la survenue du CCR est un travail réalisé pour la
première fois sur la population Algérienne.
Ce travail nous a permis de montrer qu’il n’existait aucune relation d’association entre la
plupart de ces gènes et la survenue du CCR dans notre population en dehors du
polymorphisme « délétion/ insertion de 6pb » du gène TS.
En effet, dans notre population, les individus porteurs de l’insertion de 6pb « allèle 6pb »,
avaient un plus grand risque de développer un CCR que ceux porteurs de la délétion de 6pb
« allèle 0pb ».
Les résultats obtenus dans cette étude semblent exclure donc toute implication des autres
gènes du métabolisme des xénobiotiques explorés au cours de cette étude dans le
développement du CCR dans notre population. Cependant, afin de confirmer ces
observations, il est nécessaire d’élargir l’échantillon étudié pour certains de ces
polymorphismes comme les délétions des gènes GSTM1 et GSTT1, le polymorphisme de
répétition UGT1A1*28(TA)6>(TA)7 et celui du gène TS « 2R>3R ».
La comparaison de la distribution des fréquences des différents polymorphismes étudiés avec
celle de populations d’origine ethniques différentes, nous a permis, d’une part de confronter
nos résultats car les associations peuvent varier suivant les populations et d’autre part de
contribuer à mettre à jour de nouveaux gènes de susceptibilité au CCR comme celui du gène
TS dans notre population.
Les différents gènes explorés au cours de ce travail codent pour des enzymes ayant une
fonction dans le métabolisme des xénobiotiques dont les substances carcinogènes présentes
dans l’alimentation, entre autres, mais aussi les molécules utilisées dans la chimiothérapie du
CCR comme le gène TS dans la réponse au 5FU, le gène UGT1A1 dans la toxicité à

Conclusion et perspectives
87
l’Irinotécan et le gène GSTP1 dans celle à l’Oxaliplatine. Pour cette raison, il serait
intéressant de compléter ce travail par différents volets importants :
Le premier que nous envisageons est une stratification des résultats en fonction du mode
d’alimentation et de l’exposition à d’autres facteurs favorisant le cancer comme le tabagisme.
En effet, les polymorphismes étudiés dans ce travail présentent de grandes variabilités
ethniques, et une corrélation avec ces variantes importantes dans le mode de vie serait plus
concluante pour la recherche de leurs éventuelles associations avec le risque du CCR
caractérisant notre population.
Le second volet que nous avons amorcé et que nous envisageons de poursuivre est le suivi de
la réponse à la chimiothérapie à base de 5FU, d’Irinotécan et d’Oxaliplatine des patients CCR
génotypés pour les gènes codant pour la TS, ou UGT1A1 ou encore les GST impliqués dans
la réponse et/ou la toxicité de ces molécules afin d’optimiser les dosages de ces différents
traitements et de prédire leurs éventuels effets toxiques.
Enfin, toutes les données obtenues dans cette étude sur la distribution des différents allèles de
chacun des gènes explorés dans notre population témoin, pourraient être mises à profit afin de
rechercher d’autres associations avec d’autres pathologies dans lesquelles ces gènes
interviennent.

Références bibliographiques
88
RÉFERENCES BIBLIOGRAPHIQUES
• Aaltonen LA, Peltomaki P, Leach FS. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812
• Alexandrie AK, Rannug A, Juronen E, Tasa G, Warholm M. Detection and characterization of a novel functional polymorphism in the GSTT1gene. Pharmacogenetics 2002, 12 : 613-9.
• Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase k gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem 1997;272(15): 10004-12.
• Allorge D, Loriot MA. La pharmacogénétique ou la promesse d’une médecine personnalisée: variations du métabolisme et du transport des médicaments. Ann. Biol. Clin. 2004; 62: 499-511.
• Ameziane N, Bogard M, Lamoril J, Bogard M. Principes de biologie moléculaire en biologie clinique. Publié par Elsevier Masson, 2006.
• Aparicio T, Ducreux M and Chaussade S. 5-fluorouracile : données sur le métabolisme et place actuelle dans le traitement des cancers digestifs, Gastroenterol. Clin. Biol. 26, 2002, pp. 38–47
• Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev. 2008 Jun;34(4):368-77. Review.
• Ateş NA, Tamer L, Ateş C, Ercan B, Elipek T, Ocal K, Camdeviren H. Glutathione S-transferase M1, T1, P1 genotypes and risk for development of colorectal cancer Biochem Genet. 2005. Apr;43(3-4):149-63.
• Beaune P, Loriot M. Bases moléculaires de la susceptibilité aux xénobiotiques: aspects métaboliques. Médecine/Sciences 2000 ; 16 : 1051-6.
• Beggs AD, Abulafi M, Hodgson SV.The DCC gene and colorectal cancer: the story is more complex. Colorectal Dis. 2008 Jul; 10(6):630.
• Benhamou S, Lee WJ, Alexandrie AK, et al. Meta- and pooled analyses of the effects of glutathione S-transferase M1 polymorphisms and smoking on lung cancer risk. Carcinogenesis 2002 ; 23 : 1343-50.
• Bergheim I, Bode C and Parlesak A. Decreased expression of cytochrome P450 protein in non-malignant colonic tissue of patients with colonic adenoma BMC Gastroenterology 2005, 5:34.
• Bernal ML, Sinues B, Fanlo A, Mayayo E. Frequency distribution of C3435T mutation in exon 26 of the MDR1 gene in a Spanish population Ther Drug Monit. 2003 Feb;25(1):107-11.
• Bethke L, Webb E, Sellick G, Rudd M, Penegar S, WitheyL, Qureshi M and Houlston R. Polymorphisms in the cytochrome P450 genes CYP1A2, CYP1B1, CYP3A4, CYP3A5, CYP11A1, CYP17A1, CYP19A1 and colorectal cancer risk BMC Cancer 2007, 7:123.
• Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA 1998, 95:8170–8174.
• Beutler E. Study of glucose-6-phoshate deshydrogenase: history and molecular biology. Am. J. Hematol. 1993; 42: 53-58.
• Bjork J, Akerbrant H, Iselius L, Alm T, Hultcrantz R. Epidemiology of familial adenomatous polyposis in Sweden: changes over time and differences in phenotype between males and females. Scand J Gastroenterol. 1999; 34: 1230–5.
• Board PG, Webb GC, Coggan M: Isolation of a cDNA clone and localization of the human glutathione S-transferase 3 genes to chromosome bands 11q13 and 12q13-14. Ann Hum Genet 1989, 53: 205-213.
• Bosma P.J, Chowdhury J.R, Bakker C, Gantla S, de Boer A, Oostra B.A, Lindhout D, Tytgat G.N, Jansen P.L, Oude Elferink R.P. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome, N. Engl. J. Med. 333, 1995, 1171–1175.
• Boyle P, Ferlay J, Cancer incidence and mortality in Europe 2004, Ann Oncol, 2005; 16, 481-8. • Carreras C.W and Santi D.V. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev.
Biochem 1995, 64, 721--762. • Cascorbi I, Gerloff T, Johne A, et al. Frequency of single nucleotide polymorphisms in the P-glycoprotein
drug transporter MDR1 gene in White subjects. Clin Pharmacol Ther 2001; 69:169–74. • Cavalieri E, Frenkel K, Liehr J. G, Rogan E, and Roy D. Estrogens as endogenous genotoxic agents: DNA
adducts and mutations. J. Natl. Cancer Inst. Monogr, 2000, 27: 75–93. • Ceppa F, Kenane N, Chellak S, Bigaillon C and Burnat P. Curarisation prolongée à la succinylcholine liée à
un variant silencieux de la cholinestérase plasmatique, Ann. Fr. Anesth. Reanim. 24; 2005, pp. 425–427.

Références bibliographiques
89
• Charasson V, Bellott R, Meynard D, Longy M, Gorry P and Robert J. Pharmacogenetics of human carboxylesterase 2, an enzyme involved in the activation of irinotecan into SN-38, Clin. Pharmacol. Ther. 76 2005, pp. 528–535.
• Chelbi H, Lachheb J, Ammar J, Hamzaoui K, and Hamzaoui A. Association of GST Genes Polymorphisms with Asthma in Tunisian Children Hindawi Publishing Corporation Mediators of Inflammation Volume 2007, Article ID 19564, 6 pages.
• Chen J, Hunter D.J, Stampfer M.J, Kyte C, Chan W, Wetmur J.G, Mosig R, Selhub J and Ma J. Polymorphism in the thymidylate synthase promoter enhancer region modifies the risk and survival of colorectal cancer. Cancer Epidemiol. Biomarkers Prev 2003, 12, 958--962.
• Chen K, Jiang QT, He HQ: Relationship between metabolic enzyme polymorphism and colorectal cancer. World J Gastroenterol 2005, 11:331-335.
• Chen K, Jin M, Zhu Y, Jiang Q, Yu W, Ma X and Yao K, Genetic polymorphisms of the uridine diphosphate glucuronosyltransferase 1A7 and colorectal cancer risk in relation to cigarette smoking and alcohol drinking in a Chinese population, J. Gastroenterol. Hepatol. 21; 2006, pp. 1036–1041.
• Chenevix-Trench G, Young J, Coggan M, and Board P. Glutathione S-transferase M1 and T1 polymorphisms: susceptibility to colon cancer and age of onset. Carcinogenesis (Lond) 1995; 16: 1655–1657.
• Cross AJ, Sinha R: Meat-related mutagens/carcinogens in the etiology of colorectal cancer.Environ Mol Mutagen 2004, 44(1):44-55.
• D’Errico A, Malats N, Vineis P, Boffetta P. Review of studies of selected metabolic polymorphisms and cancer. IARC Sci Publ 1999; 148 : 323-93.
• Dandara C, Ballo R and Parker M. CYP3A5 genotypes and risk of oesophageal cancer in two South African populations Cancer Lett. 2005 Jul 28; 225(2):275-82.
• Dansette PM, Bonierbale E, Minoletti C, Beaune PH, Pessayre D, Mansuy D Drug-induced immunotoxicity Eur J Drug Metab Pharmacokinet. 1998 Oct-Dec;23(4):443-51.
• De Chaisemartin L, Loriot MA. Pharmacogenetics of anticancer drugs Revue générale Pathologie Biologie 53, 2005 ; 116–124.
• De Jong F.A, De Jonge M.J.A, Verweij J and Mathijssen R.H.J. Role of pharmacogenetics in irinotecan therapy, Cancer Lett. 234, 2006, pp. 90–106.
• De Jong M.M, Nolte I.M, te Meerman G.J, Van der Graaf, W.T.A, de Vries E.G.E, Sijmons R.H, Hofstra R.M.W and Kleibeuker J.H. Low-penetrance genes and their involvement in colorectal cancer susceptibility. Cancer Epidemiol Biomarkers Prev 2002; 11, 1332--1352.
• Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 2003;43:149–73.
• Dotor E, Cuatrecases M, Martínez-Iniesta M, Navarro M, Vilardell F, Guinó E, Pareja L, Figueras A, Molleví DG, Serrano T, de Oca J, Peinado MA, Moreno V, Germà JR, Capellá G, Villanueva A. Tumor Thymidylate Synthase 1494del6 Genotype As a Prognostic Factor in Colorectal Cancer Patients Receiving Fluorouracil-Based Adjuvant Treatment. Journal of Clinical Oncology 2006, Vol 24, No 10 (April 1), pp. 1603-1611.
• Duthie, S. J. Folic acid deficiency and cancer: mechanisms of DNA instability. Br.Med. Bull 1999, 55: 578–592.
• Elexpuru-Camiruaga J, Buxton N, Kandula V, Dias PS, Campbell D, McIntosh J, Broome J, Jones P, Inskip A, Alldersea J Susceptibility to astrocytoma and meningioma: influence of allelism at glutathione S-transferase (GSTT1 and GSTM1) and cytochrome P-450 (CYP2D6) loci Cancer Res. 1995 Oct 1;55(19):4237-9
• Engel LS, Taioli E, Pfeiffer R, et al. Pooled analysis and metaanalysis of glutathione S-transferase M1 and bladder cancer : a HuGE review. Am J Epidemiol 2002 ; 156 : 95-109.
• Etienne MC, Chazal M, Laurent-Puig P, Magne N, Rosty C, Formento JL, et al. Prognostic value of tumoral thymidylate synthase and p53 in metastatic colorectal cancer patients receiving fluorouracil-based chemotherapy: phenotypic and genotypic analyses. J Clin Oncol 2002; 20: 2832-43.
• European Journal of Cancer. Volume 3, Octobre 2005. p 291 • Farnood A, Naderi N, Moghaddam SJ, Noorinayer B, Firouzi F, Aghazadeh R, daryani NE, Zali MR The
frequency of C3435T MDR1 gene polymorphism in Iranian patients with ulcerative colitis. Int J Colorectal Dis. 2007 Sep; 22(9):999-1003.
• Fenech M. The role of folic acid and Vitamin B12 in genomic stability of human cells. Mutat. Res, 2001, 475: 57–67.

Références bibliographiques
90
• Garsa A, McLeod HL and Marsh S. CYP3A4 and CYP3A5 genotyping by Pyrosequencing BMC Med Genet. 2005; 6: 19.
• Gervasini G, García-Martín E, Ladero JM, Pizarro R, Sastre J, Martínez C, García M, Diaz-Rubio M, Agúndez JA.Genetic variability in CYP3A4 and CYP3A5 in primary liver, gastric and colorectal cancer patients.BMC Cancer. 2007 Jul 2;7:118.
• Giachelia M, Palma S, Appolloni M, Padua L, Tranfo G, Spagnoli M, Tirindelli D, Cozzi R. Occupational exposure to antineoplastic agents induces a high level of chromosome damage. Lack of an effect of GST polymorphisms Antonella Testa. Toxicology and Applied Pharmacology 223, 2007; 46–55.
• Gibson P, Gill JH, Khan PA, Seargent JM, Martin SW, Batman PA, Griffith J, Bradley C, Double JA, Bibby MC, Loadman PM: Cytochrome P450 1B1 (CYP1B1) is overexpressed in human colon adenocarcinomas relative to normal colon: implications for drug development. Mol Cancer Ther 2003, 2:527-534.
• Giovannucci E, Martinez ME. Tobacco, colorectal cancer, and adenomas: a review of the evidence. J Natl Cancer Inst 1996; 88:1717 – 30.
• Giovannucci E, Rimm E. B, Ascherio A, Stampfer M. J, Colditz G. A, and Willett W. C. Alcohol, low-methionine–low-folate diets, and risk of colon cancer in men. J. Natl. Cancer Inst, 1995, 87: 265–273.
• Gosse-Brun S, Sauvaigo S, Daver A, Larra F, Kwiatkowski F, Bignon YJ, Bernard-Gallon D. Association between H-ras minisatellite and colorectal cancer risk. Anticancer Res 1998; 18: 2611–2616.
• Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet 1995;29:607–49.
• Gottesman MM, Ling V. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 2006; 580: 998–1009.
• Gottesman MM, Pastan I, Ambudkar SV. P-glycoprotein and multidrug resistance. Curr Opin Genet Dev 1996;6: 610-617.
• Gruber SB, Moreno V, Rozek LS, Rennert HS, Lejbkowicz F, Bonner JD, Greenson JK, Giordano TJ, Fearon ER, Rennert G: Genetic Variation in 8q24 Associated with Risk of Colorectal Cancer. Cancer Biol Ther 2007, 6(7).
• Habdous M, Sies Gt, Herbeth, Viry M,Visvikis S. Polymorphismes des glutathion S-transférases et pathologies humaines : bilan des études épidémiologiques Ann Biol Clin 2004, 62 : 15-24 Revue générale
• Hagler L, Herman RH Oxalate metabolism. IV Am J Clin Nutr. 1973 Oct;26(10):1073-9. • Hamilton J and Meltzer S. A Review of the Genomics of Gastric Cancer Clinical Gastroenterology AND
Hepatology 2006; 4:416–425. • Harris M.J, Coggan M, Langton L, Wilson S.R and Board P.G. Polymorphism of the Pi class glutathione S-
transferase in normal populations and cancer patients. Pharmacogenetics 1998, 8, 27—31. • Hayes JD, Flanagan JU, Jowsey IR: Glutathione transferases. Annu Rev Pharmacol Toxicol 2005, 45: 51-
88. • Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated
defence against oxidative stress. Free Radic Res 1999 ; 31 : 273-300. • Hayes JD, Strange RC. Glutathione S-transferase polymorphisms and their biological consequences.
Pharmacology 2000; 61 : 154-66. • Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J,Johne A, et al. Functional polymorphisms of
the human multidrugresistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA 2000;97:3473-3478.
• Horie N, Aiba H, Oguro K, Hojo H, Takeishi K Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5'-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct. 1995 Jun;20(3):191-197..
• Houlston R.S. and Tomlinson I.P. Polymorphisms and colorectal tumor risk. Gastroenterology 2001, 121, 282--301.
• Hustert E, Haberl M, Burk O, Wolbold R, He Y.Q and K. Klein. The genetic determinants of the CYP3A5 polymorphism, Pharmacogenetics 11, 2001, 773–779.
• Innocenti F, Grimsley C, Das S, Ramirez J, Cheng C, Kuttab-Boulos H. Haplotype structure of the UDP-glucuronosyltransferase 1A1 promoter in different ethnic groups. Pharmacogenetics 2002;12(9):725-33.
• Innocenti F, Undevia SD, Iyer L, Xian Chen P, Das S, Kocherginsky M. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predicts the risk of severe neutropenia of irinotecan. J Clin Oncol 2004;22:1382–8.
• Iravani S, Mao W, Fu L, Karl R, Yeatman T, Jove R, Coppola D. Elevated c-src protein expression is an early event in colonic neoplasia. Lab Invest 1998; 78: 365-371.

Références bibliographiques
91
• Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TJ. Activating src mutation in a subset of advanced human colon cancers. Nature genetics 1999; 21: 187-191.
• Irigaray P, Newby J.A, Clapp R, Hardell L, Howard V, Montagnier L, Epstein S, Belpomme D. Lifestyle-related factors and environmental agents causing cancer: An overview Biomedicine & Pharmacotherapy, 2007; 6 , 640-658
• James S. J, Basnakian A. G., and Miller B. J. In vitro folate deficiency induces deoxynucleotide pool imbalance, apoptosis, and mutagenesis in Chinese hamster ovary cells. Cancer Res, 54: 5075–5080, 1994.
• Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics. CA Cancer J. Clin, 2006 ; 56,106–130
• Jia Chen,2 David J. Hunter, Meir J. Stampfer, Charles Kyte, Wendy Chan, James G. Wetmur, Rebecca Mosig, Jacob Selhub, and Jing Ma Polymorphism in the Thymidylate Synthase Promoter Enhancer Region Modifies the Risk and Survival of Colorectal Cancer1 Cancer Epidemiology, Biomarkers & Prevention 2003 Vol. 12, 958–962.
• Johnston PG, Lenz HJ, Leichman CG, Danenberg KD, Allegra CJ, Danenberg PV. Thymidylate synthase gene and protein expression correlate and are associated with response to 5-fluorouracil in human colorectal and gastric tumors. Cancer Res 1995; 55: 1407-12.
• Kaderlik K.R, Mulder G.J, Shaddock J.G, Casciano D.A, Teitel C.H, Kadlubar F.F, Effect of glutathione depletion and inhibition of glucuronidation and sulfation on 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) metabolism, PhIP-DNA adduct formation and unscheduled DNA synthesis in primary rat hepatocytes, Carcinogenesis 15, 1994, 1711–1716.
• Kai-Huan Yu, Wei-Xing Wang, You-Ming Ding, Hui Li, Ze-Sheng Wang Polymorphism of thymidylate synthase gene associated with its protein expression in human colon cancer World J Gastroenterol 2008 January 28; 14(4): 617-621.
• Kato I, Dnistrian A. M, Schwartz M, Toniolo P, Koenig K, Shore R. E, Akhmedkhanov A, Zeleniuch-Jacquotte A, and Riboli E. Serum folate, homocysteine and colorectal cancer risk in women: a nested case-control study. Br. J. Cancer 1999, 79: 1917–1922.
• Katoh T, Nagata N, Kuroda Y, Itoh H, Kawahara A, Kuroki N, Ookuma R, Bell DA Glutathione S-transferase M1 (GSTM1) and T1 (GSTT1) genetic polymorphism and susceptibility to gastric and colorectal adenocarcinoma Carcinogenesis. 1996 Sep; 17(9):1855-9.
• Kawakami K, Omura K, Kanehira E and Watanabe Y. Polymorphic tandem repeats in the thymidylate synthase gene is associated with its protein expression in human gastrointestinal cancers. Anticancer Res. 1999, 19, 3249—3252.
• Kempkes M, Golka K, Reich S, Reckwitz T, Bolt HM. Glutathione S-transferase GSTM1 and GSTT1 null genotypes as potential risk factors for urothelial cancer of the bladder. Arch Toxicol 1996 ; 71 : 123-6.
• Kenji Inagaki, Kyoko Kasuya and Yoshinori Kidani Binding behavior of antitumor r,r-1,2-cyclohexanediamine platinum(ii) with oligonucleotides and DNA Chemistry letters 1984 Vol.13 , No.2 ;171-174.
• Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D Identification of FAP locus genes from chromosome 5q21 Science. 1991 Aug 9;253(5020):661-5.
• Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971 Apr;68(4):820-3.
• Kohn LT. Organizing and managing care in a changing health system Health Serv Res. 2000 Apr;35, 37-52. • Kohonen-Corish MR, Sigglekow ND, Susanto J, Chapuis PH, Bokey EL, Dent OF, Chan C, Lin BP, Seng TJ,
Laird PW, Young J, Leggett BA, Jass JR, Sutherland RL. Promoter methylation of the mutated in colorectal cancer gene is a frequent early event in colorectal cancer. Oncogene. 2007 Jun 28;26(30):4435-41.
• Komoto C, Nakamura T, Sakaeda T, Kroetz DL, Yamada T, Omatsu H, Koyama T, Okamura N, Miki I, Tamura T, Aoyama N, Kasuga M, Okumura K. MDR1 haplotype frequencies in Japanese and Caucasian, and in Japanese patients with colorectal cancer and esophageal cancer. Drug Metab Pharmacokinet. 2006 Apr;21(2):126-32.
• Kubo T, Kim SR, Sai K, Saito Y, Nakajima T, Matsumoto K, Saito H, Shirao K, Yamamoto N, and Minami H. Functional characterization of three naturally occurring single nucleotide polymorphisms in the CES2 gene encoding carboxylesterase 2 (HCE-2). Drug Metab Dispos , 2005, 33: 1482–1487.
• Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression Nat Genet. 2001 Apr;27(4):383-391.

Références bibliographiques
92
• Kweekel D, Guchelaar H-J , Gelderblom H. Clinical and pharmacogenetic factors associated with irinotecan toxicity Cancer Treatment Reviews 2008; 34, 656– 669.
• Lacopetta B, Grieu F, Joseph D, Elsaleh H. A polymorphism in the enhancer region of the thymidylate synthase promoter influences the survival of colorectal cancer patients treated with 5-fluorouracil. Br J Cancer 2001; 85: 827-30.
• Lamoril J; Deybach J-C; Bouizegarene P. L'instabilité des microsatellites dans les cancers du côlon. Immuno-analyse & biologie spécialisée 2006, vol. 21, no4, pp. 211-222.
• Lecomte T, Ferraz JM, Zinzindohoue F, Loriot MA, Tregouet DA, Landi B, et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Cancer Res 2004; 10: 5880-8.
• Lecomte T, Landi B, Beaune P, Laurent-Puig P and Loriot MA. Glutathione S-Transferase P1Polymorphism (Ile105Val) Predicts Cumulative Neuropathy in Patients Receiving Oxaliplatin-Based Chemotherapy Clin Cancer Res 2006; 12(10) May 15.
• Lee SJ, Cho SH, Park SK, et al. Combined effect of glutathione S-transferase M1 and T1 genotypes on bladder cancer risk. Cancer Lett 2002 ; 177 : 173-9.
• Lee Y.-L, Lin Y.-C, Lee Y.-C,.Wang J-Y, Hsiue T-R, and Guo Y. L. Glutathione S-transferase P1 gene polymorphism and air pollution as interactive risk factors for childhood asthma. Clinical and Experimental Allergy, 2004, vol. 34, no. 11, pp. 1707– 1713.
• Lenz HJ, Danenberg KD, Leichman CG, Florentine B,Johnston PG, Groshen S, et al. p53 and thymidylate synthase expression in untreated stage II colon cancer: associations with recurrence, survival, and site. Clin Cancer Res 1998; 4: 1227-34.
• Levine AJ; Perry ME; Chang A; Silver A; Dittmer M; Welsh D. The 1993 Walter Huber Lecture: the role of the p53 tumour-suppressor gene in tumuorigenesis. Br J Cancer. 1994;69:409–416
• Li Zhenhua, Norihiko Tsuchiya, Shintaro Narita, Takamitsu Inoue,Yohei Horikawa, Hideaki Kakinuma, Tetsuro Kato, Osamu Ogawa, Tomonori Habuchi. CYP3A5 gene polymorphism and risk of prostate cancer in a Japanese population Cancer Letters 225, 2005, 237–243.
• Lièvre A, Laurent-Puig P. Molecular biology in clinical cancer research: the example of digestive cancers Rev Epidemiol Sante Publique, 2005, 53: 267-282.
• Lin D, Meyer D. J, Ketterer B, Lang N. P, and Kadlubar F. F. Effects of human and rat glutathione S-transferases on the covalent bonding of the N-acetoxy derivatives of heterocyclic amine carcinogens in vitro: a possible mechanism of organ specificity in their carcinogenesis. Cancer Res 1994, 54: 4920–4926.
• Lin H.J, Probst-Hench N.M, Ingles S.A, Han C.Y, Lin B.K, Lee D. B, Frankl H.D, Lee E.R, Longnecker M.P, and Haile R.W. Glutathione S-transferase (GSTM1) null genotype, smoking and prevalence of colorectal adenomas. Cancer Res., 55: 1224–1226, 1995.
• Little J, Sharp L, Masson LF, Brockton NT, Cotton SC, Haites NE, Cassidy J Colorectal cancer and genetic polymorphisms of CYP1A1, GSTM1 and GSTT1: a case-control study in the Grampian region of Scotland Int J Cancer. 2006 Nov 1;119 (9):2155-64.
• Livak K.J., Marmaro J., and Todd J.A.. Towards fully automated genome-wide polymorphism screening [letter]. Nat. Genet1995. 9:341–342.
• Lo H-W and Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance Current Opinion in Pharmacology 2007, 7: 367–374
• Longley DB, Harkin P, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3:330–8.
• Longuemaux S, Delomenie C, Gallou C, Mejean A, Vincent-Viry M. Candidate genetic modifiers of individual susceptibility to renal cell carcinoma : a study of polymorphic human xenobiotic-metabolizing enzymes. Cancer Res 1999, 59: 2903-2908.
• Loriot MA, Philippe Beaune P. La pharmacogénétique : le lien entre gènes et réponse aux médicaments M/S : Médecine sciences, 2004 ; Volume 20, numéros 6-7.
• Lynch HT, Smyrk T, Lynch JF. Overview of natural history, pathology, molecular genetics and management of HNPCC (Lynch Syndrome). Int J Cancer 1996;69:38–43.
• Ma QW, Lin GF, Chen JG, Shen JH. Polymorphism of glutathione S-transferase T1, M1 and P1 genes in a Shanghai population : patients with occupational or non-occupational bladder cancer. Biomed Environ Sci 2002 ; 15 : 253-60.
• Mammano E, Belluco C, Bonafé M, Olivieri F, Mugianesi E, Barbi C, Mishto M, Cosci M, Franceschi C, Lise M and Nitti D. Association of p53 polymorphisms and colorectal cancer: Modulation of risk and progression European Journal of Surgical Oncology, 2008; 1-5.

Références bibliographiques
93
• Mandola M-V, Stoehlmacher J, Zhang W, Groshen S, Mimi C. Yu, Syma Iqbal, Heinz-Josef Lenz and Robert D. Ladner A 6 bp polymorphism in the thymidylate synthase gene causes message instability and is associated with decreased intratumoral TS mRNA levels Pharmacogenetics 2004, 14:319–327.
• Marcuello E, Altes A, Del Rio E, Cesar A, Menoyo A and Baiget M. Single nucleotide polymorphism in the tandem repeat sequences of thymidylate synthase gene predicts for response to fluorouracil-based chemotherapy in advances colorectal cancer patients, Int. J. Cancer 112 , 2004, pp. 733–737.
• Marsh S, Collie-Duguid E.S, Li T, Liu X, and McLeod, H.L. Ethnic variation in the thymidylate synthase enhancer region polymorphism among Caucasian and Asian populations. Genomics, 58: 310–312, 1999.
• Martínez C, Martín F, Fernández JM, García-Martín E, Sastre J, Díaz-Rubio M, Agúndez JA, Ladero JM. Glutathione S-transferases mu 1, theta 1, pi 1, alpha 1 and mu 3 genetic polymorphisms and the risk of colorectal and gastric cancers in humans.Pharmacogenomics 2006 Jul;7(5):711-8.
• Mattano S. S, Palella T. D, and Mitchell B. S. Mutations induced at the hypoxanthine-guanine phosphoribosyltransferase locus of human T-lymphoblasts by perturbations of purine deoxyribonucleoside triphosphate pools. Cancer Res., 50: 4566–4571, 1990.
• Mc Leod HL, Collie-Duguid ESR, Vrecken P, et al. Nomenclature for human DPYD alleles. Pharmacogenetics 1998; 8: 455–9.10.
• Meta-analysis Group In Cancer. Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors. J Clin Oncol 1998;16:3537–41.
• Milano GA, Etienne MC, Pierrefite V, Barberi-Heyob M, Deporte-Fety R, Renée N. Dihydropyrimidine deshydrogenase deficiency and fluorouracil-related tocixity. Br J Cancer 1999;79:627–30.
• Miller S.A, Dykes D and Polsky H.F. A simple salting out procedure for extracting DNA from human nucleated cell. Nucleic Acids Research 1988 ; 16 : 12.
• Mitchell R. J, Farrington
S. M, Dunlop
M. G., and Campbell H. Mismatch Repair Genes hMLH1 and hMSH2
and Colorectal Cancer: A HuGE Review American Journal of Epidemiology November 15, 2002 volume 156 numero 10.
• Moore Lee E., Huang W, Nilanjan C, Gunter M, Chanock S, Meredith Y, WelchB, PinskyP, Weissfeld J, and Hayes R. GSTM1, GSTT1, and GSTP1 Polymorphisms and Risk of Advanced Colorectal Adenoma Cancer Epidemiol Biomarkers Prev 2005; 14(7).
• Murray GI. The role of cytochrome P450 in tumour development and progression and its potential in therapy. J Pathol 2000; 192:419-26.
• Nagasubriamanian R, Innocenti F and Ratian M.J. Pharmacogenetics in cancer treatment, Annu. Rev. Med. 54 (2003), 437–452.
• Nebert DW, Adesnik M, Coon MJ, Estabrook RW, Gonzalez FJ, Guengerich FP, Gunsalus IC, Johnson EF, Kemper B, Levin W. The P450 gene superfamily: recommended nomenclature. DNA. 1987; 6(1):1-11.
• Nedelcheva Kristensen V, AndersenTI, Erikstein B,et al. Single tube multiplex polymerase chain reaction genotype analysis of GSTM1, GSTT1 and GSTP1: relation of genotypes toTP53 tumor status and clinicopathological variables in breast cancer patients. Pharmacogenetics 1998;8:441-447.
• Nijhoff W. A, Grubben M. J. A, Nagengast F. M, Jansen J. B. M. J, Verhagen H, van Poppel G, and Peters W. H. M. Effects of consumption of brussels sprouts on intestinal and lymphocytic glutathione and glutathione S transferases in humans. Carcinogenesis (Lond), 1995, 16: 2125–2128.
• Nilbert M, Kristoffersson U, Ericsson M, Johannsson O, Rambech E, and Mangell P. Broad phenotypic spectrum in familial adenomatous polyposis; from early onset and severe phenotypes to late onset of attenuated polyposis with the first manifestation at age 72 BMC Med Genet. 2008; 9: 101.
• Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, et al. Li-Fraumeni and related syndromes : correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003 ; 63 : 6643-50
• Osswald E, Johne A, Laschinski G, Arjomand-Nahad F, Malzahn U, Kirchheiner J, Gerloff T, Meisel C, Mrozikiewicz PM, Chernov J, Roots I, Köpke K.Association of MDR1 genotypes with susceptibility to colorectal cancer in older non-smokers. Eur J Clin Pharmacol. 2007 Jan;63(1):9-16.
• Ozawa S, Soyama A, Saeki M, Fukushima-Uesaka H, Itoda M, Koyano S, Sai K, Ohno Y, Saito Y, Sawada J Ethnic differences in Genetic Polymorphisms of CYP2D6, CYP2C19, CYP3As and MDR1/ABCB1. Drug Metab Pharmacokinet. 2004 Apr;19(2):83-95. Review.
• Park GT, Lee OY, Kwon SJ, Lee CG, Yoon BC, Hahm JS, Lee MH, Hoo Lee D, Kee CS, Sun HS: Analysis of CYP2E1 polymorphism for the determination of genetic susceptibility to gastric cancer in Koreans. J Gastroenterol Hepatol 2003, 18:1257-1263.
• Park JY, Schantz SP, Stern JC, Kaur T, Lazarus P. Association between glutathione p genetic polymorphisms and oral cancer risk. Pharmacogenetics 1999, 9 : 497-504

Références bibliographiques
94
• Parke DV: The cytochromes P450 and mechanisms of chemical carcinogenesis. Environ Health Perspect 1994, 102:852-853.
• Perera F.P. Environment and cancer: who are susceptible?, Science 1997, 278 , pp. 1068–1073. • Petrova DT, Nedeva P, Maslyankov S, Toshev S, Yaramov N, Atanasova S, Toncheva D, Oellerich M, von
Ahsen N. J Cancer Res Clin Oncol. (No association between MDR1 (ABCB1) 2677G>T and 3435C>T polymorphism and sporadic colorectal cancer among Bulgarian patients. 2008 Mar; 134(3):317-22.
• Petrova DT, Yaramov N, Toshev S, Nedeva P, Maslyankov S, von Ahsen N, Oellerich M, Toncheva D Genotyping of CYP3A5 polymorphisms among Bulgarian patients with sporadic colorectal cancer and controls. Onkologie. 2007 Nov;30(11):559-63.
• Plummer S.J, Conti D.V, Paris P.L , Curran A.P, Casey G and Witte J.S. CYP3A4 and CYP3A5 genotypes, haplotypes, and risk of prostate cancer, Cancer Epidemiol. Biomarkers Prev. 12 (2003), pp. 928–932
• Potter JD, Bigler J, Fosdick L, et al. Colorectal adenomatous and hyperplastic polyps: smoking and N-acetyltransferase 2 polymorphisms. Cancer Epidemiol Biomarkers Prev 1999; 8:69 – 75.
• Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359: 235–237.
• Prochaska HJ, Talalay P. Regulatory mechanisms of monofunctional and bifunctional anticarcinogenic enzyme inducers in murine liver. Cancer Res 1988; 48:4776 – 82.
• Pullarkat ST, Stoehlmacher J, Ghaderi V, Xiong YP, Ingles SA, Sherrod A, et al. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J 2001; 1: 65-70.
• Quaranta S, Chevalier D, Allorge D, Lo-Guidice JM, Migot-Nabias F, Kenani A, Imbenotte M, Broly F, Lacarelle B, Lhermitte M Ethnic differences in the distribution of CYP3A5 gene polymorphisms Xenobiotica. 2006 Dec;36(12):1191-200.
• Queneau P, Banwarth B, Carpentier F, Guliana JM, Bouget J, Trombert B, Leverve X et l’APNET. Effets indésirables médicamenteux observés dans des Services d’Accueil et d’Urgences français (Etude prospective de l’APNET et propositions pour des mesures préventives). Bull. Acad. Natle Med. 2003 ; 187 : 647-670.
• Radparvar S, Houghton P.J. and Houghton J.A. Characteristics of thymidylate synthase purified from a human colon adenocarcinoma. Arch. Biochem. Biophys 1988, 260, 342--350.
• Registre du cancer d’Oran 2006. • Rodriguez-Antona C, Ingelman-Sundberg M: Cytochrome P450 pharmacogenetics and cancer. Oncogene
2006, 25(11):1679-1691. • Ross VL, Board PG, Webb GC: Chromosomal mapping of the human Mu class glutathione S-transferases to
1p13. Genomics 1993, 18:87-91. • Rustum Y. M, Harstrick A, Cao S, Vanhoefer U, Yin M. B, Wilke H and Seeber S. Thymidylate synthase
inhibitors in cancer therapy: direct and indirect inhibitors. J. Clin. Oncol1997, 15: 389–400. • Sachse C, Smith G, Wilkie M.J, Barrett J.H, Waxman R, Sullivan F, Forman D, Bishop D.T and Wolf C.R. A
pharmacogenetic study to investigate the role of dietary carcinogens in the etiology of colorectal cancer. Carcinogenesis 2002, 23, 1839--1849.
• Sanghani SP, Quinney SK, Fredenburg TB, Davis WI, Murry DJ, and Bosron WF. Hydrolysis of irinotecan and its oxidative metabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab Dispos 2004, 32: 505–511.
• Santi D.V, McHenry C.S, and Sommer H. Mechanism of interaction of thymidylate synthetase with 5- fluorodeoxyuridylate. Biochemistry 1974. 13:471-81.
• Schiel MA, Green SL, Davis WI, Sanghani PC, Bosron WF, Sanghani SPExpression and characterization of a human carboxylesterase 2 splice variant J Pharmacol Exp Ther. 2007 Oct; 323(1):94-101.
• Schinkel AH. The physiological function of drug-transporting P glycoproteins. Semin Cancer Biol 1997;8: 161-170.
• Schneider J, Bernges U, Philipp M, Woitowitz HJ. GSTM1, GSTT1, and GSTP1 polymorphism and lung cancer risk in relation to tobacco smoking. Cancer Lett 2004; 208: 65-74.
• Schneider-Stock R, Boltze C, Peters B, Szibor R, Landt O, Meyer F, and Roessner A. Selective Loss of Codon 72 Proline p53 and Frequent Mutational Inactivation of the Retained Arginine Allele in Colorectal Cancer Neoplasia. 2004 September; 6(5): 529–535
• Sekine and Saijo N. Polymorphisms of metabolizing enzymes and transporter proteins involved in the clearance of anticancer agents, Ann. Oncol. 12, 2001, pp. 1515–1525.

Références bibliographiques
95
• Sherratt PJ, Pulford DJ, Harrison DJ, Green T, Hayes JD: Evidence that human class Theta glutathione S-transferase T1-1 can catalyse the activation of dichloromethane, a liver and lung carcinogen in the mouse. Comparison of the tissue distribution of GST T1-1 with that of classes Alpha, Mu and Pi GST in human. Biochem J 1997, 326: 837-846.
• Shi Q, Zhang Z, Neumann, Li G, Spitz M and Wei. Case--control analysis of thymidylate synthase polymorphisms and risk of lung cancer. Carcinogenesis 2005. vol.26 no.3 pp.649—656.
• Shimada T, Yamazaki H, Mimura M, Inui Y and Guengerich F.P. Interindividual variations in human liver cytochrome P450 enzymes involved in the oxidation of drugs, carcinogens, and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians, J. Pharmacol. Exp. Ther. 270 ,1994, pp. 414–423
• Siegsmund M, Brinkmann U, Schaffeler E, et al. Association of the P-glycoprotein transporter MDR1(C3435T) polymorphism with the susceptibility to renal epithelial tumors. J Am Soc Nephrol 2002;13:1847–54.
• Silverman JA Multidrug-resistance transporters. Pharm Biotechnol. 1999; 12:353-86. Review • Sinha R, Chow WH, Kulldorff M, et al. Well-done, grilled red meat increases the risk of colorectal
adenomas. Cancer Res 1999; 59: 4320 – 4. • Sinha R, Kulldorff M, Chow WH, Denobile J, Rothman N. Dietary intake of heterocyclic amines, meat-
derived mutagenic activity and risk of colorectal adenomas. Cancer Epidemiol Biomarkers Prev 2001; 10:559 – 62.
• Smith G, Stanley LA, Sim E, Strange RC, Wolf CR. Metabolic polymorphisms and cancer susceptibility. Cancer Surv 1995; 25:27–65.
• Soh K, Yanagisawa A, Hiratsuka H, et al. Variation in K-ras codon 12 point mutation rate with histological atypia within individual colorectal tumors. Jpn J Cancer Res 1993;84:388–393.
• Soussi T. Gènes et Cancers. Bulletin du cancer. Novembre 2001, Volume 88, Number 11, 1053-04. • Stieger B, Meier PJ Bile acid and xenobiotic transporters in liver. Curr Opin Cell Biol. 1998 Aug;10(4):462-
7. Review • Stoehlmacher J, Park D.J, Zhang W, Groshen S, Tsqao-Wei D.D and Yu M.C. Association between
glutathione S-transferase P1, T1, and M1 genetic polymorphism and survival of patients with metastatic colorectal cancer, J. Nat. Cancer Instit. 94, 2002, pp. 936–942.
• Tang K.S, Chiu H.F, Chen H.H, Eng H.L, Tsai C.J, Teng H.C, Huang C.S. Link between colorectal cancer and polymorphisms in the uridine diphospho glucuronosyltransferase 1A7 and 1A1 genes,World J. Gastroenterol. 11, 2005, 3250–3254.
• Taniguchi S, Mochida Y, Uchiumi T, Tahira T, Hayashi K,Takagi K et al Genetic polymorphism at the 5’ regulatory region of multidrug resistance 1 (MDR1) and its association with interindividual variation of expression level in the colon. Mol Cancer Ther. 2003. 2:1351–1359.
• Tse C and Capeau J. Quantification des acides nucléiques par PCR quantitative en temps réel. Annale de Biologie Clinique, 2003, 61 :279–293.
• Turesky R.J. Heterocyclic aromatic amine metabolism, DNA adduct formation, mutagenesis, and carcinogenesis, Drug Metab. Rev. 34, 2002, 625–650.
• Turgut S, Yaren A, Kursunluoglu R, Turgut G. MDR1 C3435T Polymorphism in Patients with Breast Cancer Archives of Medical Research 38 , 2007,539-544.
• Ulrich C.M, Bigler J, Velicer C.M, Greene E.A, Farin F.M. and Potter J.D. Searching expressed sequence tag databases: discovery and confirmation of a common polymorphism in the thymidylate synthase gene. Cancer Epidemiol. Biomarkers Prev 2000, 9, 1381--1385.
• Ulrich CM, Robien K, McLeod H. Cancer pharmacogenetics: polymorphisms, pathways and beyond. Nat Rev Cancer 2003; 3:912–20.
• Ulrich, C. M., Bigler, J., Bostick, R., Fosdick, L., and Potter, J. D. Thymidylate synthase promoter polymorphism, interaction with folate intake, and risk of colorectal adenomas. Cancer Res 2002, 62: 3361–3364,.
• Urayama KY, Wiencke JK, Buffler PA, Chokkalingam AP, Metayer C, Wiemels JL. MDR1 gene variants, indoor insecticide exposure, and the risk of childhood acute lymphoblastic leukemia, Cancer Epidemiol Biomarkers Prev. 2007 Jun; 16(6):1172-7.
• Van der Logt E.M.J, Bergevoet S.M, Roelofs H.M.J, van Hooijdonk Z, te Morsche R.H.M, Wobbes T, de Kok J.B, Nagengast F.M and Peters W.H.M. Genetic polymorphisms in UDP-glucuronosyltransferases and glutathione S-transferases and colorectal cancer risk Carcinogenesis 2004, vol.25 no.12, 2407—2415.
• Van Kuilenbourg ABP, Dobritzsch D, Meinsma R, et al. Novel diseasecausing mutations in the dihydropyrimidine dehydrogenase gene interpreted by analysis of the three-dimensional protein structure. Biochem J 2002;364:157–63

Références bibliographiques
96
• Van Kuilenbourg ABP, Haasjes J, Van Gennip H, et al. Clinical implication of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res 2000; 6: 4705–12.
• Van Kuilenbourg ABP, Meinsma JR, Zoetekouw L, Van Gennip AH. Increased risk of grade IV neutropenia after administration of 5-fluorouracil due to a dihydropyrimidine deshydrogenase deficiency: high prevalence of the IVS14 + 1G > A mutation. Int J Cancer 2002;101:253–8.
• Van Kuilenbourg ABP. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer 2004;40:935–50.
• Van Schaik RH, Van der Heiden IP, Van den Anker JN, Lindemans J. CYP3A5 variant allele frequencies in Dutch Caucasians. Clin Chem 2002, 48:1668-1671.
• Vicente J, Sinues B, Fanlo A, Vasquez P, Medina JC, Martinez-Jarreta B Polymorphism C3435T of the MDR1 gene in Central Americans and Spaniards. Mol Biol Rep 2007 Jun 19.
• Vogelstein BFE, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, et al. Genetic alterations during colorectal tumoral development. N Engl J Med 1988; 319: 525-32.
• Warmuth M, Damoiseaux R, Liu Y, Fabbro D, Gray N. SRC family kinases: potential targets for the treatment of human cancer and leukemia. Curr Pharm Des 2003; 9: 2043-2059.
• Welfare M, Monesola Adeokun A, Bassendine MF, Daly AK. Polymorphisms in GSTP1, GSTM1, and GSTT1 and susceptibility to colorectal cancer. Cancer Epidemiol Biomarkers Prev 1999; 8:289–292.
• Westlind A, Malmebo S, Johansson I, Otter C, Andersson T.B, Ingelman-Sundberg M and M. Oscarson. Cloning and tissue distribution of a novel human cytochrome P450 of the CYP3A subfamily, CYP3A43, Biochem. Biophys. Res. Commun. 281, 2001, 1349–1355.
• Whitty JP, Bjeldanes LF. The effects of dietary cabbage on xenobioticmetabolizing enzymes and the binding of aflatoxin B1 to hepatic DNA in rats. Food Chem Toxicol 1987; 25:581 – 7.
• Windmill KF, McKinnon RA, Zhu X, Gaedigk A, Grant DM, McManus ME. The role of xenobiotic metabolizing enzymes in arylamine toxicity and carcinogenesis: functional and localization studies. Mutat Res 1997;376:153–160.
• Won-Suk Lee, Gilju Seo, Hee Jung Shin, Seong Hyeon Yun, Haeran Yun, Naeyun Choi, Jinseon Lee, Daesoon Son, Jisook Cho, Jhingook Kim, Yong Beom Cho, Ho-Kyung Chun, and Woo Yong Lee. Identification of Differentially Expressed Genes in Microsatellite Stable HNPCC and Sporadic Colon Cancer Journal of Surgical Research 2008, 144, 29–35.
• Wormhoudt LW, Commandeur JNM, Vermeulen NPE. Genetic polymorphisms of human N-acetyltransferase, cytochrome P450, glutathione-S-transferase, and epoxide hydrolase enzymes : relevance to xenobiotic metabolism and toxicity. Crit RevToxicol 1999, 29 : 59-124.
• Wu MH, Chen P, Remo BF, Cook EH Jr, Das S, and Dolan ME Characterization of multiple promoters in the human carboxylesterase 2 gene. Pharmacogenetics 13: 2003, 425–435.
• Ye Z, Parry JM: Genetic polymorphisms in the cytochrome P450 1A1, glutathione S-transferase M1 and T1, and susceptibility to colon cancer. Teratog Carcinog Mutagen 2002, 22: 385-392.
• Yu J, Shannon W.D, Watson M.A., and McLeod H.L. Gene expression profiling of the irinotecan pathway in colorectal cancer. Clin Cancer Res 2005. 11:2053-2062.
• Zhang J, Cui Y, Kuang G, Li Y, Wang N, Wang R, Guo W, Wen D, Wei L, Yu F and Wang S. Association of the thymidylate synthase polymorphisms with esophageal squamous cell carcinoma and gastric cardiac adenocarcinoma. Carcinogenesis 2004, 25, 2479--2485.
• Zhong S, Wyllie A. H, Barnes D, Wolf C. R, and Spurr N. K. Relationship between the GSTM1 genetic polymorphism and susceptibility to bladder, breast and colon cancer. Carcinogenesis (Lond.), 14: 1821–1824, 1993.
• Zimniak P, Nanduri B, Pikula S, et al. Naturally occurring human glutathione S-transferase GSTP1-1 isoforms with isoleucine and valine in position 104 differ in enzymic properties. Eur J Biochem 1994; 224:893-9.

ANNEXES
97
ANNEXES
LISTE DES FIGURES
- Figure 1: Représentation schématique du métabolisme des xénobiotiques.
- Figure 2: Métabolisme du 5-fluorouracile (5FU).
- Figure 3 : Métabolisme de l’Irinotécan
- Figure 4: Principe de la technique Taqman
- Figure 5 : Graphes représentant la fluorescence émise par chacun des génotypes
- Figure 6: Détection des SNPs et constitution d’un nuage de point de fluorescence
différent correspondant aux différents génotypes en fonction de l’hybridation des
sondes VIC ou FAM à chacun des allèles du polymorphisme recherché
- Figure 7: Profils d’amplification des gènes GSTM1, GSTT1 et de digestion
enzymatique par BsmA1 du gène GSTP1 exon 5
- Figure 8: Nuages de points représentant les individus de différents génotypes
- Figure 9: Distribution de la délétion homozygote de GSTM1 entre les patients CCR et
les contrôles sur une population de l’Ouest Algérien
- Figure 10: Distribution de la fréquence de la délétion homozygote du gène GSTM1 de
la population de l’Ouest Algérien comparée à celle d’autres populations.
- Figure 11: Distribution de la délétion homozygote de GSTT1 entre les patients CCR et
les contrôles sur une population de l’Ouest Algérien.
- Figure 12: Distribution de la fréquence de la délétion homozygote du gène GSTT1 de
la population de l’Ouest Algérien comparée à celle d’autres populations.
- Figure13: Distribution de la fréquence du SNP GSTP1 313A>G entre les patients
CCR et les contrôles sur une population de l’Ouest Algérien.
- Figure 14: Distribution de la fréquence du SNP GSTP1 313A>G de la population de
l’Ouest Algérien comparée à celle d’autres populations.
- Figure 15: Distribution de la fréquence du SNP GSTP1 341C>T entre les patients
CCR et les contrôles sur une population de l’Ouest Algérien.

ANNEXES
98
- Figure 16: Distribution de la fréquence du SNP GSTP1 341C>T de la population de
l’Ouest Algérien comparée à celle de la population Caucasienne.
- Figure 17: Distribution de la fréquence du SNP MDR1 3435C>T entre les patients
CCR et les contrôles sur une population de l’Ouest Algérien
- Figure 18: Distribution de la fréquence du SNP MDR1 3435C>T dans la population
de l’Ouest Algérien comparée à celle d’autres populations.
- Figure 19: Distribution de la fréquence du SNP 6986A>G du gène CYP3A5 entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
- Figure 20: Distribution de la fréquence du SNP 6986A>G du gène CYP3A5 dans la
population de l’Ouest Algérien comparée à celle d’autres populations.
- Figure 21: Distribution de la fréquence du VNTR UGT1A1*28(TA)6>(TA)7 entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
- Figure 22: Distribution de la fréquence du SNP UGT1A1*28(TA)6>(TA)7 dans la
population de l’Ouest Algérien comparée à celle d’autres populations
- Figure 23: Distribution de la fréquence du SNP -3156 G>A du gène UGT1A1 entre
les patients CCR et les contrôles sur une population de l’Ouest Algérien.
- Figure 24: Distribution de la fréquence du SNP UGT1A1-3156G>A dans la
population de l’Ouest Algérien comparée à celle d’autres populations.
- Figure 25: Distribution de la fréquence du polymorphisme TS « 2R>3R » entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
- Figure 26: Distribution de la fréquence du polymorphisme TS « 2R>3R » dans la
population de l’Ouest Algérien comparée à celle d’autres populations.
- Figure 27: Distribution de la fréquence du polymorphisme TS « del/ins6pb » entre les
patients CCR et les contrôles sur une population de l’Ouest Algérien.
- Figure 28 : Distribution de la fréquence du polymorphisme TS « del/ins 6pb » dans la
population de l’Ouest Algérien comparée à celle d’autres populations.

ANNEXES
99
LISTE DES TABLEAUX
- Tableau 1 : Principaux polymorphismes caractérisés pour les classes GST les plus
étudiées
- Tableau 2 : Séquences des amorces utilisées pour la PCR multiplex des gènes GSTT1,
GSTM1, et GSTP1 313A>G
- Tableau 3 : Séquences des amorces et des sondes Taqman utilisées pour l’analyse des
SNPs : GSTP1 341C>T, MDR1 3435C>T, CYP3A5 6986A>G et UGT1A1 -3156 G>A
- Tableau 3: Définition des différentes allèles de GSTP1 en fonction des deux SNPs
GSTP1 313A>G et GSTP1 341C>T
- Tableau 4 : Séquences des amorces utilisées pour l’analyse des polymorphismes
UGT1A1*28 (TA)6>(TA)7, TS5’ « 2R>3R » et TS3’ « délétion/insertion de 6pb »
- Tableau 5 : Distribution des fréquences génotypiques de GSTM1 et GSTT1 dans la
population de l’Ouest Algérien.
- Tableau 6 : Distribution des fréquences allèliques et haplotypiques de GSTP1 dans la
population de l’Ouest Algérien.
- Tableau 7: Distribution des fréquences allèliques et génotypiques du SNP MDR1
3435C>T, chez des patients atteints de CCR et une population contrôle de l’Ouest
Algérien
- Tableau 8: Distribution des fréquences allèliques et génotypiques du SNP 6986>G du
gène CYP3A5 chez des patients atteints de CCR et une population contrôle de l’Ouest
Algérien.
- Tableau 9: Distribution des fréquences allèliques, génotypiques et haplotypiques du
SNP -3156 G>A et du VNTR UGT1A1*28(TA)6> (TA)7 du gène UGT1A1 chez des
patients atteints de CCR et une population contrôle de l’Ouest Algérien.
- Tableau 10: Distribution des fréquences allèliques, génotypiques et haplotypiques des
polymorphismes TS « 2R>3R » et TS « del/ins6pb » chez des patients atteints de
CCR et une population contrôle de l’Ouest Algérien.