
Inleiding tot Corrosie en Corrosiebestrijding. 5 De snelheden van elektrochemische corrosiereacties...
266
Inleiding tot Corrosie en Corrosiebestrijding. P.J. Gellings
Transcript of Inleiding tot Corrosie en Corrosiebestrijding. 5 De snelheden van elektrochemische corrosiereacties...

Inleiding tot Corrosie en Corrosiebestrijding.
P.J. Gellings


Inhoudsopgave
1 Definitie en belang van corrosie 1 1.1 Definitie van corrosie 3 1.2 Belang van corrosie 4 1.3 Corrosieweerstand en materiaalkeuze 5 1.4 Indeling van het boek 8 1.5 Opgaven 8 1.6 Literatuur 9
2 Chemische grondbegrippen 11 2.1 Inleiding 13 2.2 Elementen en verbindingen 13 2.3 Chemische reacties. 16 2.4 Chemische evenwichten en vrije-enthalpie 16 2.5 Zuren, basen en oplossingen daarvan 20 2.6 Reactiesnelheden 21 2.7 Opgaven 22 2.8 Literatuur 23
3 De drijvende kracht van corrosiereacties 25 3.1 Inleiding 27 3.2 Reacties in oplossing 27 3.3 Elektrochemische cellen 28 3.4 Elektroden en elektrodepotentialen 30 3.5 Elektrochemische reeksen en hun toepassing 32 3.6 Potentiaal-pH-diagrammen 34 3.7 Samenvatting 39 3.8 Beperkingen van thermodynamische beschouwingen. 40 3.9 Opgaven 41 3.10 Literatuur 42
4 De snelheden van elektrochemische reacties 43 4.1 Inleiding 45 4.2 Polarisatiediagrammen 46 4.3 Polarisatie van enkelvoudige elektroden 48
4.3.1 Ladingsoverdrachtpolarisatie 49 4.3.2 Diffusiepolarisatie 51
4.4 Passiviteit en polarisatie 53 4.5 Samenvatting 54 4.6 Opgaven 55 4.7 Literatuur 56

ii
5 De snelheden van elektrochemische corrosiereacties 57 5.1 Inleiding 59 5.2 Polarisatiediagrammen voor meervoudige elektroden 61 5.3 Corrosie in zure oplossingen 63 5.4 Corrosie in neutrale oplossingen: "zuurstofcorrosie" 68 5.5 Elektrolytweerstand en corrosiesnelheid 70 5.6 Contactcorrosie 73 5.7 Passiviteit 75 5.8 Opgaven 79 5.9 Literatuur 80
6 Karakteristieke verschijningsvormen van corrosie 81 6.1 Inleiding 83 6.2 Corrosie van ijzer en staal 84
6.2.1 Inleiding 84 6.2.2 Roesten en atmosferische corrosie 84
6.3 Putvormige corrosie 88 6.4 Spleet- en afzettingscorrosie 91 6.5 Microbiologisch geïnduceerde corrosie 93 6.6 Selectief oplossen van legeringen 94 6.7 Interkristallijne corrosie 96 6.8 Spanningscorrosie en corrosievermoeiing 99 6.9 Waterstofverbrossing 103 6.10 Erosie- en cavitatiecorrosie 105 6.11 Passingroest 108 6.12 Afsluitende opmerkingen 109 6.13 Literatuur 109
7 Meting van corrosie-eigenschappen van materialen 111 7.1 Inleiding 113 7.2 Vinden, bepalen en gebruiken van corrosiegegevens 113
7.2.1 Maten voor corrosiesnelheden 113 7.2.2 Corrosiegegevens verkrijgen uit de literatuur 114 7.2.3 Meten van corrosie-eigenschappen 115 7.2.4 Continue bewaking van corrosie 118
7.3 Normen en standaardvoorschriften 120 7.4 Opgaven 121 7.5 Literatuur 121
8 Voorkomen en bestrijden van elektrochemische corrosie 123 8.1 Inleiding 125 8.2 Corrosiebestrijding als onderdeel van het ontwerpproces 125 8.3 Corrosiebestrijding door waterbehandeling 127
8.3.1 Ontluchten. 127 8.3.2 Inhibitoren en passivatoren. 128

iii
8.4 Materiaalsoorten en materiaalkeuze 129 8.4.1 On- en laaggelegeerde staalsoorten 129 8.4.2 Roestvaste staalsoorten 130 8.4.3 Koper en koperlegeringen 133 8.4.4 Aluminium en aluminiumlegeringen 134 8.4.5 Nikkel en nikkellegeringen 134 8.4.6 Niet-metallische materialen 135
8.5 Kathodische en anodische bescherming 137 8.5.1 Kathodische bescherming 137 8.5.2 Anodische bescherming 141
8.6 Corrosiebestrijding met deklagen 142 8.6.1 Algemeen 142 8.6.2 Voorbehandeling 144 8.6.3 Metallische deklagen 144 8.6.4 Anorganische niet-metallische deklagen 148 8.6.5 Organische deklagen 151 8.6.6 Tijdelijke corrosiebestrijdingsmiddelen 153
8.7 Corrosiebestrijding door goede vormgeving 154 8.8 Economische aspecten van corrosiebestrijding 162 8.9 Opgaven 165 8.10 Literatuur 167
9 Hoge-temperatuuroxidatie en de bestrijding daarvan 171 9.1 Inleiding 173 9.2 Thermodynamica van oxidatiereacties 173 9.3 Kinetische vergelijkingen voor hoge-temperatuuroxidatie 175 9.4 Parabolische oxidatie 177 9.5 Snelheid van parabolische oxidatie 178 9.6 Voorkomen en bestrijden van hoge-temperatuuroxidatie 181
9.6.1 Algemeen 181 9.6.2 Materiaalkeuze 182 9.6.3 Vormgeving 184
9.7 Hoge-temperatuurcorrosie in andere gassen 185 9.8 Dauwpuntcorrosie 187 9.9 Opgaven 188 9.10 Literatuur 190
10 Epiloog: hoe kunnen we corrosie voorkomen of bestrijden? 191
11 Bijlagen 197 11.1 Potentiële besparingen van corrosiekosten 199 11.2 Constanten en conversiefactoren 200 11.3 Conversie van eenheden voor corrosiesnelheden 201 11.4 Periodiek systeem der elementen 202 11.5 Enkele algemene eigenschappen van metalen en oxiden 203

iv
11.6 Thermodynamische gegevens van oxiden 204 11.7 Celspanningen en elektrodepotentialen 205 11.8 Elektrochemische reeks 206 11.9 Standaardpotentialen van redoxreacties 207 11.10 Praktische galvanische reeks in belucht, neutraal zeewater 208 11.11 Elektrochemische equivalenten 209 11.12 Teken van de overspanning 210 11.13 Afleiding van stroom-overspanning relatie voor ladingsoverdracht-polarisatie211 11.14 Gebruik van beschermende deklagen 213 11.15 Tabel voor netto contante waarden 214 11.16 Elementen van defectchemie 216
11.16.1 Opgaven 220 11.17 Corrosiepublicaties van het Nederlands Corrosie Centrum 221 11.18 Internationale organisaties op het gebied van Corrosie en Corrosiebestrijding.227 11.19 Literatuurlijst 228 11.20 Oplossingen van opgaven 231

v

vi

vii
Ten geleide
Het is bijna een traditie te noemen; de voorzitter van het Nederlands Corrosie Centrum (Ir. E. Nagel Soepenberg) schreef in 1997 het ‘ten geleide’ en nu in 2006 valt mij de eer te beurt. Met groot genoegen leid ik de Nederlandstalige uitgave van "Inleiding tot corrosie en corrosiebestrijding" van prof.dr. P.J. (Paul) Gellings in.
2006 is sowieso een bijzonder jaar voor het NCC, we vieren dit jaar ons 50-jarige bestaan met het gastheerschap van Eurocorr 2006. Eurocorr is het jaarlijkse Europese corrosiecongres waar wetenschap, overheid en industrie elkaar ontmoeten voor het uitwisselen van kennis op het vakgebied. Een mooi moment om onze omgeving in ogenschouw te nemen. Door onder andere de globalisering van bedrijven, de grotere toegankelijkheid van kennis via internet, het wegvallen van ervaring door pensionering van specialisten, het afnemen van de corrosiekennis op en nabij de werkvloer, vinden we het als NCC noodzakelijk de bakens te verzetten.
Maar eerst het boek ….. Het boek dat voor u ligt heeft een rijke Europese historie. Deze Nederlandstalige uitgave volgde op twee Engelstalige edities en is nog steeds het enige Nederlandstalige boek na de uitgave van Dipl.Ing. H.G. Zelders "Corrosie en haar bestrijding" in 1962. In 1976 verscheen de eerste editie, "Introduction to prevention and control for engineers", onder auspiciën van de Working Party on Corrosion Education van de European Federation of Corrosion. In 1981 verscheen een door prof. K.H. Tostmann gemaakte Duitse vertaling onder de titel: "Korrosion und Korrosionsschutz von Metallen: Eine Einführung". In 1985 verscheen de tweede Engelse druk. In het boek wordt een wetenschappelijk fundament gelegd onder de praktische corrosiebestrijding. Natuurlijk is het boek bedoeld om te gebruiken in opleidingen van hogescholen en universiteiten, maar het kan ook goed gebruikt worden door materiaalkundigen en corrosie-ingenieurs. Aangezien het een breed opgezet boek is, is het ook prima te gebruiken door het management van bedrijven die te maken hebben met corrosie- en corrosiebestrijding. Er wordt veel aandacht besteed aan het begrip corrosie en het belang van corrosiebeheersing (vanuit het oogpunt van economie, energie- en grondstoffenbeheer). Ook komen de chemische grondbegrippen aan de orde. Vervolgens wordt in 3 hoofdstukken de thermodynamica en kinetiek van elektrochemische (corrosie)reacties behandeld, met speciale aandacht voor polarisatieverschijnselen. Na deze wetenschappelijke voorbereiding wordt op de fenomenologie van corrosieprocessen ingegaan. Gelukkig met veel aandacht voor de atmosferische corrosie van ijzer en staal, de microbiologisch geïnduceerde corrosie en de waterstofverbrossing. Het meten van corrosie-eigenschappen wordt in een afzonderlijk hoofdstuk behandeld. Ook het hoofdstuk over corrosiebestrijding en –preventie is zeer aan te raden aangezien er uitgebreid aandacht is voor corrosiebestrijding door waterbehandeling, materiaalsoorten en materiaalkeuze en voor kathodische en anodische bescherming. In het hoofdstuk over hoge-temperatuuroxidatie wordt verder aandacht geschonken aan corrosie door andere gassen dan zuurstof.

viii
Prof.dr. P.J. (Paul) Gellings is in staat om corrosie en corrosiebestrijding in een bredere context te plaatsen, dat blijkt ook weer uit dit boek.
- Economische context: Jaarlijks kost schade door corrosie zo’n 17,5 miljard euro. Dat is ongeveer 4% van het BNP. Dertig procent daarvan is vermijdbaar door de aanwezige kennis toe te passen! Dit boek kan daarbij ondersteunen.
- Energetische context: corrosiebeheersing draagt bij aan energiebesparing. De vervaardiging van aan corrosie onderhevige metalen gaat vaak gepaard met een hoge energie-input. Het belang van energieanalyse voor optimale corrosiebestrijding wordt in hoofdstuk 1 beschreven.
- Grondstoffen context: levensduuroptimalisering op basis van corrosie-bestrijding, gecombineerd met hergebruik, beperkt de uitputting van onze eindige voorraad grondstoffen.
Maar zonder kennis van corrosieprocessen en corrosiebeheersing is het niet mogelijk om tot optimalisering te komen van de levensduur van materialen en materiaal-systemen. Dit boek maakt de relevante basiskennis voor een breed Nederlandstalig publiek toegankelijk.
Het NCC gaat zich meer en meer positioneren als Kenniscentrum. Daarmee past het boek nog steeds erg goed bij onze eigen doelstellingen. Een kenniscentrum voor bedrijfszekerheid en corrosie, een makel en schakel functie in de kenniskolom met haar positie tussen (toegepast) wetenschappelijk onderzoek en de werkvloer. Welke kennis komt beschikbaar via het NCC? Het gaat om de praktische inzet van kennis rondom de levensduur van materialen zoals metaal, kunststof en glas en de daarmee gemoeide faalmechanismen. Deze kennis kan door bedrijven ingezet worden ter verhoging van de betrouwbaarheid, beschikbaarheid en veiligheid van technische systemen. Door de toenemende complexiteit van materiaalvraagstukken en de afnemende instroom van materiaaldeskundigen worden organisaties als het Nederlands Corrosie Centrum steeds belangrijker en daarmee “know how hubs” en “netwerk hubs”.
Prof.dr. P.J. (Paul) Gellings is in 1964 benoemd aan de Universiteit Twente tot hoogleraar Anorganische Chemie en Materiaalkunde en thans emeritus. Hij heeft tijdens het 50-jarige bestaan van het NCC een belangrijke rol gespeeld, o.a. als voorzitter. Daarom steunt het NCC deze uitgave van haar oud-voorzitter graag. We hopen dat het boek door veel mensen zal worden gebruikt en dat het jonge talent dat nu wordt opgeleid, gegrepen zal worden door ons mooie vakgebied.
Zoetermeer, 24 mei 2006 G.W. (Geert) Reitsma LIMF MICorr Voorzitter Nederlands Corrosie Centrum

ix
Voorwoord van de schrijver
Deze Nederlandse uitgave volgt in grote trekken dezelfde lijnen als de oorspron-kelijke Engelse uitgave. Behalve de verbetering van een aantal fouten en het aanbrengen van kleine aanvullingen zijn ook enkele iets belangrijker wijzigingen aangebracht die aangegeven zijn in het Ten geleide. In hoofdstuk 8 is een aparte bespreking van waterbehandeling als methode van corrosiebestrijding toegevoegd en is de paragraaf over materiaalsoorten en materiaalkeuzen uitgebreid. In enkele bijlagen zijn de Internet-adressen gegeven van het Nederlands Corrosie Centrum en van enkele andere internationale organisaties zoals de Europese Federatie Corrosie en de NACE die behulpzaam zijn bij het krijgen van aanvullende informatie van onderwerpen die in dit boek slechts beknopt zijn behandeld, onder andere via de door hen uitgegeven publicaties. Tenslotte zijn in bijlage 20 de uitwerkingen gegeven van de aan het eind van de meeste hoofdstukken geplaatste opgaven. De schrijver spreekt hierbij zijn erkentelijkheid uit aan diverse gebruikers van de vorige uitgave van dit boek voor waardevolle suggesties waarvan bij deze vertaling dankbaar gebruik is gemaakt. Verder dankt hij de heren R. Scheepmaker (VanderVelde Protection) en G.H.J. Reimerink (Stichting Doelmatig Verzinken) voor het ter beschikking stellen van illustratie- en documentatiemateriaal en voor het kritisch doorlezen van de desbetreffende paragrafen. Ir. G.A.F. Bartels (Universiteit Twente) dank ik hartelijk voor het kritisch doorlezen en het geven van waardevolle suggesties betreffende de paragraaf over economische beschouwingen van corrosie en corrosiebestrijding. De heer dr. T. Fransen van de Universitieit Twente, die dit boek veel jaren bij zijn colleges gebruikt heeft en delen van alle manuscripten heeft doorgelezen, is inmiddels helaas overleden maar ik blijf dankbaar voor zijn vele kritische en waardevolle opmerkingen. Tenslotte is de schrijver dank verschuldigd voor de steun van het Nederlands Corrosie Centrum, waardoor het totstandkomen van deze uitgave mogelijk was. Enschede, 24 mei 2006 Paul J. Gellings

x
Aanwijzingen voor het gebruik van dit boek
Dit boek is in de eerste plaats bedoeld als leerboek voor studenten in de technische studierichtingen zoals chemische technologie, procestechniek en werktuigbouwkunde op Hogescholen en Universiteiten. Daarnaast kan het gebruikt worden in chemie cursussen als een illustratie van de toepassing van chemische principes, als leerboek in andere beroepsopleidingen bijvoorbeeld in de bouwkunde, elektrotechniek, metaal- en materiaalkunde, maritieme- en luchtvaarttechniek, enzovoort. Verder is het bedoeld als een basis voor zelfstudie voor diegenen die reeds in de praktijk op dit soort gebieden werkzaam zijn. Geprobeerd is om dit boek zo te schrijven dat het redelijk volledig is en bestudeerd kan worden op basis van één à twee jaar chemie-onderwijs op HAVO- of VWO-niveau. Hoofdstuk 2 geeft voor diegenen voor wie de scheikunde-ondergrond wat beperkt is of dit lang geleden hebben gehad een kort overzicht over de belangrijkste chemische grondbegrippen die in de rest van het boek worden gebruikt. De bedoeling is dat dit boek op drie niveaus kan worden bestudeerd:
Inleidend Wanneer alleen een eerste inleiding nodig is kan de bestudering van Hoofdstukken 3 en 4 beperkt worden tot de inleidende en samenvattende paragrafen (3.1, 3.7, 3.8, 4.1, 4.5). Na bestudering van de paragrafen 5.1 en 5.2 kan verdere studie van de Hoofdstukken 5 en 6 beperkt worden tot die onderdelen die relevant zijn voor het werkgebied van de student. Afhankelijk van de detaillering komt dit overeen met 6 tot 10 lesuren.
Gemiddeld Wanneer iets meer inzicht in de achtergrondtheorie wenselijk is kan de studie uitge-breid worden tot de gehele tekst van het boek, maar zonder bestudering van Hoofd-stuk 2 en die bijlagen die dieper op de theoretische principes ingaan. Dit komt over-een met een cursus van 14 tot 18 lesuren.
Volledig Diegenen die een volledig overzicht van de principes en praktijk van corrosie en corrosiebestrijding willen krijgen kunnen het gehele boek, inclusief de meer theore-tische bijlagen bestuderen en alle opgaven uitwerken die aan het eind van de ver-schillende hoofdstukken zijn opgenomen. De meeste van deze opgaven zijn directe toepassingen van de theorie, maar sommige geven een (bescheiden) uitbreiding daarvan. Een aantal van de opgaven is zo gekozen dat ook degene die voor het inleidende of gemiddelde niveau kiest ze kan oplossen. De ervaring van de auteur is dat het oplossen van opgaven in hoge mate helpt bij het verkrijgen van inzicht en dat dit in feite veel studietijd bespaart. Maar het bestuderen van welk onderwerp dan ook, en corrosie is geen uitzondering, kost tijd en moeite. Er is dan ook geen tovermethode

xi
voor het oplossen van corrosieproblemen zonder ten minste enig begrip van hoe corrosie optreedt en van de principes van corrosiebestrijding. In bijlage 11.20 zijn numerieke antwoorden en uitwerkingen van de meeste vraagstukken gegeven. De auteur hoopt dat de grote voordelen die verbonden zijn aan een beter inzicht in de mogelijkheden van voorkomen en bestrijden van corrosie, meer dan opwegen tegen de moeite die is verricht om dat inzicht te verwerven door de bestudering van dit boek. Natuurlijk zijn er veel corrosieproblemen die zo ingewikkeld zijn dat ook een volle-dige kennis van de inhoud van dit boek onvoldoende is om die op te lossen. Maar de kennis die door bestudering van dit boek is verworven zal zeker leiden tot betere herkenning van die gevallen waar een meer deskundig advies nodig is, tot een beter begrip van de voorgestelde oplossingen en tot een meer vruchtbaar gesprek met de deskundigen die van advies dienen. De ervaring heeft geleerd dat de keuze van een praktisch toepasbare en economisch aanvaardbare oplossing van corrosieproblemen meestal alleen mogelijk is op basis van een nauwe samenwerking tussen de ontwerper, de vervaardiger, de gebruiker en de corrosiedeskundige. Voor diegenen die op bepaalde onderdelen dieper in willen gaan is in bijlage 11.19 een lijst gegeven van een groot aantal boeken op het gebied van corrosie en corrosie-bestrijding.
Verzoek aan de lezer(es)
De lezer(es) wordt verzocht eventuele (druk)fouten die zij/hij vindt aan de auteur mee te delen, liefst per e-mail aan : [email protected]. Ook andere aanmerkin-gen, opmerkingen of suggesties zijn van harte welkom.


1
Definitie en belang van corrosie


3
1.1 Definitie van corrosie
Waarschijnlijk is de reactie van gewoon staal met zijn omgeving, waarbij een volumineuze en poreuze roestlaag wordt gevormd, wel het meest bekende voorbeeld van wat corrosie wordt genoemd. Hiervoor bestaan vele bekende voorbeelden uit het dagelijks leven zoals het doorroesten van uitlaten van auto's, van autocarrosserieën en van allerlei stalen constructies. Vaak worden corrosie en roesten zelfs als syno-niemen beschouwd. Om een nauwkeuriger achtergrond te geven voor de behandeling in dit boek is het gewenst om een meer precieze en algemene definitie van corrosie te geven. In het "Corrosion Education Manual" [1] van de Europese Federatie voor Corrosie wordt de volgende definitie gegeven:
"Corrosie is de aantasting van een materiaal door reactie met de omgeving met een daardoor veroorzaakte achteruitgang van de eigenschappen. Wanneer het materiaal niet expliciet wordt genoemd is meestal een metaal bedoeld en de waardigheid daarvan wordt door de corrosie verhoogd. Een uitzondering hierop wordt gevormd door het oplossen van een metaal in een gesmolten metaal of zout. Het woord corrosie kan zowel slaan op het proces van de aantasting als op de schadelijke gevolgen daarvan. Impliciet in het begrip corrosie als proces is de aantastingssnelheid per eenheid van oppervlak; impliciet in de veroorzaakte schade is de omvang en de aard van de schade gerelateerd aan de functie van het onderdeel in kwestie."
Uit de definitie volgt in de eerste plaats dat corrosie altijd een oppervlakteverschijnsel is en noodzakelijk met de aanwezigheid van een agressief milieu is verbonden. Het corroderen van metalen is in wezen een natuurlijk proces: het metaal wordt daardoor omgezet in een stabielere toestand. Bij de bereiding van metalen worden de ertsen getransformeerd in het metaal tenkoste van veel energie. Bij het corroderen wordt het metaal, door het contact met de omgeving, spontaan weer in de stabiele vorm van zijn verbindingen omgezet. Veel van de onder atmosferische omstan-digheden ontstane corrosieproducten, zoals roest bij de corrosie van ijzer en kopergroen bij de corrosie van koper, hebben vrijwel dezelfde samenstelling als bepaalde in de natuur aangetroffen ertsen van die metalen. Uit deze definitie zien we verder dat zuiver mechanische of fysische invloeden op een materiaal, zoals slijtage, erosie, cavitatie, zwellen van polymeren en dergelijke, niet onder de definitie van corrosie vallen. Analoog beschouwen we alleen achteruitgang van eigenschappen door reactie met de omgeving als corrosie: volledig inwendige veranderingen, zoals tinpest (de verandering van de kristalstructuur van tin van wit naar grijs tin bij afkoeling beneden 15 °C) horen er niet bij. Zoals we later zullen zien (§6.10) zijn er soms gecombineerde aantastingsvormen, bijvoorbeeld corrosie plus erosie of corrosie plus cavitatie, waarbij door het gelijktijdig optreden van deze processen een veel ernstiger aantasting optreedt dan wanneer één van beide processen afzonderlijk optreedt.

Definitie en belang van corrosie
4
Duidelijk is dat uit de definitie volgt dat het doel van het voorkomen en bestrijden van corrosie is het minimaliseren van de aantasting van het materiaal of van de schade die door die aantasting wordt veroorzaakt.
1.2 Belang van corrosie
De algemene corrosie van staal en andere veel gebruikte metalen, die over hun gehele oppervlak min of meer gelijkmatig optreedt, zet enorme tonnages van deze metalen om in corrosieproducten, met als gevolg een langzame achteruitgang in de eigenschappen daarvan. Aan de andere kant zijn er verschillende soorten plaatselijke corrosie, bijvoorbeeld putvorming, die zeer ernstige gevolgen kunnen hebben ook al is de hoeveelheid materiaal dat door corrosie wordt omgezet maar klein. Bij perfo-ratie van een koelerpijp in een elektrische centrale kan bijvoorbeeld een kracht-station uitvallen, waardoor misschien een hele stad, in de winter, zonder elektriciteit en centrale verwarming komt te zitten. Analoog kan perforatie van een ondergrondse pijpleiding, die voor het transport van aardgas, water of olie dient, leiden tot grote productverliezen en een uitgebreide schade aan het milieu. Soms zal de invloed van de corrosie minder direct zijn, zoals bij het verlies van werkingsgraad door vorming van corrosieproducten die leiden tot verminderde warmteoverdracht of die de goede werking van een installatie belem-meren, bijvoorbeeld door vastlopen van bewegende onderdelen. Daarnaast zijn er nog andere belangrijke aspecten van schade door corrosie. De gelijktijdige invloed van inwendige spanning en corrosie kan soms leiden tot breuk van een metaal bij belastingen die veel lager zijn dan de gewone breuksterkte, met catastrofale ge-volgen wanneer dit vitale onderdelen betreft. Ten gevolge hiervan zijn veel doden gevallen in auto- en vliegtuigongelukken die achteraf moesten worden toegeschre-ven aan het optreden van deze zogenaamde spanningscorrosie. Het is altijd moeilijk, en zelfs onmogelijk als daarmee mensenlevens gemoeid zijn, om de kosten van corrosie en de bescherming daartegen te berekenen. Wanneer men toch kosten wil berekenen wordt geschat dat deze in geïndustrialiseerde landen van de grootte orde van 2.5 tot 3.5% van het bruto nationaal product bedragen [2, 3]. Sommige van deze kosten zijn onvermijdelijk, zoals die welke een gevolg zijn van de hogere prijs van meer corrosiebestendige materialen of van schilderen of de toepassing van andere beschermende maatregelen. Aan de andere kant is inmiddels wel duidelijk geworden dat iets tegen corrosie doen in vrijwel alle gevallen goed-koper is dan haar gewoon haar gang maar te laten gaan, al is het alleen maar door het voorkomen van onverwachte uitval of stilstand van installaties of apparaten. Volgens verschillende schattingen [2, 3] kan 20 tot 25% van de huidige corrosiekosten worden bespaard door kennis betreffende voorkomen en bestrijden van corrosie ook werkelijk toe te passen. In bijlage 11.1 wordt dit in meer detail toegelicht. Een Nederlands onderzoek over corrosie in de landbouw leidde tot soortgelijke conclusies [4]. Deze schattingen van corrosiekosten en mogelijke besparingen daarvan beperken zich tot de zogenaamde directe kosten, dat wil zeggen kosten die een direct gevolg

5
zijn van de corrosie, zoals hogere investeringen en de bijbehorende rente, verhoogd onderhoud en meer reparatie. In veel gevallen zijn de indirecte kosten, bijvoorbeeld als gevolge van productie- of productverlies, schade aan het milieu en dergelijke, veel hoger, maar nog moeilijker te schatten. Naast de onmiddellijke economische en technische gevolgen van corrosie, is corro-sie ook een ernstig probleem omdat het bijdraagt aan de uitputting van onze natuur-lijke hulpbronnen en grondstoffen. De toenemende industrialisatie van veel landen betekent dat de competitie voor en de prijs van metalen zal stijgen wanneer deze schaarser worden. Van groot belang zijn ook de gevolgen van corrosie en corrosiebestrijding voor het milieu. De ongunstige gevolgen van de corrosie zelf op het milieu: verontreiniging met corrosieproducten en met producten die bijvoorbeeld door lekkage vrijkomen krijgen van oudsher al veel belangstelling. Daarnaast ontstaat ook steeds meer aan-dacht voor de gevolgen van de corrosiebestrijding voor het milieu. Daarbij wordt steeds meer gelet op wat aan het eind van de gebruiksduur van een apparaat, constructie of transportwerktuig gebeurt, onder andere ook met het oog op herge-bruik van de daarin aanwezige materialen. Bepaalde pigmenten in verf, ook al hebben die uit het oogpunt van corrosiebestrij-ding bijzonder goede eigenschappen, zoals loodmenie en chromaten, mogen niet meer worden gebruikt, met name in verband met de milieuproblemen die zij bij onderhoud of aan het einde van de levensduur veroorzaken. Aan de andere kant wordt het gebruik van organische oplosmiddelen in verf sterk aan banden gelegd in verband met de algemene milieudoelstelling om de uitstoot van koolwaterstoffen te verminderen. In het kader hiervan wordt bijvoorbeeld veel onderzoek verricht aan de ontwikkeling van watergedragen verven [5, 6] en oplosmiddelvrije deklagen zoals poedercoatings [7]. Tenslotte moet er op gewezen worden dat corrosiebestrijding ook een grote bijdrage levert aan de energiebesparing [8]. Voor de vervaardiging van metalen uit hun ertsen zijn enorme hoeveelheden energie nodig, bijvoorbeeld voor ijzer van 55 tot 70×106 J.kg-1, aluminium 200 tot 250×106 J.kg-1 en soortgelijke hoeveelheden voor andere metalen. Een gemiddelde auto heeft een energie-inhoud van ongeveer 1.3×1011 J waarvan hoogstens 33% door hergebruik teruggewonnen kan worden. Verlenging van de levensduur van een auto maakt het dus mogelijk grote hoeveelheden energie te besparen. Het maken van metalen uit schroot kost veel minder energie dan uit erts, maar wanneer een metaal eenmaal volledig gecorrodeerd is, is het uiteraard niet meer als schroot beschikbaar!
1.3 Corrosieweerstand en materiaalkeuze
Om iets over corrosieweerstand te kunnen zeggen moeten we eerst weten welk type corrosie naar verwachting kan optreden. We spreken van chemische corrosie in alle gevallen waarin een directe chemische reactie optreedt tussen het metaal en bestanddelen van de omgeving. Dan vinden op elk punt van het oppervlak oxidatieverschijnselen van het metaal plaats (dat is per definitie gelijk aan een ver-

Definitie en belang van corrosie
6
lies van elektronen) zonder dat er sprake is van een elektrische stroom. In vrijwel alle gevallen van chemische corrosie strekt deze inwerking zich uit over het gehele metaaloppervlak dat in aanraking is met de omgeving. Er treedt dan ook meestal een tamelijk uniforme aantasting op. Meestal zijn de corrosieprocessen echter elektrochemisch van aard, in het bijzonder wanneer het metaal is blootgesteld aan een waterige oplossing. Ieder metaal-oppervlak vormt een geheel van, zich steeds verplaatsende, kortgesloten elektroden, die lokaalelementen worden genoemd en een gevolg zijn van plaatselijke verschillen in samenstelling, structuur, mechanische spanning, enzovoort. Zolang het metaal echter droog blijft worden noch corrosie, noch lokale stromen waargenomen. In waterige omgeving kunnen deze latente lokaalelementen in werking treden door het sluiten van de stroomkring via de elektrisch geleidende oplossing. Ten gevolge van deze stroom gaat (een deel van) het metaal in oplossing en treedt dus corrosie op. De corrosie van metalen in vochtige lucht (atmosferische corrosie) is in principe van ditzelfde type. Het corrosieproces is dan namelijk gelokaliseerd in een dunne vloeibare laag, die bijvoorbeeld door condensatie van het vocht uit de lucht op het metaal wordt gevormd.
Figuur 1.3.1. Factoren die de keuze van een materiaal bepalen (Met toestemming overgenomen uit [9]).
Wanneer een materiaal voor een bepaalde toepassing moet worden gekozen dan wordt deze keuze door een groot aantal factoren beïnvloed, zoals schematisch in Figuur 1.3.1 wordt getoond (zie ook [10, 11, 12, 13, 14, 15]). De corrosiebestendigheid is maar één van deze factoren. De corrosiebestendigheid hangt ook zelf weer van een aantal factoren af en de belangrijkste daarvan worden schematisch in Figuur 1.3.2 getoond.

7
Figuur 1.3.2 Factoren die van invloed zijn op de corrosiebestendigheid van een materiaal (Met toestemming overgenomen uit [16].
We zien hieruit dat de corrosiebestendigheid van een metaal geen intrinsieke eigen-schap daarvan is, zoals bijvoorbeeld zijn elasticiteitsmodulus of elektrisch gelei-dingsvermogen. Het is onmogelijk om "de" corrosiebestendigheid van een materiaal te definiëren omdat deze in hoge mate afhangt van de omgeving en de omstandig-heden. We zouden kunnen zeggen dat de corrosiebestendigheid geen materiaal- maar een systeemeigenschap is. Een metaal kan vrijwel volledig corrosiebestendig zijn in één milieu (bijvoorbeeld aluminium in de atmosfeer) terwijl het in een ander milieu snel wordt aangetast (bijvoorbeeld aluminium in een warme soda-oplossing). Roestvast staal van het type AISI 316 (18% Cr, 10% Ni, 2% Mo) is in veel milieus meer corrosiebestendig dan AISI 304 (18% Cr, 9% Ni, vaak 18-8 genoemd) en daarom wordt 316 soms veiligheidshalve gekozen in plaats van 304. Hoewel dit in chloridehoudende oplossingen in het algemeen een goede keus is, is 316 in salpeterzuur en andere zure, oxiderende oplossingen veel minder corrosiebestendig dan 304! Verder spelen ook de toepassing en de gewenste levensduur een beslissende rol. Zelfs als de levensduur van een bepaald materiaal als zodanig ruimschoots aanvaard-baar is kan de corrosiebestendigheid daarvan toch onvoldoende zijn wanneer de (kleine) hoeveelheid gevormde corrosieproducten niet toelaatbaar is. Dit kan bij-voorbeeld het geval zijn wanneer deze producten leiden tot een te sterke verontreiniging waardoor een proces of de goede werking van een apparaat worden verstoord. Sommige enzymatische processen, zoals die in gebruik zijn voor de bereiding van antibiotica, worden geheel of vrijwel geheel stopgezet door sporen van koper en andere zware metalen in concentraties lager dan 0.1 tot 1 ppm (ppm = parts per million, bijvoorbeeld 1 ppm = 1 mg per liter).

Definitie en belang van corrosie
8
1.4 Indeling van het boek
Omdat corrosieprocessen niet anders zijn dan speciale voorbeelden van che-mische of elektrochemische reacties is voor een behandeling daarvan enige basis-kennis van chemie onontbeerlijk en daarom worden in hoofdstuk 2 enkele van de belangrijkste chemische grondbegrippen kort gerecapituleerd. Zoals in §1.3 al is aangegeven zijn de gebruikelijke thermodynamische en kinetische beschouwingen ook op corrosiereacties van toepassing. De thermodynamica leert wanneer een reactie spontaan zal verlopen en de toepassing hiervan op corrosiereac-ties is het onderwerp van hoofdstuk 3. Zelfs wanneer thermodynamische argumenten tot de conclusie leiden dat een metaal zou moeten corroderen wordt in de praktijk vaak gevonden dat de corrosiesnelheid vrijwel nul is. Dat betekent dat ook de snel-heid van reacties apart moet worden bestudeerd en daaraan zijn de hoofdstukken 4 en 5 gewijd. De in de voorafgaande hoofdstukken behandelde algemene principes worden in hoofdstuk 6 toegepast op een aantal belangrijke vormen waarin corrosie kan optreden. Na een korte bespreking van belangrijke meetmethoden en andere mogelijkheden voor het verkrijgen van corrosiegegevens in hoofdstuk 7 wordt in hoofdstuk 8 een aantal van de belangrijkste manieren behandeld om corrosie te voorkomen, dan wel te bestrijden. In al deze hoofdstukken wordt de nadruk gelegd op wat meestal bekend staat als "elektrochemische corrosie" (soms, niet geheel juist, ook wel aangegeven als "natte" corrosie), ook al is een aantal van de besproken principes ook van toepassing op zuiver chemische corrosie (soms "droge" corrosie genoemd) die uitvoeriger wordt behandeld in hoofdstuk 9. Aan het eind van het boek volgt nog een aantal bijlagen, die in drie groepen kunnen worden verdeeld: a. bijlagen die numerieke gegevens bevatten; b. bijlagen die dieper ingaan op bepaalde aspecten van de theoretische
achtergrond. c. bijlagen die verwijzen naar boeken en publicaties.
Zoals in het voorwoord is aangegeven is het bestuderen van de tweede groep bijlagen niet nodig voor een eerste inleiding in het gebied van corrosie en corrosiebestrijding. In de derde groep geven bijlage 11.17 en 11.18 respectievelijk corrosiepublicaties van het Nederlands Corrosie Centrum en de Europese Federatie Corrosie, waarin veel aanvullende informatie over in dit boek behandelde onderwerpen te vinden is. Bijlage 11.19 geeft een lijst van boeken op vele gebieden van corrosie en corrosiebestrijding die vooral bedoeld is voor diegenen die dieper op bepaalde onderwerpen in willen gaan. In bijlage 11.20 zijn de uitwerkingen gegeven van de aan het eind van bijna alle hoofdstukken geplaatste opgaven.
1.5 Opgaven
Voor numerieke gegevens en omrekeningsfactoren: zie bijlagen 11.2 en 11.3.

9
1. Geef een aantal voorbeelden van corrosie die u bent tegengekomen en probeer te schatten wat de kosten zijn geweest die daaraan verbonden waren.
2. In een cilindrisch koperen vat, diameter 1 m, bevindt zich gedurende 8 uur een reactiemengsel met een dichtheid van 1 g.cm-3. Als het vat tot een hoogte van 1.5 m is gevuld en de corrosiesnelheid zodanig is dat de wanddikte van het koper met 0.1 mm/jaar afneemt, bereken dan de concentratie van koper in het reactiemengsel (in ppm) na één periode van 8 uur.
3. Wanneer het maximaal toelaatbare tingehalte in een bepaalde frisdrank 5 ppm is, bereken dan de maximaal toelaatbare corrosiesnelheid in mm/jaar wanneer het met tin bedekte blikje waarin de frisdrank bewaard wordt 10 cm diameter bij 15 cm hoogte is en de gemiddelde opslagperiode 3 maanden is. Wanneer de dikte van de tinlaag 5 μm is, hoe lang zal het dan duren voordat deze doorgecor-rodeerd is? Becommentarieer het resultaat van deze berekening.
1.6 Literatuur
1. Corrosion Education Manual. Prepared by the Working Party on Corrosion Education of the European Federation of Corrosion, 2nd International Edition, Swedish Corrosion Institute (1974)
2. Report of the Committee on Corrosion and Protection, Chairman T.P. Hoar, London, H.M.S.O. (1971)
3. Economic Effects of Metallic Corrosion in the United States, A Report to the Congress by the National Bureau of Standards, NBS Special Publication 511-1, U.S. Government Printing Office, Washington (1978)
4. J.C. Schouten, Corrosie in de landbouw, een zaak voor overheid, boeren en machine-producenten, Metaal en Techniek 29 (no.6, 1984) 40 - 42, 29 (no.7, 1984) 28 - 31
5. D.H. van der Weijde, Watergedragen verven, Afstudeerverslag TU Delft. 6. S. Smets, Overschakeling op watergedragen verf, O&C 40 (1996) 52 - 55. 7. Snel uitharden van poedercoatings, O&C 40 (1996) 188 - 190. 8. P.J. Gellings, Energieanalyse van corrosie en corrosiebestrijding, Procestechniek, ed.
Werktuigbouw 3 (1977) 545 - 551. 9. P.J. Gellings en T. Fransen, Corrosiebewust ontwerpen in: Handboek
Werktuigbouwkundig ontwerpen en construeren, Alphen aan de Rijn, Samsom BedrijfsInformatie, 1993, p. B4400-1 - 40.
10. M.G. Fontana, Corrosion Engineering, 3rd ed. New York, McGrawHill (1986) 11. J.T.N. Atkinson and H. Van Droffelaar, Corrosion and its Control. An Introduction to
the Subject. 2nd ed., Houston, NACE (1994) 12. P.J. Gellings en F. IJsseling, Corrosie en Corrosiebestrijding. Materiaalkeuze en
Constructieve Aspecten, NCC brochure 2, Delft, Waltman (1984) 13. S.A. Bradford, Corrosion Control, New York, Van Nostrand Reinhold (1993) 14. K.R. Trethewey and J. Chamberlain, Corrosion for students of science and engineering,
Longman Scientific and Technical, Harlow, (1988). 15. H.H. Uhlig, Corrosion and corrosion control – An introduction to corrosion science and
engineering, 3rd ed., NewYork, John Wiley (1985) 16. P.J. Gellings, Corrosie en Onderhoud, in: Handboek Onderhoudsmanagement, Alphen
aan de Rijn, Samsom BedrijfsInformatie, 1996, p. K3010-1 - 56.


2
Chemische grondbegrippen


13
2.1 Inleiding
In dit hoofdstuk worden zeer in het kort, voor diegenen die slechts een beperkte chemische voorkennis bezitten of waarvan de scheikundestudie allang geleden is, de belangrijkste chemische grondbegrippen behandeld. Dit kan uiteraard, in het kader van dit boek, zeker niet volledig zijn en zij die meer hierover willen weten, worden verwezen naar scheikunde boeken zoals die op het VWO worden gebruikt, bijvoorbeeld [1, 2] of inleidingen in de algemene chemie zoals het zeer goede boek van Atkins [3]. Uitgangspunt van onze beschouwingen is dat àlle stoffen zijn opgebouwd uit atomen. Deze atomen (ondanks de oorspronkelijke Griekse betekenis van het woord atomos = ondeelbaar) zijn zelf weer opgebouwd uit een zeer kleine, positief geladen kern, die vrijwel de gehele massa van het atoom bevat, met daaromheen bewegend de elektronen, waarvan het aantal juist gelijk is aan de waarde van de kernlading. De afmetingen van de atomen zoals die zich aan ons voordoen zijn in feite ongeveer gelijk aan de diameter van het gebied waarin de elektronen zich bevinden. Op de verdere details van de atoombouw gaan wij niet in en verwijzen daarvoor naar de hierboven genoemde literatuur.
2.2 Elementen en verbindingen
Alle stoffen zijn dus opgebouwd uit atomen, welke afzonderlijk aanwezig kunnen zijn maar ook als atoomgroepen die we moleculen noemen. We spreken van een verbinding wanneer niet alle atomen in zo'n molecuul gelijk zijn. In de natuur komen 92 elementen voor, dat zijn stoffen waarin maar één atoomsoort aanwezig is. Deze elementen kunnen worden ingedeeld volgens het periodiek systeem dat weergegeven is in bijlage 11.4. In dit periodiek systeem zijn de atomen gerangschikt in volgorde van het atoomnummer: dat is het aantal positieve ladingen van de kern en dus tegelijk het aantal elektronen per atoom. Iedere atoomsoort wordt aangegeven met een symbool van één of twee letters dat in het periodiek systeem onder het atoomnummer staat (deze symbolen zijn afkortingen van de Latijnse namen van de elementen). Tenslotte is, onder het symbool, de relatieve atoommassa (vroeger vaak "atoomgewicht" genoemd) gegeven. Omdat de massa van 1 atoom veel te klein is om normaal gebruikt te worden heeft men daarom de veelheid stof van 1 mol ingevoerd. De relatieve atoommassa (kortweg atoommassa) is de massa (in gram) van 1 mol van het desbetreffende element. Het aantal atomen in 1 mol van een element wordt gegeven door het getal van Avogadro en dat is 6.022 × 1023. Voorbeelden van elementen zijn ijzer (aangegeven met het symbool Fe), koper (Cu), aluminium (Al) alledrie vaste stoffen. Een ander voorbeeld is het gas zuurstof, dat bestaat uit moleculen die uit twee zuurstofatomen (met het symbool O) bestaan en daarom aangeduid worden als O2. Analoog hebben we waterstof (H2), stikstof (N2), chloor (Cl2), enzovoort. Verbindingen bestaan uit grotere of kleinere eenheden die uit meer dan één atoom-soort bestaan. Tussen de atomen heersen krachten die ervoor zorgen dat zij bij elkaar

Chemische grondbegrippen
14
blijven, hetzij in moleculen, hetzij in vaste stoffen. De elektronen spelen de hoofdrol in dit tot stand komen van bindingen tussen atomen. In veel gevallen gebeurt dit doordat elektronen als het ware door atomen "gedeeld" worden, dat wil zeggen ze horen niet meer bij één atoom, maar bij een aantal atomen tegelijk. Men spreekt in dit geval van covalente binding. In de hierboven genoemde moleculen zoals O2, N2, H2, enzovoort, zijn dit steeds twee gelijke atomen. Maar in moleculen zoals CH4 (methaan), CO (koolmonoxide), H2O (water), CH3Cl (chloroform), CH3OH (methanol of methylalcohol), enzovoort, zijn dit twee of meer verschillende atomen. Voor een verbinding geldt dat 1 mol daarvan 6.022 × 1023 moleculen bevat, dus evenveel als atomen in 1 mol. De vaste elementen zijn, op enkele uitzonderingen na metalen (koolstof, silicium en fosfor zijn enkele van de belangrijkste vaste niet-metalen), zijn zowel bij de metalen als bij de niet-metalen, de atomen in een zogenaamd kristalrooster regelmatig in de ruimte gerangschikt. Hierin zijn geen afzonderlijke moleculen te herkennen. Deze kristallen vormen als het ware zeer grote ("oneindige") moleculen. In Figuur 2.2.1 is dit voor twee gevallen getekend. Bij normale temperatuur heeft ijzer de zogenaamde ruimtelijk gecentreerde kubische structuur: de eenheidscel, dat is de kleinste eenheid waaruit men een kristal opgebouwd kan denken, bevat dan atomen op de hoekpunten van een kubus, plus één atoom in het centrum van de kubus (zie Figuur 2.2.1a). Men noemt dit wel de α-ijzer- of de ferrietstructuur. Nikkel heeft de vlakken gecentreerde kubische structuur, waarbij in de eenheidscel de hoekpunten en de middens van de 6 zijvlakken ieder met een atoom zijn bezet (zie Figuur 2.2.1b). Bij verhoogde temperatuur krijgt ijzer ook deze structuur, die daarom ook wel de γ-ijzer of de austenietstructuur wordt genoemd.
Figuur 2.2.1 Kristalstructuur van: a. ijzer (ruimtelijk gecentreerd kubisch, bcc); b. nikkel (vlakkengecentreerd kubisch, fcc).
Zuiver chroom heeft ook de ferrietstructuur, aluminium en koper hebben de austenietstructuur. Legeren kan soms tot structuurveranderingen leiden. Zo krijgt

15
ijzer waaraan ten minste 8% nikkel of mangaan is toegevoegd ook al bij kamertemperatuur de austenietstructuur. In de metallische elementen hebben de atomen één of meer elektronen afgestaan die als het ware vrij door het gehele kristal bewegen, waardoor zij dat bijeen houden. Men noemt dit wel de metallische binding. Dit verklaart tevens het grote elektrische en warmte-geleidingsvermogen van de metalen. Bij verbindingen tussen metalen en niet-metalen treedt meestal een ander soort binding op. De metaalatomen staan één of meer elektronen af en deze worden dan opgenomen door de atomen van het niet-metaal. Tussen de nu geladen atomen, die men ionen noemt, ontstaat een elektrische aantrekkingskracht (volgens de wet van Coulomb) die de ionen, meestal in een kristalrooster, bijeen houdt. Men noemt dit de ionogene binding. Een zeer veel voorkomende structuur is die van natriumchloride (NaCl, keukenzout) waarvan de eenheidscel geschetst is in Figuur 2.2.2. Hierin zien we dat de ene soort ionen (bijvoorbeeld Na+) op dezelfde plaatsen zitten als de ato-men in het hierboven besproken vlakkengecentreerde kubische rooster, terwijl de an-dere ionen (in dit geval dus Cl-) op de middens van de ribben en in het centrum van de kubus geplaatst zijn. In feite vormen de Cl--ionen ook een vlakkengecentreerd rooster dat een halve ribbe van de kubus verschoven is ten opzichte van het rooster van de Na+-ionen.
Figuur 2.2.2 Natriumchloride structuur.
Er zijn zeer veel verbindingen met deze structuur, bijvoorbeeld nikkeloxide (NiO), ijzeroxide (FeO), mangaanoxide (MnO), kobalt oxide (CoO), kaliumchloride (KCl), enzovoort. Zouten zijn ionogene verbindingen (waarbij oxiden, sulfiden, en dergelijke meestal worden uitgezonderd) die dikwijls in water oplosbaar zijn. Omdat deze oplossingen (en ook de gesmolten zouten) de elektrische stroom geleiden, wordt aangenomen dat bij het oplossen of smelten het zout in de ionen uiteenvalt, of met andere woorden

Chemische grondbegrippen
16
dissocieert. Zo wordt dan het oplossen of smelten van keukenzout ofwel natrium-chloride (NaCl) geschreven als chemische reactie:
−+ +→ Cl Na NaCl (2.2.1) Zouten zijn in het algemeen zoals men dat noemt "volledig" gedissocieerd, dat wil zeggen dat (vrijwel) geen ongedissocieerde moleculen (in dit geval NaCl moleculen) in de oplossing aanwezig zijn.
2.3 Chemische reacties.
Tussen elementen en verbindingen kunnen allerlei reacties optreden, waarbij nieuwe verbindingen kunnen ontstaan. Zo reageren zuurstof en waterstof onder vorming van water:
OH 2 O H 2 222 →+
en ijzer met zuurstof onder vorming van ijzeroxide: FeO 2 O Fe 2 2 →+
Soms ontstaan bij een reactie meerdere producten, bijvoorbeeld bij de verbranding van zwavelwaterstof (H2S) met zuurstof ontstaan water en zwaveldioxide:
2222 SO 2 OH 2 O 3 SH 2 +→+
Dit zijn allemaal reacties in gassen of van gassen met vaste stoffen, maar we zullen hieronder zien dat ook in oplossingen allerlei reacties kunnen optreden en juist deze zijn voor de corrosie van bijzonder belang. Wanneer een reactie zo verloopt dat (vrijwel) alle reactanten worden omgezet en na afloop (vrijwel) alleen de producten aanwezig zijn spreken we van een aflopende reactie. Heel vaak wordt na enige tijd een evenwichtstoestand bereikt waarin eindige hoeveelheden van zowel de reactanten als de producten aanwezig zijn. In zo'n geval spreekt men van een evenwichtsreactie.
2.4 Chemische evenwichten en vrije-enthalpie
Van zeer groot belang is natuurlijk de tendens van reacties om te verlopen en men zegt wel dat een verbinding die gemakkelijk wordt gevormd stabiel is, terwijl een verbinding die spontaan uiteen wil vallen instabiel is. Dit nogal vage begrip "stabiliteit" kan met behulp van de thermodynamica wat beter gepreciseerd worden. In de natuur kennen we de tendens van veel systemen om spontaan naar een toestand van minimale energie te streven. Een eenvoudig voorbeeld is een vallend voorwerp: de val leidt tot een verlaging van de potentiële energie. Analoog wordt bij het verbranden van een brandstof de energie verlaagd door de vorming van sterke chemische bindingen tussen de atomen van de brandstof en van zuurstof, en deze energie wordt als warmte afgegeven. Strikt genomen moet voor processen die bij constante druk verlopen in plaats van de energie U de zogenaamde enthalpie H = U

17
+ pV (p = druk, V = volume) gebruikt worden. In de praktijk kan het verschil tussen H en U echter meestal worden verwaarloosd (zie opgave 1.). Aan de andere kant kennen we ook spontane processen waarin de inwendige energie toeneemt, bijvoorbeeld het oplossen of smelten van een kristal of het verdampen van een vloeistof: in al deze gevallen moet energie worden toegevoerd. In al deze geval-len gaat aantrekkingsenergie tussen atomen of moleculen verloren. Deze veranderin-gen corresponderen met en kunnen worden opgebracht door een tweede tendens in de natuur: het streven naar een toestand van maximale wanorde. In een kristal is de ordening bijna ideaal, maar in de opgeloste of gesmolten toestand kunnen de atomen of moleculen vrij bewegen waardoor de wanorde sterk is toegenomen. Dit streven naar wanorde krijgt een toenemende invloed bij hogere temperatuur. De wanorde in een systeem wordt uitgedrukt met behulp van de entropie S. Deze is klein in geordende systemen, zoals kristallen, groter in vloeistoffen, en nog groter in verdunde oplossingen en gassen. In werkelijkheid is de drijvende kracht voor een proces nooit de enthalpie of de entropie afzonderlijk. Alle processen, waaronder chemische reacties, worden beheerst door een combinatie van deze twee grootheden. Daarom is een gecom-bineerde grootheid ingevoerd, de Gibbs vrije-energie of vrije-enthalpie G = H - T.S. Wanneer we de verandering van een grootheid X ten gevolge van het verlopen van een reactie aangeven met ΔX dan geldt voor de vrije-enthalpieverandering voor een bij constante druk en temperatuur verlopende reactie:
STHG Δ−Δ=Δ (2.4.1)
Algemeen geldt dat een proces, bij constante temperatuur en druk, spontaan zal trachten te verlopen wanneer:
0G <Δ (2.4.2)
Als ΔG = 0 dan zeggen we dat de reactie in evenwicht is. Samen met vergelijking (2.4.2) betekent dit dat de vrije-enthalpie van een systeem in evenwicht minimaal is. We zien ook dat de hierboven genoemde voorbeelden grensgevallen van vergelij-king (2.4.2) zijn. Als T of ΔS klein zijn dan is de tweede term in vergelijking (2.4.1) verwaarloosbaar en overheerst het streven naar minimale energie. Omgekeerd overheerst het streven naar wanorde wanneer T of ΔS groot zijn. De studie van consequenties en toepassingen van de vergelijkingen (2.4.1) en (2.4.2) is het onderwerp van de chemische thermodynamica [3, 4, 5] en in het volgende hoofdstuk passen we dit toe op corrosiereacties. Maar eerst geven we hier nog enkele algemene beschouwingen als achtergrond voor de later te bespreken meer specifieke reacties. We beschouwen daartoe eerst als voorbeeld de reactie tussen ijzer en zuurstof onder vorming van het zogenaamde hematiet, het oxide van driewaardig ijzer:
(s)OFe2(g)O3Fe(s)4 322 →+ (2.4.3)
IJzer en ijzeroxide zijn aanwezig als zuivere vaste stoffen (aangegeven met s van "solid") en zuurstof is een gas (aangegeven met g). De verandering in vrije-enthalpie bij een reactie is natuurlijk gelijk aan het verschil tussen de vrije-enthalpie van de producten en de reactanten:

Chemische grondbegrippen
18
322 OFeOFe 234G μ+μ−μ−=Δ (2.4.4)
In deze vergelijking is μi de vrije-enthalpie van 1 mol van de stof i. Meestal wordt deze de thermodynamische of chemische potentiaal van de stof i genoemd. In een bepaald opzicht is dit vergelijkbaar met de elektrische of mechanische potentiaal: een meer negatieve waarde van deze potentiaal wijst op een meer stabiele toestand. Per definitie geldt voor iedere stof X:
XXX Tsh −=μ (2.4.5)
waarin hX en sX de enthalpie en de entropie per mol X zijn en de molaire enthalpie en de molaire entropie worden genoemd. Voor zuivere vaste stoffen zijn deze maar weinig afhankelijk van temperatuur en druk. Zij kunnen dan ook meestal gelijk gesteld worden aan de waarden 0
Xh en 0Xs bij een standaard temperatuur en druk
(meestal 25 °C = 298 K en 1 atm). Uit vergelijking (2.4.5) krijgen we: 0Xμ (298 K, 1 atm) = 0
Xh - 298 0Xs
en voor iedere andere temperatuur T definiëren we dan: 0Xμ (T) = 0
Xμ (298) - (T - 298) 0Xs (2.4.6).
Voor ideale gassen (in corrosiewerk kunnen we altijd aannemen dat gassen zich ideaal gedragen) is de enthalpie per mol onafhankelijk van de druk en vrijwel onafhankelijk van de temperatuur. De entropie is echter sterk afhankelijk van de druk, of in een mengsel van ideale gassen van de partiële druk, van het beschouwde gas en daarvoor geldt:
sX(T, gas) = 0Xs (T) - R ln pX (2.4.7)
waarin pX de (partiële) druk is van het gas X. De standaard molaire entropie 0Xs is
de entropie van 1 mol gas bij een standaarddruk, meestal 1 atm (voor definitie en notatie van standaardgrootheden zie ook bijlage 11.6) en temperatuur T. Deze vergelijking kan kwalitatief worden begrepen door te bedenken dat bij lage druk een groter volume bezet wordt door hetzelfde aantal moleculen en dit betekent dat de wanorde dus groter is. Vergelijkingen (2.4.5), (2.4.6). en (2.4.7) geven dan tezamen voor een gas:
μX(T, gas) = 0Xμ (T) + RT ln pX (2.4.8)
Invullen hiervan in vergelijking (2.4.4) geeft:
2322 O0
OFe0O
0Fe plnRT3)234(G −μ+μ−μ−=Δ
of:
2O0 plnRT3GG −Δ=Δ (2.4.9)
Voor reacties waaraan meer dan één gas meedoet krijgen we natuurlijk overeenkom-stig meer termen waarin de partiële druk voorkomt in de vergelijking voor de ΔG voor die reactie. Bijvoorbeeld voor de reactie:
)g(H3)s(OFe)g(OH3)s(Fe2 2322 +→+ (2.4.10)

19
is de vrije-enthalpieverandering:
⎟⎟⎠
⎞⎜⎜⎝
⎛−μ+μ+μ−μ−=Δ
2
2
2322H
OH0H
0OFe
0OH
0Fe p
plnRT3)332(G
of 3
OH
H0
2
2
pp
lnRTGG ⎟⎟⎠
⎞⎜⎜⎝
⎛+Δ=Δ (2.4.11)
In evenwicht is ΔG = 0 en daaruit volgt dat:
03
OH
H Gpp
lnRT2
2 Δ−=⎟⎟⎠
⎞⎜⎜⎝
⎛ (2.4.12)
Omdat Δ 0Xg alleen een functie van de temperatuur is geldt dat ook voor de logarit-
mische term en voor de daarin voorkomende verhouding. Deze verhouding wordt meestal de evenwichtsconstante K genoemd, die in dit geval dus gelijk is aan:
( )RTGexppp
K 03
OH
H
2
2 Δ−=⎟⎟⎠
⎞⎜⎜⎝
⎛= (2.4.13)
Tenslotte staan we nog even stil bij reacties waarbij één of meer componenten in oplossing voorkomen, zoals bijvoorbeeld:
(g)H (aq)FeClHCl(aq)2 Fe(s) 22 +→+ (2.4.14)
waarin (aq) aangeeft dat de desbetreffende stof in waterige oplossing aanwezig is. De thermodynamische potentiaal voor in oplossing aanwezige stoffen is analoog aan die voor gassen (vergelijking (2.4.8)) en wel geldt daarvoor:
μX(T, aq) = 0Xμ (T, aq) + RT ln [X] (2.4.15)
waarin [X] de concentratie van de stof X in mol.l-1 aangeeft en 0Xμ (T, aq) gedefini-
eerd is voor een bepaalde standaardtoestand en standaardconcentratie, meestal 298 K en 1 mol.l-1 (voor definitie en notatie van standaardgrootheden zie ook bijlage 11.6). Wanneer een andere concentratiemaat wordt gekozen verandert ook de numerieke waarde van 0
Xμ (zie opgave 2.).
2H20
H20H
0FeCl
0HCl
0Fe
[HCl]
]p[FeClln RTG
pRTln]RTln[FeCl[HCl]RTln2)2(G
2
222
+Δ=
++−++−−=Δ μμμμ(2.4.16)
Volkomen analoog aan wat hierboven is gezegd, kunnen we ook hier een even-wichtsconstante definiëren die dan gelijk is aan:
)RTGexp(]HCl[
p]FeCl[K 0
2H2 2 Δ−== (2.4.17)

Chemische grondbegrippen
20
Hierin komen in dit geval zowel de concentraties van de opgeloste stoffen als de partiële druk van een gas voor.
2.5 Zuren, basen en oplossingen daarvan
Een zuur wordt klassiek gedefinieerd als een verbinding die bij oplossen en dis-sociëren in water H+-ionen geeft. Omgekeerd geeft een base OH--ionen, of het is een verbinding die H+-ionen opneemt. Water, zelfs wanneer het volledig zuiver is, bevat altijd ook een zekere hoeveelheid van die ionen, ten gevolge van wat men de zelfio-nisatie van water noemt:
−++→ OHHOH2 (2.5.1)
Het dissociatieproduct wordt gedefinieerd als het product van de concentraties van de H+- en de OH--ionen:
]OH][H[Kw−+= (2.5.2)
In feite is dit de evenwichtsconstante van reactie (2.5.1) vermenigvuldigd met de concentratie van water zelf. Aangezien deze in dit verband als constant kan worden beschouwd wordt deze in de evenwichtsconstante opgenomen. Bij kamertempera-tuur is Kw = 10-14 mol2l-2. Dit betekent dat in zuiver water, waarin vanzelfsprekend de concentraties van H+ en van OH- gelijk moeten zijn:
[H+] = [OH-] = 10-7 mol.l-1 (2.5.3) We kunnen hiervan gebruik maken door een zure oplossing te definiëren als een oplossing waarin [H+] > 10-7 mol.l-1 en een basische oplossing waarin [OH-] > 10-7 mol.l-1. Uit vergelijking (2.5.2) volgt dat in zure oplossingen [OH-] < 10-7 mol.l-1 en in basische oplossingen [H+] < 10-7 mol.l-1. Omdat het werken met machten vaak onhandig is wordt het zure of basische karakter van een oplossing meestal aangegeven met de zogenaamde pH-waarde, die gedefinieerd is als:
pH = - 10log [H+] (2.5.4) Voor een neutrale oplossing is dus pH = 7, voor een zure oplossing pH < 7, voor een basische pH > 7. Zuren zijn lang niet altijd volledig gedissocieerd, zoals dat met zouten wel het geval is. Zuren die niet volledig gedissocieerd zijn worden zwakke zuren genoemd, terwijl volledig gedissocieerde zuren sterke zuren worden genoemd. In dit verband defini-eert men de zuursterkte als de evenwichtsconstante van de zuurdissociatiereactie:
−++= AHHA (2.5.5)
namelijk:
]HA[]A][H[Ka−+= (2.5.6)
Sterke zuren hebben dus een grote, zwakke een kleine Ka. De meeste oxiden van niet-metalen zoals zwavel, koolstof, enzovoort lossen in water op onder vorming van zuren. Metaaloxiden daarentegen hebben in het algemeen een basisch karakter: zij geven een basische oplossing in water en neigen ertoe in zuren

21
op te lossen. Enkele oxiden, zoals bijvoorbeeld aluminiumoxide (Al2O3) en chroomoxide (Cr2O3) lossen zowel op in zuren als in basen en men noemt dit amfotere oxiden. Voor een meer diepgaande behandeling, met name ook van de begrippen zwakke zuren en basen, wordt naar de reeds eerder genoemde boeken [1, 2, 3] verwezen.
2.6 Reactiesnelheden
Bij chemische reacties is een zeer belangrijk aspect de snelheid waarmee zij verlopen. Een mengsel van waterstof en zuurstof is, zoals uit de thermodynamica blijkt, instabiel ten opzichte van water. Hetzelfde geldt bijvoorbeeld voor ijzer in lucht ten opzichte van ijzeroxide. Toch vindt onder normale omstandigheden geen merkbare reactie plaats: de reactiesnelheid is onmeetbaar klein. Pas door externe invloeden, bijvoorbeeld verwarming of het toevoegen van een katalysator (gedefi-nieerd als een stof die de reactiesnelheid van een bepaalde reactie versnelt) kunnen deze reacties verlopen. In het algemeen geldt dat de reactiesnelheid een functie is van de concentraties van de reactanten. Dit is begrijpelijk omdat een reactie natuurlijk alleen kan verlopen wanneer de reactant-moleculen met elkaar in aanraking komen en het aantal aanrakingen per tijdseenheid zal in het algemeen toenemen met de concentratie. Daarnaast speelt ook de temperatuur een essentiële rol. Het is namelijk gebleken dat moleculen alleen tot reactie komen wanneer zij voldoende energie bezitten en de kinetische energie van een molecuul neemt sterk toe met de temperatuur. In het algemeen kan de reactiesnelheid r dan ook geschreven worden als een product van 2 functies:
)c(f)T(kr = (2.6.1)
waarin de term k(T) de snelheidsconstante wordt genoemd omdat deze onafhanke-lijk is van de concentraties. De functie f(c) drukt juist de concentratieafhankelijkheid uit. Voor de snelheidsconstante geldt de zogenaamde Arrhenius-vergelijking:
)RTEexp(kk(T) act0 −= (2.6.2)
Hierin is Eact de zogenaamde activeringsenergie die een maat is voor de energie die de moleculen moeten bezitten om te kunnen reageren en k0 is de pre-exponentiële factor die onafhankelijk is van de concentraties en van de temperatuur. Men kan zich voorstellen dat de situatie bij een reactie is zoals geschetst in Figuur 2.6.1 waar de energie is getekend als functie van de afstand tussen twee reagerende moleculen. De energie van die moleculen moet ten minste Eact zijn om te kunnen reageren. We zien hier tevens dat voor de teruggaande reactie de activeringsenergie hoger is dan voor de heengaande.
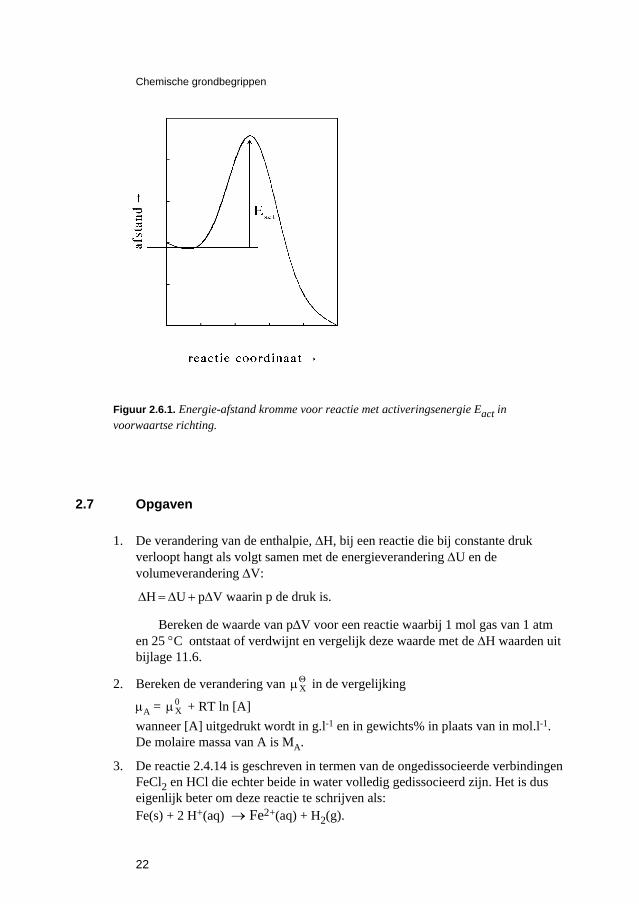
Chemische grondbegrippen
22
Figuur 2.6.1. Energie-afstand kromme voor reactie met activeringsenergie Eact in voorwaartse richting.
2.7 Opgaven
1. De verandering van de enthalpie, ΔH, bij een reactie die bij constante druk verloopt hangt als volgt samen met de energieverandering ΔU en de volumeverandering ΔV:
VpUH Δ+Δ=Δ waarin p de druk is.
Bereken de waarde van pΔV voor een reactie waarbij 1 mol gas van 1 atm en 25 °C ontstaat of verdwijnt en vergelijk deze waarde met de ΔH waarden uit bijlage 11.6.
2. Bereken de verandering van ΘμX in de vergelijking
μA = 0Xμ + RT ln [A]
wanneer [A] uitgedrukt wordt in g.l-1 en in gewichts% in plaats van in mol.l-1. De molaire massa van A is MA.
3. De reactie 2.4.14 is geschreven in termen van de ongedissocieerde verbindingen FeCl2 en HCl die echter beide in water volledig gedissocieerd zijn. Het is dus eigenlijk beter om deze reactie te schrijven als: Fe(s) + 2 H+(aq) → Fe2+(aq) + H2(g).

23
De ionenconcentraties worden net zo aangegeven als in §2.5 dus [Fe2+] en [H+]. Leid de vergelijking voor de vrije-enthalpieverandering van deze reactie geschreven in ionvorm af en bepaal daaruit ook de evenwichtsconstante.
4. Als vuistregel wordt wel eens gezegd dat een chemische reactie bij verhoging van de temperatuur met 10 °C tweemaal zo snel gaat verlopen. Wanneer dat voor een bepaalde reactie bij kamertemperatuur ( ≈ 300 K) geldt, hoe groot is dan de activeringsenergie? Wat is de factor voor de snelheid van deze reactie bij verhoging van de temperatuur van 400 tot 410 °C en van 1000 tot 1010 °C?
2.8 Literatuur
1. F.J. Carelsen, W.K.M. Engbers, F. Gierveld, en H.A.W.M. van Harssel, Scheikunde voor nu en straks (4 HV), Thieme, Zutphen (1992)
2. H.P. van Keulen, L.F.J. van Gastel en R.H. Smit, Chemie in theorie en praktijk, Den Haag, Nijgh en van Ditmar (1988) (deel 2V voor 4-VWO en 3V voor 5-VWO).
3. P.W. Atkins and J.A. Beran, General Chemistry, New York, Scientific American Books (1992)
4. O. Kubaschewski and C.B. Alcock, Metallurgical Thermochemistry, 5th ed., Oxford/New York, Pergamon Press (1979)
5. C.H.P. Lupis, Chemical thermodynamics of materials, New York/Amsterdam, North Holland (1983)


3
De drijvende kracht van corrosiereacties


27
3.1 Inleiding
Het is een bekend feit dat een ijzeren voorwerp, wanneer dat in water wordt geplaatst, gaat corroderen waarbij roest wordt gevormd. Aan de andere kant zal een edel metaal, zoals zilver, onder dezelfde omstandigheden niet reageren, dat is juist de reden dat het edel wordt genoemd. Dus is ijzer niet stabiel onder deze omstandigheden, terwijl zilver dat wèl is. Wanneer echter een zilveren voorwerp in een oplossing wordt geplaatst waarin zich sulfiden bevinden (bijvoorbeeld een ei) wordt het snel bedekt met een zwarte laag zilversulfide. Onder die omstandigheden is zilver dus niet stabiel en corrodeert wèl. Er zijn veel meer van dergelijke waarnemingen betreffende de stabiliteit van metalen onder verschillende omstandigheden. Ook andere metalen dan zilver, zijn vaak stabiel, maar worden instabiel onder bepaalde omstandigheden. Zeer weinig metalen, praktisch alleen goud en platina, zijn stabiel onder vrijwel alle omstandigheden. Maar zelfs die metalen kunnen onder zeer speciale omstandigheden in oplossing worden gebracht. Met behulp van de in het vorige hoofdstuk besproken grondslagen van de thermodynamica behandelen we in dit hoofdstuk de drijvende kracht van corrosiereacties om een beter inzicht te krijgen in de omstandigheden waaronder corrosie verwacht kan worden. Bij een eerste lezing van dit boek en voor diegenen die slechts een beperkte kennis van de theorie nodig hebben kan de bestudering van dit hoofdstuk worden beperkt tot §3.8 en §0, waarin een samenvatting van de belangrijkste punten wordt gegeven. Voor een meer uitvoerige behandeling zie bijvoorbeeld de boeken van Kaesche [1] en van Piron [2].
3.2 Reacties in oplossing
Wanneer een stuk ijzer, bijvoorbeeld een ijzeren schroevendraaier, in een waterige oplossing van kopersulfaat wordt gedompeld zien we dat op het ijzer een rood neerslag van koper wordt gevormd. Bij analyse van de oplossing blijken daarin nu ook ijzerionen aanwezig te zijn, die uit het ijzer zijn opgelost. De reactie die heeft plaatsgevonden kunnen we schrijven als:
Cu(s) + )aq(Fe (aq)Cu + Fe(s) +2+2 → (3.2.1)
De verandering van de vrije-enthalpie van deze reactie kan, net als in hoofdstuk 2 geschreven worden als het verschil van de chemische potentialen van de producten en de uitgangsstoffen:
++ μ+μ+μ−μ−=Δ 22 FeCuCuFeg (3.2.2)
De kleine letter g geeft aan dat 1 mol Cu wordt gevormd wanneer de reactie éénmaal verloopt zoals in (201H3.2.1) geschreven. De verdere notatie is gelijk aan die welke in §2.4 is beschreven. Met de daar behandelde theorie leiden we af:

De drijvende kracht van corrosiereacties
28
( )][Cu][FelnRTgg 220 +++Δ=Δ (3.2.3)
en Δg moet dan negatief zijn omdat reactie (201H3.2.1) spontaan naar rechts verloopt. De standaard vrije-enthalpieverandering Δg0 is alleen maar afhankelijk van de temperatuur. In evenwicht is Δg = 0, zodat bij een bepaalde temperatuur de evenwichtsverhouding van de concentraties van de ijzer- en de koperionen constant is. Dit is de in §2.4 gedefinieerde evenwichtsconstante en vergelijking (204H3.2.3) geeft daarvoor:
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ−==
+
+
RTgexp
][Cu][FeK
0
2
2 (3.2.4)
Voor deze reactie is Δg0 = - 151 kJ.mol-1 waaruit volgt dat bij kamertemperatuur pas evenwicht heerst bij de extreem grote verhouding 1026. In alle praktische omstandigheden is deze verhouding veel kleiner, zodat reactie (201H3.2.1) altijd naar rechts zal verlopen. Belangrijk is dat alleen de verhouding van de twee concentraties de drijvende kracht van deze reactie, en natuurlijk ook van andere analoge reacties, beïnvloedt, maar niet de absolute waarde van de afzonderlijke concentraties.
3.3 Elektrochemische cellen
De reactie die in §3.2 is besproken kan in twee deelreacties worden gesplitst: Fe → Fe2+ + 2 e- (3.3.1.a)
Cu2+ + 2 e- → Cu (210H3.3.1.b) waarin de elektronen die worden geproduceerd door de eerste reactie worden verbruikt door de tweede. Wanneer we deze twee reacties optellen komen we terug bij reactie (201H3.2.1). De eerste reactie wordt een oxidatie genoemd, de tweede een reductie. Dit is een algemene afspraak in de chemie, namelijk dat bij een oxidatie elektronen worden afgestaan, terwijl bij een reductie elektronen worden opgenomen. Hoewel deze splitsing zuiver formeel is wanneer die wordt toegepast op de reactie zoals beschreven in §3.2, namelijk door het dompelen van een stuk ijzer in een koperionen bevattende oplossing, is het wèl mogelijk deze reactie zo uit te voeren dat de oxidatie en de reductie op verschillende plaatsen optreden. We plaatsen dan een ijzeren staaf in een oplossing van ijzerionen en een koperen staaf in een oplossing van koperionen. De oplossingen worden dan via een poreus membraan verbonden zodat ze zich niet met elkaar mengen, maar dat stroomdoorgang wèl mogelijk is. Wanneer de ijzeren en koperen staaf dan via een metalen draad worden verbonden gaat een stroom lopen waarbij ijzer oplost en koper (nu op de koperen staaf) neerslaat. Deze opstelling noemt men een elektrochemische cel die schematisch in (210H3.3.1.a) is getekend. De twee metalen worden de elektroden genoemd. De elektrode waaraan de oxidatie plaatsvindt, dat wil zeggen waar de elektronen worden geproduceerd, noemt men de anode en de reactie die daaraan

29
optreedt de anodische reactie. De elektrode waaraan elektronen worden verbruikt, dus waar de reductie optreedt, noemt men de kathode en de daar optredende reactie de kathodische reactie. In het hierboven gegeven voorbeeld is ijzer de anode, terwijl koper de kathode is.
a. b.
Figuur 3.3.1. Elektrochemische cel bestaande uit een koper- en een ijzerelektrode: a. elektroden verbonden via een geleidende draad, dat betekent met een uitwendige stroom; b. elektroden verbonden via een voltmeter met hoge inwendige weerstand, dus zonder uitwendige stroom.
Wanneer de elektroden niet door een draad maar door een voltmeter worden verbonden, zoals getekend in Figuur (211H210H3.3.1.b), wordt een elektrisch potentiaalverschil tussen de elektroden gemeten en dat wordt de celspanning genoemd. De oorsprong hiervan kan als volgt duidelijk worden gemaakt. Wanneer een metaal, bijvoorbeeld ijzer, in een oplossing van zijn ionen wordt geplaatst zal dat in het algemeen niet in evenwicht zijn. Dit betekent dat òf metaalionen in oplossing gaan onder achterlating van een equivalente hoeveelheid elektronen in het metaal, òf metaalionen slaan neer uit de oplossing op het metaal, onder achterlating van een equivalente hoeveelheid negatieve ionen in de oplossing. In beide gevallen ontstaat een elektrisch potentiaalverschil tussen het metaal en de oplossing, in het eerste geval is het metaal negatief, in het tweede positief ten opzichte van de oplossing. De metaalionen zullen verder oplossen of neerslaan tot de neiging hiertoe precies in balans is met het elektrische potentiaalverschil, dat ze immers juist de andere kant op trekt, zodat dan evenwicht is bereikt. In het geval van het hier gegeven voorbeeld heeft koper een kleinere neiging om in oplossing te gaan, of een grotere om neer te slaan, dan ijzer. Dat wil zeggen dat de

De drijvende kracht van corrosiereacties
30
koperelektrode positief wordt ten opzichte van de ijzerelektrode. In §3.2 zagen we dat reactie (201H3.2.1) spontaan naar rechts verliep. Dit betekent dat de elektronen van het ijzer naar het koper lopen en de stroom, volgens de in de elektriciteitsleer geldende afspraak, dus van koper naar ijzer. Dit is juist wat we verwachten: de stroom loopt van de positieve naar de negatieve elektrode in de uitwendige stroomkring. Een elektrochemische cel levert energie wanneer de reactie verloopt. Thermodynamisch kan worden aangetoond dat de maximale hoeveelheid energie die geleverd kan worden bij het verlopen van één mol reactie Δg is (vandaar de naam vrije-enthalpie). Maar die energie is ook gelijk aan de celspanning vermenigvuldigd met de getransporteerde lading, die gelijk is aan nF, waarin n het aantal deelnemende elektronen is en F de Faraday = 96487 C, dat is de absolute waarde van de lading van 1 mol elektronen. Hieruit volgt:
Δg = - nFE (3.3.2) Per definitie geldt dit voor evenwicht en is E = Eev. We definiëren nu de standaardcelspanning E0 = - Δg0 en dat geeft dan voor de celspanning in het hier beschouwde geval
])[Cu][Fe(ln)2FRT(EE 220 ++−= (3.3.3)
en dit wordt de Nernst-vergelijking genoemd. In de praktijk wordt deze meestal in tientallige logaritmen geschreven en bij 25 °C krijgen we dan:
(Volt)])[Cu][Fe(log2
0.059EE 220 ++−= (3.3.4)
Dit is een zeer belangrijke vergelijking, onder andere omdat door metingen aan galvanische cellen, met behulp van vergelijking (3.3.2), de vrije-enthalpieveran-deringen van de bij de cellen behorende reacties kunnen worden bepaald.
3.4 Elektroden en elektrodepotentialen
Een elektrochemische cel bestaat uit twee elektroden. Wanneer we een enkele elektrode beschouwen, die ook wel een halfcel wordt genoemd, kunnen we proberen de elektrodepotentiaal daarvan te definiëren als de spanning van de halfcel. Het is echter helaas onmogelijk die te meten: we kunnen alleen de spanning van een volledige cel meten, of: we kunnen alleen potentiaalverschillen meten. Daarom introduceert men een zogenaamde standaardelektrode en definieert dan "de" elektrodepotentiaal van een enkele elektrode als de elektrische spanning van een cel die opgebouwd is uit de standaardelektrode en de beschouwde enkele elektrode. Op deze manier hebben we, bijvoorbeeld voor 100 elektroden, maar 100 elektrode potentialen nodig in plaats van 4950 celspanningen. (Er zijn 100 × 99 / 2 = 4950 mogelijkheden om verschillende paren uit 100 objecten te kiezen). Voor theoretisch werk is de standaard referentie elektrode de waterstofelektrode. Dit is een elektrode van een inert (dat wil zeggen niet reagerend) metaal, meestal platina, dat geplaatst wordt in een zure oplossing met een waterstofionenconcentratie

31
[H+] = 1 mol.l-1, dus pH = 1 (zie §2.5) terwijl langs het metaal waterstofgas wordt geborreld. De elektrode reactie van de waterstofelektrode is:
2 H+ + 2 e- = H2 (3.4.1) We kunnen deze nu combineren met een andere elektrode, bijvoorbeeld de ijzerelektrode, en zo een cel vormen:
H2 (g) | H+(aq) | Fe2+ | Fe (3.4.2) In overeenstemming met de hiervoor gemaakte afspraken, zie bijlage 11.7, betekent dit dat de linker elektrode de waterstof- en de rechter de ijzerelektrode is. Iedere verticale lijn geeft een fasegrens aan. Figuur 3.4.1 geeft hiervan een schematische weergave.
Figuur 3.4.1. Elektrochemische cel bestaande uit een waterstof en een ijzer elektrode die verbonden zijn via een voltmeter met hoge inwendige weerstand.
De celreactie die in cel (3.4.2) verloopt is: Fe2+ + H2 = Fe + 2 H+ (3.4.3)
Voor de verandering van de vrije-enthalpie ten gevolge van deze reactie leiden we af:
( )][Fep]H[lnRTgg 2H
202
+++Δ=Δ (3.4.4)
en met vergelijking (3.3.2) geeft dit voor de celspanning de vergelijking:
]Fe[ln)F2RT(]H[ln)FRT(pln)F2RT(EE 2H
02
+−−= + (3.4.5)

De drijvende kracht van corrosiereacties
32
Wanneer we de standaard waterstofelektrode gebruiken is 2Hp = 1 atm en [H+] = 1
mol.l-1. We krijgen dan, bij 25 °C : E = E0 + (0.059/2) log [Fe2+] (Volt) (3.4.60
In deze vergelijking is nu E per definitie de evenwichtspotentiaal van de ijzerelek-trode. Wanneer deze beschouwd wordt onder standaardomstandigheden, dat wil zeggen met [Fe2+] = 1 mol.l-1 en T = 298 K is E = E0 en dit wordt de standaard-potentiaal van de ijzerelektrode genoemd. Hetzelfde wat we hier deden voor de ijzerelektrode kan natuurlijk ook voor iedere andere elektrode worden gedaan. Voor praktische toepassingen worden dikwijls andere standaardelektroden toegepast dan de waterstofelektrode. Enkele belangrijke voorbeelden daarvan zijn de verzadigde calomel-elektrode (potentiaal + 0.241 V ten opzichte van de waterstofelektrode), de Ag/AgCl-elektrode (+ 0.222 V) en de Cu/CuSO4-elektrode (+ 0.32 V).
3.5 Elektrochemische reeksen en hun toepassing
Voor iedere halfcel reactie Mn+ + n e- = M (3.5.1)
luidt de Nernst-vergelijking: E = E0 + (0.059/n) log[Mn+] (Volt bij 25 °C) (3.5.2)
In bijlage 11.8 zijn de standaardpotentialen gegeven voor een aantal reacties van dit type in volgorde van toenemende E0. Dit wordt meestal de elektrochemische reeks van de metalen genoemd. Zoals hierboven besproken zijn deze potentialen gedefinieerd ten opzichte van de standaard waterstofelektrode. Daaruit volgt dat voor alle metalen met een negatieve standaardpotentiaal de reactie:
(n/2) H2 + Mn+ + n e- = n H+ + M (3.5.3) een positieve Δg0 (= - nFE0) heeft en dus spontaan naar links wil verlopen: het metaal wil corroderen. Daarom worden deze metalen onedel genoemd, in tegenstelling tot de edele metalen, die juist een positieve E0 hebben. Bij deze definitie, die uitgaat van een sterk zure oplossing (pH = 0) en 1 molair in metaalionen, is deze reeks alleen van theoretische betekenis. Uit de elektrochemische reeks volgt ook dat, wanneer we twee metalen, bijv. zink en cadmium, met elkaar verbinden en in een oplossing plaatsen die beide metaalionen bevat, dat met de laagste standaardpotentiaal de neiging heeft om te corroderen, terwijl dat met de hoogste potentiaal juist de neiging heeft om neer te slaan. Zo zal een onedel metaal, wanneer dat geplaatst wordt in een oplossing de ionen van een meer edel metaal, de neiging hebben om te corroderen onder neerslaan van het meer edele, juist zoals §3.2 is besproken voor het geval van ijzer en koper. Wanneer de oplossingen niet 1 molair zijn kunnen we toch bepalen wat er gebeurt met behulp van vergelijking (3.5.2) voor de twee metalen en toepassen van regel

33
nummer 5 uit bijlage 11.7. Wanneer we bijvoorbeeld een stuk ijzer plaatsen in een oplossing die 0.1 mol.l-1 Cd2+-ionen en 10-6 mol.l-1 Fe2+-ionen bevat is de celreactie:
Cd2+ + Fe = Cd + Fe2+ (3.5.4) met
E = ECd - EFe = = ECd
0 + 0.03 log [Cd2+] - EFe0 - 0.03 log[Fe2+]
= - 0.40 + 0.03(-1) + 0.44 - 0.03(-6) = + 0.19 Volt. Omdat dit positief is, is Δg voor reactie (3.5.4) negatief onder deze omstandigheden, zodat ijzer de neiging heeft om te corroderen. Wanneer we echter nemen [Cd2+] = 10-6 mol.l-1 en [Fe2+] = 10-1 mol.l-1 dan krijgen we E = - 0.11 Volt zodat dan juist het cadmium de neiging tot corrosie heeft. Behalve voor de waterstofelektrode beperkten we ons tot nog toe tot reacties waarbij één van de componenten een vast metaal was. Het is echter ook mogelijk dat zowel de gereduceerde als de geoxideerde vorm van een stof die aan een elektrode reactie deelneemt in oplossing aanwezig zijn, of dat één ervan een ander gas dan waterstof is. In zo'n geval spreken we van een redox-reactie (of van een redox-koppel). De potentiaal van een redox-reactie wordt gemeten door een inert metaal, heel dikwijls platina, in de oplossing te plaatsen. Deze neemt dan de potentiaal van het redox-koppel aan. In bijlage 11.9. zijn de standaardpotentialen van een aantal van deze redox-reacties gegeven. Evenals een metaal neigt tot corroderen onder waterstofontwikkeling wanneer zijn evenwichtspotentiaal lager is dan die van de waterstofelektrode in de desbetreffende oplossing, toont het dezelfde neiging bij plaatsing in een oplossing waarin een redox-reactie mogelijk is met een hogere evenwichtspotentiaal dan die van het metaal. Één van de meest gebruikelijke en belangrijke reacties van dit type is de reductie van zuurstof:
O2 + 2 H2O + 4 e- = 4 OH- , E0 = 0.401 V (3.5.5) met de Nernst-vergelijking:
E = E0 + (RT/4F)ln{pO2/[OH-]4}
= 0.401 + 0.015 log pO2 - 0.06 log[OH-].
Omdat [OH-] = 10-14/[H+] (zie §2.5) kan dit ook worden geschreven als: E = 0.401 + 0.015 log pO2
- 0.06(pH - 14). (3.5.6)
In een neutrale oplossing is pH = 7 en onder atmosferische omstandigheden is pO2
= 0.2 bar waaruit volgt dat E = 0.804 Volt. Door dit te vergelijken met de elektrochemische reeks zien we dat veel metalen die zelfs in een sterk zure oplossing geen neiging vertonen om te corroderen, zoals koper en zilver, in een neutrale oplossing die zuurstof bevat, hetgeen vrijwel altijd het geval is, wèl deze neiging vertonen. Tot nog toe gingen we er steeds van uit dat het metaal in de vorm van zijn ionen oploste. Er zijn echter vaak componenten in een oplossing aanwezig die met metaalionen onoplosbare verbindingen vormen. Zo zijn vele oxiden, hydroxiden,

De drijvende kracht van corrosiereacties
34
sulfiden, enzovoort. niet of nauwelijks in water oplosbaar. Deze oplosbaarheid wordt meestal uitgedrukt in het oplosbaarheidsproduct, een grootheid die afgeleid is uit de evenwichtsconstante voor de oplosreactie. Het oplosbaarheidsproduct voor zilversulfide, met de oplosreactie:
Ag2S = 2 Ag+ + S2- (3.5.7) is dan:
Kopl = [Ag+]2[S2-] (3.5.8) Wanneer we nu de Nernst-vergelijking schrijven voor de zilverelektrode geplaatst in een oplossing die sulfide-ionen bevat dan geeft dit:
E = E0 + 0.059 log[Ag+] = E0 + 0.0295 log (Kopl/[S2-]) (3.5.9) Het oplosbaarheidproduct van Ag2S is 1.9×10-49 mol3.l-3 en in een oplossing die 1 ppm S2--ionen bevat (dat wil zeggen [S2-] = 3×10-5 mol.l-1) wordt voor de evenwichtspotentiaal van zilver E =– 0.55 V gevonden. In een neutrale oplossing is de evenwichtspotentiaal van de waterstofelektrode E = – 0.42 V. We zien dat zo'n kleine hoeveelheid zwavel al voldoende is om zilver in een neutrale oplossing de neiging tot corrosie te geven! Dit verklaart het welbekende zwart worden van zilver aan de atmosfeer of in bepaalde zwavel bevattende levensmiddelen (eieren), hetgeen genoemd werd in de inleiding van dit hoofdstuk. Een analoge verlaging van de evenwichtspotentiaal van metalen treedt op in oplossingen van verbindingen die stabiele, oplosbare complexen vormen met de metaalionen. Een bekend voorbeeld hiervan is dat van koper in ammoniak houdende oplossingen. Ook dan kan een corrosiereactie de neiging hebben spontaan te verlopen, terwijl dat in een oplossing zonder die complexvormer niet het geval is. Deze voorbeelden tonen dat de elektrochemische reeks van de metalen, hoewel op zich erg belangrijk, in de praktijk toch met grote voorzichtigheid moet worden gehanteerd.
3.6 Potentiaal-pH-diagrammen
De bovenstaande beschouwingen kunnen heel goed worden samengevat in grafische vorm. Dit werd door Pourbaix in 1945 getoond, in wat genoemd worden potentiaal-pH of Pourbaix-diagrammen. Een vereenvoudigde vorm van het E-pH-diagram van water [3] (waarbij weinig voorkomende molecuul- en ionsoorten zoals H2O2, O3, HO2
-, H-, enzovoort, buiten beschouwing worden gelaten) is getoond in Figuur 3.6.1. Lijn a in het diagram geeft het evenwicht weer van de reactie:
O2 + 4 H+ + 4 e- = 2 H2O (3.6.1) met de Nernst-vergelijking
Ea = Ea0 + (0.059/4) log (
2Op .[H+]4) (3.6.2)
of bij pO2 = 1 bar:
Ea = 1.228 - 0.059.pH (3.6.3)
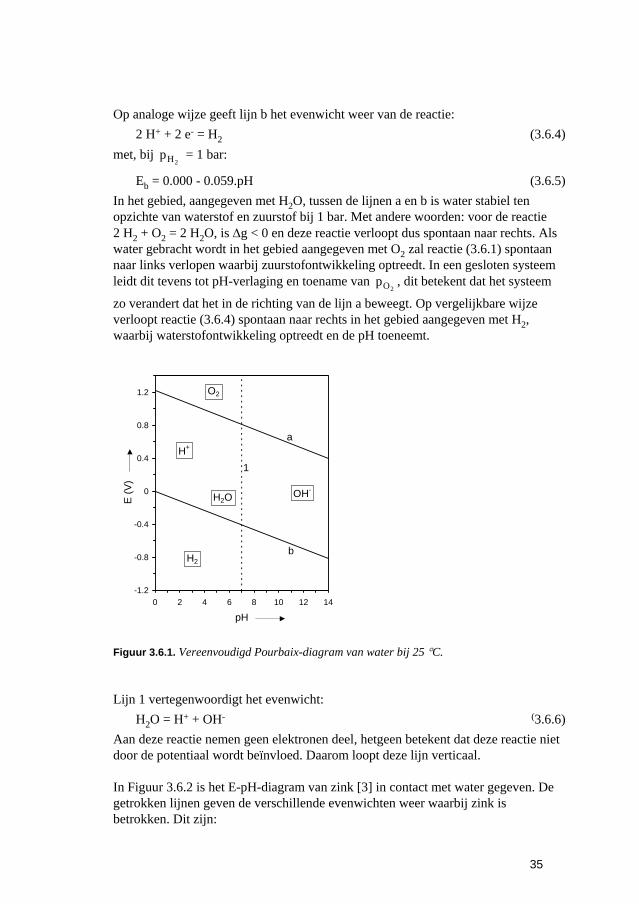
35
Op analoge wijze geeft lijn b het evenwicht weer van de reactie: 2 H+ + 2 e- = H2 (3.6.4)
met, bij 2Hp = 1 bar:
Eb = 0.000 - 0.059.pH (3.6.5) In het gebied, aangegeven met H2O, tussen de lijnen a en b is water stabiel ten opzichte van waterstof en zuurstof bij 1 bar. Met andere woorden: voor de reactie 2 H2 + O2 = 2 H2O, is Δg < 0 en deze reactie verloopt dus spontaan naar rechts. Als water gebracht wordt in het gebied aangegeven met O2 zal reactie (3.6.1) spontaan naar links verlopen waarbij zuurstofontwikkeling optreedt. In een gesloten systeem leidt dit tevens tot pH-verlaging en toename van
2Op , dit betekent dat het systeem
zo verandert dat het in de richting van de lijn a beweegt. Op vergelijkbare wijze verloopt reactie (3.6.4) spontaan naar rechts in het gebied aangegeven met H2, waarbij waterstofontwikkeling optreedt en de pH toeneemt.
-1.2
-0.8
-0.4
0
0.4
0.8
1.2
0 2 4 6 8 10 12 14
pH
E (V
)
H2
H2O
O2
H+
OH-
1
a
b
Figuur 3.6.1. Vereenvoudigd Pourbaix-diagram van water bij 25 °C.
Lijn 1 vertegenwoordigt het evenwicht: H2O = H+ + OH- (3.6.6)
Aan deze reactie nemen geen elektronen deel, hetgeen betekent dat deze reactie niet door de potentiaal wordt beïnvloed. Daarom loopt deze lijn verticaal. In Figuur 3.6.2 is het E-pH-diagram van zink [3] in contact met water gegeven. De getrokken lijnen geven de verschillende evenwichten weer waarbij zink is betrokken. Dit zijn:

De drijvende kracht van corrosiereacties
36
I Zn2+ + 2 e- = Zn met
EI = - 0.76 + (0.059/2) log [Zn2+] II ZnO + 2 H+ + 2 e- = Zn + H2O met
EII = - 0.44 - 0.059 pH III ZnO2
2- + 4 H+ + 2 e- = Zn + 2 H2O met
EIII = 0.44 - 0.118 pH + (0.059/2) log [ZnO22-]
i Zn2+ + H2O = ZnO + 2 H+ met
Ki = [H+]2/[Zn2+] = 1.1×10-11 mol.l-1 ii ZnO + H2O = ZnO2
2- + 2 H+ met
Kii = [ZnO22-].[H+]2 = mol3.l-3.
-1.6
-1.2
-0.8
-0.4
0
0.4
0.8
1.2
0 2 4 6 8 10 12 14
pH
E (V
)
Zn
ZnO
Zn2+
ZnO22-
a
b
III
III
i ii
Figuur 3.6.2. Vereenvoudigd Pourbaix-diagram voor zink in water bij 25 °C.
Alle lijnen voor de zinkevenwichten zijn getekend voor [Zn2+], respectievelijk [ZnO2
2-] = 10-6 mol.l-1. Deze concentratie wordt voor corrosiewerk vaak gekozen omdat deze zo laag is dat deze representatief is voor "normale" omstandigheden. We bespreken nu de toepassing van dit type diagram op de corrosie van zink. Om te beginnen zien we dat in zure oplossingen lijn I onder de lijn b van de

37
waterstofontwikkelingsreactie ligt. Dit betekent dat reactie I dan niet in evenwicht kan zijn in water omdat de totale reactie
Zn + 2 H+ = Zn2+ + H2 spontaan naar rechts verloopt zodat zink in oplossing gaat. Alleen wanneer, met behulp van een uitwendige spanningsbron, de potentiaal van het zink zo laag wordt gemaakt dat die onder de lijn I ligt zal de corrosie ophouden. In dit gebied zegt men dat het zink immuun is en we spreken van kathodische bescherming, die vollediger wordt besproken in §8.5. De waterstofontwikkeling gaat natuurlijk wel, zelfs versneld, door. Wanneer de pH wordt verhoogd komen we bij de lijn II. Ook hier zal tussen deze lijn en lijn b waterstofontwikkeling optreden en de totale reactie:
Zn + H2O = ZnO + H2 heeft daar de neiging om spontaan naar rechts te verlopen. Nu hangt het echter van de vorm af waarin het vaste zinkoxide wordt geproduceerd of deze reactie werkelijk doorloopt. Wanneer het oxide een gesloten, hechtende laag op het zinkoppervlak vormt zal de reactie, hoewel deze thermodynamisch gezien spontaan wil blijven verlopen, toch stoppen omdat door de oxidelaag het contact tussen het metaal en de oplossing wordt verbroken. Men zegt dan dat het zink passief is geworden. Wanneer het oxide poreus en niet-hechtend is dan treedt passiviteit niet op en gaat de corrosie gewoon door. Het E-pH-diagram toont wèl de gebieden waar passiviteit op kan treden, maar niet of dat inderdaad gebeurt. Dat hangt namelijk af van de vorm waarin het oxide ontstaat. Passiviteit wordt uitvoeriger besproken in §4.4 en §5.7. Bij nog hogere pH begint het zink weer te corroderen volgens de reactie:
Zn + 2 OH- = ZnO22- + H2.
Alle oplosreacties van zink liggen uiteraard onder de lijn a van de zuurstofreductie reactie. In een zuurstofhoudende oplossing zullen we dus tegelijk met de hiervoor genoemde reacties ook reacties hebben zoals:
2 Zn + O2 + 4 H+ = 2 Zn2+ + 2 H2O, enzovoort. die dan spontaan naar rechts verlopen.
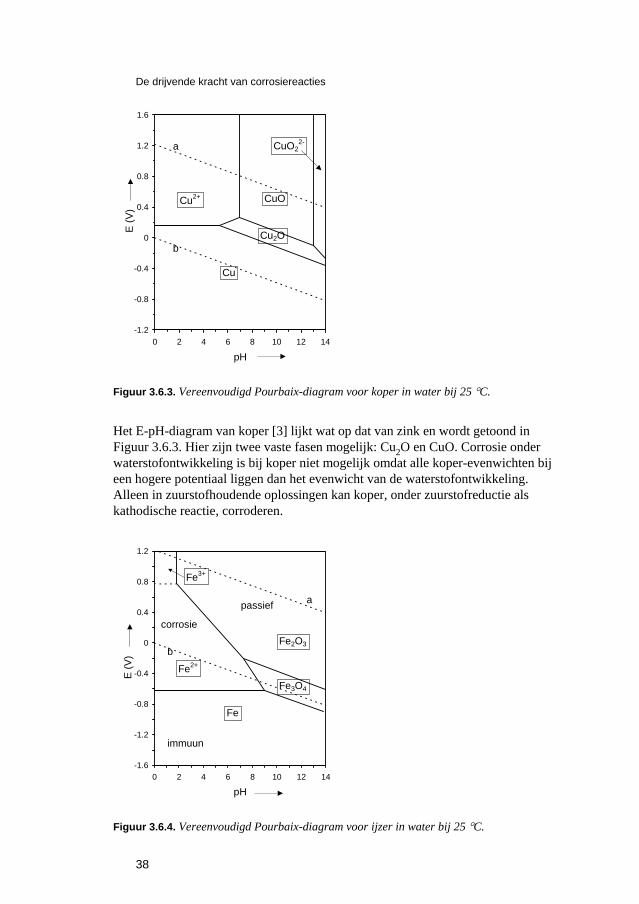
De drijvende kracht van corrosiereacties
38
-1.2
-0.8
-0.4
0
0.4
0.8
1.2
1.6
0 2 4 6 8 10 12 14
pH
E (V
)
Cu
CuOCu2+
CuO22-a
bCu2O
Figuur 3.6.3. Vereenvoudigd Pourbaix-diagram voor koper in water bij 25 °C.
Het E-pH-diagram van koper [3] lijkt wat op dat van zink en wordt getoond in Figuur 3.6.3. Hier zijn twee vaste fasen mogelijk: Cu2O en CuO. Corrosie onder waterstofontwikkeling is bij koper niet mogelijk omdat alle koper-evenwichten bij een hogere potentiaal liggen dan het evenwicht van de waterstofontwikkeling. Alleen in zuurstofhoudende oplossingen kan koper, onder zuurstofreductie als kathodische reactie, corroderen.
-1.6
-1.2
-0.8
-0.4
0
0.4
0.8
1.2
0 2 4 6 8 10 12 14
pH
E (V
)
Fe
Fe2O3
Fe2+
a
b
Fe3O4
Fe3+
immuun
passief
corrosie
Figuur 3.6.4. Vereenvoudigd Pourbaix-diagram voor ijzer in water bij 25 °C.

39
Het E-pH-diagram van ijzer [3] in water wordt getoond in Figuur 3.6.4. Dit is ingewikkelder dan de hiervoor getoonde diagrammen omdat ijzer twee belangrijke ladingstoestanden: Fe2+ en Fe3+ heeft, zowel in oplossing als in vaste verbindingen. We kunnen echter ook hier de verschillende gebieden onderscheiden waar corrosie te verwachten is, waar passiviteit op kan treden als de gevormde vaste verbindingen een gesloten en hechtende laag vormen en waar immuniteit op kan treden.
3.7 Samenvatting
Het centrale punt in dit hoofdstuk is dat bij het plaatsen van een metaal M in een oplossing die zijn eigen ionen Mn+ bevat het dynamische evenwicht:
MenMn ←→
−++
ingesteld wordt. Dit veroorzaakt een potentiaalverschil tussen het metaal en de oplossing. Wanneer het evenwicht is ingesteld wordt deze potentiaal gegeven door de Nernst-vergelijking:
]M[ln)nFRT(EE n0 ++=
of, bij 25 °C:
Volt]M[log)n059.0(EE n0 ++=
E0 wordt de standaardpotentiaal genoemd. Wanneer de metalen geplaatst worden in volgorde van toenemende E0 krijgen we de elektrochemische reeks van de metalen die gegeven is in bijlage 11.8. Het nulpunt van deze reeks is zo gekozen dat dit de standaardpotentiaal van de waterstofelektrode is. Wanneer twee elektroden met verschillende evenwichtspotentiaal met elkaar worden verbonden met een geleidende draad (zoals in Figuur 3.3.1.a) gaat een stroom lopen in zo'n richting dat het potentiaalverschil verkleind wordt. Bij één van de elektroden vindt reductie plaats en die wordt de kathode genoemd, aan de andere treedt oxidatie op en dat heet de anode. Het bovenstaande is van toepassing op alle redox-reacties van het algemene type:
dRebenOxa ←→
−+
met de overeenkomstige Nernst-vergelijking: ba0 [Red][Ox]ln)nFRT(EE +=
Voor een aantal van dergelijke reacties zijn de standaardpotentialen gegeven in bijlage 11.9. Twee belangrijke voorbeelden, waar men bij corrosiereacties vaak te maken heeft, zijn de waterstofontwikkelingsreactie:
2 H+ + 2 e- = H2 met E = 0.0 + (RT/2F) ln [H+]2/
2Hp en de zuurstof reductie reactie:
O2 + 4 H+ + 4 e- = 2 H2O met E = 0.401 + (RT/4F) ln
2Op /[OH-]4.

De drijvende kracht van corrosiereacties
40
Een metaal heeft de neiging om te corroderen wanneer het in een omgeving wordt geplaatst waarin de geoxideerde vorm van het redox-koppel (Ox in het algemene voorbeeld) aanwezig is met een evenwichtspotentiaal hoger dan van het beschouwde metaal. Zo heeft ieder metaal met een evenwichtspotentiaal lager dan 0.0 Volt de neiging tot oplossen in een sterk zuur en deze worden daarom onedel genoemd. Aan veel elektrodereacties wordt deelgenomen door H+- of OH--ionen. In zo'n geval wordt de potentiaal van de desbetreffende reactie beïnvloed door de pH en kan het gedrag van het systeem weergegeven worden met behulp van een E-pH- of Pourbaix-diagram. Figuur 3.6.1 tot Figuur 3.6.4 geven hiervan enkele voorbeelden. Wanneer, in plaats van opgeloste ionen, vaste verbindingen kunnen worden gevormd kan het gebeuren dat het metaal daar volledig mee wordt bedekt en verdere reacties daardoor onmogelijk wordt gemaakt. Dit wordt passiviteit genoemd. Thermodynamische overwegingen leiden alleen tot conclusies over de richting van mogelijke reacties en niet over de snelheid waarmee ze verlopen. Reactiesnelheden worden daarom afzonderlijk besproken in de volgende hoofdstukken.
3.8 Beperkingen van thermodynamische beschouwingen.
De thermodynamica van corrosiereacties, die in dit hoofdstuk besproken, is een belangrijke gids bij de bepaling van de reacties die in principe mogelijk zijn onder gegeven omstandigheden. Samenvattend kan gezegd worden dat een metaal neigt tot oplossen in of reageren met een oplossing waarin een kathodische reactie mogelijk is met een evenwichtspotentiaal hoger dan die van de metaalreactie in die oplossing. Maar er zijn ook enkele ernstige beperkingen die het onmogelijk maken om alleen op de thermodynamica te vertrouwen. In bijlage 11.10 wordt bijvoorbeeld een zogenaamde "praktische galvanische reeks" gegeven die de potentialen van een aantal metalen en legeringen geeft onder praktisch belangrijke omstandigheden. Ofschoon er zeker een gelijkenis is met de elektrochemische reeks, gegeven in bijlage 11.8, zijn er ook grote verschillen voor sommige metalen. Bijvoorbeeld voor roestvast staal of aluminium, is dit een gevolg van het optreden van passiviteit. In andere gevallen echter worden deze verschillen veroorzaakt, zoals we in het volgende hoofdstuk bespreken, doordat de metaal-oplosreactie niet in evenwicht is maar met een eindige snelheid verloopt. Verder is het nauwelijks te verwachten dat de oplosreacties van de verschillende componenten van een legering dezelfde evenwichtspotentiaal hebben. Per definitie is het onmogelijk om met behulp van de thermodynamica inzicht te krijgen in de snelheden van de beschouwde reacties. Zelfs als een reactie thermodynamisch mogelijk is kan de snelheid daarvan zo laag zijn dat hij in de praktijk eigenlijk in het geheel niet verloopt. Een bekend voorbeeld is de reactie tussen waterstof en zuurstof bij kamertemperatuur. Wanneer een mengsel van deze gassen ongestoord blijft kan het onbeperkt worden bewaard, ondanks het feit dat de drijvende kracht om water te vormen erg hoog is: ΔgΘ = - 237 kJ.mol-1. Alleen door ontsteking met een vlam of vonk, of door het toevoegen van een katalysator, kan

41
deze reactie op gang worden gebracht, maar dan verloopt hij ook explosief (vandaar de naam knalgas voor een waterstof-zuurstof mengsel). Tenslotte kan de vorm van de reactieproducten, zoals we bij de passiviteit hebben gezien, een grote invloed uitoefenen op het verlopen van reacties. Ook dit is weer een kwestie van kinetiek en niet van thermodynamica.
3.9 Opgaven
1. Geef de afleiding voor de celspanning behorend bij reactie (3.5.4)
2. Een stuk ijzer wordt in een oplossing geplaatst die Ag+, Fe2+ en Fe3+-ionen bevat. Schrijf de vergelijking op van de reactie die naar verwachting zal optreden. Als [Ag+] = 10-6 mol.l-1 wanneer verwacht u dan dat deze reactie juist in evenwicht zal zijn? Wat gebeurt er in dat geval wanneer [Fe2+] = [Fe3+] = 0.1 mol.l-1? En wat wanneer beide gelijk zijn aan 10-7 mol.l-1? Wat verwacht u dat er zal gebeuren wanneer [Ag+] = 1 mol.l-1 bij de eerder genoemde concentraties van de Fe2+- en Fe3+-ionen?
3. Welke van de metalen in bijlage 11.8 verwacht u dat zal corroderen onder waterstofontwikkeling bij pH = 7? En welke bij pH = 12?
4. Welke van de metalen in bijlage 11.8 verwacht u dat zal corroderen onder zuurstofreductie bij pH = 0, 7 en 14?
5. Goud-ionen vormen zeer stabiele complexen met cyanide ionen volgens de reactievergelijking:
Au+ + 2 CN- = Au(CN)2-
De evenwichtsconstante van deze reactie is: K = [Au(CN)2
-]/[ Au+][ CN-]2 = 3 × 1038. Bereken de evenwichtspotentiaal van goud in een oplossing die 10-6 mol.l-1
goud- en 0.1 mol.l-1 CN—ionen bevat. Geef aan wat dit betekent. 6. Het oplosbaarheidsproduct van kopersulfide is:
Ksol = [Cu2+][S2-] = 10-44. Bereken de evenwichtspotentiaal van koper dat staat in een oplossing die 1
mg per liter sulfide ionen bevat. Geef aan wat dit betekent. 7. Een, gesloten, koperen voorraad vat bevat verdund, zuurstofvrij zwavelzuur met
een pH = 0.1. De gasatmosfeer boven het zuur bestaat uit waterstof bij een druk van 1 atm. 1. bereken de maximale evenwichtsconcentratie van koper in het zuur in
mol.l−1 en in ppm 2. wat zal het maximale kopergehalte zijn wanneer de partiële druk van
waterstof in het gas boven het zuur 10-8 atm is?
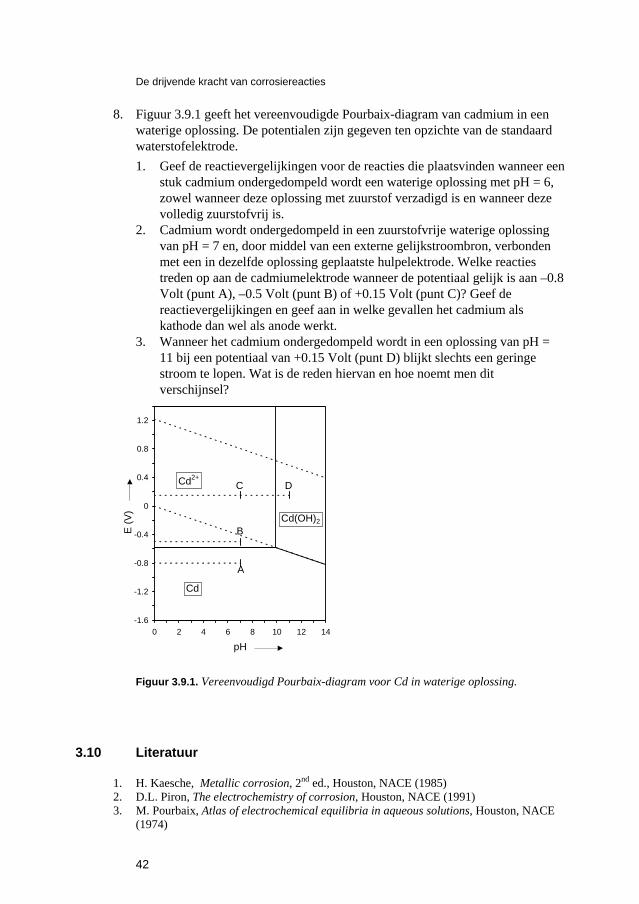
De drijvende kracht van corrosiereacties
42
8. Figuur 3.9.1 geeft het vereenvoudigde Pourbaix-diagram van cadmium in een waterige oplossing. De potentialen zijn gegeven ten opzichte van de standaard waterstofelektrode. 1. Geef de reactievergelijkingen voor de reacties die plaatsvinden wanneer een
stuk cadmium ondergedompeld wordt een waterige oplossing met pH = 6, zowel wanneer deze oplossing met zuurstof verzadigd is en wanneer deze volledig zuurstofvrij is.
2. Cadmium wordt ondergedompeld in een zuurstofvrije waterige oplossing van pH = 7 en, door middel van een externe gelijkstroombron, verbonden met een in dezelfde oplossing geplaatste hulpelektrode. Welke reacties treden op aan de cadmiumelektrode wanneer de potentiaal gelijk is aan –0.8 Volt (punt A), –0.5 Volt (punt B) of +0.15 Volt (punt C)? Geef de reactievergelijkingen en geef aan in welke gevallen het cadmium als kathode dan wel als anode werkt.
3. Wanneer het cadmium ondergedompeld wordt in een oplossing van pH = 11 bij een potentiaal van +0.15 Volt (punt D) blijkt slechts een geringe stroom te lopen. Wat is de reden hiervan en hoe noemt men dit verschijnsel?
-1.6
-1.2
-0.8
-0.4
0
0.4
0.8
1.2
0 2 4 6 8 10 12 14
pH
E (V
)
Cd
Cd(OH)2
Cd2+
A
B
C D
Figuur 3.9.1. Vereenvoudigd Pourbaix-diagram voor Cd in waterige oplossing.
3.10 Literatuur
1. H. Kaesche, Metallic corrosion, 2nd ed., Houston, NACE (1985) 2. D.L. Piron, The electrochemistry of corrosion, Houston, NACE (1991) 3. M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions, Houston, NACE
(1974)

4
De snelheden van elektrochemische reacties


45
4.1 Inleiding
In §3.8 is aangegeven dat het niet voldoende is om te weten dat een metaal onder bepaalde omstandigheden kan corroderen. Het is noodzakelijk om te weten hoe snel een mogelijke reactie kan verlopen. Er zijn vele, welbekende, voorbeelden van metalen, zoals aluminium en zink, die volgens evenwichtsberekeningen zeer instabiel zijn, maar die onder veel omstandigheden veel minder snel corroderen dan een relatief gezien meer stabiel metaal, bijvoorbeeld ijzer. Wij beperken ons in dit hoofdstuk tot elektrochemische corrosie hetgeen betekent dat we speciaal in de snelheden van elektrodereacties geïnteresseerd zijn. Voor een koperelektrode is de elektrodereactie:
Cu
aI
cI
2eCu 2⎯⎯⎯ ⎯←←
⎯⎯⎯ →⎯→
+ −+ (4.1.1).
In evenwicht zijn de snelheden van de heengaande en teruggaande reacties, uitgedrukt als de kathodische stroom Ic en de anodische stroom Ia gelijk en tegengesteld. Dit betekent dat er geen netto stroom loopt en:
Ia + Ic = 0 (4.1.2). De anodische stroom wordt positief gerekend, de kathodische stroom negatief. Wanneer er geen evenwicht heerst stroomt er een netto stroom I naar of van de elektrode, omdat dan de deelstromen niet meer gelijk zijn en we hebben:
I = Ia + Ic (4.1.3). Wanneer |Ia| > |Ic| dan is de stroom positief en koper gaat in oplossing, wanneer |Ia| < |Ic| is de stroom negatief en koper slaat neer. Dit is in overeenstemming met de gebruikelijke tekenafspraak voor de stroom: deze is positief voor de richting waarin positieve ladingen bewegen. De uitwendige stroom komt overeen met een netto chemische reactie. Volgens de wet van Faraday is de stroom evenredig met de reactiesnelheid en dit wordt uitgedrukt door de vergelijking:
Ietm ×=ΔΔ (4.1.4).
Hierin is Δm de massa van de stof die gevormd wordt of wegreageert door de reactie in de tijd Δt en e wordt het elektrochemische equivalent van deze stof genoemd.Voor metalen wordt dit gegeven door:
FnMe XXX = (4.1.5)
waarin MX de atomaire massa en nX de waardigheid van het metaal X zijn. Een tabel met elektrochemische equivalenten is gegeven in bijlage 11.11. Wanneer geen uitwendige stroom loopt is een elektrode niet noodzakelijkerwijs in evenwicht: er kunnen twee of meer reacties tegelijk aan het elektrodeoppervlak verlopen. We spreken dan van een meervoudige elektrode. De anodische reacties aan zo'n elektrode produceren gezamenlijk hetzelfde aantal elektronen als door de kathodische reacties worden verbruikt. Een elektrode waaraan, zoals hierboven

De snelheden van elektrochemische reacties
46
besproken, maar één reactie verloopt wordt een enkelvoudige elektrode genoemd. In het geval van een meervoudige elektrode blijft vergelijking (4.1.2) blijft geldig maar nu zijn de anodische en kathodische stromen het gevolg van verschillende reacties. Een voorbeeld is het, in hoofdstuk 3 al genoemde, geval van een ijzerelektrode in een oplossing die koper-ionen bevat. De twee naast elkaar, tegelijk optredende reacties zijn dan:
Cue2Cu
Fee2Fe+2
+2
→+
←+−
−
(4.1.6)
met als totale elektrodereactie:
Cu+FeCuFe +2+2 →+ (4.1.7)
Met de vergelijkingen (4.1.4) en (4.1.5) vinden we dat het oplossen van 1 gram ijzer in een uur overeenkomt met een stroom van ongeveer 1 A. De corrosiesnelheid van een metaal kan op deze manier dus uitgedrukt worden in de daarmee equivalente stroom. In het algemeen verdient het de voorkeur om de stroomdichtheid, dat is de stroom per oppervlakte-eenheid, te gebruiken omdat deze, zoals ook blijkt uit bijlage 11.3, direct evenredig is met de dikteafname of penetratie door de corrosie. Wanneer een enkelvoudige elektrode niet in evenwicht is verschilt de potentiaal daarvan van die welke volgens de vergelijking van Nernst in de desbetreffende oplossing wordt verwacht. Dit noemt men polarisatie en de verandering in potentiaal is de overspanning η = E - Eev. De richting van deze potentiaalverandering is zodanig dat het product van overspanning en stroom positief is:
0 I >×η (4.1.8)
Voor een netto anodische reactie hebben we dus een positieve overspanning, voor een netto kathodische reactie een negatieve overspanning (voor een bewijs hiervan zie bijlage 11.12). In dit hoofdstuk bespreken we de kinetiek van elektrochemische reacties die aan enkelvoudige elektroden optreden. Voor een uitvoeriger behandeling zie bijvoorbeeld [1, 2, 3, 4]. Het gedrag van meervoudige elektroden vormt het onderwerp van hoofdstuk 5. Bij eerste lezing en voor diegenen die alleen een beperkte kennis van de theorie nodig hebben kan de bestudering van dit hoofdstuk beperkt blijven tot §4.5 die een samenvatting van de belangrijkste punten geeft.
4.2 Polarisatiediagrammen
We beschouwen een elektrochemische cel, zoals weergegeven in Figuur 4.2.1, die bestaat uit een zinkelektrode, staande in een oplossing met [Zn2+] = 1 mol.l-1 en een standaard waterstofelektrode.

47
Figuur 4.2.1. Elektrochemische cel die bestaat uit een zink- en een waterstofelektrode en welke stroom levert aan een uitwendige weerstand R
De elektroden zijn verbonden door een variabele weerstand R, die in serie staat met een ampèremeter, waarmee de door de weerstand en de cel lopende stroom wordt gemeten. Tegelijk wordt de celspanning gemeten met een voltmeter V met een hoge inwendige weerstand. Zoals uit de elektrochemische reeks blijkt is de celspanning -0.76 V wanneer de weerstand R oneindig groot is.Wanneer R een eindige waarde heeft loopt een stroom en de celspanning daalt totdat die bij kortsluiting (R = 0) nul wordt, wanneer de inwendige weerstand van de cel zelf kan worden verwaarloosd. Door gebruik te maken van aparte standaardelektroden waar géén stroom door loopt kunnen we de potentialen van de twee elektroden afzonderlijk meten. Op die manier is het mogelijk een stroom-spanningsdiagram of, zoals dat vaak wordt genoemd, een polarisatiediagram te construeren. In Figuur 4.2.2 is een zeer schematische weergave hiervan gegeven. In Figuur 4.2.2a is de stroom uitgezet met het overeenkomstige teken: een kathodische stroom naar links, een anodische stroom naar rechts. In veel gevallen is het echter gemakkelijker om beide stromen naar rechts uit te zetten, zoals getoond in Figuur 4.2.2b, en men noemt dit meestal een Evans-diagram. Hieruit zien we direct dat bij een stroom I de celspanning gelijk is aan:
2HZn EEE ′−′=′ . Dit komt overeen met de overspanningen:
0EE 0ZnZnZn >−′=η en 0EE 0
HHH 222<−′=η
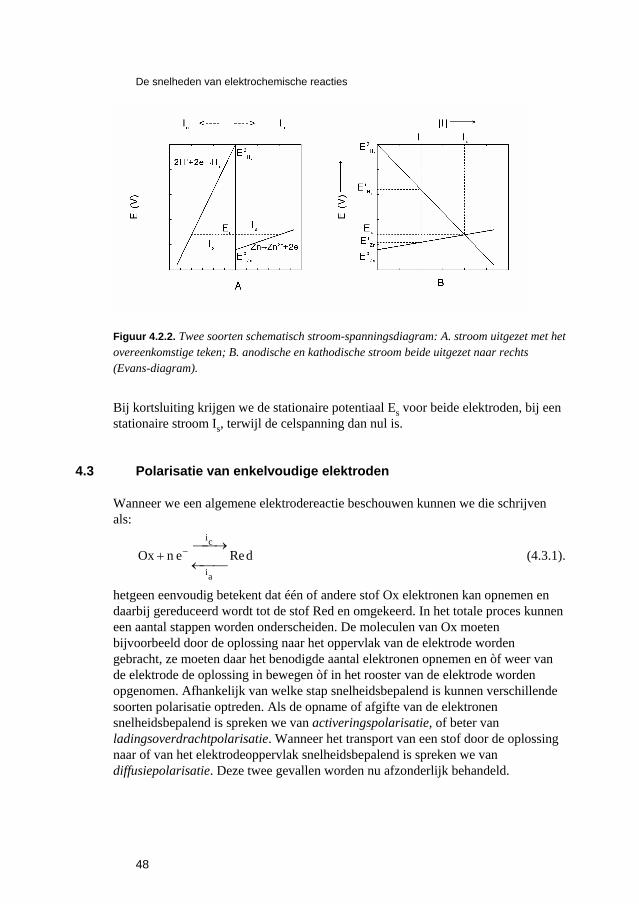
De snelheden van elektrochemische reacties
48
Figuur 4.2.2. Twee soorten schematisch stroom-spanningsdiagram: A. stroom uitgezet met het overeenkomstige teken; B. anodische en kathodische stroom beide uitgezet naar rechts (Evans-diagram).
Bij kortsluiting krijgen we de stationaire potentiaal Es voor beide elektroden, bij een stationaire stroom Is, terwijl de celspanning dan nul is.
4.3 Polarisatie van enkelvoudige elektroden
Wanneer we een algemene elektrodereactie beschouwen kunnen we die schrijven als:
dReenOxai
ci
⎯⎯←⎯→⎯
+ − (4.3.1).
hetgeen eenvoudig betekent dat één of andere stof Ox elektronen kan opnemen en daarbij gereduceerd wordt tot de stof Red en omgekeerd. In het totale proces kunnen een aantal stappen worden onderscheiden. De moleculen van Ox moeten bijvoorbeeld door de oplossing naar het oppervlak van de elektrode worden gebracht, ze moeten daar het benodigde aantal elektronen opnemen en òf weer van de elektrode de oplossing in bewegen òf in het rooster van de elektrode worden opgenomen. Afhankelijk van welke stap snelheidsbepalend is kunnen verschillende soorten polarisatie optreden. Als de opname of afgifte van de elektronen snelheidsbepalend is spreken we van activeringspolarisatie, of beter van ladingsoverdrachtpolarisatie. Wanneer het transport van een stof door de oplossing naar of van het elektrodeoppervlak snelheidsbepalend is spreken we van diffusiepolarisatie. Deze twee gevallen worden nu afzonderlijk behandeld.

49
4.3.1 Ladingsoverdrachtpolarisatie
Zoals al besproken in §2.6 geldt voor de snelheden van chemische reacties de Arrhenius-vergelijking. We schrijven deze nu in de vorm:
RTEacte.Ar −= (4.3.2)
waarin r de reactiesnelheid is en A een pre-exponentiële factor, waarin naast de k0 ook andere grootheden, zoals concentraties kunnen voorkomen. Eact is de activeringsenergie en geeft de hoogte van de energiebarrière die de moleculen moeten passeren om te kunnen reageren. Wanneer we met geladen deeltjes te maken hebben wordt deze barrière beïnvloed door een elektrisch veld. Zoals aangetoond in bijlage 11.13. betekent dit dat de partiële stroomdichtheden voor de twee richtingen van een reactie zoals (279H4.3.1) gegeven worden door:
]RT)nFexp[(ii:anodisch 0a ηα= (4.3.3.a)
]RT))nF((1exp[ii:kathodisch 0c ηα−−−= (282H4.3.3.b)
In deze vergelijkingen is i0 de uitwisselingsstroomdichtheid, dat is de stroomdichtheid waarmee de reactie tegelijkertijd in beide richtingen verloopt bij evenwicht, dus wanneer de totale stroomdichtheid i = ia + ic = 0. De constante α wordt de overdrachtscoëfficiënt genoemd. Deze ligt altijd tussen 0 en 1 en is in veel gevallen ongeveer 0.5. Wanneer geen evenwicht heerst is de nettostroom gelijk aan:
)]RT)nF)(1(exp(- - )RT)nF[exp((i i 0 ηα−ηα= (4.3.4).
Deze polarisatiekromme is geschetst in Figuur 4.3.1 samen met de afzonderlijke polarisatiekrommen van de twee deelreacties. Voor kleine overspanning ( |η| < 0.01 V) krijgen we een vereenvoudigde vergelijking:
00 nFiRTi = of (nF/RT)i i ηη= (4.3.5)
Deze vergelijking is geldig dicht bij de oorsprong in Figuur 4.3.1a. De factor RT/nFi0 heeft de dimensie van een weerstand en wordt de polarisatieweerstand (Rp) genoemd. Bij grote overspanning (|η| > 0.1 V) mag één van de twee termen in vergelijking (4.3.4). worden verwaarloosd en we krijgen dan de vergelijking:
η = β log|i/i0| (4.3.6). met β = 2.3RT/αnF voor een anodische en β = -2.3RT/(1-α)nF voor een kathodische reactie. Deze vorm staat bekend als de Tafel-vergelijking. Wanneer de overspanning tamelijk groot is wordt de overspanning vaak tegen log|i| uitgezet zoals getoond in Figuur 4.3.1b. Bij grote overspanning krijgen we rechte lijnen die, bij extrapolatie naar η = 0 elkaar snijden bij i = i0.

De snelheden van elektrochemische reacties
50
a.
η
i0
ic
ia
i
ic i0ia
b.
η
log|i|
i0
Figuur 4.3.1. Polarisatiediagram voor een enkelvoudige elektrode met ladingsoverdrachtpolarisatie: a. overspanning uitgezet tegen stroomdichtheid; b. overspanning uitgezet tegen log|stroomdichtheid| (Tafel-diagram).
a.
η
ic ia
b.
η
log|i|
i0
c.
η
icia
d.
η
log|i|
i0
Figuur 4.3.2. Polarisatiediagrammen voor enkelvoudige elektroden met ladingsoverdrachtpolarisatie: a., b. kleine uitwisselingsstroomdichtheid; c.,d. grote uitwisselingsstroomdichtheid.
De uitwisselingsstroomdichtheid is een belangrijk kenmerk van een elektrodereactie en de invloed op het polarisatiediagram wordt schematisch getoond in Figuur 4.3.2. Voor reacties met een grote uitwisselingsstroomdichtheid, hetgeen overeenkomt met een kleine polarisatieweerstand, is een veel kleinere overspanning nodig om een bepaalde stroomdichtheid te bereiken dan voor reacties met een kleine

51
uitwisselingsstroomdichtheid. In een grafiek van η tegen I geeft dit een minder steile kromme bij grotere i0, in een grafiek van η tegen i0 een verschuiving naar rechts.
4.3.2 Diffusiepolarisatie
Wanneer de snelheid van ladingsoverdracht erg groot is, dat wil zeggen bij grote i0 en/of grote overspanning, kan de snelheid van het transport van moleculen of ionen naar of van de elektrode snelheidsbepalend worden. Dit transport kan in principe via drie mechanismen plaats vinden: − migratie in een elektrisch veld (voor geladen deeltjes) − convectie ten gevolge van beweging van de vloeistof − diffusie als gevolg van concentratieverschillen in de oplossing. Migratie kan in vrijwel gevallen waar we in dit boek mee te maken hebben worden verwaarloosd omdat er bijna altijd een overmaat is aan zogenaamde "neutrale elektrolyt", dat wil zeggen van ionen die niet aan de eigenlijke elektrodereactie deelnemen. Convectie veroorzaakt homogenisatie van de oplossing, maar, afhankelijk van vloeistofsnelheid en meetkundige factoren, blijven concentratieverschillen bestaan in een dunne grenslaag van 0.01 tot 0.5 mm dikte aan het oppervlak van de elektrode. Daar treedt dus altijd diffusie op. Nernst voerde een eenvoudig model in om het effect van de diffusie te berekenen. In dit model wordt verondersteld dat in de oplossing op voldoend grote afstand van de elektrode de concentratie c0 overal gelijk is. In een grenslaag van dikte δ direct aan de elektrode veronderstelde Nernst de aanwezigheid van een lineaire concentratiegradiënt met een concentratie c aan het elektrodeoppervlak. Dit wordt schematisch getoond in Figuur 4.3.3.
Figuur 4.3.3. Concentratieprofiel in diffusielaag volgens het model van Nernst: a: geen stroom, b: geringe diffusiepolarisatie, c: sterke diffusiepolarisatie.
Volgens de eerste wet van Fick is de flux van een stof naar de elektrode evenredig met de concentratiegradiënt, met de diffusiecoëfficiënt D als

De snelheden van elektrochemische reacties
52
evenredigheidsconstante. Gecombineerd met de wet van Faraday (vergelijking (4.1.4)) geeft dit voor de stroomdichtheid de vergelijking:
δ−
−=cc
nFDi 0 (4.3.7).
Omdat de concentratie c natuurlijk nooit negatief kan worden is de maximale stroomdichtheid, die meestal de grensstroomdichtheid ig wordt genoemd, gelijk aan:
δ−= 0
gc
nFDi (4.3.8).
voor c = 0. Wanneer we dit combineren met de Nernst-vergeljking vinden we: ( ) ( )00 cclogn059.0)E(cE(c) =−=η (4.3.9).
Met de vergelijkingen (4.3.7). en (4.3.8). geeft dit: ( ) ( )gii1logn059.0 −=η (4.3.10)
In Figuur 4.3.4 is de hiermee overeenkomende polarisatiekromme voor een kathodische reactie schematisch weergegeven.
Figuur 4.3.4. Polarisatiekromme voor een kathodische reactie met volledige diffusiepolarisatie.
Uit de vergelijking voor de grensstroomdichtheid volgt dat deze toeneemt met toenemende concentratie c0. Vergroten van de stromingssnelheid van de vloeistof leidt tot een afname van de dikte van de diffusiegrenslaag δ en dus ook tot een vergroting van ig. Omdat de diffusiecoëfficiënt sterk toeneemt met stijgende temperatuur is dat ook het geval met de grensstroomdichtheid.
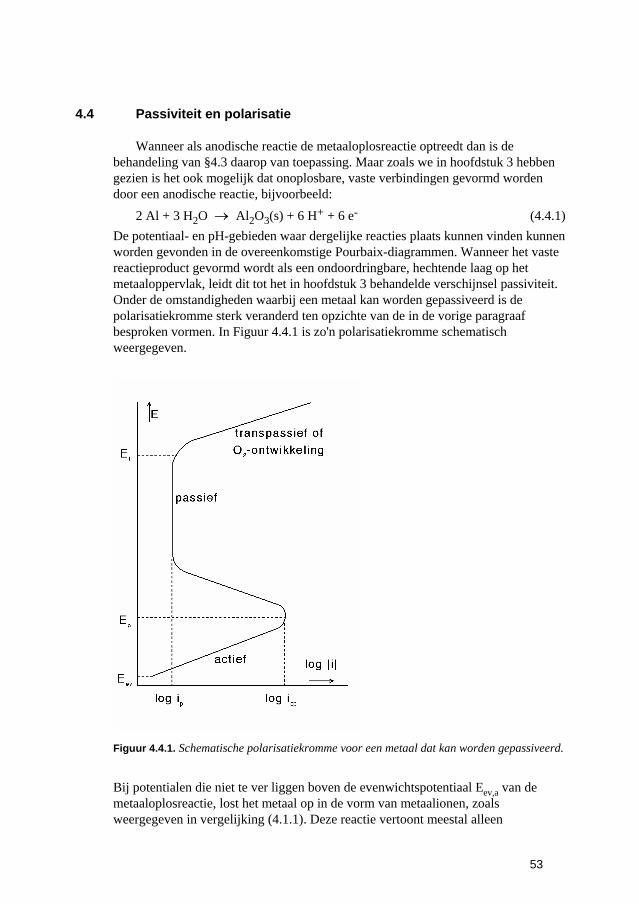
53
4.4 Passiviteit en polarisatie
Wanneer als anodische reactie de metaaloplosreactie optreedt dan is de behandeling van §4.3 daarop van toepassing. Maar zoals we in hoofdstuk 3 hebben gezien is het ook mogelijk dat onoplosbare, vaste verbindingen gevormd worden door een anodische reactie, bijvoorbeeld:
2 Al + 3 H2O → Al2O3(s) + 6 H+ + 6 e- (4.4.1) De potentiaal- en pH-gebieden waar dergelijke reacties plaats kunnen vinden kunnen worden gevonden in de overeenkomstige Pourbaix-diagrammen. Wanneer het vaste reactieproduct gevormd wordt als een ondoordringbare, hechtende laag op het metaaloppervlak, leidt dit tot het in hoofdstuk 3 behandelde verschijnsel passiviteit. Onder de omstandigheden waarbij een metaal kan worden gepassiveerd is de polarisatiekromme sterk veranderd ten opzichte van de in de vorige paragraaf besproken vormen. In Figuur 4.4.1 is zo'n polarisatiekromme schematisch weergegeven.
Figuur 4.4.1. Schematische polarisatiekromme voor een metaal dat kan worden gepassiveerd.
Bij potentialen die niet te ver liggen boven de evenwichtspotentiaal Eev,a van de metaaloplosreactie, lost het metaal op in de vorm van metaalionen, zoals weergegeven in vergelijking (4.1.1). Deze reactie vertoont meestal alleen

De snelheden van elektrochemische reacties
54
ladingsoverdracht polarisatie (Tafel-gedrag). Wanneer de potentiaal voldoende wordt verhoogd bereikt de stroomdichtheid een maximum: de kritische passiveringsstroomdichtheid icp bij de passiveringspotentiaal Ep. Wanneer de potentiaal boven deze waarde komt neemt de stroomdichtheid snel af tot een zeer lage waarde: de passieve stroomdichtheid ip. Zo is voor ijzer in neutrale waterige oplossing de icp ongeveer 10-3 A.cm-2 bij de passiveringspotentiaal van ongeveer +0.1 V. De passieve stroomdichtheid ip daarentegen is slechts ongeveer 10-8 A.cm-2. Boven de passiveringspotentiaal is het metaal bedekt met een ondoordringbare en hechtende laag van reactieproducten, in de meeste gevallen een oxide of hydroxide, die de oorzaak is van de passiviteit. Bij een veel hogere potentiaal, de zogenaamde transpassieve potentiaal Et, zien we soms opnieuw een toename van de stroomdichtheid, zoals in Figuur 4.4.1 is aangegeven. De oorzaak hiervan is het gaan optreden van een secondaire anodische reactie. Soms heeft deze niets met het metaal zelf te maken, bijvoorbeeld bij de zuurstofontwikkelingsreactie:
V 1.23E ,e 4H 4O OH 2 022 =++→ −+ (4.4.2)
Aan de andere kant is het echter ook mogelijk dat een nieuwe corrosiereactie gaat verlopen, zoals bijvoorbeeld bij chroom en chroomhoudende legeringen. Daar kunnen boven E = 0.8 tot 1 V chromaationen worden gevormd volgens de vergelijking:
V 0.71E ,e 6H8CrO OH 4 +Cr 0-242 =++→ −+ (4.4.3)
In het algemeen geldt dus dat, wanneer we willen dat een metaal zich passief gedraagt, ervoor moet worden gezorgd dat de potentiaal tussen Ep en Et wordt gehouden.
4.5 Samenvatting
In §4.1 hebben we gezien dat er een direct verband is tussen de snelheid van een elektrochemische reactie en de stroom of de stroomdichtheid aan de elektrode waaraan die reactie verloopt. Wanneer een stroom loopt dan verschilt de potentiaal van de elektrode van de evenwichtspotentiaal: dit noemen we polarisatie. Het gedrag van elektrode wordt dan beschreven met de relatie tussen de potentiaalverandering of overspanning enerzijds en de stroomdichtheid anderzijds. Dit wordt vaak grafisch weergegeven in de polarisatiekromme. Voor anodische reacties zijn overspanning en stroom positief, voor kathodische reacties negatief. De vorm van de polarisatiekromme wordt bepaald door de snelheidsbepalende stap van het elektrodeproces. Wanneer de ladingsoverdracht aan de elektrode snelheidsbepalend is spreken we van ladingsoverdrachtpolarisatie en de polarisatiekromme heeft de in de Figuur 4.3.1 en Figuur 4.3.2 getekende vorm. Een belangrijke parameter die de reactie in dit geval karakteriseert is de uitwisselingsstroomdichtheid die gelijk is aan de snelheid waarmee de reactie in evenwicht tegelijkertijd in tegengestelde richtingen verloopt. Bij kleine

55
uitwisselingsstroomdichtheid is een grote overspanning nodig om een bepaalde stroomdichtheid te krijgen, bij grote is een kleine overspanning voldoende. Speciaal wanneer de ladingsoverdrachtreactie snel is (grote stroomdichtheid) kan het transport van reactanten of producten naar of van het elektrodeoppervlak snelheidsbepalend worden en dan spreken we van diffusiepolarisatie. De polarisatiekromme hiervoor is geschetst in Figuur 4.3.4. In dit geval is er een grensstroomdichtheid die overeenkomt met de maximale snelheid waarmee de reactie kan verlopen. Verhoging van de concentratie van de reagerende stof(fen) leidt tot een verhoging van de grensstroomdichtheid en omgekeerd. Tenslotte is er nog het geval dat een metaal passief kan worden ten gevolge van de vorming van een ondoordringbare en hechtende laag van onoplosbare reactieproducten van de anodische reactie. Dan heeft de polarisatiekromme de in Figuur 4.4.1 schematisch weergegeven vorm. In dit geval gedraagt het metaal zich passief wanneer de potentiaal daarvan ligt tussen de potentialen Ep en Et. In hoofdstuk 5 passen we deze concepten toe op de corrosie van metalen, waarbij het metaal zich als een meervoudige elektrode gedraagt, dat wil zeggen dat dan minstens twee verschillende elektrochemische reactie aan het oppervlak plaatsvinden. Dit zijn (ten minste) één anodische reactie waarin het metaal wordt geoxideerd en één kathodische reactie waarin een of andere component uit de omgeving wordt gereduceerd.
4.6 Opgaven
1. Bereken de hoeveelheid ijzer (in gram) die in de vorm van Fe2+-ionen oplost wanneer gedurende 24 uur een anodische stroom van 1 mA door een ijzerelektrode loopt.
2. Bereken de hoeveelheid zink (in gram) die neerslaat wanneer een kathodische stroom van 10 mA gedurende 5 uur door een elektrode loopt. Wat is de dikte van het neerslag wanneer dat gelijkmatig wordt gevormd en de oppervlakte van de elektrode 15 cm2 is?
3. Bereken de hoeveelheid waterstof (in ml bij 25 °C en 1 atm) die wordt ontwikkeld door een kathodische stroom van 5 mA gedurende 15 uur.
4. Bereken de hoeveelheid zuurstof (in ml bij 25 °C en 1 atm) die wordt verbruikt door een kathodische stroom van 2 mA gedurende 48 uur.
5. Teken het polarisatiediagram voor een elektrodereactie welke alleen ladingsoverdrachtpolarisatie vertoont, met een uitwisselingsstroomdichtheid van 10-5 A.cm-2, overdrachtscoëfficiënt α = 0.45 en aantal elektronen n = 2.
6. Bereken de overspanning η in Volt voor een elektrodereactie die alleen ladingsoverdrachtpolarisatie vertoont met een uitwisselingsstroomdichtheid van 5 × 10-7 A.cm-2, overdrachtscoëfficiënt α = 0.5 en aantal elektronen n = 1, bij een netto stroomdichtheid van 10 mA.cm-2.

De snelheden van elektrochemische reacties
56
7. Bereken de grensstroomdichtheid voor zuurstofreductie aan een elektrode staande in een oplossing die 8 mg.l-1 zuurstof bevat. De diffusiecoëfficiënt van zuurstof is 10-5 cm2s-1 en de dikte van de diffusielaag is 0.5 mm.
4.7 Literatuur
1. J. Albery, Electrode Kinetics, Oxford Chemistry Series no. 14, Oxford, Clarendon Press (1975).
2. A.J. Bard and L.R. Faulkner, Electrochemical Methods, Fundamentals and Applications, New York, John Wiley (1980)
3. D.L. Piron, The electrochemistry of corrosion, Houston, NACE (1991) 4. H. Kaesche, Metallic corrosion, 2nd ed., Houston, NACE (1985)

5
De snelheden van elektrochemische corrosiereacties

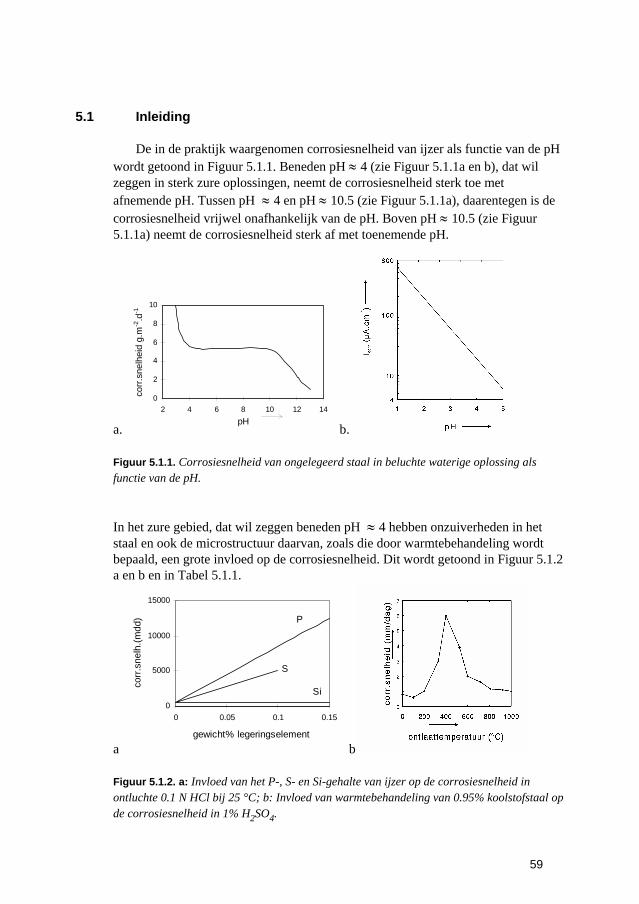
59
5.1 Inleiding
De in de praktijk waargenomen corrosiesnelheid van ijzer als functie van de pH wordt getoond in Figuur 5.1.1. Beneden pH ≈ 4 (zie Figuur 5.1.1a en b), dat wil zeggen in sterk zure oplossingen, neemt de corrosiesnelheid sterk toe met afnemende pH. Tussen pH ≈ 4 en pH ≈ 10.5 (zie Figuur 5.1.1a), daarentegen is de corrosiesnelheid vrijwel onafhankelijk van de pH. Boven pH ≈ 10.5 (zie Figuur 5.1.1a) neemt de corrosiesnelheid sterk af met toenemende pH.
a.
0
2
4
6
8
10
2 4 6 8 10 12 14pH
corr.
snel
heid
g.m
-2.d
-1
b.
Figuur 5.1.1. Corrosiesnelheid van ongelegeerd staal in beluchte waterige oplossing als functie van de pH.
In het zure gebied, dat wil zeggen beneden pH ≈ 4 hebben onzuiverheden in het staal en ook de microstructuur daarvan, zoals die door warmtebehandeling wordt bepaald, een grote invloed op de corrosiesnelheid. Dit wordt getoond in Figuur 5.1.2 a en b en in Tabel 5.1.1.
a
0
5000
10000
15000
0 0.05 0.1 0.15
gewicht% legeringselement
corr
.sne
lh.(m
dd) P
Si
S
b
Figuur 5.1.2. a: Invloed van het P-, S- en Si-gehalte van ijzer op de corrosiesnelheid in ontluchte 0.1 N HCl bij 25 °C; b: Invloed van warmtebehandeling van 0.95% koolstofstaal op de corrosiesnelheid in 1% H2SO4.
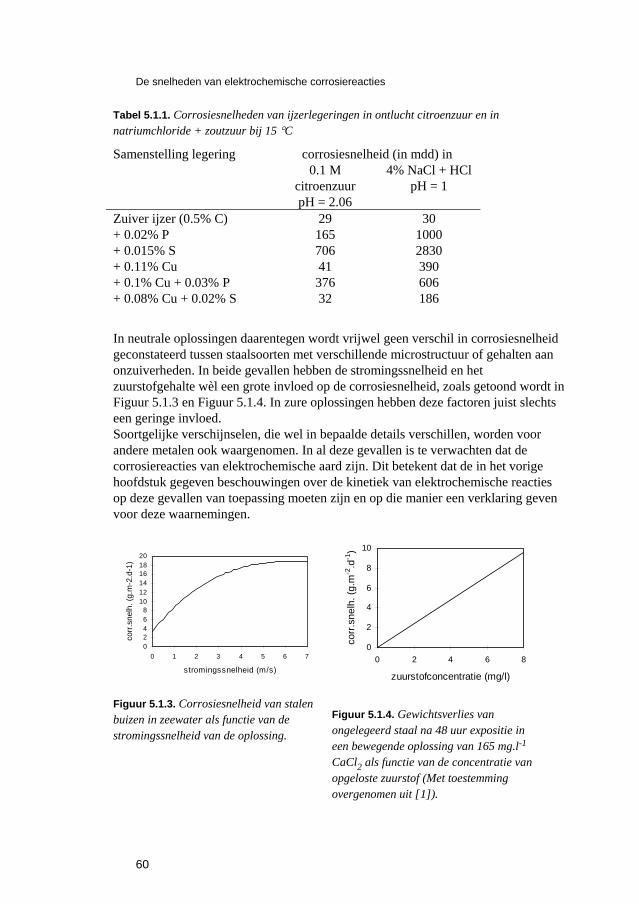
De snelheden van elektrochemische corrosiereacties
60
Tabel 5.1.1. Corrosiesnelheden van ijzerlegeringen in ontlucht citroenzuur en in natriumchloride + zoutzuur bij 15 °C
Samenstelling legering corrosiesnelheid (in mdd) in 0.1 M
citroenzuur pH = 2.06
4% NaCl + HClpH = 1
Zuiver ijzer (0.5% C) 29 30 + 0.02% P 165 1000 + 0.015% S 706 2830 + 0.11% Cu 41 390 + 0.1% Cu + 0.03% P 376 606 + 0.08% Cu + 0.02% S 32 186
In neutrale oplossingen daarentegen wordt vrijwel geen verschil in corrosiesnelheid geconstateerd tussen staalsoorten met verschillende microstructuur of gehalten aan onzuiverheden. In beide gevallen hebben de stromingssnelheid en het zuurstofgehalte wèl een grote invloed op de corrosiesnelheid, zoals getoond wordt in Figuur 5.1.3 en Figuur 5.1.4. In zure oplossingen hebben deze factoren juist slechts een geringe invloed. Soortgelijke verschijnselen, die wel in bepaalde details verschillen, worden voor andere metalen ook waargenomen. In al deze gevallen is te verwachten dat de corrosiereacties van elektrochemische aard zijn. Dit betekent dat de in het vorige hoofdstuk gegeven beschouwingen over de kinetiek van elektrochemische reacties op deze gevallen van toepassing moeten zijn en op die manier een verklaring geven voor deze waarnemingen.
02468
101214161820
0 1 2 3 4 5 6 7
stromingssnelheid (m/s)
corr.
snel
h. (g
.m-2
.d-1
)
Figuur 5.1.3. Corrosiesnelheid van stalen buizen in zeewater als functie van de stromingssnelheid van de oplossing.
0
2
4
6
8
10
0 2 4 6 8
zuurstofconcentratie (mg/l)
corr
.sne
lh. (
g.m
-2.d
-1)
Figuur 5.1.4. Gewichtsverlies van ongelegeerd staal na 48 uur expositie in een bewegende oplossing van 165 mg.l-1
CaCl2 als functie van de concentratie van opgeloste zuurstof (Met toestemming overgenomen uit [1]).

61
In §4.1 is al gesteld dat een corroderend metaal een meervoudige elektrode vormt en we beginnen daarom in §5.2 met een beschouwing van polarisatiediagrammen van meervoudige elektroden. We bespreken daarna achtereenvolgens corrosie in zure en neutrale oplossingen, de invloed van de weerstand van de elektrolyt op de corrosie, de effecten van de koppeling van ongelijke metalen en de omstandigheden waaronder passiviteit optreedt. Voor een iets uitvoeriger behandeling van de elektrochemische theorie van corrosie verwijzen we naar de boeken van West [2], Bradford [3], Kaesche [4] en Piron [5].
5.2 Polarisatiediagrammen voor meervoudige elektroden
Een corroderend metaal vormt een meervoudige elektrode omdat aan het oppervlak daarvan ten minste twee elektrodereacties tegelijk optreden. Dit zijn de anodische metaaloplosreactie, bijvoorbeeld:
−+→ e2Fe Fe +2 (5.2.1)
en één of andere kathodische reactie, bijvoorbeeld:
2+ He 2H 2 →+ − (5.2.2)
Deze twee reacties zijn hier geschreven in de richting in welke ze in de praktijk verlopen gedurende corrosie. Zij kunnen natuurlijk ook, maar dan niet tegelijkertijd, als evenwichtsreacties optreden bij de bijbehorende evenwichtspotentialen Eev,a en Eev,c, met de uitwisselingsstroomdichtheden i0a en i0c. Zoals in hoofdstuk 3 is aangetoond is corrosie alleen mogelijk wanneer Eev,a < Eev,c. De netto stroomdichtheid wordt volgens vergelijking (4.1.3) gegeven door:
ca ii = i + (5.2.3)
waarbij is aangenomen dat de anodische en kathodische reacties allebei plaatsvinden op het gehele elektrodeoppervlak. In principe wordt de polarisatie van beide reacties, wanneer we ons voorlopig tot ladingsoverdrachtpolarisatie beperken, gegeven door een vergelijking van de vorm (4.3.4). Het volledige polarisatiediagram is nu een combinatie van twee diagrammen van het type getoond in Figuur 4.3.1 en is weergegeven in Figuur 5.2.1. Hieruit zien we dat wanneer het verschil tussen de evenwichtspotentialen, dat is: Eev,c - Eev,a, groot genoeg is alleen rekening gehouden hoeft te worden met de anodische kromme van de anodische reactie en de kathodische kromme van de kathodische reactie.
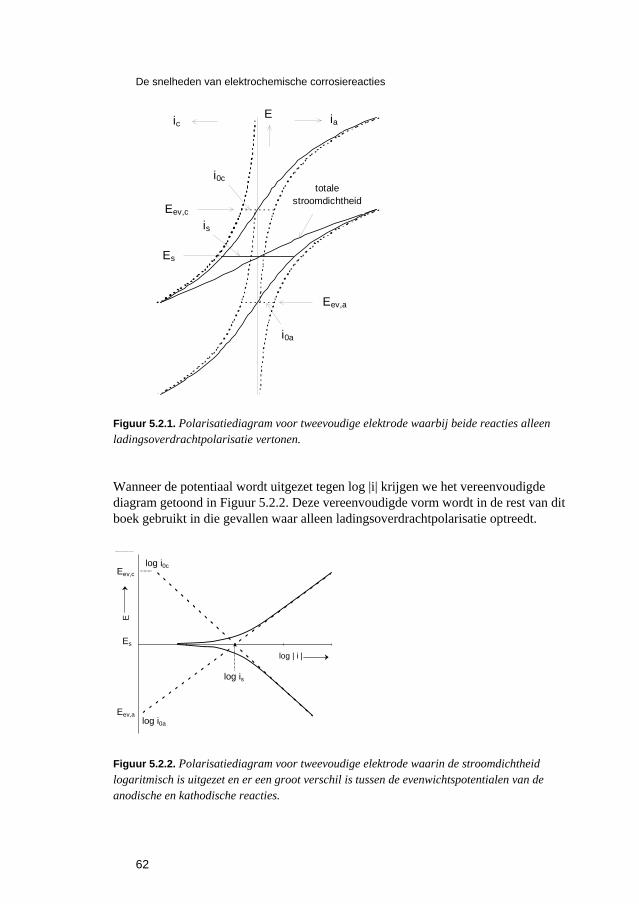
De snelheden van elektrochemische corrosiereacties
62
Eic ia
totale stroomdichtheid
Es
isEev,c
Eev,a
i0c
i0a
Figuur 5.2.1. Polarisatiediagram voor tweevoudige elektrode waarbij beide reacties alleen ladingsoverdrachtpolarisatie vertonen.
Wanneer de potentiaal wordt uitgezet tegen log |i| krijgen we het vereenvoudigde diagram getoond in Figuur 5.2.2. Deze vereenvoudigde vorm wordt in de rest van dit boek gebruikt in die gevallen waar alleen ladingsoverdrachtpolarisatie optreedt.
log | i |
E
log is
log i0c
log i0a
Eev,a
Eev,c
Es
Figuur 5.2.2. Polarisatiediagram voor tweevoudige elektrode waarin de stroomdichtheid logaritmisch is uitgezet en er een groot verschil is tussen de evenwichtspotentialen van de anodische en kathodische reacties.
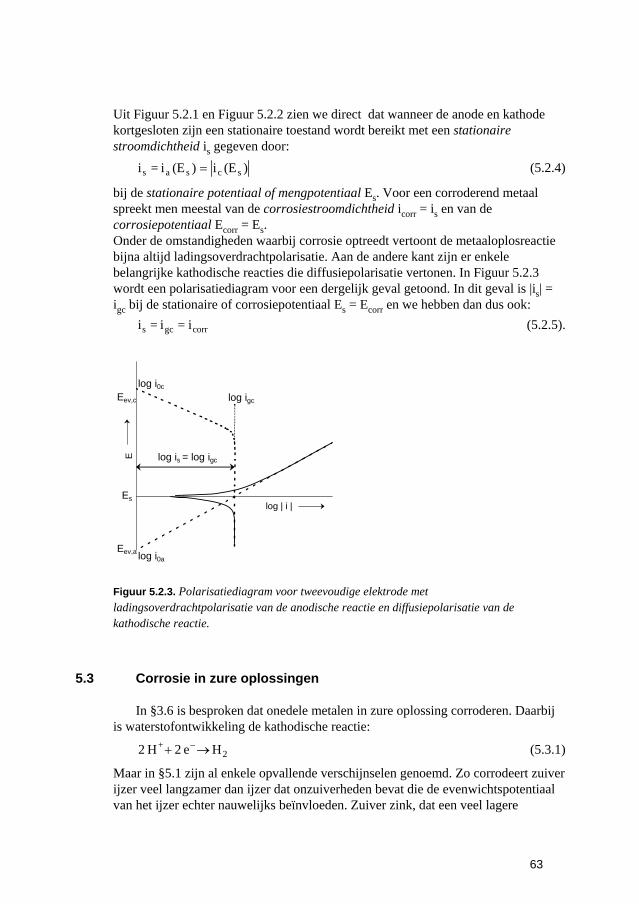
63
Uit Figuur 5.2.1 en Figuur 5.2.2 zien we direct dat wanneer de anode en kathode kortgesloten zijn een stationaire toestand wordt bereikt met een stationaire stroomdichtheid is gegeven door:
)(Ei)(Ei = i scsas = (5.2.4)
bij de stationaire potentiaal of mengpotentiaal Es. Voor een corroderend metaal spreekt men meestal van de corrosiestroomdichtheid icorr = is en van de corrosiepotentiaal Ecorr = Es. Onder de omstandigheden waarbij corrosie optreedt vertoont de metaaloplosreactie bijna altijd ladingsoverdrachtpolarisatie. Aan de andere kant zijn er enkele belangrijke kathodische reacties die diffusiepolarisatie vertonen. In Figuur 5.2.3 wordt een polarisatiediagram voor een dergelijk geval getoond. In dit geval is |is| = igc bij de stationaire of corrosiepotentiaal Es = Ecorr en we hebben dan dus ook:
corrgcs i = i = i (5.2.5).
log | i |
E
log igc
log i0a
log i0c
log is = log igc
Eev,c
Es
Eev,a
Figuur 5.2.3. Polarisatiediagram voor tweevoudige elektrode met ladingsoverdrachtpolarisatie van de anodische reactie en diffusiepolarisatie van de kathodische reactie.
5.3 Corrosie in zure oplossingen
In §3.6 is besproken dat onedele metalen in zure oplossing corroderen. Daarbij is waterstofontwikkeling de kathodische reactie:
2+ He 2H 2 →+ − (5.3.1)
Maar in §5.1 zijn al enkele opvallende verschijnselen genoemd. Zo corrodeert zuiver ijzer veel langzamer dan ijzer dat onzuiverheden bevat die de evenwichtspotentiaal van het ijzer echter nauwelijks beïnvloeden. Zuiver zink, dat een veel lagere

De snelheden van elektrochemische corrosiereacties
64
evenwichtspotentiaal heeft dan ijzer corrodeert onder overigens gelijke omstandigheden maar weinig sneller. Een aanwijzing voor de verklaring van deze waarnemingen wordt door het volgende experiment geleverd. Zuiver zink in een zure oplossing corrodeert langzaam, maar wanneer het wordt verbonden met een stuk platina dat in dezelfde oplossing staat zien we sterke waterstofontwikkeling op het platinaoppervlak en tegelijkertijd begint het zink snel op te lossen. Blijkbaar is het gemak waarmee de waterstofontwikkelingsreactie verloopt bepalend voor de corrosiesnelheid. Reactie (5.3.1) vertoont altijd ladingsoverdrachtpolarisatie bij voldoend lage pH (≤ 4). Verder is gebleken dat de uitwisselingsstroomdichtheid zeer gevoelig is voor het type en de oppervlaktetoestand van het metaal waaraan deze reactie verloopt. Dit blijkt duidelijk uit de in Tabel 5.3.1 verzamelde gegevens over de waterstofontwikkelingsreactie aan verschillende metalen.
Tabel 5.3.1. Parameters van ladingsoverdrachtpolarisatie voor waterstofontwikkeling aan verschillende metalen (uit: H.H.Uhlig, Corrosion and Corrosion Control, New York, Wiley, 2nd ed. 1972)
Metaal oplossing - β (Volt) i0 (A.cm-2) η voor i = 1 mA.cm-2
Pt 1 N HCl 0.03 10-3 0.00 Au 1 N HCl 0.05 10-6 0.15 Ag 0.1 N HCl 0.10 5 × 10-7 0.30 Ni 0.1 N HCl 0.15 8 × 10-7 0.31 Fe 1 N HCl 0.10 10-6 0.45 Fe 4% NaCl,
pH = 1 - 4 0.12 10-7 0.40
Cu 0.1 N HCl 0.10 2 × 10-7 0.44 Al 2 N H2SO4 0.10 10-10 0.70 Sn 1 N HCl 0.15 10-8 0.75 Cd 1 N HCl 0.20 10-7 0.80 Zn 1 N H2SO4 0.12 10-11 0.94 Mg 0.1 N HCl 0.12 7 × 10-13 1.10 Pb 0.01 - 8 N HCl 0.12 2 × 10-13 1.16
Voor de anodische oplosreacties van ijzer en zink, welke ook ladingsoverdrachtpolarisatie vertonen zijn de overeenkomstige parameters:
−+→ e2Fe Fe +2 : βa = 0.12 V, i0a = 10-6 A.cm-2
−+→ e2ZnZn +2 : βa = 0.12 V, i0a = 10-11 A.cm-2.
Met deze gegevens kunnen we het polarisatiediagram van Figuur 5.3.1 construeren. We zien daaruit dat zink inderdaad bijna dezelfde corrosiesnelheid vertoont als ijzer ondanks zijn veel lagere evenwichtspotentiaal.
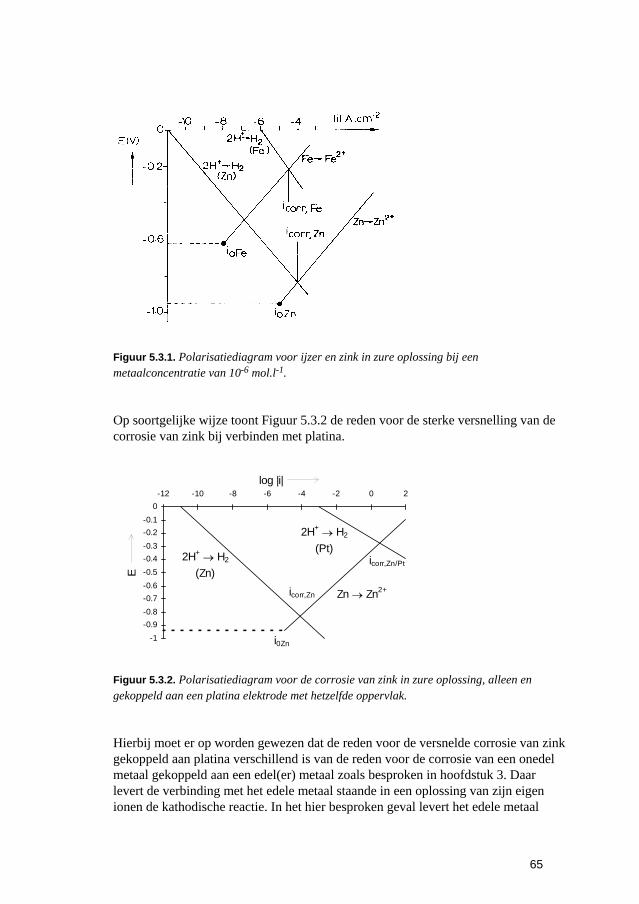
65
Figuur 5.3.1. Polarisatiediagram voor ijzer en zink in zure oplossing bij een metaalconcentratie van 10-6 mol.l-1.
Op soortgelijke wijze toont Figuur 5.3.2 de reden voor de sterke versnelling van de corrosie van zink bij verbinden met platina.
-1-0.9-0.8-0.7-0.6-0.5-0.4-0.3-0.2-0.1
0-12 -10 -8 -6 -4 -2 0 2
log |i|
E
Zn → Zn2+
2H+ → H2
(Zn)
2H+ → H2
(Pt)
i0Zn
icorr,Zn
icorr,Zn/Pt
Figuur 5.3.2. Polarisatiediagram voor de corrosie van zink in zure oplossing, alleen en gekoppeld aan een platina elektrode met hetzelfde oppervlak.
Hierbij moet er op worden gewezen dat de reden voor de versnelde corrosie van zink gekoppeld aan platina verschillend is van de reden voor de corrosie van een onedel metaal gekoppeld aan een edel(er) metaal zoals besproken in hoofdstuk 3. Daar levert de verbinding met het edele metaal staande in een oplossing van zijn eigen ionen de kathodische reactie. In het hier besproken geval levert het edele metaal

De snelheden van elektrochemische corrosiereacties
66
alleen het oppervlak waaraan de kathodische reactie, die ook al aan het oppervlak van het zink zelf verloopt, met een veel hogere snelheid kan verlopen. Het sneller worden van corrosie in aanwezigheid van verontreinigingen in het metaal wordt op dezelfde manier verklaard als die ten gevolge van het koppelen aan platina. De waterstofontwikkeling verloopt namelijk in veel gevallen veel sneller aan dergelijke onzuiverheden, dat wil zeggen dat de waterstofontwikkelingsreactie daaraan een veel hogere uitwisselingsstroomdichtheid heeft. Wanneer deze verhoging voldoende groot is kunnen zelfs kleine hoeveelheden verontreiniging een sterk verhoogde corrosiesnelheid veroorzaken (zie opgave 1 aan het eind van dit hoofdstuk). Dit wordt ook geïllustreerd met de gegevens in Tabel 5.3.2 voor de invloed van legeringselementen op de corrosiesnelheid van zink in zure oplossing. We zien hier dezelfde volgorde als in Tabel 5.3.1 voor de uitwisselingsstroomdichtheid van de waterstofontwikkeling, hetgeen het boven gegeven beeld ondersteunt.
Tabel 5.3.2. Tijd die nodig is voor de ontwikkeling van 40 ml H2 bij blootstellen van zink en zinklegeringen aan 0.25 M H2SO4.
Legeringselement gehalte (gewichts%)
tijd (uur)
Cu 0.97 0.3 Fe 1.2 0.55 Sb 1.07 1.0 As 1.1 3.0 Sn 1.03 3.5 Cd 1.0 4.0 Zuiver zink - 6.5
log |i|
E
is5 is4
is0
is1is2
is3
i0a
i0c
0
12
34
5
pH
Figuur 5.3.3. Schematisch polarisatiediagram van een metaal dat onder waterstofontwikkeling corrodeert in zure oplossingen van verschillende pH.
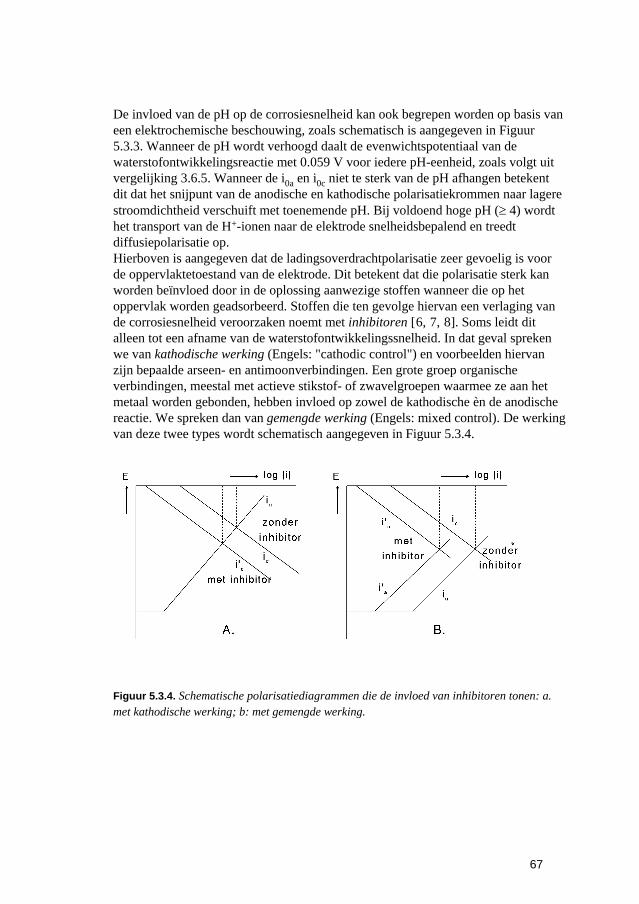
67
De invloed van de pH op de corrosiesnelheid kan ook begrepen worden op basis van een elektrochemische beschouwing, zoals schematisch is aangegeven in Figuur 5.3.3. Wanneer de pH wordt verhoogd daalt de evenwichtspotentiaal van de waterstofontwikkelingsreactie met 0.059 V voor iedere pH-eenheid, zoals volgt uit vergelijking 3.6.5. Wanneer de i0a en i0c niet te sterk van de pH afhangen betekent dit dat het snijpunt van de anodische en kathodische polarisatiekrommen naar lagere stroomdichtheid verschuift met toenemende pH. Bij voldoend hoge pH (≥ 4) wordt het transport van de H+-ionen naar de elektrode snelheidsbepalend en treedt diffusiepolarisatie op. Hierboven is aangegeven dat de ladingsoverdrachtpolarisatie zeer gevoelig is voor de oppervlaktetoestand van de elektrode. Dit betekent dat die polarisatie sterk kan worden beïnvloed door in de oplossing aanwezige stoffen wanneer die op het oppervlak worden geadsorbeerd. Stoffen die ten gevolge hiervan een verlaging van de corrosiesnelheid veroorzaken noemt met inhibitoren [6, 7, 8]. Soms leidt dit alleen tot een afname van de waterstofontwikkelingssnelheid. In dat geval spreken we van kathodische werking (Engels: "cathodic control") en voorbeelden hiervan zijn bepaalde arseen- en antimoonverbindingen. Een grote groep organische verbindingen, meestal met actieve stikstof- of zwavelgroepen waarmee ze aan het metaal worden gebonden, hebben invloed op zowel de kathodische èn de anodische reactie. We spreken dan van gemengde werking (Engels: mixed control). De werking van deze twee types wordt schematisch aangegeven in Figuur 5.3.4.
Figuur 5.3.4. Schematische polarisatiediagrammen die de invloed van inhibitoren tonen: a. met kathodische werking; b: met gemengde werking.
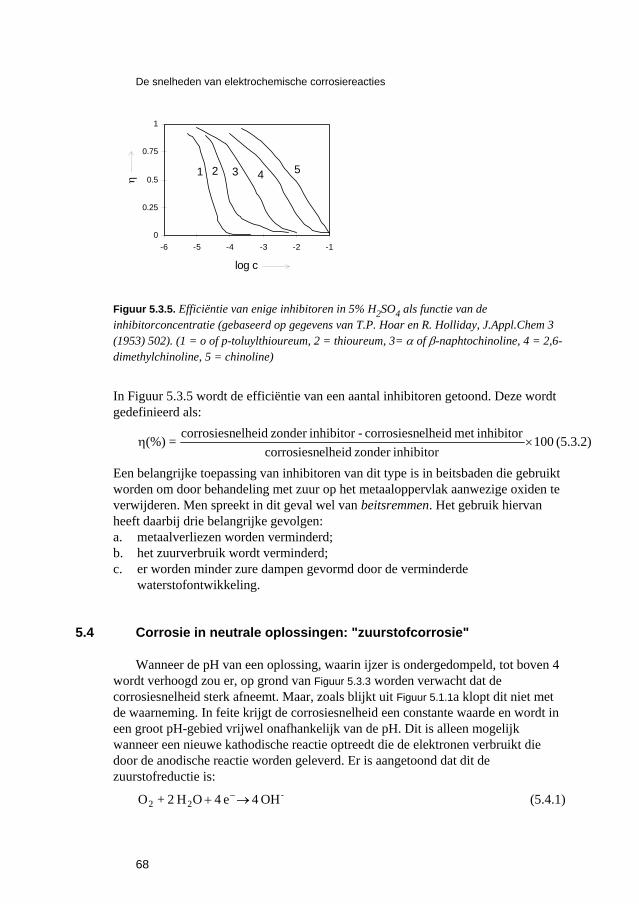
De snelheden van elektrochemische corrosiereacties
68
0
0.25
0.5
0.75
1
-6 -5 -4 -3 -2 -1
log c
η
1 2 3 4 5
Figuur 5.3.5. Efficiëntie van enige inhibitoren in 5% H2SO4 als functie van de inhibitorconcentratie (gebaseerd op gegevens van T.P. Hoar en R. Holliday, J.Appl.Chem 3 (1953) 502). (1 = o of p-toluylthioureum, 2 = thioureum, 3= α of β-naphtochinoline, 4 = 2,6-dimethylchinoline, 5 = chinoline)
In Figuur 5.3.5 wordt de efficiëntie van een aantal inhibitoren getoond. Deze wordt gedefinieerd als:
100inhibitorzonder elheidcorrosiesn
inhibitormet elheidcorrosiesn -inhibitor zonder elheidcorrosiesn=(%) ×η (5.3.2)
Een belangrijke toepassing van inhibitoren van dit type is in beitsbaden die gebruikt worden om door behandeling met zuur op het metaaloppervlak aanwezige oxiden te verwijderen. Men spreekt in dit geval wel van beitsremmen. Het gebruik hiervan heeft daarbij drie belangrijke gevolgen: a. metaalverliezen worden verminderd; b. het zuurverbruik wordt verminderd; c. er worden minder zure dampen gevormd door de verminderde
waterstofontwikkeling.
5.4 Corrosie in neutrale oplossingen: "zuurstofcorrosie"
Wanneer de pH van een oplossing, waarin ijzer is ondergedompeld, tot boven 4 wordt verhoogd zou er, op grond van Figuur 5.3.3 worden verwacht dat de corrosiesnelheid sterk afneemt. Maar, zoals blijkt uit Figuur 5.1.1a klopt dit niet met de waarneming. In feite krijgt de corrosiesnelheid een constante waarde en wordt in een groot pH-gebied vrijwel onafhankelijk van de pH. Dit is alleen mogelijk wanneer een nieuwe kathodische reactie optreedt die de elektronen verbruikt die door de anodische reactie worden geleverd. Er is aangetoond dat dit de zuurstofreductie is:
-22 OH 4e 4OH 2 + O →+ − (5.4.1)
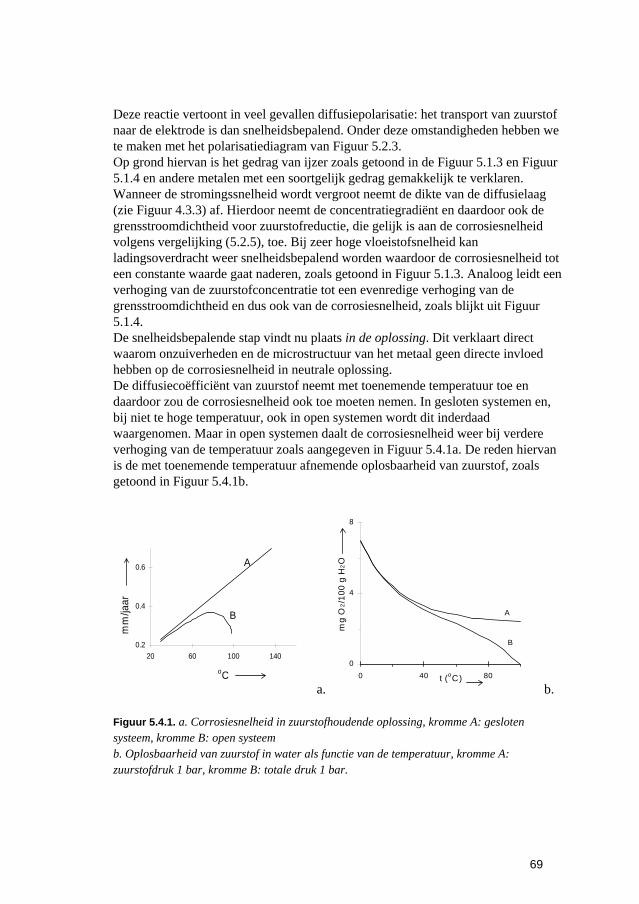
69
Deze reactie vertoont in veel gevallen diffusiepolarisatie: het transport van zuurstof naar de elektrode is dan snelheidsbepalend. Onder deze omstandigheden hebben we te maken met het polarisatiediagram van Figuur 5.2.3. Op grond hiervan is het gedrag van ijzer zoals getoond in de Figuur 5.1.3 en Figuur 5.1.4 en andere metalen met een soortgelijk gedrag gemakkelijk te verklaren. Wanneer de stromingssnelheid wordt vergroot neemt de dikte van de diffusielaag (zie Figuur 4.3.3) af. Hierdoor neemt de concentratiegradiënt en daardoor ook de grensstroomdichtheid voor zuurstofreductie, die gelijk is aan de corrosiesnelheid volgens vergelijking (5.2.5), toe. Bij zeer hoge vloeistofsnelheid kan ladingsoverdracht weer snelheidsbepalend worden waardoor de corrosiesnelheid tot een constante waarde gaat naderen, zoals getoond in Figuur 5.1.3. Analoog leidt een verhoging van de zuurstofconcentratie tot een evenredige verhoging van de grensstroomdichtheid en dus ook van de corrosiesnelheid, zoals blijkt uit Figuur 5.1.4. De snelheidsbepalende stap vindt nu plaats in de oplossing. Dit verklaart direct waarom onzuiverheden en de microstructuur van het metaal geen directe invloed hebben op de corrosiesnelheid in neutrale oplossing. De diffusiecoëfficiënt van zuurstof neemt met toenemende temperatuur toe en daardoor zou de corrosiesnelheid ook toe moeten nemen. In gesloten systemen en, bij niet te hoge temperatuur, ook in open systemen wordt dit inderdaad waargenomen. Maar in open systemen daalt de corrosiesnelheid weer bij verdere verhoging van de temperatuur zoals aangegeven in Figuur 5.4.1a. De reden hiervan is de met toenemende temperatuur afnemende oplosbaarheid van zuurstof, zoals getoond in Figuur 5.4.1b.
0.2
0.4
0.6
20 60 100 140
oC
mm
/jaar
A
B
a.
0
4
8
0 40 80t (oC)
mg
O2/1
00 g
H2O
A
B
b.
Figuur 5.4.1. a. Corrosiesnelheid in zuurstofhoudende oplossing, kromme A: gesloten systeem, kromme B: open systeem b. Oplosbaarheid van zuurstof in water als functie van de temperatuur, kromme A: zuurstofdruk 1 bar, kromme B: totale druk 1 bar.

De snelheden van elektrochemische corrosiereacties
70
Dit alles leidt direct tot een methode om dit soort corrosie te bestrijden: het verwijderen van zuurstof, hetgeen vaak ontluchten wordt genoemd. Dit kan chemisch worden gedaan met stoffen die zuurstof verwijderen (zuurstofbinders of Engels: "scavengers"). De belangrijkste hiervan zijn hydrazine (N2H4) en sulfieten (met de SO3
2--groep) die als volgt reageren: N2H4 + O2 → N2 + 2 H2O; 2 SO3
2- + O2 → 2 SO42-.
Tegenwoordig wordt ontluchten nog vaker toegepast om zuurstof te verwijderen. Dit wordt gedaan door een vacuumbehandeling of door doorblazen van een inert gas, bijna altijd gecombineerd met verwarming, bijvoorbeeld met stoom. Dat centrale verwarmingssystemen meestal verbazend weinig corrosie vertonen is een gevolg van het feit dat in deze, praktisch volledig gesloten, systemen het zuurstofgehalte al snel heel laag wordt. Dit is zowel een gevolg van het verwarmen als van reactie met het (zeer grote) ijzeroppervlak. Maar wanneer zo'n systeem een lek vertoont en regelmatig vers water wordt toegevoegd kan ernstige corrosie een gevolg zijn van de toevoer van verse zuurstof en een te korte tijd voor volledige verwijdering.
5.5 Elektrolytweerstand en corrosiesnelheid
Corrosie vindt meestal plaats in oplossingen die zogenaamde "neutrale" elektrolyten bevatten, die noch de pH beïnvloeden, noch deelnemen aan de elektrodereacties. Maar zij maken het wel mogelijk dat ladingen door de oplossing van anode naar kathode en omgekeerd worden getransporteerd. Wanneer de oplossing een eindige weerstand heeft is er ook een eindig potentiaalverschil ΔE (vaak genoemd de potentiaalval) tussen de anodische en kathodische plaatsen op het oppervlak nodig om een stroom door de oplossing te laten lopen. Dit heeft een directe invloed op de corrosiesnelheid, zoals schematisch getoond wordt in Figuur 5.5.1.
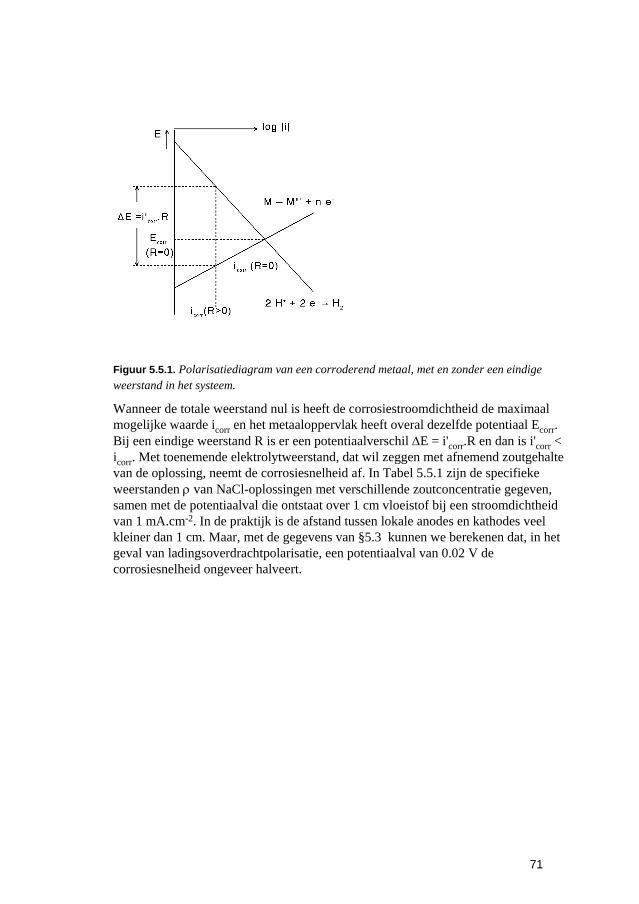
71
Figuur 5.5.1. Polarisatiediagram van een corroderend metaal, met en zonder een eindige weerstand in het systeem.
Wanneer de totale weerstand nul is heeft de corrosiestroomdichtheid de maximaal mogelijke waarde icorr en het metaaloppervlak heeft overal dezelfde potentiaal Ecorr. Bij een eindige weerstand R is er een potentiaalverschil ΔE = i'corr.R en dan is i'corr < icorr. Met toenemende elektrolytweerstand, dat wil zeggen met afnemend zoutgehalte van de oplossing, neemt de corrosiesnelheid af. In Tabel 5.5.1 zijn de specifieke weerstanden ρ van NaCl-oplossingen met verschillende zoutconcentratie gegeven, samen met de potentiaalval die ontstaat over 1 cm vloeistof bij een stroomdichtheid van 1 mA.cm-2. In de praktijk is de afstand tussen lokale anodes en kathodes veel kleiner dan 1 cm. Maar, met de gegevens van §5.3 kunnen we berekenen dat, in het geval van ladingsoverdrachtpolarisatie, een potentiaalval van 0.02 V de corrosiesnelheid ongeveer halveert.
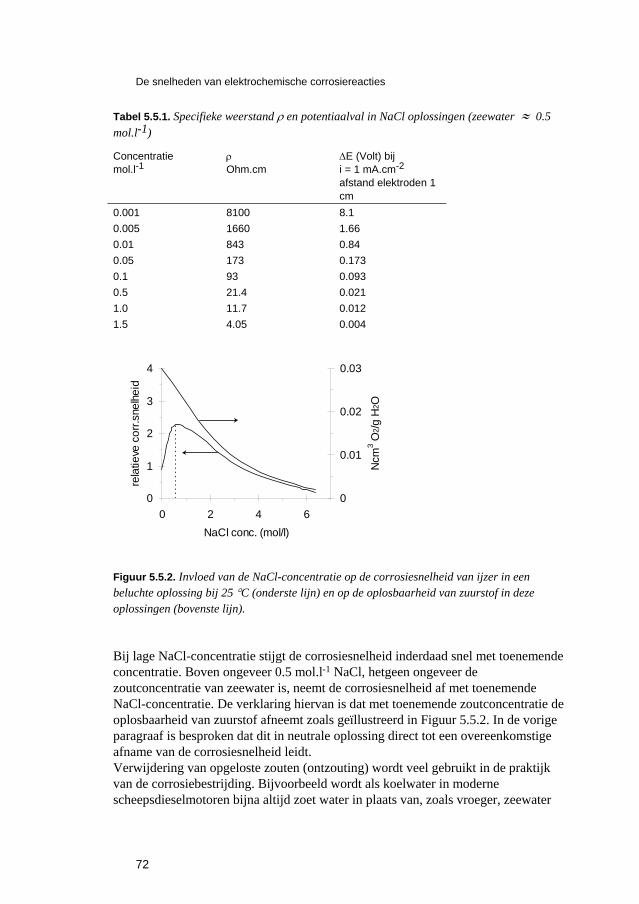
De snelheden van elektrochemische corrosiereacties
72
Tabel 5.5.1. Specifieke weerstand ρ en potentiaalval in NaCl oplossingen (zeewater ≈ 0.5 mol.l-1)
Concentratie mol.l-1
ρ Ohm.cm
ΔE (Volt) bij i = 1 mA.cm-2 afstand elektroden 1 cm
0.001 8100 8.1 0.005 1660 1.66 0.01 843 0.84 0.05 173 0.173 0.1 93 0.093 0.5 21.4 0.021 1.0 11.7 0.012 1.5 4.05 0.004
0
1
2
3
4
0 2 4 6NaCl conc. (mol/l)
rela
tieve
cor
r.sne
lhei
d
0
0.01
0.02
0.03N
cm3 O
2 /g H
2 O
Figuur 5.5.2. Invloed van de NaCl-concentratie op de corrosiesnelheid van ijzer in een beluchte oplossing bij 25 °C (onderste lijn) en op de oplosbaarheid van zuurstof in deze oplossingen (bovenste lijn).
Bij lage NaCl-concentratie stijgt de corrosiesnelheid inderdaad snel met toenemende concentratie. Boven ongeveer 0.5 mol.l-1 NaCl, hetgeen ongeveer de zoutconcentratie van zeewater is, neemt de corrosiesnelheid af met toenemende NaCl-concentratie. De verklaring hiervan is dat met toenemende zoutconcentratie de oplosbaarheid van zuurstof afneemt zoals geïllustreerd in Figuur 5.5.2. In de vorige paragraaf is besproken dat dit in neutrale oplossing direct tot een overeenkomstige afname van de corrosiesnelheid leidt. Verwijdering van opgeloste zouten (ontzouting) wordt veel gebruikt in de praktijk van de corrosiebestrijding. Bijvoorbeeld wordt als koelwater in moderne scheepsdieselmotoren bijna altijd zoet water in plaats van, zoals vroeger, zeewater

73
gebruikt. Op analoge manier wordt ketelvoedingswater bijna altijd ontzout door middel van ionenwisseling of destillatie.
5.6 Contactcorrosie
Verschillende keren is al genoemd dat het koppelen van twee verschillende metalen die in dezelfde oplossing staan versnelde corrosie van het onedeler metaal veroorzaakt. Dit wordt galvanische corrosie, bimetallische corrosie of contactcorrosie genoemd. Een speciale vorm hiervan, namelijk wanneer de waterstofontwikkelingsreactie sneller aan het edele dan aan het onedele metaal verloopt is al besproken in §5.3. In dat geval is het niet de evenwichtspotentiaal van het edele metaal zelf, maar die van de waterstofontwikkelingsreactie die, samen met de kinetische parameters daarvan, bepalend is voor de versnelling van de corrosie.
-6 -5 -4 -3 -2log | i |
E icorr (Ac=10Aa)
2H++2e-→H2
M→M2++2e-
op M op N
icorr (Ac=Aa)icorr
(alleen M)
Figuur 5.6.1. Polarisatiediagram voor bimetallische corrosie van een onedel metaal M met waterstofontwikkeling aan een edel metaal N.
De verhouding van de oppervlakten van de twee metalen is ook van grote betekenis. In Figuur 5.6.1 wordt dit getoond voor een kathodische reactie die ladingsoverdrachtpolarisatie vertoont. Wanneer de oppervlakten van de anode (Aa) en de kathode (Ac) gelijk zijn wordt de corrosiesnelheid gegeven door het snijpunt van de polarisatiekrommen, wanneer de zeer geringe waterstofontwikkeling aan de anode verwaarloosd wordt. In deze beschouwingen houden we ook geen rekening met de elektrolietweerstand. Wanneer de oppervlakte van de kathode 10 maal zo groot is als van de anode moet de stroomdichtheid aan de kathode een tiende zijn van die aan de anode (behoud van lading). In Figuur 5.6.1 zien we dat dit bij een hogere potentiaal het geval is en dus bij een hogere corrosiesnelheid.
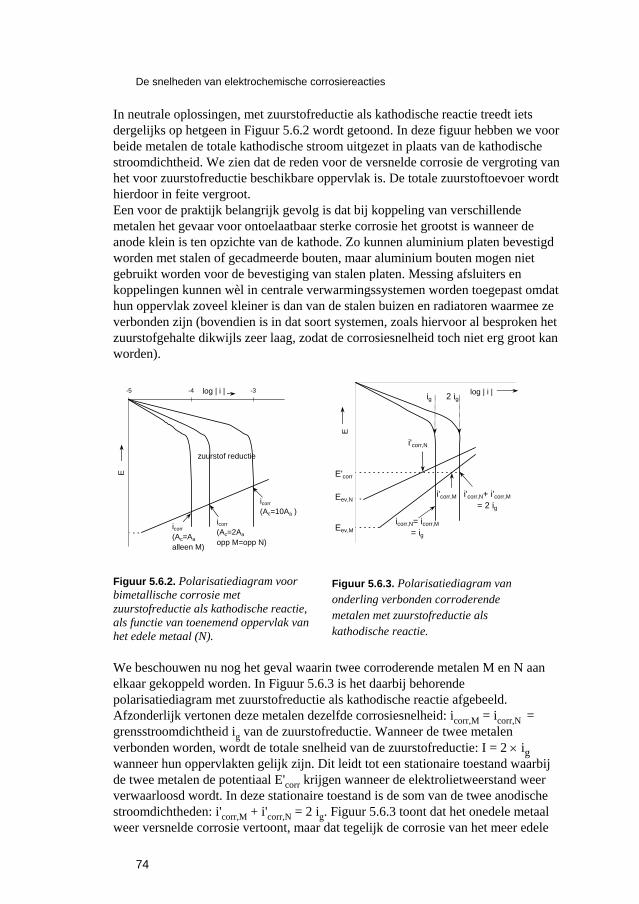
De snelheden van elektrochemische corrosiereacties
74
In neutrale oplossingen, met zuurstofreductie als kathodische reactie treedt iets dergelijks op hetgeen in Figuur 5.6.2 wordt getoond. In deze figuur hebben we voor beide metalen de totale kathodische stroom uitgezet in plaats van de kathodische stroomdichtheid. We zien dat de reden voor de versnelde corrosie de vergroting van het voor zuurstofreductie beschikbare oppervlak is. De totale zuurstoftoevoer wordt hierdoor in feite vergroot. Een voor de praktijk belangrijk gevolg is dat bij koppeling van verschillende metalen het gevaar voor ontoelaatbaar sterke corrosie het grootst is wanneer de anode klein is ten opzichte van de kathode. Zo kunnen aluminium platen bevestigd worden met stalen of gecadmeerde bouten, maar aluminium bouten mogen niet gebruikt worden voor de bevestiging van stalen platen. Messing afsluiters en koppelingen kunnen wèl in centrale verwarmingssystemen worden toegepast omdat hun oppervlak zoveel kleiner is dan van de stalen buizen en radiatoren waarmee ze verbonden zijn (bovendien is in dat soort systemen, zoals hiervoor al besproken het zuurstofgehalte dikwijls zeer laag, zodat de corrosiesnelheid toch niet erg groot kan worden).
-5 -4 -3log | i |
E
zuurstof reductie
icorr
(Ac=Aa
alleen M)
icorr
(Ac=2Aa
opp M=opp N)
icorr
(Ac=10Aa )
Figuur 5.6.2. Polarisatiediagram voor bimetallische corrosie met zuurstofreductie als kathodische reactie, als functie van toenemend oppervlak van het edele metaal (N).
log | i |
E
2 igig
i'corr,N
i'corr,M
icorr,N= icorr,M
= ig
i'corr,N+ i'corr,M
= 2 ig
E'corr
Eev,N
Eev,M
Figuur 5.6.3. Polarisatiediagram van onderling verbonden corroderende metalen met zuurstofreductie als kathodische reactie.
We beschouwen nu nog het geval waarin twee corroderende metalen M en N aan elkaar gekoppeld worden. In Figuur 5.6.3 is het daarbij behorende polarisatiediagram met zuurstofreductie als kathodische reactie afgebeeld. Afzonderlijk vertonen deze metalen dezelfde corrosiesnelheid: icorr,M = icorr,N = grensstroomdichtheid ig van de zuurstofreductie. Wanneer de twee metalen verbonden worden, wordt de totale snelheid van de zuurstofreductie: I = 2 × ig wanneer hun oppervlakten gelijk zijn. Dit leidt tot een stationaire toestand waarbij de twee metalen de potentiaal E'corr krijgen wanneer de elektrolietweerstand weer verwaarloosd wordt. In deze stationaire toestand is de som van de twee anodische stroomdichtheden: i'corr,M + i'corr,N = 2 ig. Figuur 5.6.3 toont dat het onedele metaal weer versnelde corrosie vertoont, maar dat tegelijk de corrosie van het meer edele
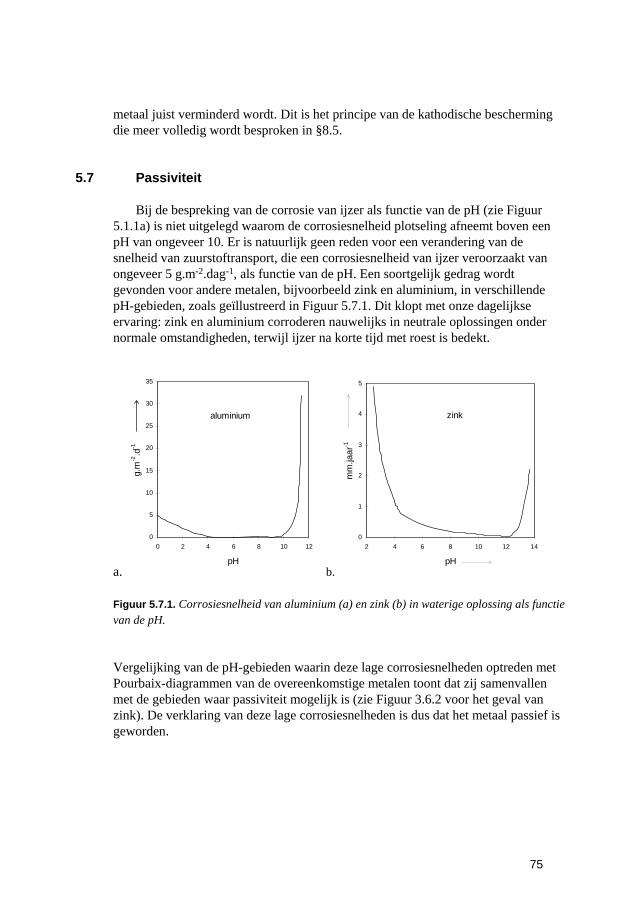
75
metaal juist verminderd wordt. Dit is het principe van de kathodische bescherming die meer volledig wordt besproken in §8.5.
5.7 Passiviteit
Bij de bespreking van de corrosie van ijzer als functie van de pH (zie Figuur 5.1.1a) is niet uitgelegd waarom de corrosiesnelheid plotseling afneemt boven een pH van ongeveer 10. Er is natuurlijk geen reden voor een verandering van de snelheid van zuurstoftransport, die een corrosiesnelheid van ijzer veroorzaakt van ongeveer 5 g.m-2.dag-1, als functie van de pH. Een soortgelijk gedrag wordt gevonden voor andere metalen, bijvoorbeeld zink en aluminium, in verschillende pH-gebieden, zoals geïllustreerd in Figuur 5.7.1. Dit klopt met onze dagelijkse ervaring: zink en aluminium corroderen nauwelijks in neutrale oplossingen onder normale omstandigheden, terwijl ijzer na korte tijd met roest is bedekt.
a.
0
5
10
15
20
25
30
35
0 2 4 6 8 10 12
pH
g.m
-2.d
-1
aluminium
b.
0
1
2
3
4
5
2 4 6 8 10 12 14
pH
mm
.jaar
-1
zink
Figuur 5.7.1. Corrosiesnelheid van aluminium (a) en zink (b) in waterige oplossing als functie van de pH.
Vergelijking van de pH-gebieden waarin deze lage corrosiesnelheden optreden met Pourbaix-diagrammen van de overeenkomstige metalen toont dat zij samenvallen met de gebieden waar passiviteit mogelijk is (zie Figuur 3.6.2 voor het geval van zink). De verklaring van deze lage corrosiesnelheden is dus dat het metaal passief is geworden.
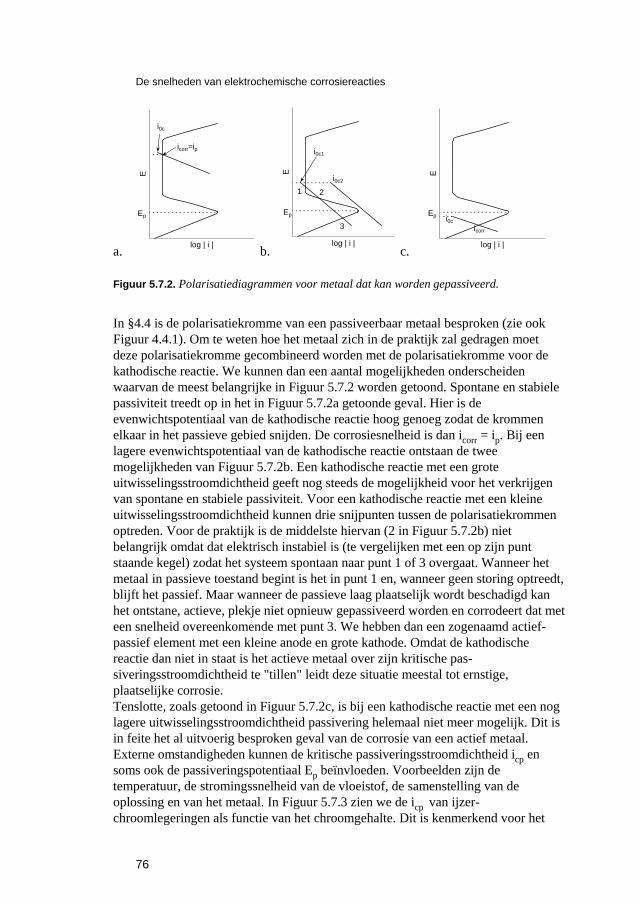
De snelheden van elektrochemische corrosiereacties
76
a. log | i |
E
Ep
icorr=ip
i0c
b.log | i |
E
Ep
1 2
3
i0c2
i0c1
c. log | i |
E
Ep i0cicorr
Figuur 5.7.2. Polarisatiediagrammen voor metaal dat kan worden gepassiveerd.
In §4.4 is de polarisatiekromme van een passiveerbaar metaal besproken (zie ook Figuur 4.4.1). Om te weten hoe het metaal zich in de praktijk zal gedragen moet deze polarisatiekromme gecombineerd worden met de polarisatiekromme voor de kathodische reactie. We kunnen dan een aantal mogelijkheden onderscheiden waarvan de meest belangrijke in Figuur 5.7.2 worden getoond. Spontane en stabiele passiviteit treedt op in het in Figuur 5.7.2a getoonde geval. Hier is de evenwichtspotentiaal van de kathodische reactie hoog genoeg zodat de krommen elkaar in het passieve gebied snijden. De corrosiesnelheid is dan icorr = ip. Bij een lagere evenwichtspotentiaal van de kathodische reactie ontstaan de twee mogelijkheden van Figuur 5.7.2b. Een kathodische reactie met een grote uitwisselingsstroomdichtheid geeft nog steeds de mogelijkheid voor het verkrijgen van spontane en stabiele passiviteit. Voor een kathodische reactie met een kleine uitwisselingsstroomdichtheid kunnen drie snijpunten tussen de polarisatiekrommen optreden. Voor de praktijk is de middelste hiervan (2 in Figuur 5.7.2b) niet belangrijk omdat dat elektrisch instabiel is (te vergelijken met een op zijn punt staande kegel) zodat het systeem spontaan naar punt 1 of 3 overgaat. Wanneer het metaal in passieve toestand begint is het in punt 1 en, wanneer geen storing optreedt, blijft het passief. Maar wanneer de passieve laag plaatselijk wordt beschadigd kan het ontstane, actieve, plekje niet opnieuw gepassiveerd worden en corrodeert dat met een snelheid overeenkomende met punt 3. We hebben dan een zogenaamd actief-passief element met een kleine anode en grote kathode. Omdat de kathodische reactie dan niet in staat is het actieve metaal over zijn kritische pas-siveringsstroomdichtheid te "tillen" leidt deze situatie meestal tot ernstige, plaatselijke corrosie. Tenslotte, zoals getoond in Figuur 5.7.2c, is bij een kathodische reactie met een nog lagere uitwisselingsstroomdichtheid passivering helemaal niet meer mogelijk. Dit is in feite het al uitvoerig besproken geval van de corrosie van een actief metaal. Externe omstandigheden kunnen de kritische passiveringsstroomdichtheid icp en soms ook de passiveringspotentiaal Ep beïnvloeden. Voorbeelden zijn de temperatuur, de stromingssnelheid van de vloeistof, de samenstelling van de oplossing en van het metaal. In Figuur 5.7.3 zien we de icp van ijzer-chroomlegeringen als functie van het chroomgehalte. Dit is kenmerkend voor het
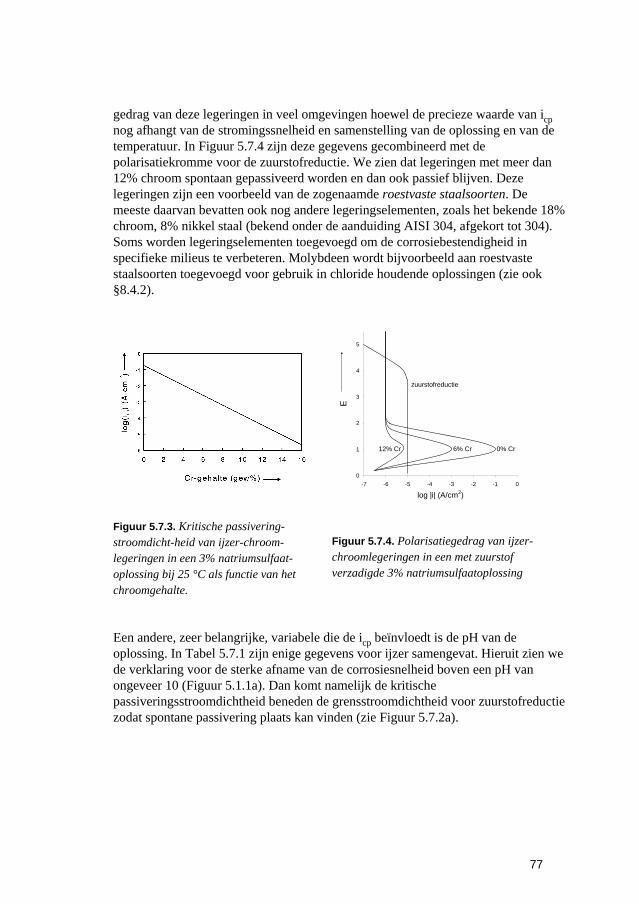
77
gedrag van deze legeringen in veel omgevingen hoewel de precieze waarde van icp nog afhangt van de stromingssnelheid en samenstelling van de oplossing en van de temperatuur. In Figuur 5.7.4 zijn deze gegevens gecombineerd met de polarisatiekromme voor de zuurstofreductie. We zien dat legeringen met meer dan 12% chroom spontaan gepassiveerd worden en dan ook passief blijven. Deze legeringen zijn een voorbeeld van de zogenaamde roestvaste staalsoorten. De meeste daarvan bevatten ook nog andere legeringselementen, zoals het bekende 18% chroom, 8% nikkel staal (bekend onder de aanduiding AISI 304, afgekort tot 304). Soms worden legeringselementen toegevoegd om de corrosiebestendigheid in specifieke milieus te verbeteren. Molybdeen wordt bijvoorbeeld aan roestvaste staalsoorten toegevoegd voor gebruik in chloride houdende oplossingen (zie ook §8.4.2).
Figuur 5.7.3. Kritische passivering-stroomdicht-heid van ijzer-chroom-legeringen in een 3% natriumsulfaat-oplossing bij 25 °C als functie van het chroomgehalte.
0
1
2
3
4
5
-7 -6 -5 -4 -3 -2 -1 0
log |i| (A/cm2)
E
zuurstofreductie
0% Cr6% Cr12% Cr
Figuur 5.7.4. Polarisatiegedrag van ijzer-chroomlegeringen in een met zuurstof verzadigde 3% natriumsulfaatoplossing
Een andere, zeer belangrijke, variabele die de icp beïnvloedt is de pH van de oplossing. In Tabel 5.7.1 zijn enige gegevens voor ijzer samengevat. Hieruit zien we de verklaring voor de sterke afname van de corrosiesnelheid boven een pH van ongeveer 10 (Figuur 5.1.1a). Dan komt namelijk de kritische passiveringsstroomdichtheid beneden de grensstroomdichtheid voor zuurstofreductie zodat spontane passivering plaats kan vinden (zie Figuur 5.7.2a).

De snelheden van elektrochemische corrosiereacties
78
Tabel 5.7.1. Passiveringsparameters voor ijzer als functie van de pH.
pH icp A.cm-2 Ep Volt ip A.cm-2
0.3 2 × 10-1 0.4 8 × 10-6 3 6 × 10-2 0.4 3 × 10-6 4 8 × 10-3 0.35 1.5 × 10-6 5 3 × 10-3 0.3 6 × 10-7 6 1.5 × 10-3 0.25 2 × 10-7 8.4 10-5 -0.4 - 9.3 10-7 -0.5 7 × 10-9
Een andere mogelijkheid om passiviteit te verkrijgen is het toevoegen van passivatoren aan de oplossing. Dit zijn meestal anorganische, oxiderende verbindingen met een hoge evenwichtspotentiaal en een hoge uitwisselings-stroomdichtheid zodat daarmee het polarisatiediagram van Figuur 5.7.2a wordt verkregen. Twee van de meest gebruikte zijn natrium- (of kalium-)chromaat en natrium nitriet. In neutrale oplossing is een concentratie van ongeveer 10-3 mol.l-1 meestal voldoende om ongelegeerd staal te passiveren. Bij lagere pH en/of hoog chloridegehalte zijn hogere concentraties nodig. Daarom wordt tegelijk met de passivator vaak een basische verbinding zoals natriumbenzoaat of natriumcarbonaat toegevoegd. Wanneer, onder andere door verbruik van de passivator, de concentratie van een passivator daalt kan de situatie van Figuur 5.7.2b ontstaan ten gevolge van het omlaag verschuiven van de evenwichtspotentiaal en daarmee van de polarisatiekromme van de kathodische reactie. Ook kan eventueel diffusiepolarisatie op gaan treden. Omdat passivatoren tijdens hun beschermende werking worden verbruikt moeten, om de beschermende werking te behouden en plaatselijke corrosie te voorkomen, deze regelmatig worden aangevuld. Sommige verbindingen, zoals bijvoorbeeld natriumbenzoaat en natriumpolyfosfaat, zijn niet in staat om een ijzeroppervlak direct te passiveren. Maar wèl leiden ze tot sterk verminderde corrosie doordat zij met het ijzeroppervlak reageren waardoor de kritische passiveringsstroomdichtheid veel kleiner wordt, zodat zuurstofreductie voldoende is om passiviteit van staal te veroorzaken. Chromaat wordt vooral gebruikt in gesloten koelwatersystemen, zoals van verbrandingsmotoren en procesinstallaties in de chemische industrie. Gebruikelijk is toevoeging van 0.04 tot 0.1 gewichts% natriumchromaat terwijl de pH op 7.5 tot 9.5 wordt gebracht. Een praktisch nadeel is de giftigheid van chromaten terwijl sommige mensen er allergisch voor zijn. Verder worden veel organische verbindingen, zoals glycol dat dikwijls als anti-vriestoevoeging wordt toegepast, tot zuren geoxideerd. Hierdoor wordt het verbruik van chromaat vergroot, neemt de pH af en wordt de antivries vernietigd. Hierom, maar zeker ook om redenen van milieubescherming (lekkage!) is het gebruik van chromaten de laatste tijd sterk afgenomen. Een gevolg van deze nadelen is een toenemend gebruik van natriumnitriet, in het bijzonder in koelsystemen waarin ook antivries aanwezig is. Verder wordt zowel in

79
open als in gesloten systemen natriummolybdaat als een zeer effectieve en niet giftige passivator van staal gebruikt. Voor een verdere bespreking van deze vorm van corrosiebestrijding zie §8.3.2. Tenslotte noemen we nog een andere mogelijkheid om gebruik te maken van de mogelijkheid om een metaal passief te maken en dat is de zogenaamde anodische bescherming. In zekere zin is dit eigenlijk het omgekeerde van kathodische bescherming: het te beschermen metaal wordt hierbij verbonden met een hulpelektrode via een uitwendige stroombron en door verhoging van de potentiaal in het passieve gebied gebracht. Zie hiervoor verder §8.5.2.
5.8 Opgaven
1. IJzer wordt in een zure oplossing met pH = 2 geplaatst terwijl [Fe2+] = 10-5 mol.l-1. a. Wat zijn de anodische en kathodische reacties die aan het ijzer-oppervlak
verlopen? Aangenomen mag worden dat beide reacties ladingsoverdrachtpolarisatie
vertonen met als parameters: anodische reactie: βa = 0.12 V, i0a = 10-8 A.cm-2 kathodische reactie: βc = - 0.1 V, i0c = 10-7 A.cm-2. b. Construeer het volledige polarisatiediagram en bepaal daarmee de
corrosiesnelheid van het ijzer wanneer de elektrolietweerstand 0.1 Ohm is. Wat wordt de corrosiesnelheid wanneer de elektrolietweerstand 10000 Ohm is?
We veronderstellen nu dat het ijzer een onzuiverheid bevat zodanig dat 1% van het ijzeroppervlak uit een component bestaat waaraan de kathodische reactie veel sneller verloopt en de volgende kinetische parameters vertoont: β'c = - 0.1 V, i'0c = 10-3 A.cm-2. c. Teken de hiermee overeenkomende polarisatiekromme in het diagram b. en
houdt daarbij rekening met de oppervlakte van de verontreiniging. De verandering van het ijzeroppervlak mag worden verwaarloosd. Wat wordt in dit geval de corrosiesnelheid van het ijzer bij een elektrolietweerstand van 0.1 Ohm?
2. Leidt een vergelijking af voor de corrosiestroomdichtheid van een metaal wanneer de anodische en kathodische reactie beide ladingsoverdrachtpolarisatie vertonen. (Aanwijzing: gebruik vergelijking (4.3.6) en bedenk dat ηc = E - Eev,c voor de kathodische reactie en ηa = E - Eev,a voor de anodische en dat E = Es in de stationaire toestand).
3. Van een aantal ijzermonsters met dezelfde oppervlakte worden verschillende fracties van het oppervlak bedekt met een edel metaal. Wanneer deze monsters in een beluchte waterige elektrolietoplossing worden geplaatst vertonen ze na een bepaalde expositietijd allemaal precies hetzelfde gewichtsverlies,

De snelheden van elektrochemische corrosiereacties
80
onafhankelijk van de grootte van het door het edele metaal bedekte oppervlak. Leg dit uit en geef commentaar!
4. IJzer wordt geplaatst in de waterige oplossing zoals gespecificeerd in vraagstuk 7 van §3.6. Bij welke pH zal het ijzer spontaan passief worden?
5. In een aantal figuren in dit hoofdstuk wordt het gedrag van metalen in de praktijk onder verschillende omstandigheden geïllustreerd. Hierbij worden verschillende eenheden voor de corrosiesnelheid gebruikt. Bereken deze in een gemeenschappelijke eenheid, bijvoorbeeld in mm.jaar-1.
6. Om de aantasting van staal tijdens het beitsen in zuur te beperken wordt dikwijls een beitsrem aan het zuur toegevoegd. Één van de manieren waarop de effectiviteit van zo'n inhibitor kan worden bepaald is het meten van de hoeveelheid gas dat wordt ontwikkeld in zuur mèt en zònder beitsrem. a. Welk gas wordt in dit geval gevormd? Geef de reactievergelijking van het
gehele proces. In een bepaald onderzoek worden testplaatjes van 100 × 50 mm2 en 2 mm dik
gebruikt voor deze metingen. In zuur met beitsrem werd in 10 min 256.7 ml gas (25 °C, 1 atm) ontwikkeld. Met beitsrem was dit 37.4 ml (25 °C, 1 atm) in 1 uur. b. Bereken de corrosiesnelheid van het ijzer in mm per dag en in A.cm-2 voor
deze twee gevallen. In beide gevallen vertonen zowel de anodische als de kathodische reactie
ladingsoverdrachtpolarisatie. Voor de anodische reactie is de uitwisselingsstroomdichtheid 10-5 A.cm-2 en de Tafel helling βa = 0.1 V, onafhankelijk van het al of niet aanwezig zijn van de beitsrem. Voor de kathodische reactie is de Tafel helling βc = - 0.1 V. De pH van de oplossing is 1 en de oplossing bevat 10 g Fe per liter. c. Schets het polarisatiediagram. Bereken grafisch of algebraïsch wat de
uitwisselingsstroomdichtheid van de kathodische reactie is mèt en zònder beitsrem.
5.9 Literatuur
1. P.J. Gellings, Corrosie en Onderhoud, in: Handboek Onderhoudsmanagement, Alphen aan de Rijn, Samsom BedrijfsInformatie, 1996, p. K3010-1 - 56.
2. J.M. West, Basic Corrosion and Oxidation, Ellis Horwood (1980) 3. S.A. Bradford, Corrosion Control, New York, Van Nostrand Reinhold (1993) 4. H. Kaesche, Metallic corrosion, 2nd ed., Houston, NACE (1985) 5. D.L. Piron, The electrochemistry of corrosion, Houston, NACE (1991) 6. Corrosion inhibition – Theory and Practice, NACE Reference 7, Ed. by R.H. Hausler,
Houston, NACE (1988) 7. A. Snel en E.D.D. During, Corrosiebestrijding door waterbehandeling, NCC brochure 5,
Delft, Waltman (1993) 8. Reviews on corrosion inhibitor science and technology, Ed. by A. Raman and P. Labine,
Houston, NACE (1991)

6
Karakteristieke verschijningsvormen van corrosie


83
6.1 Inleiding
Corrosie manifesteert zich in een aantal karakteristieke vormen. Het basisonderscheid is tussen algemene aantasting en plaatselijke aantasting. Zoals de naam aangeeft vindt algemene (of gelijkmatige) aantasting (vrijwel) gelijkelijk over het hele metaaloppervlak plaats. Dit wordt gevonden bij metalen in actieve toestand en bij passieve metalen, maar in dit laatste geval is de aantasting uiteraard extreem langzaam. Dit type corrosie is, in het algemeen, niet een groot probleem vanuit een technisch standpunt omdat de snelheid daarvan met een redelijke nauwkeurigheid kan worden bepaald. Daardoor is een schatting van de te verwachten levensduur van een apparaat of installatie meestal niet moeilijk en kan, indien nodig, een corrosie-toeslag worden toegepast. De meeste in hoofdstuk 5 besproken methoden van corrosiebestrijding kunnen hierbij ook worden gebruikt, samen met andere metho-den, zoals bijvoorbeeld de toepassing van beschermende deklagen, hetgeen uit-voeriger wordt besproken in hoofdstuk 8. Een speciale soort algemene corrosie is het atmosferisch roesten van ijzer en staal, dat uitvoeriger wordt behandeld in §6.2 Veel gevaarlijker en veel minder voorspelbaar zijn de verschillende vormen van plaatselijke corrosie die in de volgende paragrafen van dit hoofdstuk worden besproken. Hierbij gaat het over: putcorrosie, spleetcorrosie en corrosie onder afzettingen, selectief oplossen, interkristallijne corrosie, spanningscorrosie en corrosievermoeidheid, erosie-corrosie en tenslotte passingroest. Deze vormen leiden meestal tot onverwacht, plaatselijk falen. Sommige hiervan zijn niet van zuiver elektrochemische aard maar worden sterk beïnvloed door mechanische en/of metallurgische factoren. Een aantal specifieke corrosiegevallen, met hun oplossingen en economische gevolgen, worden uitvoerig besproken en geïllustreerd in de Corrosion Atlas [1]. Iets minder uitvoerig is de uitgave [2].
Figuur 6.1.1. Schematische weergave van algemene en plaatselijke corrosie. Het metaalverlies bij algemene corrosie is veel groter dan bij plaatselijke corrosie, hetgeen meestal gepaard gaat met een aanzienlijk langere levensduur (Met toestemming overgenomen uit [3].

Karakteristieke verschijningsvormen van corrosie
84
In Figuur 6.1.1 wordt schematisch het verschil in de omvang van de corrosie getoond bij falen van een materiaal dat aan een mechanische belasting is onderworpen, wanneer algemene, dan wel plaatselijke corrosie, hier in de vorm van scheuren, optreedt. Duidelijk is het veel grotere materiaalverlies dat nodig is bij algemene corrosie om een bepaalde doorsnedeverkleining van het materiaal te veroorzaken. Dit betekent dat bij algemene corrosie de levensduur meestal groter is dan bij plaatselijke.
6.2 Corrosie van ijzer en staal
6.2.1 Inleiding
IJzer en staal zijn ongetwijfeld het meest gebruikte metallische constructiemateriaal. Transportmiddelen (auto's, schepen, treinen, enzovoort), staalconstructies (bijvoorbeeld gebouwen, bruggen, kranen), vele andere soorten industriële installaties, ketels en verwarming- en koelapparatuur, enzovoort. De corrosie daarvan vindt in veel gevallen in de atmosfeer plaats zodat we daaraan apart aandacht besteden, ook al gaat het daarbij vrijwel altijd om algemene corrosie.
6.2.2 Roesten en atmosferische corrosie
In (ongeveer) neutrale oplossingen en bij blootstelling aan de atmosfeer vindt corrosie van ijzer en staal plaats onder vorming van roest [4]. Dit is een verzamelwoord voor een aantal gehydrateerde ijzeroxiden en hydroxiden met de algemene samenstelling FeO(OH).nH2O, waarin ijzer in driewaardige vorm aanwezig is.
Figuur 6.2.1 Schematische weergave van de vorming van roest in een waterdruppel op een staaloppervlak (Met toestemming overgenomen uit [3]) .
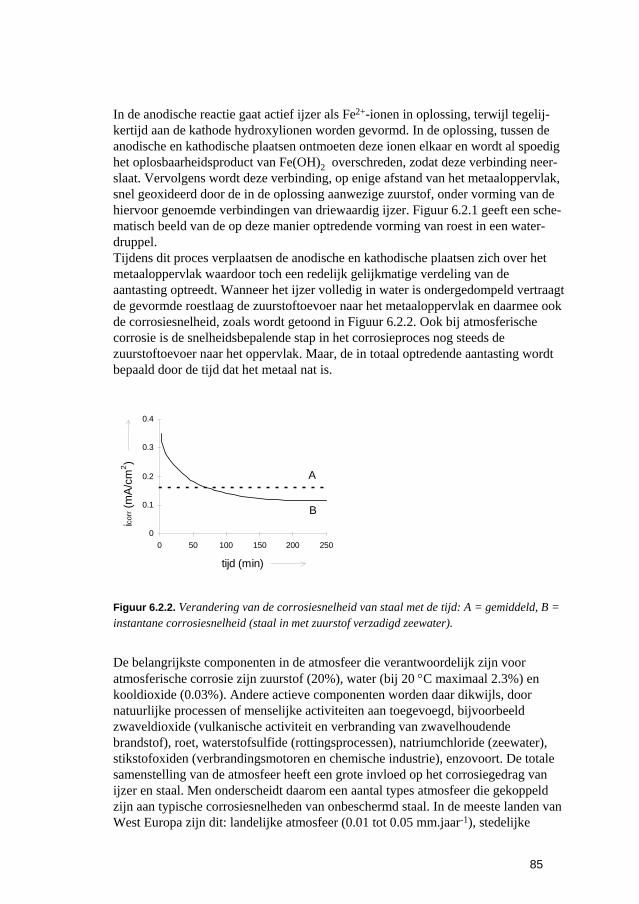
85
In de anodische reactie gaat actief ijzer als Fe2+-ionen in oplossing, terwijl tegelij-kertijd aan de kathode hydroxylionen worden gevormd. In de oplossing, tussen de anodische en kathodische plaatsen ontmoeten deze ionen elkaar en wordt al spoedig het oplosbaarheidsproduct van Fe(OH)2 overschreden, zodat deze verbinding neer-slaat. Vervolgens wordt deze verbinding, op enige afstand van het metaaloppervlak, snel geoxideerd door de in de oplossing aanwezige zuurstof, onder vorming van de hiervoor genoemde verbindingen van driewaardig ijzer. Figuur 6.2.1 geeft een sche-matisch beeld van de op deze manier optredende vorming van roest in een water-druppel. Tijdens dit proces verplaatsen de anodische en kathodische plaatsen zich over het metaaloppervlak waardoor toch een redelijk gelijkmatige verdeling van de aantasting optreedt. Wanneer het ijzer volledig in water is ondergedompeld vertraagt de gevormde roestlaag de zuurstoftoevoer naar het metaaloppervlak en daarmee ook de corrosiesnelheid, zoals wordt getoond in Figuur 6.2.2. Ook bij atmosferische corrosie is de snelheidsbepalende stap in het corrosieproces nog steeds de zuurstoftoevoer naar het oppervlak. Maar, de in totaal optredende aantasting wordt bepaald door de tijd dat het metaal nat is.
0
0.1
0.2
0.3
0.4
0 50 100 150 200 250
tijd (min)
icorr
(mA
/cm
2 )
A
B
Figuur 6.2.2. Verandering van de corrosiesnelheid van staal met de tijd: A = gemiddeld, B = instantane corrosiesnelheid (staal in met zuurstof verzadigd zeewater).
De belangrijkste componenten in de atmosfeer die verantwoordelijk zijn voor atmosferische corrosie zijn zuurstof (20%), water (bij 20 °C maximaal 2.3%) en kooldioxide (0.03%). Andere actieve componenten worden daar dikwijls, door natuurlijke processen of menselijke activiteiten aan toegevoegd, bijvoorbeeld zwaveldioxide (vulkanische activiteit en verbranding van zwavelhoudende brandstof), roet, waterstofsulfide (rottingsprocessen), natriumchloride (zeewater), stikstofoxiden (verbrandingsmotoren en chemische industrie), enzovoort. De totale samenstelling van de atmosfeer heeft een grote invloed op het corrosiegedrag van ijzer en staal. Men onderscheidt daarom een aantal types atmosfeer die gekoppeld zijn aan typische corrosiesnelheden van onbeschermd staal. In de meeste landen van West Europa zijn dit: landelijke atmosfeer (0.01 tot 0.05 mm.jaar-1), stedelijke
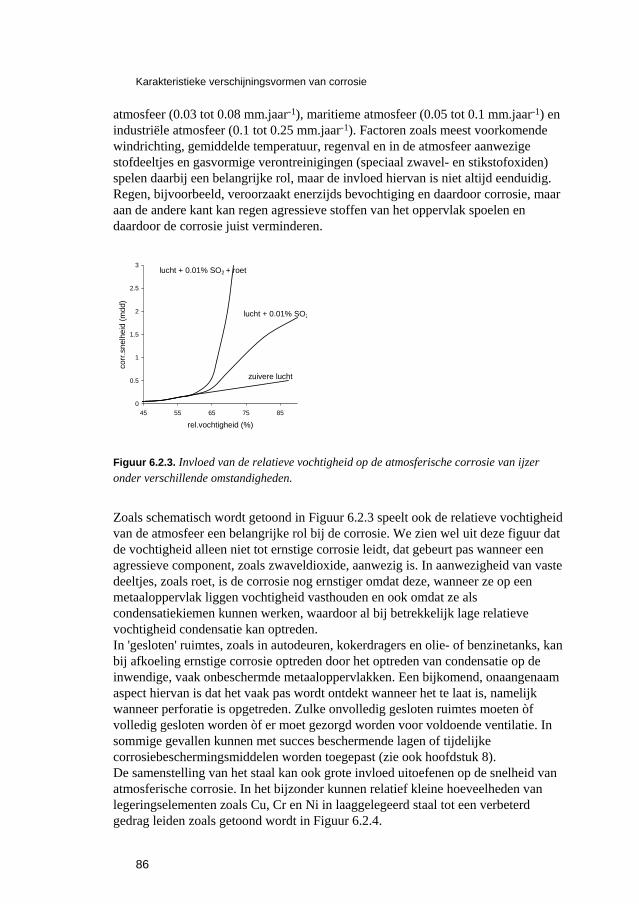
Karakteristieke verschijningsvormen van corrosie
86
atmosfeer (0.03 tot 0.08 mm.jaar-1), maritieme atmosfeer (0.05 tot 0.1 mm.jaar-1) en industriële atmosfeer (0.1 tot 0.25 mm.jaar-1). Factoren zoals meest voorkomende windrichting, gemiddelde temperatuur, regenval en in de atmosfeer aanwezige stofdeeltjes en gasvormige verontreinigingen (speciaal zwavel- en stikstofoxiden) spelen daarbij een belangrijke rol, maar de invloed hiervan is niet altijd eenduidig. Regen, bijvoorbeeld, veroorzaakt enerzijds bevochtiging en daardoor corrosie, maar aan de andere kant kan regen agressieve stoffen van het oppervlak spoelen en daardoor de corrosie juist verminderen.
0
0.5
1
1.5
2
2.5
3
45 55 65 75 85
rel.vochtigheid (%)
corr.
snel
heid
(mdd
)
lucht + 0.01% SO2 + roet
lucht + 0.01% SO2
zuivere lucht
Figuur 6.2.3. Invloed van de relatieve vochtigheid op de atmosferische corrosie van ijzer onder verschillende omstandigheden.
Zoals schematisch wordt getoond in Figuur 6.2.3 speelt ook de relatieve vochtigheid van de atmosfeer een belangrijke rol bij de corrosie. We zien wel uit deze figuur dat de vochtigheid alleen niet tot ernstige corrosie leidt, dat gebeurt pas wanneer een agressieve component, zoals zwaveldioxide, aanwezig is. In aanwezigheid van vaste deeltjes, zoals roet, is de corrosie nog ernstiger omdat deze, wanneer ze op een metaaloppervlak liggen vochtigheid vasthouden en ook omdat ze als condensatiekiemen kunnen werken, waardoor al bij betrekkelijk lage relatieve vochtigheid condensatie kan optreden. In 'gesloten' ruimtes, zoals in autodeuren, kokerdragers en olie- of benzinetanks, kan bij afkoeling ernstige corrosie optreden door het optreden van condensatie op de inwendige, vaak onbeschermde metaaloppervlakken. Een bijkomend, onaangenaam aspect hiervan is dat het vaak pas wordt ontdekt wanneer het te laat is, namelijk wanneer perforatie is opgetreden. Zulke onvolledig gesloten ruimtes moeten òf volledig gesloten worden òf er moet gezorgd worden voor voldoende ventilatie. In sommige gevallen kunnen met succes beschermende lagen of tijdelijke corrosiebeschermingsmiddelen worden toegepast (zie ook hoofdstuk 8). De samenstelling van het staal kan ook grote invloed uitoefenen op de snelheid van atmosferische corrosie. In het bijzonder kunnen relatief kleine hoeveelheden van legeringselementen zoals Cu, Cr en Ni in laaggelegeerd staal tot een verbeterd gedrag leiden zoals getoond wordt in Figuur 6.2.4.
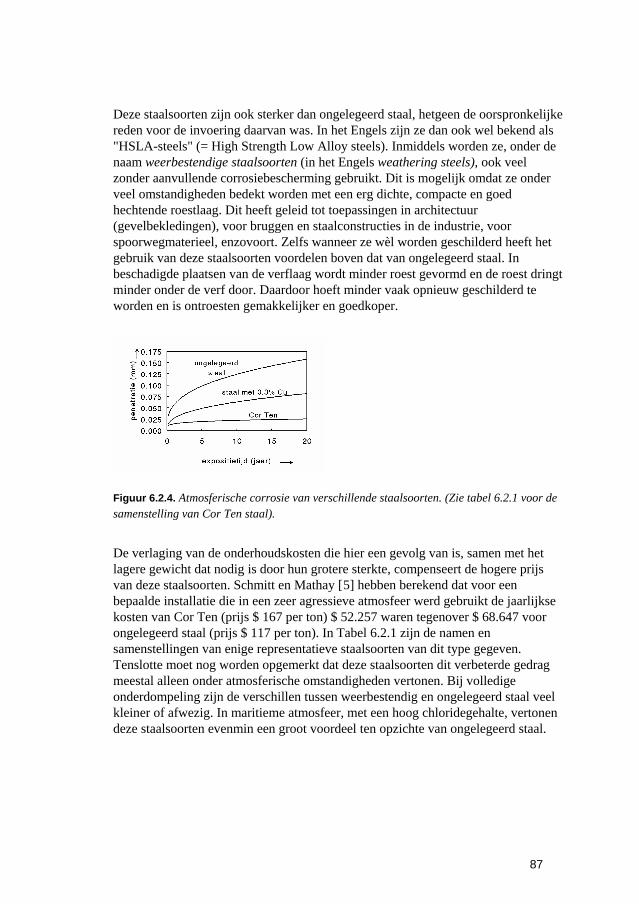
87
Deze staalsoorten zijn ook sterker dan ongelegeerd staal, hetgeen de oorspronkelijke reden voor de invoering daarvan was. In het Engels zijn ze dan ook wel bekend als "HSLA-steels" (= High Strength Low Alloy steels). Inmiddels worden ze, onder de naam weerbestendige staalsoorten (in het Engels weathering steels), ook veel zonder aanvullende corrosiebescherming gebruikt. Dit is mogelijk omdat ze onder veel omstandigheden bedekt worden met een erg dichte, compacte en goed hechtende roestlaag. Dit heeft geleid tot toepassingen in architectuur (gevelbekledingen), voor bruggen en staalconstructies in de industrie, voor spoorwegmaterieel, enzovoort. Zelfs wanneer ze wèl worden geschilderd heeft het gebruik van deze staalsoorten voordelen boven dat van ongelegeerd staal. In beschadigde plaatsen van de verflaag wordt minder roest gevormd en de roest dringt minder onder de verf door. Daardoor hoeft minder vaak opnieuw geschilderd te worden en is ontroesten gemakkelijker en goedkoper.
Figuur 6.2.4. Atmosferische corrosie van verschillende staalsoorten. (Zie tabel 6.2.1 voor de samenstelling van Cor Ten staal).
De verlaging van de onderhoudskosten die hier een gevolg van is, samen met het lagere gewicht dat nodig is door hun grotere sterkte, compenseert de hogere prijs van deze staalsoorten. Schmitt en Mathay [5] hebben berekend dat voor een bepaalde installatie die in een zeer agressieve atmosfeer werd gebruikt de jaarlijkse kosten van Cor Ten (prijs $ 167 per ton) $ 52.257 waren tegenover $ 68.647 voor ongelegeerd staal (prijs $ 117 per ton). In Tabel 6.2.1 zijn de namen en samenstellingen van enige representatieve staalsoorten van dit type gegeven. Tenslotte moet nog worden opgemerkt dat deze staalsoorten dit verbeterde gedrag meestal alleen onder atmosferische omstandigheden vertonen. Bij volledige onderdompeling zijn de verschillen tussen weerbestendig en ongelegeerd staal veel kleiner of afwezig. In maritieme atmosfeer, met een hoog chloridegehalte, vertonen deze staalsoorten evenmin een groot voordeel ten opzichte van ongelegeerd staal.
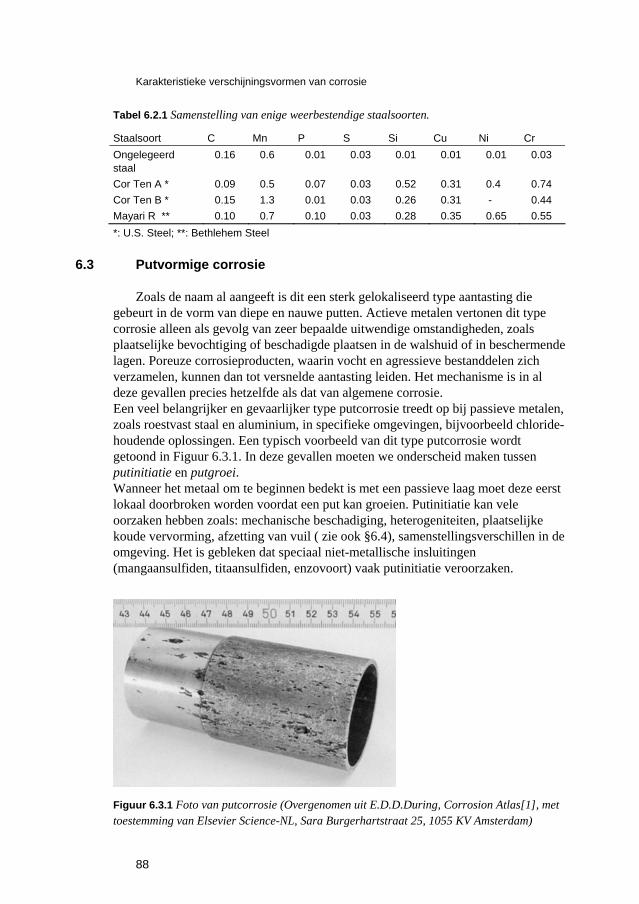
Karakteristieke verschijningsvormen van corrosie
88
Tabel 6.2.1 Samenstelling van enige weerbestendige staalsoorten.
Staalsoort C Mn P S Si Cu Ni Cr Ongelegeerd staal
0.16 0.6 0.01 0.03 0.01 0.01 0.01 0.03
Cor Ten A * 0.09 0.5 0.07 0.03 0.52 0.31 0.4 0.74 Cor Ten B * 0.15 1.3 0.01 0.03 0.26 0.31 - 0.44 Mayari R ** 0.10 0.7 0.10 0.03 0.28 0.35 0.65 0.55 *: U.S. Steel; **: Bethlehem Steel
6.3 Putvormige corrosie
Zoals de naam al aangeeft is dit een sterk gelokaliseerd type aantasting die gebeurt in de vorm van diepe en nauwe putten. Actieve metalen vertonen dit type corrosie alleen als gevolg van zeer bepaalde uitwendige omstandigheden, zoals plaatselijke bevochtiging of beschadigde plaatsen in de walshuid of in beschermende lagen. Poreuze corrosieproducten, waarin vocht en agressieve bestanddelen zich verzamelen, kunnen dan tot versnelde aantasting leiden. Het mechanisme is in al deze gevallen precies hetzelfde als dat van algemene corrosie. Een veel belangrijker en gevaarlijker type putcorrosie treedt op bij passieve metalen, zoals roestvast staal en aluminium, in specifieke omgevingen, bijvoorbeeld chloride- houdende oplossingen. Een typisch voorbeeld van dit type putcorrosie wordt getoond in Figuur 6.3.1. In deze gevallen moeten we onderscheid maken tussen putinitiatie en putgroei. Wanneer het metaal om te beginnen bedekt is met een passieve laag moet deze eerst lokaal doorbroken worden voordat een put kan groeien. Putinitiatie kan vele oorzaken hebben zoals: mechanische beschadiging, heterogeniteiten, plaatselijke koude vervorming, afzetting van vuil ( zie ook §6.4), samenstellingsverschillen in de omgeving. Het is gebleken dat speciaal niet-metallische insluitingen (mangaansulfiden, titaansulfiden, enzovoort) vaak putinitiatie veroorzaken.
Figuur 6.3.1 Foto van putcorrosie (Overgenomen uit E.D.D.During, Corrosion Atlas[1], met toestemming van Elsevier Science-NL, Sara Burgerhartstraat 25, 1055 KV Amsterdam)
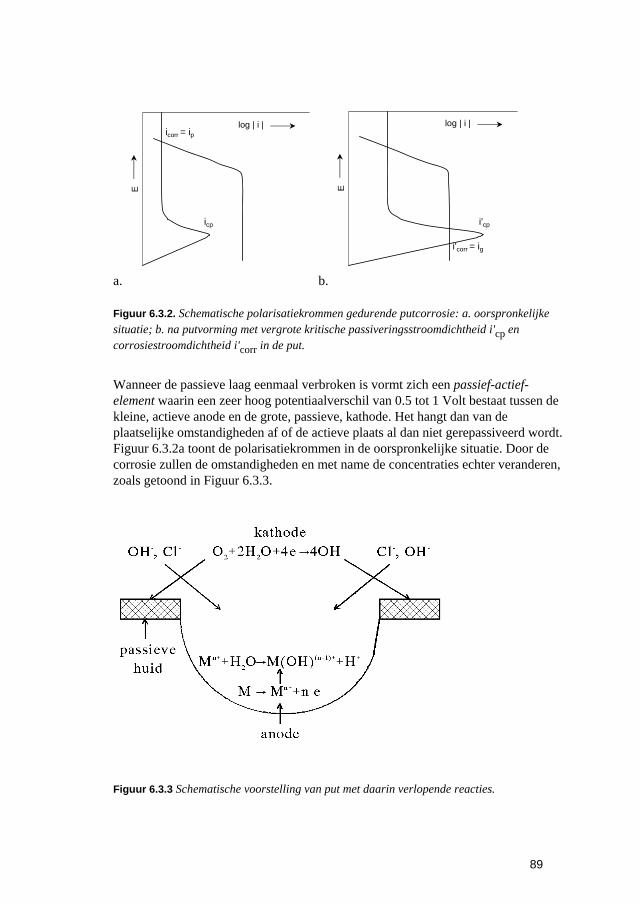
89
a.
log | i |E
icorr = ip
icp
b.
log | i |
E
i'corr = ig
i'cp
Figuur 6.3.2. Schematische polarisatiekrommen gedurende putcorrosie: a. oorspronkelijke situatie; b. na putvorming met vergrote kritische passiveringsstroomdichtheid i'cp en corrosiestroomdichtheid i'corr in de put.
Wanneer de passieve laag eenmaal verbroken is vormt zich een passief-actief-element waarin een zeer hoog potentiaalverschil van 0.5 tot 1 Volt bestaat tussen de kleine, actieve anode en de grote, passieve, kathode. Het hangt dan van de plaatselijke omstandigheden af of de actieve plaats al dan niet gerepassiveerd wordt. Figuur 6.3.2a toont de polarisatiekrommen in de oorspronkelijke situatie. Door de corrosie zullen de omstandigheden en met name de concentraties echter veranderen, zoals getoond in Figuur 6.3.3.
Figuur 6.3.3 Schematische voorstelling van put met daarin verlopende reacties.

Karakteristieke verschijningsvormen van corrosie
90
Één van die veranderingen is een verhoging van de chloride ionenconcentratie als gevolg van de bijdrage die zij leveren aan het stroomtransport door de oplossing. Verder daalt in het algemeen de pH van de oplossing in de put ten gevolge van de hydrolyse van de opgeloste metaalionen, volgens een reactie zoals:
22
52362 H(OH)]O)[Cr(HO)Cr(H +→ ++
waarin driewaardig chroom als voorbeeld is gebruikt. Onder deze omstandigheden krijgen we dan de situatie van Figuur 6.3.2b. Beide samenstellingsveranderingen leiden namelijk tot een sterke verhoging van de kritische passiveringsstroomdichtheid i'cp waardoor spontane repassivering onmogelijk wordt. In de put corrodeert het metaal dan met de zeer grote, locale stroomdichtheid i'corr. In de praktijk wordt de bruikbare levensduur van een apparaat natuurlijk bepaald door de maximum putdiepte. Experimenteel is gevonden dat putdieptes ongeveer volgens een Gauss-verdeling rond de gemiddelde putdiepte zijn verdeeld. Dit betekent automatisch dat de verwachte levensduur, bij een gegeven gemiddelde putdiepte, korter is wanneer er meer putten zijn of, onder overigens gelijke omstandigheden, wanneer het totale oppervlak groter is. Een belangrijke consequentie hiervan is dat het erg gevaarlijk is om conclusies over praktisch putvormingsgedrag te trekken uit experimenten aan proefplaatjes, omdat die uiteraard altijd een veel kleiner totaal oppervlak hebben dan het apparaat zelf. Enige methoden die kunnen worden toegepast om putcorrosie te bestrijden of verminderen zijn: 1. Verhoging van de pH. Hierdoor is er een grotere bijdrage van de hydroxylionen
aan de stroom zodat in de put zowel de pH hoger en de chloride-ionenconcen-tratie kleiner blijven. Voor metalen zoals aluminium, met een amfoteer karakter, dat wil zeggen waarvan de oxiden zowel in zure als in basische oplossingen oplossen (zie Figuur 5.7.1) is deze methode natuurlijk maar beperkt toepasbaar.
2. Kathodische bescherming. In het geval van passieve metalen, zoals aluminium of roestvast staal, wordt de potentiaal dan niet in het immuniteitsgebied gebracht, maar alleen zover verlaagd dat stabiele passiviteit wordt verkregen.
3. Het gebruik van legeringen met speciale toevoegingen. Molybdeen verhoogt bijvoorbeeld de bestendigheid tegen putcorrosie van roestvast staal. De kritische passiveringsstroomdichtheid neemt voor molybdeenhoudend roestvast staal minder toe met toenemend chloorionengehalte en verlaging van de pH.
4. Toevoeging van passivatoren. Het is belangrijk dat voldoende passivator wordt toegevoegd omdat bij te lage concentratie de putcorrosie zelfs versneld kan worden (zie §5.7).
5. Vergroten van de stromingssnelheid van de oplossing. Dit verkleint het gevaar voor putcorrosie door afzettingen, maar nog belangrijker is dat de vloeistof in en bij de put beter gemengd wordt zodat de plaatselijke concentratieverschillen kleiner worden.
6. Verlaging van de temperatuur. Voor de meeste metalen is er een kritische temperatuur, afhankelijk van de samenstelling van het metaal en die van de oplossing, waar beneden geen putcorrosie optreedt. Voor roestvast staal wordt deze kritische temperatuur hoger met toenemend molybdeengehalte.
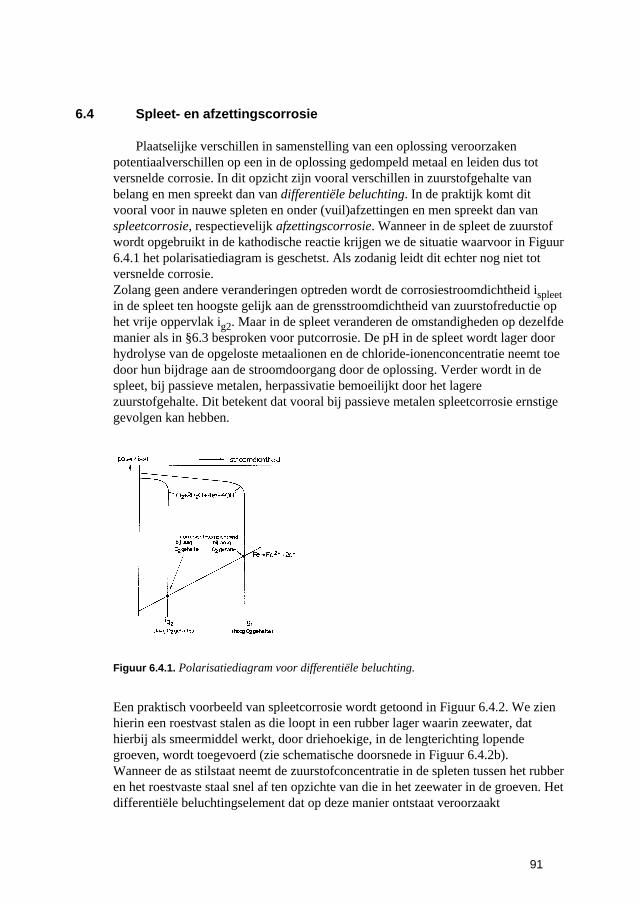
91
6.4 Spleet- en afzettingscorrosie
Plaatselijke verschillen in samenstelling van een oplossing veroorzaken potentiaalverschillen op een in de oplossing gedompeld metaal en leiden dus tot versnelde corrosie. In dit opzicht zijn vooral verschillen in zuurstofgehalte van belang en men spreekt dan van differentiële beluchting. In de praktijk komt dit vooral voor in nauwe spleten en onder (vuil)afzettingen en men spreekt dan van spleetcorrosie, respectievelijk afzettingscorrosie. Wanneer in de spleet de zuurstof wordt opgebruikt in de kathodische reactie krijgen we de situatie waarvoor in Figuur 6.4.1 het polarisatiediagram is geschetst. Als zodanig leidt dit echter nog niet tot versnelde corrosie. Zolang geen andere veranderingen optreden wordt de corrosiestroomdichtheid ispleet in de spleet ten hoogste gelijk aan de grensstroomdichtheid van zuurstofreductie op het vrije oppervlak ig2. Maar in de spleet veranderen de omstandigheden op dezelfde manier als in §6.3 besproken voor putcorrosie. De pH in de spleet wordt lager door hydrolyse van de opgeloste metaalionen en de chloride-ionenconcentratie neemt toe door hun bijdrage aan de stroomdoorgang door de oplossing. Verder wordt in de spleet, bij passieve metalen, herpassivatie bemoeilijkt door het lagere zuurstofgehalte. Dit betekent dat vooral bij passieve metalen spleetcorrosie ernstige gevolgen kan hebben.
Figuur 6.4.1. Polarisatiediagram voor differentiële beluchting.
Een praktisch voorbeeld van spleetcorrosie wordt getoond in Figuur 6.4.2. We zien hierin een roestvast stalen as die loopt in een rubber lager waarin zeewater, dat hierbij als smeermiddel werkt, door driehoekige, in de lengterichting lopende groeven, wordt toegevoerd (zie schematische doorsnede in Figuur 6.4.2b). Wanneer de as stilstaat neemt de zuurstofconcentratie in de spleten tussen het rubber en het roestvaste staal snel af ten opzichte van die in het zeewater in de groeven. Het differentiële beluchtingselement dat op deze manier ontstaat veroorzaakt

Karakteristieke verschijningsvormen van corrosie
92
spleetcorrosie onder het rubber, naast de groeven. Van belang is vooral dat deze aantasting optreedt wanneer de as niet draait!
a b
Figuur 6.4.2 Spleetcorrosie van roestvast stalen as in rubber lager: a. foto van gecorrodeerde as; b. schematische doorsnede van as in lager (bij de met x aangegeven punten treedt corrosie op. De spleet is ter wille van de duidelijkheid overdreven wijd getekend).
a b
Figuur 6.4.3. Spleetcorrosie onder pakking in een flens: a. foto (Overgenomen uit E.D.D.During, Corrosion Atlas[1], met toestemming van Elsevier Science-NL, Sara Burgerhartstraat 25, 1055 KV Amsterdam); b. schematische doorsnede (Met toestemming overgenomen uit [3]).
Een ander voorbeeld is een flens waarin spleetcorrosie is opgetreden onder de pakking, zoals getoond in Figuur 6.4.3. Een schematische doorsnede is gegeven in Figuur 6.4.3b.

93
De reden hiervoor kan verschillend zijn: poreuze in plaats van dichte pakking, oneffenheden van flens en pakking die door een te harde pakking niet worden afgedicht, dikteverschil in de pakking en tenslotte scheef aantrekken van de flens waardoor een spleet overblijft tussen flens en pakking. Dit zijn punten die zowel bij de fabricage als bij onderhoud, bijvoorbeeld bij vervanging van pakkingen, van belang zijn. De meest belangrijke methode om spleetcorrosie te voorkomen is natuurlijk het volledig vermijden van spleten, door ze òf volledig af te sluiten òf zo wijd te maken dat verversing van de oplossing mogelijk wordt. Voor enkele voorbeelden van dit type ontwerpaanpassing zie ook §8.7. Wanneer nauwe spleten onvermijdelijk zijn kan de ernst van eventuele corrosie verminderd worden door verhoging van de pH van de oplossing en door verlaging van de chloride-ionenconcentratie. Een andere mogelijkheid is natuurlijk het toepassen van materialen die minder gevoelig zijn voor spleetcorrosie. In maritieme omgeving en andere chloridehoudende oplossingen zijn bijvoorbeeld monel en speciale roestvaste staalsoorten met verhoogd molybdeen- en stikstofgehalte zeer resistent tegen spleetcorrosie. Wanneer afzettingen de oorzaak van spleetcorrosie zijn kunnen een verhoogde stromingssnelheid of reiniging door filtratie of centrifugeren van de oplossing effectieve methoden zijn om de afzettingscorrosie te voorkomen.
6.5 Microbiologisch geïnduceerde corrosie
Wanneer een metaal aan een waterige oplossing wordt blootgesteld beginnen direct allerlei elektrochemische reacties te verlopen, zoals hiervoor uitvoerig is besproken. In bepaalde gevallen kunnen micro-organismen het resulterende corrosieproces beïnvloeden en we spreken dan van microbiologisch geïnduceerde corrosie of van microbiologisch beïnvloede corrosie. De micro-organismen die in water aanwezig zijn veroorzaken op het metaaloppervlak vaak de vorming van een zogenaamde biofilm die het metaal afsluit van direct contact met de omringende vloeistof en waaronder de samenstelling van het milieu door diverse oorzaken kan gaan veranderen en daardoor tot verhoging van de corrosiesnelheid kan leiden, ook zonder directe invloed van de micro-organismen op het corrosieproces. In feite hebben we dan een vorm van spleetcorrosie (zie §6.4). Maar er zijn ook gevallen waar de micro-organismen tot specifieke vormen van corrosie leiden, waarbij met name de volgende groepen bacteriën een rol spelen: − sulfaat reducerende bacteriën; − ijzer- en mangaan bacteriën; − zwavel-oxiderende bacteriën. Verder onderscheidt men vaak aërobe versus anaërobe bacteriën, die gedeeltelijk met de eerder genoemde categorieën overlappen. Verder spreekt men nog van slijmvormende tegenover zuurvormende bacteriën.

Karakteristieke verschijningsvormen van corrosie
94
Vooral bij ondergrondse corrosie van pijpleidingen of tanks kan de aanwezigheid van sulfaatreducerende bacteriën een belangrijke rol spelen. Dit zijn anaërobe bacteriën die sulfaat reduceren tot sulfide volgens:
SO42- + 8 H+ + 8 e- → S2- + 4 H2O.
Enerzijds kan de vorming van het sulfide tot versnelde corrosie aanleiding geven, onder andere omdat de waterstofontwikkeling aan ijzersulfide veel sneller verloopt dan aan metallisch ijzer (zie ook §5.3). Anderzijds kan ook op het oppervlak geadsorbeerde waterstof in deze reactie worden verbruikt, waardoor de waterstofontwikkelingsreactie, die door de geadsorbeerde waterstof wordt geremd, versneld wordt. IJzer- en mangaan oxiderende bacteriën spelen een directe rol in het corrosieproces, met name door het verder oxideren van het in oplossing gaande Fe2+ en Mn2+ tot hogere valenties waarbij enerzijds meer (poreuze) corrosieproducten worden gevormd en anderzijds de locale metaalionenconcentraties laag blijven waardoor de oplosreactie beter zal verlopen. In feite krijgen we het mechanisme dat is afgebeeld in Figuur 6.3.3, maar dan versneld door de aanwezigheid van de bacteriën. Typerend voor microbiologisch geïnduceerde corrosie is de vorming van diepe putten, vaak met een opening die nauwer is dan hun doorsnede op enige afstand van het oppervlak. Voor een meer diepgaande en volledige behandeling van deze vorm van corrosie wordt naar de literatuur [6] verwezen.
6.6 Selectief oplossen van legeringen
Figuur 6.6.1 Laagvormige ontzinking van messing. De donkere buitenlaag is poreus rood koper, de lichte binnenkant onaangetast messing (Overgenomen uit E.D.D.During, Corrosion Atlas[1], met toestemming van Elsevier Science-NL, Sara Burgerhartstraat 25, 1055 KV Amsterdam)

95
Bepaalde legeringen die bestaan uit twee componenten met een groot verschil in edelheid kunnen het verschijnsel van de selectieve aantasting vertonen. Hierbij gaat dan de minst edele component van het metaal selectief in oplossing, terwijl de meer edele rest achterblijft als een poreuze, mechanisch zeer zwakke massa. Het meest bekende voorbeeld is de ontzinking van bepaalde messingsoorten. Laagvormige ontzinking is een speciaal type algemene corrosie omdat deze gelijkmatig over het gehele oppervlak optreedt. Figuur 6.6.1 toont een doorsnede van een messing onderdeel waarin laagvormige ontzinking is opgetreden. Propvormige ontzinking treedt op in de vorm van kegelvormige putten, die gevuld zijn met een poreus residu van rood koper. Figuur 6.6.2 en Figuur 6.6.3 tonen een voorbeeld van propvormige ontzinking in een messing pijp.
a. b.
Figuur 6.6.2 Propvormige ontzinking van messing, a. schematische doorsnede van prop; b. foto van propvormige ontzinking in messingpijp (Met toestemming overgenomen uit [3]).
Figuur 6.6.3 Microscopische doorsnede van propvormige ontzinking (Met toestemming overgenomen uit [3]).
Deze corrosie treedt bij voorkeur op in spleten en onder vuilafzettingen. Er zijn hiervoor twee mechanismen voorgesteld. Volgens het eerste mechanisme lost de minst edele component selectief op uit de legering onder achterlating van de meer edele component. Hieraan is in feite de naam van dit type corrosie ontleend. Volgens

Karakteristieke verschijningsvormen van corrosie
96
het tweede mechanisme gaat de legering als geheel in oplossing waarna de meer edele component neerslaat op de achtergebleven legering. Deze heeft namelijk een potentiaal die ligt beneden de evenwichtspotentiaal van de zuivere edele component (zie hiervoor ook §3.2). De tweede verklaring wordt ondersteund door de, hiervoor reeds genoemde, waarneming dat dit type corrosie bijna altijd onder afzettingen optreedt. Er is dan minder verversing van de oplossing mogelijk waardoor neerslaan van de opgeloste edeler component waarschijnlijker wordt. Deze afzettingen kunnen een gevolg zijn van vuil in het water, kalkafzettingen uit hard water (ketelsteen), poreuze corrosieproducten, enzovoort. Wanneer de meer edele component eenmaal is neergeslagen wordt het proces versneld door het potentiaalverschil tussen het neerslag en de nog niet aangetaste legering. Uiteraard is een belangrijke factor bij de bestrijding van deze vorm van corrosie het voorkomen van vuilafzettingen, bijvoorbeeld door vergroting van de vloeistofsnelheid, maar dat is lang niet altijd mogelijk (centrale verwarmingssystemen!). Verder kan geprobeerd worden de vloeistof minder agressief te maken door verhoging van de pH en/of verlaging van de chlorideconcentratie. Aan de andere kant moet wel gewaakt worden voor de vorming van ketelsteen. Het zachter maken (verlagen van het calciumgehalte) van water is in dit opzicht gunstig door de verminderde kans op neerslaan van ketelsteen. Tenslotte leidt toevoegen van kleine hoeveelheden tin, of nog beter van arsenicum, aan messing tot een aanzienlijke verbetering van de bestendigheid daarvan tegen ontzinking.
6.7 Interkristallijne corrosie
Alle in de praktijk gebruikte metalen zijn polykristallijn, hetgeen betekent dat ze bestaan uit een groot aantal, meestal willekeurig georiënteerde kristallijne gebiedjes: kristallieten of korrels genoemd. In het geval van interkristallijne corrosie worden alleen de dunne grensgebieden tussen deze korrels aangetast. Ondanks het hierbij meestal betrekkelijk kleine gewichtsverlies treedt toch snel falen op doordat de samenhang tussen de kristallieten verdwijnt. Het meest belangrijke voorbeeld is de interkristallijne corrosie van roestvast staal waarvan een voorbeeld wordt getoond in Figuur 6.7.1. In de microscopische doorsnede van Figuur 6.7.2 is duidelijk te zien hoe de corrosie langs de korrelgrenzen diep in het materiaal dringt. Bij roestvast staal is de oorzaak van interkristallijne corrosie het neerslaan, langs de korrelgrenzen, van chroomcarbiden ten gevolge van verhitting in een bepaald temperatuurgebied, zoals dat bij warmtebehandeling of lassen gebeurt, volgens de reactie:
23 Cr + 6 C → Cr23C6. (6.7.1) Deze reactie verloopt met een aanzienlijke snelheid tussen ongeveer 600 en 750 °C. De precipitatie gebeurt bij voorkeur langs de korrelgrenzen omdat nucleatie van een nieuwe fase gemakkelijker is aan een grensvlak dan in het binnenste van een kristal.
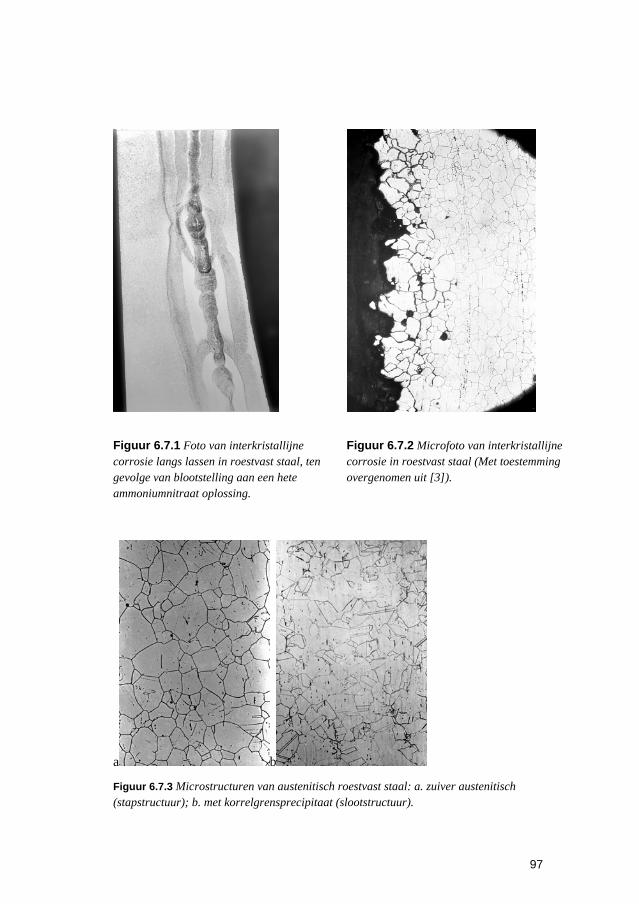
97
Figuur 6.7.1 Foto van interkristallijne corrosie langs lassen in roestvast staal, ten gevolge van blootstelling aan een hete ammoniumnitraat oplossing.
Figuur 6.7.2 Microfoto van interkristallijne corrosie in roestvast staal (Met toestemming overgenomen uit [3]).
a b
Figuur 6.7.3 Microstructuren van austenitisch roestvast staal: a. zuiver austenitisch (stapstructuur); b. met korrelgrensprecipitaat (slootstructuur).
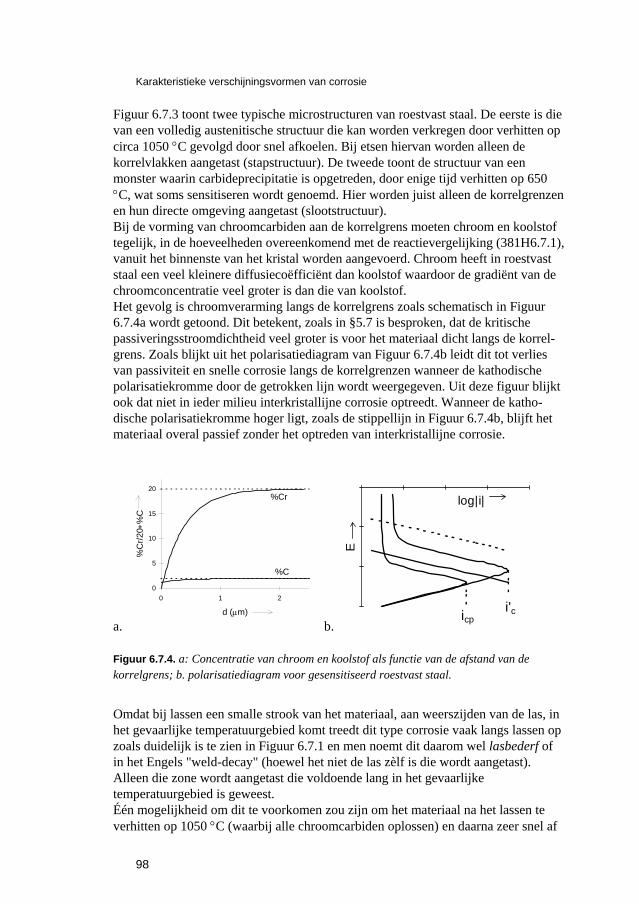
Karakteristieke verschijningsvormen van corrosie
98
Figuur 6.7.3 toont twee typische microstructuren van roestvast staal. De eerste is die van een volledig austenitische structuur die kan worden verkregen door verhitten op circa 1050 °C gevolgd door snel afkoelen. Bij etsen hiervan worden alleen de korrelvlakken aangetast (stapstructuur). De tweede toont de structuur van een monster waarin carbideprecipitatie is opgetreden, door enige tijd verhitten op 650 °C, wat soms sensitiseren wordt genoemd. Hier worden juist alleen de korrelgrenzen en hun directe omgeving aangetast (slootstructuur). Bij de vorming van chroomcarbiden aan de korrelgrens moeten chroom en koolstof tegelijk, in de hoeveelheden overeenkomend met de reactievergelijking (381H6.7.1), vanuit het binnenste van het kristal worden aangevoerd. Chroom heeft in roestvast staal een veel kleinere diffusiecoëfficiënt dan koolstof waardoor de gradiënt van de chroomconcentratie veel groter is dan die van koolstof. Het gevolg is chroomverarming langs de korrelgrens zoals schematisch in Figuur 6.7.4a wordt getoond. Dit betekent, zoals in §5.7 is besproken, dat de kritische passiveringsstroomdichtheid veel groter is voor het materiaal dicht langs de korrel-grens. Zoals blijkt uit het polarisatiediagram van Figuur 6.7.4b leidt dit tot verlies van passiviteit en snelle corrosie langs de korrelgrenzen wanneer de kathodische polarisatiekromme door de getrokken lijn wordt weergegeven. Uit deze figuur blijkt ook dat niet in ieder milieu interkristallijne corrosie optreedt. Wanneer de katho-dische polarisatiekromme hoger ligt, zoals de stippellijn in Figuur 6.7.4b, blijft het materiaal overal passief zonder het optreden van interkristallijne corrosie.
a.
0
5
10
15
20
0 1 2
d (μm)
%C
r/20 ∗
%C
%C
%Cr
b.
log|i|
E
icpi'c
Figuur 6.7.4. a: Concentratie van chroom en koolstof als functie van de afstand van de korrelgrens; b. polarisatiediagram voor gesensitiseerd roestvast staal.
Omdat bij lassen een smalle strook van het materiaal, aan weerszijden van de las, in het gevaarlijke temperatuurgebied komt treedt dit type corrosie vaak langs lassen op zoals duidelijk is te zien in Figuur 6.7.1 en men noemt dit daarom wel lasbederf of in het Engels "weld-decay" (hoewel het niet de las zèlf is die wordt aangetast). Alleen die zone wordt aangetast die voldoende lang in het gevaarlijke temperatuurgebied is geweest. Één mogelijkheid om dit te voorkomen zou zijn om het materiaal na het lassen te verhitten op 1050 °C (waarbij alle chroomcarbiden oplossen) en daarna zeer snel af

99
te koelen. Maar dit is natuurlijk alleen mogelijk voor relatief kleine en niet al te ingewikkelde onderdelen. De twee meest belangrijke methodes die in de praktijk worden toegepast zijn het gebruik van laag-koolstof roestvast staal met een koolstofgehalte onder de 0.03% en het gebruik van gestabiliseerd roestvaste staal, dat wil zeggen waaraan een koolstofbindend element, bijvoorbeeld titaan of niobium, is toegevoegd. In het eerste geval wordt de vorming van chroomcarbiden zodanig vertraagd dat zij tijdens normaal lassen niet worden gevormd. In het tweede geval worden zeer stabiele titaan- of niobiumcarbiden gevormd zodat de koolstof niet meer beschikbaar is voor de vorming van chroomcarbiden. Ook in omgevingen waar interkristallijne corrosie als zodanig geen probleem is, bijvoorbeeld in zeewater, kan het gebruik van deze roestvaste staalsoorten toch nuttig zijn omdat putinitiatie (zie §6.3) veel sneller is in gesensitiseerde gebieden. Ook met behulp van de toegepaste lastechniek kan de kans op interkristallijne corrosie van dit type worden verminderd. Bij het uitvoeren van reparatielassen bij onderhoudswerkzaamheden moet men hierop speciaal attent zijn omdat de lasomstandigheden dan meestal veel minder gunstig zijn dan tijdens de fabricage. Sommige andere materialen zoals de nikkel-chroomlegeringen bekend als Hastelloy en bepaalde aluminium-koper legeringen kunnen soortgelijke corrosieverschijnselen vertonen. Ook in die gevallen is aangetoond dat precipitaten gevormd langs de kor-relgrenzen verantwoordelijk zijn voor deze plaatselijke aantasting.
6.8 Spanningscorrosie en corrosievermoeiing
Zoals door de naam wordt aangegeven zijn dit twee onderling samenhangende vormen van corrosie die veroorzaakt worden door in het materiaal aanwezige spanningen. Bij wisselende spanningen spreekt men van corrosievermoeiing. De in het materiaal aanwezige trekspanningen leiden in beide gevallen, samen met de corrosieve aantasting, tot het optreden van scheuren [7]. Dit kunnen transkristallijne scheuren zijn, zoals weergegeven in Figuur 6.8.1a, of interkristallijne scheuren, zoals getoond in Figuur 6.8.1b. In beide gevallen staat de richting van de scheuren ongeveer loodrecht op die van de aanwezige trekspanning. Interkristallijne spanningscorrosie is duidelijk verschillend van gewone interkristallijne corrosie, zoals beschreven in §6.7, hetgeen direct blijkt uit een vergelijking van Figuur 6.7.2 en Figuur 6.8.1b. Bijna alle bekende legeringen zijn gevoelig voor spanningscorrosie in een beperkt aantal, zeer specifieke omgevingen. Voorbeelden zijn: koperlegeringen in vochtige atmosfeer waarin ammoniak of zwaveldioxide aanwezig is (interkristallijn), on- en laaggelegeerd staal in nitraten en geconcentreerde loogoplossingen (interkristallijn), roestvaste staalsoorten in hete chloride-oplossingen (transkristallijn) en in met chloriden verontreinigde stoom (trans- en interkristallijn), aluminium legeringen in zeewater (interkristallijn). Een vollediger overzicht wordt gegeven door Logan [8].
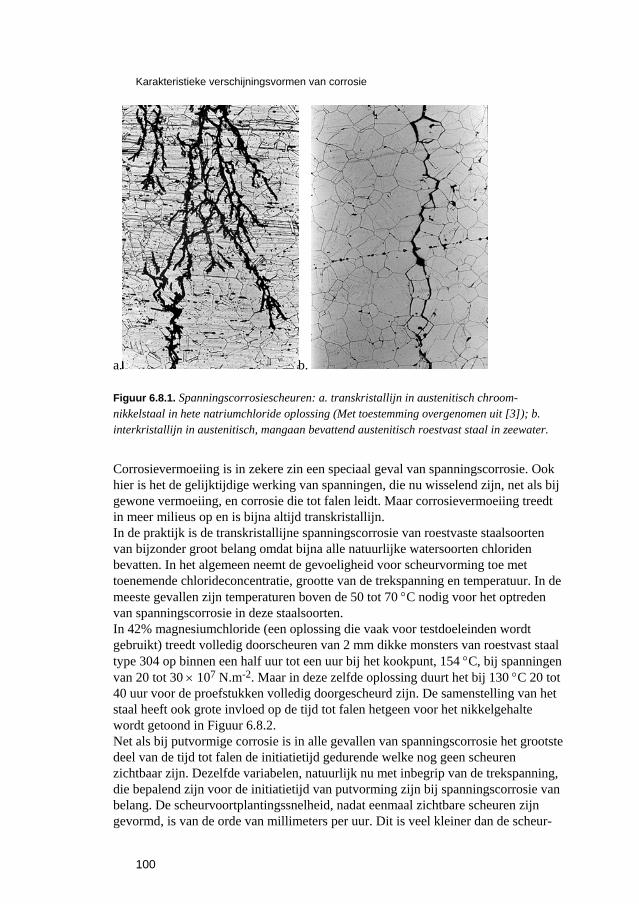
Karakteristieke verschijningsvormen van corrosie
100
a. b.
Figuur 6.8.1. Spanningscorrosiescheuren: a. transkristallijn in austenitisch chroom-nikkelstaal in hete natriumchloride oplossing (Met toestemming overgenomen uit [3]); b. interkristallijn in austenitisch, mangaan bevattend austenitisch roestvast staal in zeewater.
Corrosievermoeiing is in zekere zin een speciaal geval van spanningscorrosie. Ook hier is het de gelijktijdige werking van spanningen, die nu wisselend zijn, net als bij gewone vermoeiing, en corrosie die tot falen leidt. Maar corrosievermoeiing treedt in meer milieus op en is bijna altijd transkristallijn. In de praktijk is de transkristallijne spanningscorrosie van roestvaste staalsoorten van bijzonder groot belang omdat bijna alle natuurlijke watersoorten chloriden bevatten. In het algemeen neemt de gevoeligheid voor scheurvorming toe met toenemende chlorideconcentratie, grootte van de trekspanning en temperatuur. In de meeste gevallen zijn temperaturen boven de 50 tot 70 °C nodig voor het optreden van spanningscorrosie in deze staalsoorten. In 42% magnesiumchloride (een oplossing die vaak voor testdoeleinden wordt gebruikt) treedt volledig doorscheuren van 2 mm dikke monsters van roestvast staal type 304 op binnen een half uur tot een uur bij het kookpunt, 154 °C, bij spanningen van 20 tot 30 × 107 N.m-2. Maar in deze zelfde oplossing duurt het bij 130 °C 20 tot 40 uur voor de proefstukken volledig doorgescheurd zijn. De samenstelling van het staal heeft ook grote invloed op de tijd tot falen hetgeen voor het nikkelgehalte wordt getoond in Figuur 6.8.2. Net als bij putvormige corrosie is in alle gevallen van spanningscorrosie het grootste deel van de tijd tot falen de initiatietijd gedurende welke nog geen scheuren zichtbaar zijn. Dezelfde variabelen, natuurlijk nu met inbegrip van de trekspanning, die bepalend zijn voor de initiatietijd van putvorming zijn bij spanningscorrosie van belang. De scheurvoortplantingssnelheid, nadat eenmaal zichtbare scheuren zijn gevormd, is van de orde van millimeters per uur. Dit is veel kleiner dan de scheur-
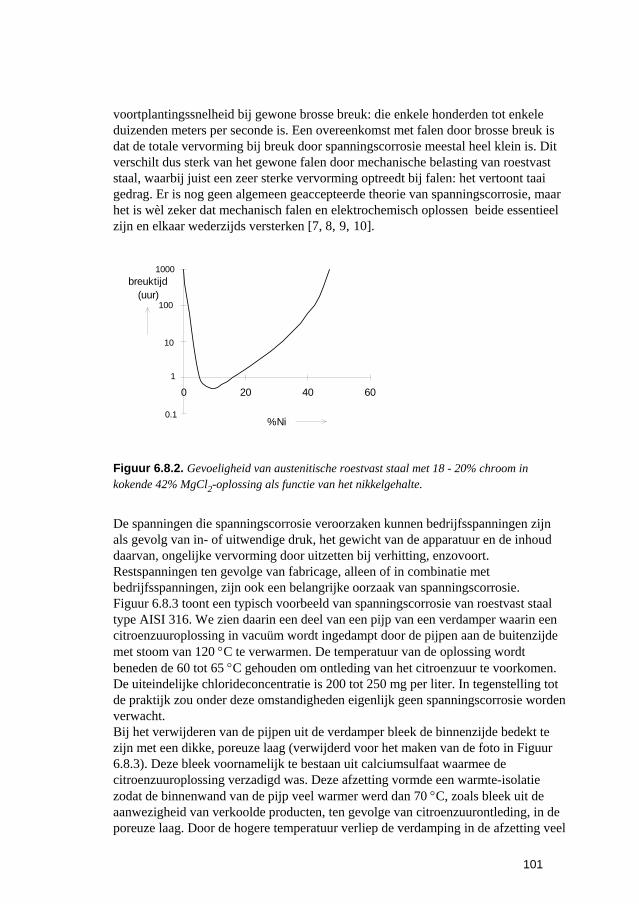
101
voortplantingssnelheid bij gewone brosse breuk: die enkele honderden tot enkele duizenden meters per seconde is. Een overeenkomst met falen door brosse breuk is dat de totale vervorming bij breuk door spanningscorrosie meestal heel klein is. Dit verschilt dus sterk van het gewone falen door mechanische belasting van roestvast staal, waarbij juist een zeer sterke vervorming optreedt bij falen: het vertoont taai gedrag. Er is nog geen algemeen geaccepteerde theorie van spanningscorrosie, maar het is wèl zeker dat mechanisch falen en elektrochemisch oplossen beide essentieel zijn en elkaar wederzijds versterken [7, 8, 9, 10].
0 20 40 60
%Ni
1000breuktijd
(uur)100
10
1
0.1
Figuur 6.8.2. Gevoeligheid van austenitische roestvast staal met 18 - 20% chroom in kokende 42% MgCl2-oplossing als functie van het nikkelgehalte.
De spanningen die spanningscorrosie veroorzaken kunnen bedrijfsspanningen zijn als gevolg van in- of uitwendige druk, het gewicht van de apparatuur en de inhoud daarvan, ongelijke vervorming door uitzetten bij verhitting, enzovoort. Restspanningen ten gevolge van fabricage, alleen of in combinatie met bedrijfsspanningen, zijn ook een belangrijke oorzaak van spanningscorrosie. Figuur 6.8.3 toont een typisch voorbeeld van spanningscorrosie van roestvast staal type AISI 316. We zien daarin een deel van een pijp van een verdamper waarin een citroenzuuroplossing in vacuüm wordt ingedampt door de pijpen aan de buitenzijde met stoom van 120 °C te verwarmen. De temperatuur van de oplossing wordt beneden de 60 tot 65 °C gehouden om ontleding van het citroenzuur te voorkomen. De uiteindelijke chlorideconcentratie is 200 tot 250 mg per liter. In tegenstelling tot de praktijk zou onder deze omstandigheden eigenlijk geen spanningscorrosie worden verwacht. Bij het verwijderen van de pijpen uit de verdamper bleek de binnenzijde bedekt te zijn met een dikke, poreuze laag (verwijderd voor het maken van de foto in Figuur 6.8.3). Deze bleek voornamelijk te bestaan uit calciumsulfaat waarmee de citroenzuuroplossing verzadigd was. Deze afzetting vormde een warmte-isolatie zodat de binnenwand van de pijp veel warmer werd dan 70 °C, zoals bleek uit de aanwezigheid van verkoolde producten, ten gevolge van citroenzuurontleding, in de poreuze laag. Door de hogere temperatuur verliep de verdamping in de afzetting veel

Karakteristieke verschijningsvormen van corrosie
102
sneller terwijl verversing van de oplossing daar juist veel langzamer is. Dit leidde tot een daar sterk verhoogde chlorideconcentratie die uit het chloridegehalte van de laag geschat werd op 100 tot 200 gram per liter. Deze verhoogde chlorideconcentratie, samen met de verhoogde temperatuur leidden tot de waargenomen spanningscorrosie. Het voorkomen van afzettingsvorming door een hogere vloeistofsnelheid samen met enkele kleine veranderingen van het ontwerp van de verdamper leidden tot effectieve bestrijding van de spanningscorrosie.
Figuur 6.8.3. Spanningscorrosie in pijp uit citroenzuurverdamper. Materiaal: roestvast staal type AISI 316.
Er zijn verschillende gevallen geconstateerd van spanningscorrosie van de onderkanten van ophangkabels van hangbruggen. Aangetoond kon worden dat dit werd veroorzaakt door de aanwezigheid van nitraten in regenwater, in het bijzonder tijdens of na onweer. De regen verzamelt zich aan de onderkant van de ophangkabels en wanneer de afvoer van het water onvoldoende is kunnen door verdamping hoge, plaatselijke nitraatconcentraties ontstaan, terwijl trekspanningen uiteraard daar aanwezig zijn. Volledige en snelle afvoer van het regenwater is dus nodig om dit type spanningscorrosie te voorkomen. Een ander voorbeeld van spanningscorrosie is de zogenaamde seizoenziekte (Engels: "season cracking") van messing. Dit werd al aan het eind van de vorige eeuw waargenomen in messing granaathulzen. Aangetoond werd dat dit een gevolg was van een vochtige atmosfeer waarin ammoniak aanwezig was (paardenstallen!), gecombineerd met zeer hoge inwendige spanningen ten gevolge van de koude vervorming. Spanningsvrij gloeien van messing is een effectieve bestrijding van deze vorm van spanningscorrosie. Figuur 6.8.4. toont een slecht gelegde las in roestvast staal waar hoge, plaatselijke spanningen tot zeer snelle spanningscorrosie hebben geleid. Soms kan in dit soort gevallen ook spanningsvrij gloeien worden toegepast, maar wèl moet worden bedacht dat ongelijkmatige afkoeling toch weer aanleiding kan geven tot grote restspanningen. Bovendien kan spanningsvrij gloeien van roestvast staal leiden tot

103
carbideprecipitatie waardoor het materiaal gevoelig wordt voor interkristallijne corrosie, afhankelijk van temperatuur en tijd van de behandeling.
Figuur 6.8.4. Spanningscorrosie in en naast een verkeerd gelegde las in roestvast staal na 600 uur in hete chlorideoplossing (Met toestemming overgenomen uit [3]).
De belangrijkste methoden om spanningscorrosie en vermoeiingscorrosie te bestrijden of voorkomen zijn: 1. voorkomen van hoge trekspanningen en in het bijzonder van
spanningsconcentraties; 2. verwijderen van agressieve componenten uit het milieu; 3. in sommige gevallen toepassen van kathodische bescherming, inhibitoren of
beschermende deklagen. Wanneer geen van deze methoden kan worden toegepast is de enige oplossing het kiezen van een ander materiaal dat in het gegeven milieu bestand is tegen spanningscorrosie. Bijvoorbeeld kunnen in warm zeewater legeringen zoals cupronikkel of monel worden gekozen in plaats van roestvast staal.
6.9 Waterstofverbrossing
Atomaire waterstof, die bijvoorbeeld geproduceerd wordt door de kathodische reactie bij corrosie van een metaal in zuur kan in principe in dat metaal oplossen en naar binnen diffunderen en dan leiden tot verbrossing, blaasvorming of zelfs scheuren. In aanwezigheid van trekspanningen kan volledig falen van het metaal onverwacht en al na korte tijd optreden. Ook andere processen dan corrosie in zuur kunnen aanleiding geven tot de vorming van waterstof zoals beitsen van metaal in geconcentreerd zuur, kathodische bescherming en galvanisch bedekken met erg onedele metalen. In al deze gevallen is de eerste stap de vorming van geadsorbeerde waterstof:
H+ + e- → Hads (6.9.1) Normaal gesproken recombineren geadsorbeerde waterstofatomen tot waterstofmoleculen die als gas ontwijken:

Karakteristieke verschijningsvormen van corrosie
104
Hads + Hads→ H2 (6.9.2) maar de geadsorbeerde waterstof kan ook in het metaal oplossen volgens:
Hads → H(metaal) (6.9.3) Dit kan bevorderd worden door oppervlaktevergiftiging waardoor diffusie van de geadsorbeerde waterstof over het oppervlak bemoeilijkt wordt. Het kleine waterstofatoom diffundeert tussen de atomen van het metaal door (interstitiële diffusie). De waterstof kan in principe worden verwijderd door "uitstoken", dat wil zeggen gedurende 3 uur verhitten op 170 tot 180 °C. Bij hoge temperaturen, bijvoorbeeld in chemische processen of bij lassen met beklede elektroden, kan moleculaire waterstof op een metaaloppervlak dissociëren en dan ook als atomaire waterstof in het metaal doordringen, waarbij soortgelijke problemen optreden als bij de opname van waterstof door kathodische processen. Bij de diffusie door het metaal kan waterstof zich verzamelen bij inwendige holtes, verontreinigingen en dergelijke waarbij de aparte atomen zich weer tot moleculen verenigen. Hierdoor kunnen binnen in het metaal enorm hoge inwendige drukken optreden, soms hoger dan 300 tot 400 atm, waardoor blazen op het oppervlak of scheuren in het metaal kunnen ontstaan. Hiervan wordt een voorbeeld getoond in Figuur 6.9.1.
Figuur 6.9.1. Foto van waterstofblazen in ongelegeerd staal (Overgenomen uit E.D.D.During, Corrosion Atlas[1], met toestemming van Elsevier Science-NL, Sara Burgerhartstraat 25, 1055 KV Amsterdam)
Bij de scheurvorming is speciaal van belang dat bij de vorming van een nieuw oppervlak de waterstof daarop kan adsorberen waardoor de oppervlakte-energie wordt verlaagd en de scheurvorming nog gemakkelijker verloopt. Bestrijding van schade door waterstofopname kan gebeuren door het hiervoor al genoemde uitstoken, door ervoor te zorgen dat geen onnodig hoge kathodische stroom loopt (voorkomen van overbescherming bij kathodische bescherming) door

105
het kiezen van speciale, hiertegen beter resistente legeringen en door het vermijden van onnodige trekspanningen in het metaal.
6.10 Erosie- en cavitatiecorrosie
Wanneer de gelijktijdige werking van corrosie en erosie door een snelstromende vloeistof tot een onverwacht hevige aantasting leidt, die veel groter is dan de som van de afzonderlijke verschijnselen spreken we van erosie-corrosie.
Dit verschijnsel wordt in het bijzonder waargenomen op plaatsen waar sterke turbulentie in de vloeistof optreedt zoals bij scherpe bochten, vernauwingen, inlaateinden van nauwe pijpen, pompwaaiers, enzovoort. De algemeen aanvaarde verklaring is dat de erosieve werking een op het metaal aanwezige beschermende laag vernield, waardoor direct contact tussen onbedekt metaal en vloeistof mogelijk wordt. Twee typische voorbeelden van erosie-corrosie worden getoond in Figuur 6.10.1 en Figuur 6.10.2. Naast de "gewone" erosie-corrosie zijn er nog twee andere, hiermee nauw verwante vormen van gecombineerde mechanische en corrosieve aantasting, namelijk cavitatie-corrosie en inslag-aantasting.
Figuur 6.10.1. Erosie-corrosie in bocht van pijp (Met toestemming overgenomen uit [3]).
Cavitatie-corrosie is een gevolg van cavitatie, dat is belvorming in de vloeistof door sterke onderdruk, die vervolgens imploderen in de onmiddellijke omgeving van het metaaloppervlak dat daardoor wordt beschadigd. Dit kan bijvoorbeeld een gevolg zijn van een snelle beweging van het metaal in een richting loodrecht op het oppervlak daarvan waardoor in de vloeistof plaatselijk een vacuüm ontstaat. In de meeste gevallen worden hierbij dicht bijeen geplaatste putten met scherpe randen in het metaal gevormd. In de praktijk wordt dit gevonden bij scheepsschroeven, pompwaaiers en soms, zoals getoond in Figuur 6.10.3, aan de koelwaterzijde van cilinders van dieselmotoren. Inslag-aantasting (Engels: impingement attack) is een vorm van putcorrosie als gevolg van de vernieling van een beschermende laag door het inslaan op het oppervlak van luchtbellen die met hoge snelheid in (of met) de vloeistof bewegen,

Karakteristieke verschijningsvormen van corrosie
106
bijvoorbeeld in condensorpijpen. Een soortgelijke aantasting kan ook optreden bij het inslaan van een sterke vloeistofstraal op een metaaloppervlak.
Figuur 6.10.2. Erosie-corrosie van een gietijzeren pompwaaier na een bedrijfstijd van minder dan een jaar in een basische permanganaatoplossing waarin vast mangaandioxide was gesuspendeerd.
Enige mogelijkheden om deze vormen van aantasting te voorkomen zijn: 1. verandering van het ontwerp die leiden tot lagere vloeistofsnelheden en een
meer “gestroomlijnde” beweging, bijvoorbeeld door het vergroten van de buigstraal in bochten (zie ook §8.7);
2. toepassing van kathodische bescherming, inhibitoren of passivatoren; 3. filteren of op andere wijze reinigen van de vloeistof door vaste deeltjes en/of
luchtbellen te verwijderen; 4. toepassing van beschermende lagen, waarvan een speciaal voorbeeld (zie Figuur
6.10.4) is het aanbrengen van plastic inzetstukken. Dit wordt toegepast in condensors en warmtewisselaars waar dikwijls in de eerste centimeters aan de intreezijde van de pijpen erosie-corrosie optreedt door de sterke turbulentie dicht bij de inlaat. Het inzetstuk moet dan zolang zijn dat de vloeistof pas in contact komt met het metaal nadat een laminaire grenslaag is ontstaan.

107
5. toepassing van materialen met een sterkere beschermende huid. In het in Figuur 6.10.2 getoonde voorbeeld werd het grijze gietijzer vervangen door Ni-resist dat een levensduur van 7 jaar gaf onder overigens dezelfde omstandigheden. Een ander voorbeeld is de toepassing van cupronikkel inplaats van messing in condensors en zeewaterverdampers.
Figuur 6.10.3. Cavitatie-corrosie aan de koelwaterzijde van een cilinder van een dieselmotor.
Figuur 6.10.4. Kunststof inzetstuk (gearceerd in de figuur) aan de inlaatzijde van een koeler- of condensorpijp.

Karakteristieke verschijningsvormen van corrosie
108
6.11 Passingroest
Wanneer slijtage en corrosie tegelijk optreden kan er sprake zijn van het optreden van tribochemische reacties. Hierbij treden tijdens het slijtageproces chemische reacties op waardoor materiaal wordt omgezet in reactieproducten. Wanneer de reactieproducten oxiden zijn die ontstaan door reactie met zuurstof uit de omgeving spreekt men van oxidatieslijtage. De natuurlijke, dunne oxidelaag die op metaaloppervlakken gevormd wordt kan hiertegen dikwijls voldoende bescherming geven wanneer die laag niet afbrokkelt en niet poreus is. Maar wanneer deze laag door de voortdurende wrijving steeds wordt verwijderd en de vorming van een nieuwe laag versneld optreedt, bijvoorbeeld door hoge temperatuur, wrijvingswarmte en/of een agressieve omgeving kan toch oxidatieslijtage optreden. Wanneer de onderlinge beweging van de metaaloppervlakken niet continu is, maar bestaat uit oscillerende bewegingen met kleine amplitude kan een ander verschijnsel optreden dat bekend staat als passingroest (Engels: "fretting corrosion") [11]. Door de kleine bewegingen kunnen de slijtageproducten niet van het contactoppervlak verwijderd worden. Deze worden dan ter plaatse geoxideerd en omdat oxiden vaak harder zijn dan het metaal zelf treedt versnelde slijtage op. Hierdoor kan de passing-roest onverwacht snel verlopen. Gevolgen kunnen zijn het optreden van verstoppingen, vreten van de langs elkaar bewegende oppervlakken en volledig vastlopen. Verder kan als gevolg hiervan ook vermoeiing van het metaal optreden met name door de spanningsconcentratie als gevolg van de gevormde, vaak zeer scherpe putten. Figuur 6.11.1 toont als praktisch voorbeeld drukringen die zijn gemonteerd tussen de krukas van een dieselmotor en de daardoor aangedreven schroefas. Een belangrijke oorzaak van passingroest is de vibratie van onderdelen tijdens transport en is bijvoorbeeld waargenomen bij assen in lagers, bij kogel-, rol- en naaldlagers, enzovoort. Onder normale bedrijfsomstandigheden vertoont dit type onderdelen een continue beweging waarbij passingroest niet optreedt.
Figuur 6.11.1. Passingroest van drukringen.

109
Belangrijke bestrijdingsmethoden van passingroest zijn: 1. smering met speciale oliesoorten met een lage viscositeit en grote hechting die
zo mogelijk bovendien inhibitoren bevatten, zodat de wrijving wordt voorkomen en agressieve stoffen zoals vocht en zuurstof buitengesloten worden;
2. fosfateren van de onderdelen en impregneren van de poreuze fosfaatlaag met olie;
3. toepassen van materialen met verschillende hardheid voor de onderdelen; 4. gebruik maken van rubber of plastic tussenmateriaal om vibratie te verminderen
en agressieve componenten buiten te sluiten; 5. toepassen van speciale keramische deklagen, zoals nitriden of MoS2; 6. gebruik van tijdelijke corrosiebeschermingsmiddelen (zie ook §8.6.6) in
gevallen waar tijdens transport gevaar voor passingroest bestaat.
6.12 Afsluitende opmerkingen
In dit hoofdstuk zijn de meest voorkomende en belangrijkste vormen van corrosie besproken. Daarbij werd tevens ingegaan op de omstandigheden waaronder de verschillende types corrosie op kunnen treden en op enkele hiervoor specifieke bestrijdingsmogelijkheden. Het is duidelijk dat dit overzicht zeer onvolledig is. Daarom wordt voor verdere, meer uitgebreide, beschouwingen naar de literatuur verwezen en wel in het bijzonder naar referenties 1 en 2 bij dit hoofdstuk.
6.13 Literatuur
1. E.D.D. During, Corrosion Atlas. A collection of illustrated case histories. 3rd expanded and revised edition, Amsterdam, Elsevier (1997)
2. Controlling Corrosion. 5. Case Studies, Department of Industry, London, HMSO (1979). 3. P.J. Gellings, Corrosie en Onderhoud, in: Handboek Onderhoudsmanagement, Alphen
aan de Rijn, Samsom BedrijfsInformatie, 1996, p. K3010-1 - 56. 4. K. Barton, Protection against atmospheric corrosion, New York, John Wiley (1976). 5. Schmitt and Mathay, Materials Protection 6 (sept. 1967) p.37. 6. S.W. Borenstein, Microbiologically influenced corrosion handbook, Cambridge,
Woodhead Publishing Ltd. (1994). 7. Stress corrosion, Guides to practice in corrosion control, no.4, Department of Industry,
London. HMSO (1980). 8. H.L. Logan, The stress corrosion of metals, New York, John Wiley (1966). 9. M.G. Fontana, Corrosion Engineering, 3rd ed. New York, McGrawHill (1986) 10. S.A. Bradford, Corrosion Control, New York, Van Nostrand Reinhold, 1993. 11. R.B. Waterhouse, Fretting corrosion, Oxford, Pergamon Press (1972).


7
Meting van corrosie-eigenschappen van materialen


113
7.1 Inleiding
Om te kunnen beoordelen of in een bepaalde situatie corrosie te verwachten is en de in de voorgaande hoofdstukken besproken principes toe te passen zijn numerieke gegevens nodig. In dit hoofdstuk worden een aantal aspecten verbonden aan het verkrijgen en meten van corrosie-eigenschappen besproken. Hierbij wordt ook aandacht geschonken aan het toepassingsgebied van de verschillende methoden en aan normalisatie en standaardproeven.
7.2 Vinden, bepalen en gebruiken van corrosiegegevens
In §1.3 is al gesproken over corrosiebestendigheid van materialen en aangege-ven dat dit eigenlijk geen materiaaleigenschap maar een systeemeigenschap is. Zoals besproken wordt in hoofdstuk 8 en is aangegeven in het schema van Figuur 8.2.1 moeten we toch het gedrag van een materiaal onder bepaalde omstandigheden en in het gegeven milieu kennen om tot een verantwoorde keuze te komen. In deze paragraaf wordt daarom op een aantal aspecten die hierbij van belang zijn ingegaan.
7.2.1 Maten voor corrosiesnelheden
7.2.1.1 Algemene corrosie
De snelheid waarmee een materiaal corrodeert kan op verschillende manieren worden aangegeven, bijvoorbeeld als de dikte- of de gewichtsafname per tijdseen-heid. Een aantal veel gebruikte eenheden zijn gegeven in bijlage 11.3, waarbij ook de omrekeningsfactoren zijn opgenomen. Hierbij vinden we ook een eenheid om de corrosiesnelheid als stroomdichtheid uit te drukken. De achtergrond hiervan is be-sproken in §4.1 en berust op de toepassing van de wet van Faraday. De corrosie-snelheid kan ook worden uitgedrukt in de zogenaamde polarisatieweerstand die is genoemd in §4.3.1 en uitvoeriger aan de orde komt in §7.2.3. Bij geschilderde of op andere wijze van een deklaag voorziene constructies wordt als maat voor de corrosiesnelheid dikwijls gebruikt het percentage van het oppervlak dat in een bepaalde tijd door zichtbare roestvorming is aangetast.
7.2.1.2 Plaatselijke corrosie
Voor de verschillende vormen van plaatselijke corrosie, zoals besproken in hoofdstuk 6, moeten uiteraard andere grootheden worden gebruikt als voor algemene corrosie ook al zijn de eenheden soms wèl hetzelfde. In het geval van putcorrosie neemt men als maat voor de snelheid de maximale putdiepte die in een bepaalde tijd wordt bereikt. Zoals in §6.3 is besproken vertonen putdieptes meestal ongeveer een normale verdeling rond het gemiddelde zodat de maximale waargenomen putdiepte afhankelijk is van de grootte van de steekproef. Een ander belangrijk punt is dat putcorrosie vrijwel altijd eerst een initiatietijd vertoont, waarvan de lengte kan variëren van enkele dagen tot enkele maanden.

Meting van corrosie-eigenschappen van materialen
114
Deze wordt dan gevolgd door een periode van putgroei. Meestal wordt echter bij de vermelding van putcorrosie verzuimd te vermelden of dit de snelheid van de putgroei zèlf is, dus de tijd vanaf het ogenblik waarop de put is gevormd, of de gemiddelde snelheid, dus vanaf het begin van de expositie. Aangezien dit vrijwel nooit wordt aangegeven is het merendeel van de in de literatuur gegeven snelheden van putcorrosie onbruikbaar als hulpmiddel voor de materiaalkeuze. Voor scheurvormende corrosie, dus bij spanningscorrosie en corrosievermoeiing, wordt niet een snelheid opgegeven, maar de tijd vanaf het begin van de proef tot de vorming van de eerste waarneembare scheur of de tijd tot breuk. In het eerste geval wordt er meestal van uitgegaan dat de scheurvoortplanting zo snel is dat de tijd daarvan te verwaarlozen is ten opzichte van de tijd tot vorming van de scheur (initiatietijd). Voor het aangeven van de snelheid van interkristallijne corrosie wordt in veel gevallen het gewichtsverlies per tijdseenheid gekozen. Dit is natuurlijk alleen mogelijk wanneer de aantasting gelijkmatig over het hele oppervlak plaatsvindt. Wanneer de interkristallijne corrosie plaatselijk optreedt, bijvoorbeeld in de omgeving van een las, kan dat niet meer en kiest men liever de penetratiediepte, bijvoorbeeld in μm per proefperiode van 24 of 48 uur. In principe kan deze laatste microscopisch worden bepaald mits er voor gezorgd wordt dat ook na de corrosie de plaats van het oorspronkelijke buitenoppervlak van het proefstuk nog kan worden vastgesteld.
7.2.2 Corrosiegegevens verkrijgen uit de literatuur
Veel experimentele gegevens verkregen uit laboratoriumproeven of praktijkexperimenten zijn in getabelleerde vorm gepubliceerd [1, 2, 3, 4]. De meeste van dit soort tabellen zijn alfabetisch ingedeeld op corrosief milieu. Bij ieder daarvan wordt dan het gedrag van een aantal materialen gegeven in de vorm van getallen, symbolen, of soms een meer uitvoerige beschrijving. Men moet zeer voorzichtig en kritisch zijn bij het gebruik van de uit dit type tabellen verkregen corrosiegegevens. Veel van deze gegevens zijn namelijk afkomstig van laboratoriumproeven, waardoor zij niet altijd representatief zijn voor de praktijksituatie waarin men de gegevens wil toepassen. Kleine hoeveelheden verontreinigingen kunnen een grote invloed uitoefenen op de corrosiesnelheid of op het type corrosie dat kan optreden. Bij laboratoriumproeven wordt bijvoorbeeld bijna altijd gedestilleerd water gebruikt voor het bereiden van de oplossingen waarin het materiaal wordt onderzocht. In de praktijk worden echter altijd natuurlijke watersoorten gebruikt die 200 tot 5000 mg per liter, of soms nog meer, chloride-ionen kunnen bevatten. Voor metalen die in chloridehoudend milieu gevoelig zijn voor putcorrosie, zoals de meeste roestvaste staalsoorten en aluminium, kan dit tot zeer onaangename verrassingen leiden. De tabellen geven meestal alleen gegevens betreffende algemene corrosie, bovendien bijna altijd op grond van de impliciete veronderstelling dat de corrosiesnelheid onafhankelijk is van de tijd. Dikwijls worden de gegevens slechts in zeer globale termen gegeven zoals: bestand (corrosiesnelheid minder dan 0.1 mm/jaar), tamelijk bestand (0.1 tot 0.5 mm/jaar),

115
niet erg bestand (0.5 tot 2.5 mm/jaar) en onbruikbaar (meer dan 2.5 mm/jaar). Verder zijn deze aanduidingen meestal alleen gebaseerd op de bereikbare levensduur en niet op andere factoren. In hoofdstuk 1 is al aangegeven dat het zeker kan voorkomen dat uit het oogpunt van levensduur een aantasting van 0.2 mm/jaar alleszins acceptabel is, maar dat de daarbij gevormde hoeveelheid corrosieproducten zo groot is dat die tot onaanvaardbare storingen leidt of tot productverontreiniging, die bijvoorbeeld in de levensmiddelen- of geneesmiddelenindustrie zeker ontoelaatbaar is. In sommige gevallen worden dergelijke aanwijzingen wèl gegeven, zoals bijvoorbeeld op het blad "Ammoniumnitraat" van de Dechema Werkstoff Tabelle [3]. Daar wordt namelijk aangegeven dat de aantasting van koper- en koperlegeringen tamelijk gering is. Desondanks kunnen deze volgens een nadere toelichting niet worden gebruikt, omdat kleine hoeveelheden koper in het product de ontleding daarvan katalytisch versnellen en daardoor tot vergroot explosiegevaar leiden. Maar als een dergelijke aanduiding ontbreekt mag men er zeker niet van uitgaan dat dan geen problemen van dit type op kunnen treden. Een belangrijke factor, die meestal niet wordt aangegeven, is de invloed van de stromingssnelheid. Vooral zachte metalen, die volgens zo'n tabel goed bruikbaar lijken, zijn soms al bij matige stromingssnelheden van 0.5 tot 1 m/sec gevoelig voor erosie-corrosie. Omgekeerd kan een te lage stromingssnelheid, onder andere als gevolg van vuilafzetting, bij bepaalde metalen tot een verhoogde kans op putcorrosie leiden. In de meeste gevallen is de enige betrouwbare conclusie die uit dit soort tabellen kan worden getrokken welke materialen zeker niet bruikbaar zijn zonder aanvullende bescherming. Over de toepasbaarheid en het effect van de verschillende mogelijkheden van corrosiebestrijding zoals kathodische of anodische bescherming, verandering van de pH of toevoeging van inhibitoren, enzovoort, geven deze tabellen hoogstens in een klein aantal incidentele gevallen uitsluitsel. Dit betekent dus dat de gegevens uit deze tabellen altijd moeten worden aangevuld met andere gegevens die òf uit ervaring òf uit speciaal daarvoor uitgevoerde experimenten afkomstig zijn.
7.2.3 Meten van corrosie-eigenschappen
Wanneer corrosiegegevens niet in de literatuur beschikbaar zijn is het nodig om corrosiemetingen uit te voeren om beslissingen over bruikbaarheid van materialen of corrosiebestrijdingsmethoden te kunnen nemen. Corrosiebeproeving is een uitvoerig gebied met veel gevaren en valkuilen en de resultaten moeten zeer kritisch worden bekeken voor ze kunnen worden toegepast (zie bijvoorbeeld [5, 6, 7]). Om te beginnen is het al zo dat de tijd die beschikbaar is voor het onderzoek meestal veel korter is dan de gewenste levensduur van de apparatuur of constructie waarvoor het te onderzoeken materiaal is bedoeld. Iedere onnauwkeurigheid in de meetresultaten wordt dan enorm sterk vergroot door de zeer grote benodigde extrapolatie. Bovendien kan de corrosiesnelheid sterk met de tijd variëren en dit kan, als functie van de aard van deze tijdsafhankelijkheid, zowel tot een zeer grote onderschatting als overschatting van de te verwachten corrosie leiden. Dit betekent in ieder geval

Meting van corrosie-eigenschappen van materialen
116
dat het in hoge mate wenselijk is niet alleen statische metingen uit te voeren, maar vooral ook het gedrag als functie van de tijd te bepalen. In veel gevallen worden testplaatjes beproefd in een omgeving waarin verwacht wordt dat het materiaal moet worden gebruikt. Dit kan in het laboratorium worden gedaan of, bij voorkeur, onder praktijkomstandigheden. In dat geval moet er op worden gelet dat contact met het materiaal van de apparatuur waarin de experimenten worden uitgevoerd niet tot foute resultaten leidt door contactcorrosie of kathodische bescherming! Bovendien mogen eventuele in het milieu aanwezige corrosieproducten van het voor de apparatuur gebruikte materiaal niet de corrosie van de proefplaatjes beïnvloeden. Een groot probleem bij de schijnbaar eenvoudige methode om corrosiesnelheden te bepalen uit gewichtsverliesmetingen aan testplaatjes is het verwijderen van de gevormde corrosieproducten van het oppervlak zonder tegelijk ook iets van het materiaal zelf af te halen. Een ander belangrijk punt is dat het in de meeste gevallen niet mogelijk is om een idee van het verloop van de corrosie met de tijd te krijgen door één enkel proefplaatje, door herhaalde expositie, te onderzoeken. Omdat de gevormde corrosieproducten meestal een aanzienlijke invloed op de corrosiesnelheid hebben, bijvoorbeeld door zuurstoftransport naar het metaaloppervlak te belemmeren, wordt het gedrag na schoonmaken en wegen volledig anders dan wanneer het plaatje langer in de oplossing was gebleven. Tenslotte worden gewichtsverliesmetingen zinloos wanneer plaatselijke corrosie, zoals putvormige of scheurvormende corrosie optreden. Dan moet de indringdiepte van de putten of scheuren worden bepaald, bijvoorbeeld door doorsneden van het proefstuk microscopisch te onderzoeken. In veel gevallen worden versnelde corrosieproeven gebruikt, in het bijzonder bij laboratoriumproeven. Hierbij moet worden opgemerkt dat veel van de standaardproeven van dit type (zie §7.3) in de eerste plaats zijn ontwikkeld om te kunnen bepalen of een bepaald materiaal aan de daaraan gestelde eisen voldoet. Ze zijn meestal totaal ongeschikt om de bruikbaarheid van een materiaal voor een nieuwe toepassing vast te stellen. Zie voor een uitvoerige bespreking van versnelde corrosieproeven referentie 8. Dit laatste geldt niet voor de, vaak ook snelle, elektrochemische meetmethoden die beschikbaar zijn voor het meten van corrosiesnelheden van metalen die blootgesteld zijn aan elektrolietoplossingen. Een eerste voorbeeld berust op de in §5.2 besproken situatie, waarvoor het polarisatiediagram in Figuur 7.2.1 wordt gegeven. Door de stroom-spanningskrommen te meten (kruisjes in de figuur) en terug te extrapoleren naar de corrosiepotentiaal Ecorr wordt uit het snijpunt de corrosiestroomdichtheid icorr gevonden. Een bezwaar van deze methode is dat door de grote externe stroom de vorming van (eventueel beschermende) corrosieproducten kan worden beïnvloed, waardoor de bepaalde corrosiesnelheid verschilt van die welke in de ongestoorde toestand gevonden zou worden. Daarnaast zijn de polarisatiekrommen niet altijd over een voldoend groot gebied recht, waardoor de extrapolatie erg onzeker wordt.
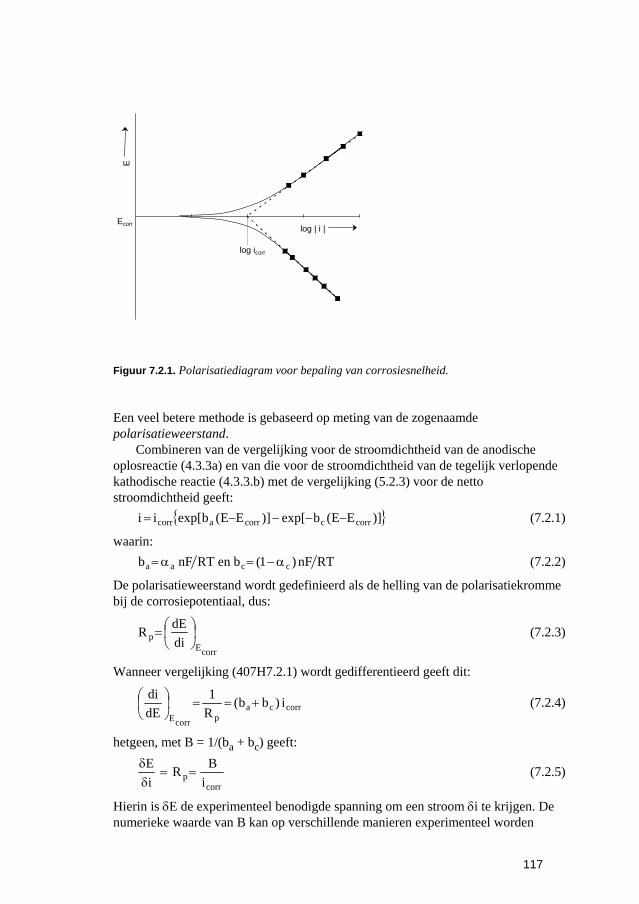
117
log | i |
E
log icorr
Ecorr
Figuur 7.2.1. Polarisatiediagram voor bepaling van corrosiesnelheid.
Een veel betere methode is gebaseerd op meting van de zogenaamde polarisatieweerstand.
Combineren van de vergelijking voor de stroomdichtheid van de anodische oplosreactie (4.3.3a) en van die voor de stroomdichtheid van de tegelijk verlopende kathodische reactie (4.3.3.b) met de vergelijking (5.2.3) voor de netto stroomdichtheid geeft:
{ })]EE(bexp[)]EE(bexp[ii corrccorracorr −−−−= (7.2.1)
waarin: RTnF)1(benRTnFb ccaa α−=α= (7.2.2)
De polarisatieweerstand wordt gedefinieerd als de helling van de polarisatiekromme bij de corrosiepotentiaal, dus:
corrEp di
dER ⎟⎠⎞
⎜⎝⎛= (7.2.3)
Wanneer vergelijking (407H7.2.1) wordt gedifferentieerd geeft dit:
corrcapcorrE
i)bb(R1
dEdi
+==⎟⎠⎞
⎜⎝⎛ (7.2.4)
hetgeen, met B = 1/(ba + bc) geeft:
corrp i
BRiE
==δδ (7.2.5)
Hierin is δE de experimenteel benodigde spanning om een stroom δi te krijgen. De numerieke waarde van B kan op verschillende manieren experimenteel worden

Meting van corrosie-eigenschappen van materialen
118
bepaald. Op theoretische gronden is te verwachten dat de waarde van 2.303 × B tussen 6.5 en 52 mV ligt. De in de praktijk gevonden waarden liggen in het algemeen niet al te ver van 20 mV en die waarde wordt daarom vaak in commerciële apparatuur gebruikt. De meting van de polarisatieweerstand kan gedurende expositie van een materiaal worden uitgevoerd zonder het corrosieproces te verstoren omdat de benodigde spanning δE hoogstens enkele mV is. Op deze manier kan, door herhaald meten als functie van de tijd, ook informatie over de tijdsafhankelijkheid van de corrosiesnelheid worden verkregen. De gegevens gepresenteerd in figuur 6.2.2 zijn bijvoorbeeld op deze wijze bepaald. Deze methode geeft ook de mogelijkheid om snel te toetsen of een materiaal bruikbaar kan zijn voor een bepaalde toepassing onder bepaalde omstandigheden. Voor een vollediger behandeling van de mogelijkheden, maar ook van de grenzen van de toepasbaarheid van deze methode moet naar de literatuur, bijvoorbeeld het overzicht van Mansfeld [9] worden verwezen. Er zijn ook verschillende andere belangrijke elektrochemische meetmethoden ontwikkeld, zowel voor het meten van corrosie-eigenschappen als voor het onderzoek van corrosieprocessen en middelen voor corrrosiebestrijding (zie bijvoorbeeld de corrosiepublicaties 19 en 30 van het Nederlands Corrosie Centrum, bijlage 11.17 en publicatie 4 van de Europese Federatie Corrosie, bijlage 11.18). Speciaal moet hierbij genoemd worden de elektrochemische impedantie spectroscopie. Behandeling van deze methoden voert in dit kader te ver en daarom wordt hiervoor naar de literatuur verwezen [10, 11, 12].
7.2.4 Continue bewaking van corrosie
Van toenemend belang is continue corrosiebewaking (Engels: corrosion monitoring) hetgeen gedefinieerd kan worden als: "de systematische en (semi-)continue meting van de corrosie van een onderdeel van een apparaat of constructie om informatie te verkrijgen over de geschiktheid van een materiaal gedurende het gebruik". Het wordt toegepast voor het bewaken van pijpleidingen, reactievaten in de chemische industrie, verwarming- en koelsystemen, enzovoort. Toepassing hiervan maakt het mogelijk om de omstandigheden te wijzigen (bijvoorbeeld toevoegen van een inhibitor of verwijderen van een agressieve component) of onverwacht falen te voorkomen. Er zijn drie hoofdmethoden voor continue corrosiebewaking: - metaalverliesmetingen: hierbij wordt het verlies van materiaal door erosie of
corrosie langs fysische of elektrische weg gemeten; - elektrochemische metingen: hier wordt de agressiviteit van een milieu bepaald
onafhankelijk van feitelijk materiaalverlies; - aanvullende analyses: hierbij behoren onder andere metaalconcentratie-
bepalingen in de oplossing, micro-organisme tellingen, meting van gasontwikkeling of -verbruik, enzovoort.
In dit verband kunnen ook nog niet-destructieve testmethoden, zoals röntgenonderzoek en ultrasone diktemetingen worden genoemd. Deze geven echter

119
meestal alleen informatie achteraf: dus of en zo ja hoeveel corrosie er op een bepaald ogenblik is opgetreden en niet het verloop van de corrosie met de tijd. Voor continue metaalverliesmetingen worden dikwijls elektrische weerstandproefstukken gebruikt. Deze bestaan uit een dunne draad of strip van een materiaal dat in het milieu wordt geplaatst. Wanneer corrosie optreedt neemt de elektrische weerstand, die continu kan worden gemeten, door de diktevermindering toe. Omdat de weerstand ook een functie van de temperatuur is gebruikt men dikwijls twee parallel geplaatste, identieke proefstukken waarvan er één wordt blootgesteld aan het agressieve milieu en beide op dezelfde temperatuur worden gehouden. Door het verschil in weerstand continu te meten wordt de meting niet door temperatuurveranderingen gestoord en kan zeer gevoelig alleen de mate van corrosie worden bepaald. In een recente variant van deze methode wordt de verandering van de inductieve weerstand van een corroderend monster gemeten. De massa daarvan verhoogt de intensiteit het magnetische veld in een spoel, wat op zijn beurt de inductieve weerstand daarvan verhoogt. De gevoeligheid wordt hierdoor sterk vergroot, het signaal van deze methode is 100 tot 2500 maal zo groot als van een normaal weerstandsproefstuk [13]. Bovendien is de temperatuurgevoeligheid een factor 10 kleiner, zodat temperatuurcorrectie veel eenvoudiger is. De weerstandmethode heeft verder als voordeel dat zij niet afhankelijk is van het geleidingsvermogen van de oplossing en dat bijvoorbeeld erosie (mits die gelijkmatig optreedt) ook wordt gemeten. Een veel gebruikte3 elektrochemische methode is die van de meting van de polarisatieweerstand. Figuur 7.2.1 toont een meetsonde met verwisselbare elektroden dat voor dit type metingen kan worden gebruikt.
Figuur 7.2.2. Drie elektrode meetsonde, met verwisselbare elektroden voor polarisatieweerstandsmetingen (Foto ter beschikking gesteld door Landré Intechmij BV).
Tenslotte kunnen continue analyses worden uitgevoerd van het milieu, bijvoorbeeld door meting van het ijzergehalte, van de pH of van het zuurstofgehalte. Ook kan

Meting van corrosie-eigenschappen van materialen
120
bijvoorbeeld in de gasruimte van een tank of reactievat het waterstofgehalte van het daarin aanwezige gas worden gemeten in gevallen waar corrosie onder waterstofontwikkeling kan worden verwacht. Voor een uitvoeriger bespreking van de verschillende methoden, hun toepassingsgebieden en dergelijke meer zie bijvoorbeeld [14].
7.3 Normen en standaardvoorschriften
De verschillende instellingen die zich met standaardisatie en normalisatie bezighouden hebben ook veel standaardvoorschriften voor het uitvoeren van allerlei corrosieproeven gepubliceerd. Een uitvoerig overzicht hiervan is gegeven in de Corrosiewijzer van het NCC [15]. Twee typische voorbeelden uitgegeven door de British Standard Institution op dit gebied zijn: BS 3745 "Method for the evaluation of results of accelerated corrosion tests on metallic coatings" en BS 2011 "Environmental testing tests". In deze gevallen gaat het meer om het bepalen van de effectiviteit van corrosiebescherming dan om het meten van de corrosiesnelheid van een bepaald materiaal als zodanig. Zeer uitgebreid zijn, ook op het gebied van corrosie en corrosiebestrijding, de activiteiten van de American Society for Testing and Materials (ASTM). Enkele typische voorbeelden van door de ASTM uitgegeven voorschriften op verschillende gebieden zijn: ASTM-G36 "Practice for performing stress corrosion test in boiling magnesium chloride solution"; ASTM-B287 "Acetic acid salt spray (fog) testing"; ASTM-G31 "Practice for laboratory immersion corrosion testing of metals"; ASTM-G59 "Practice for conducting potentiodynamic polarization resistance measure-ments"; ASTM-G4 "Recommended practice for conducting plant corrosion tests"; ASTM-G16 "Recommended practice for applying statistics to the analysis of corro-sion data". Vooral de laatste twee zijn van belang als voorbeelden van voorschriften die nuttig zijn bij het opzetten van een proevenprogramma omdat een goed gebruik hiervan het mogelijk maakt bij dezelfde inspanning en kosten meer en vooral betrouwbaarder resultaten te verkrijgen. Door de Deutscher Normen Ausschusz (DNA) is onder andere uitgegeven de norm DIN 50905 "Korrosion der Metalle" die uit 4 uitvoerige delen bestaat. Hiervoor geldt hetzelfde als hierboven gezegd ten aanzien van de laatste twee voorbeelden van ASTM-voorschriften. Ook de International Standards Organisation (ISO) heeft een groot aantal normen op dit gebied uitgegeven, bijvoorbeeld: ISO 8565 "Metals and alloys – Atmospheric corrosion testing. General requirements for field tests" en ISO 7539 "Corrosion of metals and alloys – Stress corrosion testing". Deze laatste bestaat uit 8 delen die ingaan op vele aspecten van het testen van materialen op gevoeligheid voor spanningscorrosie. Ook de activiteiten van de National Association of Corrosion Engineers (NACE) mogen zeker niet onvermeld blijven. Voorbeelden zijn TM-01-69 "Laboratory test-ing of metals for the process industries" en TM-02-74 "Dynamic corrosion testing of metals in high temperature water". Alle door de NACE uitgegeven standaarden zijn in één band verzameld [16].

121
In dit kader moet wel op de beperkingen van veel standaardvoorschriften worden gewezen: in volledig nieuwe situaties zijn ze slechts ten dele of vaak helemaal niet bruikbaar. De bedoeling van die voorschriften is juist om daar waar al een zekere overeenstemming bestaat, de mogelijkheid te scheppen goed vergelijkbare proeven te doen in verschillende laboratoria. Maar ze zijn zeker niet in de eerste plaats bedoeld of geschikt om nieuwe materialen te onderzoeken of materialen voor nieuwe toepassingen te selecteren. Bovendien moet men steeds bedenken dat het gedrag, zoals dat bepaald is aan de hand van proefstukken, niet met het praktijkgedrag behoeft overeen te komen. In ieder geval is het nodig om de verschillende toestanden waarin het metaal wordt gebruikt: gelast, koud en/of warm vervormd, geslepen, enzovoort, allemaal door geschikte proefstukken te onderzoeken. Het is duidelijk onvoldoende en zeer riskant, om alleen op grond van het gedrag van een materiaal in leveringstoestand conclusies over de bruikbaarheid te trekken.
7.4 Opgaven
1. In een bepaald type corrosiemeetapparaat wordt een proefstuk gebruikt dat bestaat uit een roestvast stalen draad van 0.5 mm diameter en 20 cm lengte. De corrosiviteit van het medium wordt dan gecontroleerd door continu de weerstand van de draad te meten. Wanneer de specifieke weerstand van het roestvaste staal 80 × 10-6 Ohm.cm is wat is dan de verandering van de weerstand per 24 uur bij een corrosiesnelheid van 0.25 mm/jaar?
2. Voor welke corrosievormen is controle van de snelheid mogelijk met behulp van lineaire polarisatie of weerstandsmetingen? Leg uit waarom dat volgens u zo is en bespreek in het bijzonder waarom deze methoden bij andere vormen van corrosie niet kunnen worden toegepast.
3. Een warmtewisselaar bestaat uit 175 koperen pijpen met een inwendige diameter van 3 cm en een lengte van 4 m (totaal oppervlak 70 m2 , totaal volume 0.5 m3) waardoor water stroomt met een snelheid van 1 m/s. Bij de uitgang van de warmtewisselaar wordt het kopergehalte bepaald. Wanneer het kopergehalte in het binnenkomende water te verwaarlozen en dat bij het verlaten van de warmtewisselaar 15 mg/l is, hoe groot is dan de corrosiesnelheid van de pijpen?
7.5 Literatuur
1. Corrosion Data Survey – Metals Section, ed. by .L. Graver, 6th ed., Houston, NACE (1985).
2. ASM Handbook, Vol 13: Corrosion, Materials Park, Ohio, ASM International (1987).

Meting van corrosie-eigenschappen van materialen
122
3. Dechema Werkstoff Tabelle, (losbladig, wordt regelmatig aangevuld en verbeterd),
Frankfurt am M., Dechema. 4. P.A. Schweitzer, Corrosion Resistance Tables, 4th ed., 3 volumes, New York, Marcel
Dekker (1995). 5. Handbook on corrosion testing and evaluation, ed. by W.H. Ailor, New York, John
Wiley (1971). 6. Techniques for corrosion measurement, ed. by A. Bronson, G. Warren, Houston, NACE
(1992). 7. R. Baboian, Corrosion tests and standards – Application and interpretation,
Philadelphia, ASTM (1995). 8. Application of accelerated corrosien tests to service life prediction of materials, ed. by
G.A. Cragnolino and N. Sridhar, Philadelphia, ASTM (1994) 9. F. Mansfeld, The polarization technique for measuring corrosion currents, in:
Adv.Corrosion Science and Technology, vol. VI, New York, Plenum Press (1976) 10. Electrochemical Impedance – Analysis and interpretation, ed. by J.R. Scully, D.C.
Silverman, and W.M. Koenig, Philadelphia, ASTM (1990) 11. Electrochemical Techniques for Corrosion, ed. byR. Baboian, Houston, NACE (1977) 12. A.J. Bard and L.R. Faulkner, Electrochemical Methods, Fundamentals and Applications,
New York, John Wiley (1980) 13. A.F. Denzine, M.S. Reading, A new, rapid corrosion rate measurement technique for all
process environments, Paper 97287 presented at Symposium Corrosion Measurement Technology, NACE Corrosion/97
14. Controlling Corrosieon 6: Monitoring, Department of Industry, HMSO (1977). 15. Corrosiewijzer, Informatiegids voor de nederlandse Industrie, Bilthoven, NCC (1996) p.
32 – 36. 16. NACE Book of Standards, Houston, NACE.

8
Voorkomen en bestrijden van elektrochemische corrosie


125
8.1 Inleiding
In de voorafgaande hoofdstukken is speciaal aandacht besteed aan hoe elektrochemische corrosie optreedt, waarbij zowel de uiterlijke verschijningsvorm als de meer fundamentele mechanismen zijn besproken. Op een aantal punten werd ook aandacht geschonken aan de mogelijkheden om de besproken corrosie te voorkomen of te bestrijden. Het doel van dit hoofdstuk is om een overzicht te geven van de verschillende methoden die kunnen worden toegepast om corrosie te voorkomen en te bestrijden. Daarbij wordt ook gelet op de factoren die van belang zijn bij de keuze tussen de verschillende methoden. Om corrosie te voorkomen of te bestrijden, kan uit een groot aantal mogelijkheden worden gekozen, zoals: − materiaalkeuze − wijziging van het milieu of van de samenstelling daarvan − wijziging van omstandigheden (temperatuur, stromingssnelheid, enzovoort) − ingrijpen in reacties (vooral met elektrochemische methoden) − scheiden van materiaal en milieu door een deklaag − aanpassen van ontwerp, bewerking en constructie. In wezen moet aan al deze punten tijdens het ontwerpstadium aandacht worden geschonken., waarbij bedacht dient te worden dat ze bovendien nog onderling afhankelijk zijn. Hierop komen we in de volgende paragrafen uitvoeriger terug. Sommige van deze punten spelen ook een belangrijke rol bij het onderhoud [1]. In ieder geval is het om te beginnen nodig dat bij onderhoud ook aandacht wordt geschonken aan de bij het ontwerp gekozen corrosiebestrijdingsmethoden. Op zijn minst dient daarbij te worden nagegaan in hoeverre die methoden nog goed functioneren en moet, indien dat niet (meer) het geval is, worden bekeken hoe verdere aantasting door toepassing van andere maatregelen kan worden voorkomen.
8.2 Corrosiebestrijding als onderdeel van het ontwerpproces
Het grote belang van het voorkomen van corrosie betekent dat hieraan al in het ontwerpstadium aandacht moet worden besteed [2, 3, 4]. In Figuur 8.2.1 is een schema gegeven van het ontwerpproces, voorzover dat met corrosiebestrijding te maken heeft. Hoewel de vormgeving, waarop in §8.7 wordt ingegaan, zeer belangrijk is, is dit zeker niet het enige waarop moet worden gelet. In hoofdstuk 7 is al afzonderlijk ingegaan op de mogelijkheden om de corrosiegegevens te verkrijgen die nodig zijn om tot goede beslissingen en keuzen te komen. Aan de andere onderdelen van het schema wordt in dit hoofdstuk aandacht geschonken. De methoden van corrosiebestrijding die in de voorafgaande hoofdstukken aan de orde kwamen hebben vooral te maken met verandering van het materiaal òf met verandering van het milieu of de omstandigheden. Deze punten worden verder uitgewerkt voor wat betreft wijziging van het milieu in §8.3 en voor wat betreft

Voorkomen en bestrijden van elektrochemische corrosie
126
materiaalkeuze in §8.4. Kathodische en anodische bescherming komen aan de orde in §8.5. Zoals hierboven al aangegeven is een volgende belangrijke mogelijkheid om corrosie te voorkomen of bestrijden het aanbrengen van een deklaag. De belangrijkste soorten deklagen en een aantal aspecten verbonden aan het aanbrengen daarvan worden besproken in §8.6. We wijzen er hier nog speciaal op dat de in het schema getoonde samenhang en het gelijktijdig in beschouwing nemen van de verschillende aspecten essentieel is om te komen tot een, uit corrosieoogpunt, goed ontwerp. Hoewel dit vanzelfsprekend lijkt, is in de praktijk helaas maar al te vaak te zien dat pas aan de corrosiebestrijding wordt gedacht op het moment dat het ontwerp eigenlijk al klaar is. In zo'n geval is het dan vrijwel nooit meer mogelijk de optimale methode van corrosiebestrijding toe te passen. Dat kan alleen wanneer de corrosiebestrijding een integraal onderdeel is van het gehele ontwerpproces [2, 5, 6].
Figuur 8.2.1. Ontwerpschema met aantal aspecten van corrosiebestrijding, -preventie en onderhoud (Met toestemming overgenomen uit [1]).
Verder is ook de in het schema aangegeven terugkoppeling vanuit gebruik en onderhoud essentieel [1, 6]. Alleen op die manier kan rekening worden gehouden met de achtergronden van de in het ontwerpproces genomen beslissingen en kan anderzijds de verkregen ervaring bij toekomstige ontwerpen worden toegepast. Zowel de corrosie zelf als de methoden die worden toegepast om die te voorkomen of bestrijden leiden tot verhoogde kosten. Het belangrijkste doel van corrosiebestrijding is juist het zoeken naar een methode om deze kosten te

127
minimaliseren! Daarom wordt in §8.8 op enkele economische aspecten van corrosiebestrijding ingegaan. Bij het gehele ontwerpproces moeten ook de economische aspecten mee in beschouwing worden genomen. Van groot belang hierbij is dat men niet alleen op de aanschaffing (= investering) let, maar tevens op de gebruiks- en onderhoudskosten. Figuur 8.2.2 geeft hiervan een schematische weergave. Bij korte levensduur kan een lage investering gekoppeld aan veel onderhoud voordelig zijn, maar bij langere levensduur wordt meestal een hogere investering gekoppeld aan lagere onderhoudskosten gunstiger. Gewaakt moet worden tegen "overdesign" waar zo'n hoge investering wordt gekozen dat de totale kosten onnodig hoog zijn.
Figuur 8.2.2. Corrosiekosten als functie van de levensduur (Met toestemming overgenomen uit [1].
8.3 Corrosiebestrijding door waterbehandeling
In een aantal gevallen is het mogelijk corrosie te bestrijden door aanpassing van het milieu. Dit betreft in het bijzonder waterige systeem, zoals koelcircuits, verwarmingsinstallaties, ketelinstallaties, enzovoort. Voor een algemene behandeling hiervan zie [7]. Twee voorbeelden worden hier kort besproken.
8.3.1 Ontluchten.
Omdat in neutrale oplossing de corrosiereactie alleen kan verlopen indien voldoende zuurstof aanwezig is voor de kathodische reactie kan corrosie worden bestreden door het water te ontluchten. De corrosiesnelheid neemt hierbij evenredig met het zuurstofgehalte af. In §5.4 is besproken dat ontluchten fysisch kan gebeuren door verwarmen, eventueel onder verlaagde druk, anderzijds chemisch door stoffen toe te voegen, zoals

Voorkomen en bestrijden van elektrochemische corrosie
128
hydrazine (N2H4) of sulfieten (bijv. Na2SO3), die met zuurstof reageren. Het hangt van het type installatie en van de omstandigheden af welk van deze methoden het beste is. In gesloten systemen, zoals bijvoorbeeld verwarmingsinstallaties, is afzonderlijk ontluchten dikwijls niet nodig, hetgeen in §5.4 al is besproken. Dit komt omdat de betrekkelijk geringe hoeveelheid zuurstof die aanwezig is in leidingwater snel is opgebruikt door de kathodische zuurstofreductie waarbij slechts een geringe hoeveelheid van het metaal wordt omgezet. Benadrukt moet hierbij worden dat de installatie goed dicht is en geen regelmatige toevoeging van vers water nodig is. Daarmee wordt namelijk ook steeds opnieuw zuurstof toegevoegd hetgeen dan toch tot ernstige corrosie kan leiden.
8.3.2 Inhibitoren en passivatoren.
In §5.3 is de toepassing van inhibitoren in zure oplossingen besproken, waarbij speciaal is ingegaan op de toepassing van beitsremmen voor de bescherming van staal bij het beitsen ter verwijdering van oxidehuiden. De elektrochemische reactie van de metaalcorrosie wordt door dit type verbindingen wèl vertraagd, met name omdat de waterstofontwikkelingsreactie door de adsorptie van de inhibitor op het metaaloppervlak wordt belemmerd. De zuiver chemische reactie van het oplossen van oxiden wordt door de inhibitor niet beïnvloed. In §5.7 is besproken dat, bijvoorbeeld in koelwatersystemen, soms inhibitoren of passivatoren kunnen worden toegevoegd die de corrosie sterk verminderen. Deze maken òf zelf dat het metaal passief wordt, zoals natriumnitriet en natriumchromaat, òf ze maken het mogelijk dat opgeloste zuurstof voor passivering zorgt, bijvoorbeeld natriumbenzoaat. Daar is tevens aangegeven dat het noodzakelijk is steeds te controleren wordt of nog voldoende inhibitor in het systeem aanwezig is. Wanneer de concentratie namelijk te laag wordt bestaat het gevaar voor versnelde corrosie! Inhibitoren worden ook veel gebruikt in olie-water emulsies zoals die bij de verspanende bewerking (draaien, frezen, boren, enzovoort) van metalen worden gebruikt. Dit is vooral belangrijk omdat de bij deze bewerkingen nieuw ontstane metaaloppervlakken extreem gevoelig zijn voor corrosie omdat zich daarop nog geen natuurlijk beschermende oxidehuid heeft kunnen vormen. Een andere belangrijke toepassing is in pijpleidingen voor aardolieproducten. Hoewel deze zèlf meestal niet agressief zijn bevatten zij dikwijls water dat, vooral bij lage temperatuur, afgescheiden kan worden. Ten gevolge van de hoge oplosbaarheid van zuurstof in vele aardolieproducten (ongeveer 6 maal zo groot als in water) kan dit aanleiding geven tot sterke roestvorming. Om dit te voorkomen wordt continu een geconcentreerde (5 tot 30%) waterige oplossing van natriumnitriet geïnjecteerd, samen met een basische verbinding omdat nitriet beneden pH ongeveer 6 niet meer werkzaam is. Voor een meer diepgaande behandeling van toepassingen en eigenschappen van inhibitoren en passivatoren wordt verwezen naar de literatuur [8, 9].

129
8.4 Materiaalsoorten en materiaalkeuze
Het is natuurlijk onmogelijk om hier een volledig overzicht te geven van alle materialen en hun corrosie-eigenschappen. We beperken ons daarom in deze paragraaf tot enkele van de meest belangrijke en veel toegepaste materiaalsoorten en de bespreking van hun karakteristieke eigenschappen en toepassingen. Voor enkele andere punten verwijzen we naar de literatuur [10, 11]
8.4.1 On- en laaggelegeerde staalsoorten
Ongelegeerd staal is in eerste instantie, door zijn lage prijs, het economisch meest aantrekkelijke materiaal is voor veel constructies. Daarbij kan worden aangetekend, dat ook de grote beschikbaarheid en de constantheid van het prijsniveau van groot belang zijn. Ook de gunstige mechanische eigenschappen en de goede verwerkbaarheid en lasbaarheid spelen een belangrijke rol. Vanuit het oogpunt van corrosieweerstand onder atmosferische omstandigheden is de keuze van ongelegeerd staal veelal een slechte, zoals iedereen uit ervaring weet: slecht onderhouden stalen tuinstoelen, fietsen, en niet goed gelakte en/of getectyleerde auto's zijn slechts enige uit de grote reeks van voorbeelden, die in dit verband kunnen worden genoemd. De oorzaak hiervan is, dat ijzer een nogal onedel element is, hetgeen wil zeggen, dat het gemakkelijk reageert met waterstofionen (dus met zuren), maar ook met de zuurstofmoleculen, die in water opgelost zijn. In het laatste geval worden corrosieproducten gevormd, die wij kennen als roest. Dit is een ingewikkeld mengsel van nogal complexe volumineuze oxidatieproducten, die veel water bevatten. Het gevolg is, dat ongelegeerd staal onder atmosferische omstandigheden "doorroest", zelfs wanneer het droog is, omdat de met roest bedekte plekken veel langer nat blijven dan de niet aangetaste delen. Zoals in §6.2 is besproken vormen sommige laaggelegeerde staalsoorten hierop een uitzondering. Men treft ze tegenwoordig in ongeverfde toestand aan in vele parken en vijvers, als constructiemateriaal van kunstvoorwerpen, maar ook worden zij toegepast bij de bouw van bruggen, als beplating van gevels, voor schoorstenen van verwarmingsinstallaties, enzovoort. Zij bevatten meestal enkele tienden van procenten koper, nikkel en/of chroom. Als gevolg daarvan wordt onder atmosferische omstandigheden een compacte, goed hechtende roestlaag gevormd, die bovendien veel minder vocht opneemt dan die op ongelegeerd staal, waardoor verdere aantasting wordt geremd. Deze materialen zijn bekend als "weathering steels" of weerbestendige staalsoorten. Zoals al genoemd heeft het gebruik van deze staalsoorten ook duidelijke voordelen wanneer wèl wordt geschilderd. Aan één kant onbeschermd ongelegeerd staal wordt daarentegen in veel gebouwen toegepast in de vorm van buizen en radiatoren als onderdeel van centrale-verwar-mingsystemen. De binnenkant van deze onderdelen is niet geverfd, noch op een andere wijze tegen corrosie beschermd. Desondanks functioneren dergelijke systemen vaak vele jaren achtereen zonder noemenswaardige storingen. De reden hiervan is (zie ook §5.4), dat men in dit geval te maken heeft met een gesloten systeem: de zuurstof in het daarin aanwezige water reageert snel met het ijzer aan de binnenkant onder de vorming van een zwart, dun, compact laagje ijzeroxide, waarna

Voorkomen en bestrijden van elektrochemische corrosie
130
de aantasting stopt bij gebrek aan meer zuurstof. Dit aan de binnenzijde gevormde laagje bestaat uit een ijzeroxide dat bekend staat als magnetiet of ijzerhamerslag (Fe3O4). Als de verwarmingsinstallatie lek is, moet op gezette tijden de hoeveelheid water worden aangevuld, en het corrosieproces zet zich dan voort door de regelma-tige verse aanvoer van water met daarin opgeloste zuurstof en ook wel van zuurstof via het lek. Aan de binnenkant van de leidingen vormt zich nu geleidelijk een volu-mineuze, bruine roestlaag, en na verloop van tijd zal het verwarmingssysteem hier en daar "doorroesten".
8.4.2 Roestvaste staalsoorten
Door het legeren van ijzer met ten minste 12-14% chroom ontstaan legeringen met de verzamelnaam roestvast staal, die in vele milieus een goede corrosieweerstand vertonen door de vorming van een gesloten, chroomrijke oxidelaag. Dat wil dus zeggen dat het materiaal zich passief gedraagt. Met nadruk dient gesteld te worden dat dit gunstige gedrag alleen bereikt wordt bij een oppervlak dat volledig met zo'n oxidelaag is bedekt of, met andere woorden, volledig is gepassiveerd. Roestvaste staalsoorten zijn, afhankelijk van het milieu, gevoelig voor de in Hoofdstuk 6 besproken lokale corrosievormen, als putvormige-, spleet-, interkristallijne- en spanningscorrosie. Wanneer deze verschijnselen optreden, is dit te wijten aan het zeer lokaal doorbreken van de passieve laag. Afhankelijk van de verdere samenstelling kunnen we binnen de groep van de roest-vaste staalsoorten een vijftal typen onderscheiden, namelijk: de martensitische, de ferritische, de austenitische, de duplex- en de precipitatiehardende soorten (zie Tabel 8.4.1).
8.4.2.1 Martensitisch roestvast staal
Het samenstellingsgebied hiervan is 12-18% chroom, 0-2% nikkel, 0-2% molybdeen, 0.1-0.5% koolstof. Het koolstofgehalte bepaalt vooral de maximaal bereikbare sterkte en hardheid. Deze soorten zijn magnetisch en kunnen gehard worden, hetgeen wil zeggen dat door warmtebehandeling hun mechanische eigenschappen kunnen worden beïnvloed. Met de hardbaarheid van deze soorten hangt samen dat ze moeilijk of niet lasbaar zijn: de daarbij optredende warmteinvloeden veroorzaken ongewenste sterkte-eigenschappen. De sterkte en slijtvastheid zijn hoger dan die van de ferritische soorten, de corrosievastheid echter wat minder. Deze kan door toevoegen van nikkel en molybdeen worden verbeterd voor toepassing in maritiem milieu. Martensitische roestvaste staalsoorten worden veel toegepast ter vervaardiging van: snijwerktuigen (zowel huishoudelijk als industrieel), gereedschappen (tangen, schroevendraaiers), schoepen voor gas- en stoomturbines. Bij dit laatste speelt ook de geringe gevoeligheid voor erosie-corrosie een rol. In het algemeen worden deze staalsoorten gebruikt voor de vervaardiging van onderdelen en niet voor con-structies.

131
8.4.2.2 Ferritisch roestvast staal
Het samenstellingsgebied hiervan is 12-28% chroom en minder dan 0.25% koolstof. Door het hoge chroom-gehalte in combinatie met het relatief lage koolstof-gehalte is geen harding mogelijk: bij verhitting blijft tot aan het smeltpunt de ferrietstructuur behouden. De ferritische staalsoorten zijn magnetisch en vrij kerfge voelig. Dit type is in het algemeen niet erg goed lasbaar, vooral omdat in de las en in de naaste omgeving daarvan sterke korrelgroei optreedt die tot slechte mechanische eigenschappen leidt. Als toch moet worden gelast, gebruikt men meestal austenitisch toevoegmateriaal. De oxidatiebestendigheid is uitstekend, zeker bij legeringen met meer dan 20% chroom. De corrosiebestendigheid in de atmosfeer is goed. Men treft ferritisch roestvast staal aan voor: versiering en andere onderdelen (zoals knalpotten) van auto's, als constructiemateriaal voor salpeterzuurfabrieken, in de voedingsmiddelenindustrie en voor scheepsschroeven.
8.4.2.3 Austenitisch roestvast staal
Dit is wel het meest gebruikte type. Het samenstellingsgebied hiervan is: 18-25% chroom, 8-20% nikkel, 0-3% molybdeen, eventueel titaan of niobium als stabilisator en minder dan 0.15% koolstof, bij de meeste typen nog aanzienlijk lager. Het meest bekende staal wordt vaak aangeduid met "18/8" staal, ook wel: staal 304. Bij austenitisch roestvast staal is warmtebehandeling ter verkrijging van andere mechanische eigenschappen evenmin mogelijk als bij ferritisch, nu omdat bij alle temperaturen tot aan het smeltpunt de austenietstructuur behouden blijft. Ze zijn niet magnetisch, goed lasbaar (indien maatregelen worden genomen om lasbederf te voorkomen) en zeer ductiel. De corrosiebestendigheid is in veel gevallen beter dan die van de ferritische en de martensitische soorten, vooral in de atmosfeer en in organische zuren. Wel zijn sommige soorten gevoelig voor put- en spleetcorrosie. In chloridehoudend milieu kan spanningscorrosie optreden, een verschijnsel dat de ferritische en de martensitische soorten in veel mindere mate vertonen. Austenitisch roestvast staal wordt zeer veel toegepast als constructiemateriaal voor apparaten in de chemische, voedingsmiddelen- en farmaceutische industrie. Verder wordt het op grote schaal gebruikt voor laboratorium en huishoudelijke apparatuur, als versiering voor (dure) auto's en in de architectuur, bijvoorbeeld voor gevelelementen, stootplaten op deuren, hekken en leuningen, enzovoort.
8.4.2.4 Duplex staalsoorten.
Deze staalsoorten, ook wel austenitisch-ferritische staalsoorten genoemd, hebben een gemengde austeniet-ferriet structuur. De belangrijkste legeringselementen zijn chroom (20-30%) en nikkel (4-10%). In verband met hun geringere gevoeligheid voor spanningscorrosie dan de volledig austenitische roestvast staalsoorten worden zij vaak toegepast in warme, chloride-houdende
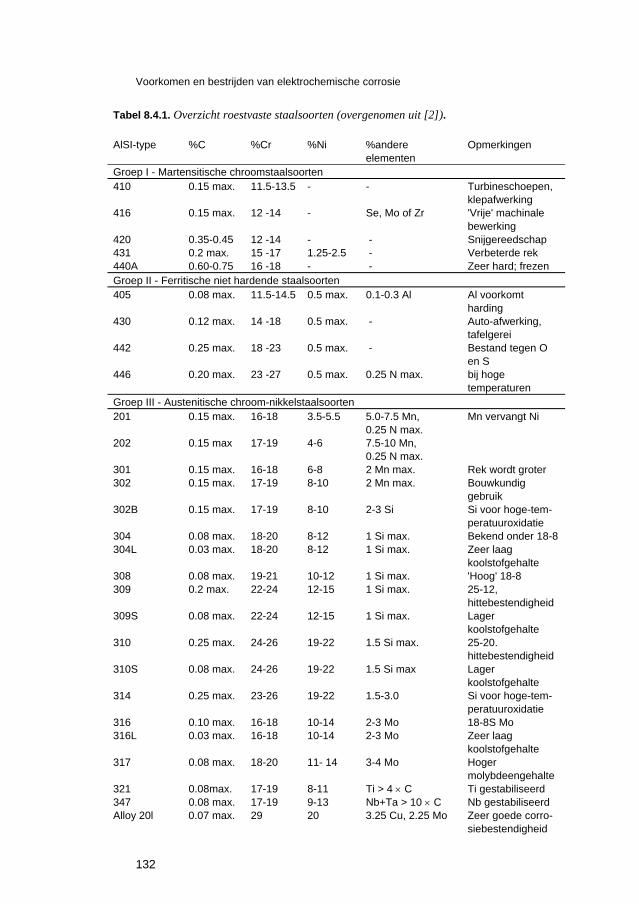
Voorkomen en bestrijden van elektrochemische corrosie
132
Tabel 8.4.1. Overzicht roestvaste staalsoorten (overgenomen uit [2]).
AlSI-type %C %Cr %Ni %andere elementen
Opmerkingen
Groep I - Martensitische chroomstaalsoorten 410 0.15 max. 11.5-13.5 - - Turbineschoepen,
klepafwerking 416 0.15 max. 12 -14 - Se, Mo of Zr 'Vrije' machinale
bewerking 420 0.35-0.45 12 -14 - - Snijgereedschap 431 0.2 max. 15 -17 1.25-2.5 - Verbeterde rek 440A 0.60-0.75 16 -18 - - Zeer hard; frezen Groep II - Ferritische niet hardende staalsoorten 405 0.08 max. 11.5-14.5 0.5 max. 0.1-0.3 Al Al voorkomt
harding 430 0.12 max. 14 -18 0.5 max. - Auto-afwerking,
tafelgerei 442 0.25 max. 18 -23 0.5 max. - Bestand tegen O
en S 446 0.20 max. 23 -27 0.5 max. 0.25 N max. bij hoge
temperaturen Groep III - Austenitische chroom-nikkelstaalsoorten 201 0.15 max. 16-18 3.5-5.5 5.0-7.5 Mn,
0.25 N max. Mn vervangt Ni
202 0.15 max 17-19 4-6 7.5-10 Mn, 0.25 N max.
301 0.15 max. 16-18 6-8 2 Mn max. Rek wordt groter 302 0.15 max. 17-19 8-10 2 Mn max. Bouwkundig
gebruik 302B 0.15 max. 17-19 8-10 2-3 Si Si voor hoge-tem-
peratuuroxidatie 304 0.08 max. 18-20 8-12 1 Si max. Bekend onder 18-8 304L 0.03 max. 18-20 8-12 1 Si max. Zeer laag
koolstofgehalte 308 0.08 max. 19-21 10-12 1 Si max. 'Hoog' 18-8 309 0.2 max. 22-24 12-15 1 Si max. 25-12,
hittebestendigheid 309S 0.08 max. 22-24 12-15 1 Si max. Lager
koolstofgehalte 310 0.25 max. 24-26 19-22 1.5 Si max. 25-20.
hittebestendigheid 310S 0.08 max. 24-26 19-22 1.5 Si max Lager
koolstofgehalte 314 0.25 max. 23-26 19-22 1.5-3.0 Si voor hoge-tem-
peratuuroxidatie 316 0.10 max. 16-18 10-14 2-3 Mo 18-8S Mo 316L 0.03 max. 16-18 10-14 2-3 Mo Zeer laag
koolstofgehalte 317 0.08 max. 18-20 11- 14 3-4 Mo Hoger
molybdeengehalte 321 0.08max. 17-19 8-11 Ti > 4 × C Ti gestabiliseerd 347 0.08 max. 17-19 9-13 Nb+Ta > 10 × C Nb gestabiliseerd Alloy 20l 0.07 max. 29 20 3.25 Cu, 2.25 Mo Zeer goede corro-
siebestendigheid

133
oplossingen, waar een groot risico bestaat voor spanningscorrosie van austenitische materialen. Zij worden verder gekenmerkt door een hoge vloeigrens, een grote taaiheid en een goede gietbaarheid. De lasbaarheid is goed, mits men de lasinstructies nauwkeurig navolgt.
8.4.2.5 Precipitatie-hardend roestvast staal.
Hieronder verstaat men staalsoorten met ongeveer dezelfde samenstelling als de austenitische, maar waaraan bovendien kleine hoeveelheden aluminium, koper, of fosfor zijn toegevoegd. Geschikte warmtebehandeling veroorzaakt de uitscheiding van een zeer fijn verdeelde, nieuwe fase, waardoor een grote sterkte en hardheid kan worden verkregen in combinatie met een redelijke ductiliteit en corrosiebestendigheid, waarbij de laatste eigenschap overeen komt met die van het austenitische type 304. Door de hoge sterkte-gewichtverhouding worden deze precipitatiehardende stalen nogal eens toegepast in de lucht- en ruimtevaart.
8.4.3 Koper en koperlegeringen
Koper is een relatief edel metaal en kan daarom in sterk zure waterige oplossing niet corroderen onder waterstofontwikkeling (zie ook §3.6). Zodra echter zuurstof of andere oxidatiemiddelen aanwezig zijn die een hogere potentiaal ten opzichte van de oplossing hebben dan koper, bestaat de mogelijkheid van corrosie wèl. In het algemeen zijn koper en koperlegeringen redelijk tot goed bestendig in niet al te sterk oxiderende milieus, mits geen complexvormers aanwezig zijn. Ammoniak- en cyanideoplossingen bijvoorbeeld zijn meestal zeer agressief. Een bezwaar van koper zelf is dat het zwak en zacht is en daardoor erg gevoelig voor erosie-corrosie (de maximaal toelaatbare stromingssnelheid in koperen pijpen is van de orde van 0.5 m.sec-1). De oudst bekende legeringen van koper (namelijk met tin: brons en met zink: messing) zijn in dit opzicht beter. Door hun grotere sterkte zijn ze minder gevoelig voor erosie-corrosie. Brons is meestal zeer goed corrosiebestendig en zelfs iets minder gevoelig voor oxiderende componenten dan koper zelf. Daarentegen ver-toont het relatief goedkope en goed bewerkbare messing, vooral bij toenemend zinkgehalte, meestal een geringere weerstand tegen algemene corrosie. Messing is bovendien gevoelig voor interkristallijne spanningscorrosie in een ammoniak-houdende omgeving en voor ontzinking. Speciaal in koelers en condensors wordt wel gebruik gemaakt van pijpen van het zogenaamde marinemessing (70% koper, 29% zink, 1% Sn) en nog meer van alumi-niummessing (76% koper, 22% zink, 2% aluminium). Vooral dank zij hun behoor-lijke weerstand tegen erosie-corrosie zijn daarbij grotere watersnelheden toelaatbaar (maximaal van de orde van 1.5 m.sec-1). Wanneer nog zwaardere eisen worden gesteld, met name indien bij hoge temperatuur en/of stroomsnelheden moet worden gewerkt, gaat men meestal over op de kopernikkellegeringen zoals cupronikkel 90-10 of 70-30 (samenstelling: koper met 10, respectievelijk 30 gewichts% nikkel). Vooral als hierin ook nog 1-3% ijzer en mangaan aanwezig zijn, is de weerstand tegen erosie-corrosie hoog en kunnen zeer hoge snelheden worden toegepast (van de

Voorkomen en bestrijden van elektrochemische corrosie
134
orde van 2 à 2.5 m.sec-1 ). Bovendien zijn deze legeringen bestand tegen spannings-corrosie in ammoniakale milieus. Het 90-10 materiaal (en in mindere mate 70-30) is gevoelig voor sulfide dat in vervuild zeewater voor kan komen, maar dat onder anaërobe omstandigheden ook in stilstaand, relatief schoon zeewater kan worden ge-vormd. Voor speciale toepassingen gebruikt men wel aluminium-koperlegeringen die 7 tot 12% aluminium bevatten plus enige procenten ijzer, mangaan en soms nikkel, vaak als aluminiumbrons aangeduid. Vooral in sterk oxiderende milieus vertonen deze legeringen een uitstekende corrosieweerstand door de vorming van een aluminiumrijke beschermende oxidelaag. In zwavelzuurkoelers, waarin door de aanwezigheid van chloride in het koelwater roestvast staal minder geschikt is, ten gevolge van het gevaar voor putvormige en/of spanningscorrosie wordt dit soort legeringen vaak toegepast. De goede bestendigheid van koperlegeringen tegen chloridehoudende milieus, met de goede weerstand tegen aangroei van plantaardige en dierlijke organismen (fouling), is vaak de reden van hun uitgebreide toepassing in zeewater en andere natuurlijke watersoorten.
8.4.4 Aluminium en aluminiumlegeringen
Aluminium is een onedel metaal, dat dank zij de vorming van een beschermende oxidehuid, goed bestand is tegen atmosferische corrosie. Door kunstmatige oxidatie langs chemische of elektrochemische weg (anodiseren of eloxeren) kan de oxidelaag verder versterkt worden. Het metaal is niet bestand tegen chemicaliën die de oxidehuid aantasten, zoals sterke zuren en basen. Het is in zuivere vorm daarentegen wèl bestand tegen zeewater, neutrale en oxiderende zoutoplossingen, salpeterzuur en de meeste organische zuren. Vooral om de mechanische eigenschappen te verbeteren, is een aantal legeringen ontwikkeld. Behalve in het geval van aluminium- magnesium legeringen gaat dit echter meestal gepaard met een achteruitgang van de corrosiebestendigheid. Vaak zal het toch gewenst zijn constructies/onderdelen te beschermen tegen corrosie. Het aanbrengen van een kunstmatige oxidelaag biedt goede mogelijkheden. Bij een aantal legeringen kunnen in dit verband problemen optreden bij aanwezig-heid van bepaalde legeringselementen. Ook toepassing van organische deklagen is mogelijk, evenals kathodische bescherming. In het laatste geval is een nauwkeurige potentiaalcontrole noodzakelijk om overbescherming te voorkomen.
8.4.5 Nikkel en nikkellegeringen
De corrosieweerstand van nikkel is belangrijk groter dan die van staal, maar het heeft een veel geringere sterkte en is erg duur. De oxidatiesnelheid van nikkel bij hogere temperatuur is kleiner dan dat van staal. Een zwak punt is hierbij echter de sterke aantasting door reducerende, zwavelhoudende gassen. Hierbij treedt een soort interkristallijne aantasting op, doordat een laagsmeltend mengsel van nikkel en nikkelsulfide langs de korrelgrenzen het materiaal binnendringt, waardoor de samenhang tussen de kristallieten verloren gaat.

135
Het metaal is bestendig in vochtige lucht en in water, maar in zeewater vertoont het neiging tot putvorming. Het wordt niet of nauwelijks aangetast door basische oplossingen, ammoniak en de meeste organische zuren. In corrosiebestendige nikkellegeringen worden voornamelijk koper, chroom, ijzer en molybdeen als legeringselementen toegepast. Voor speciale toepassingen zijn de zogenaamde "superlegeringen" ontwikkeld die een hoge oxidatiebestendigheid paren aan een ook bij hoge temperaturen nog acceptabele tot goede (kruip)sterkte.
8.4.6 Niet-metallische materialen
Van deze grote en zeer gevarieerde groep materialen zullen we er slechts twee, en dan nog zeer beknopt, bespreken: kunststoffen en beton.
8.4.6.1 Kunststoffen
Ten opzichte van metalen zijn kunststoffen minder sterk en slechts bruikbaar tot relatief lage temperaturen. Door toevoegen van geschikte vulstoffen en in meerdere mate door toepassing van een wapening, zoals in de glasvezel-versterkte kunststoffen, kan de mechanische sterkte echter aanzienlijk worden opgevoerd. Daartegenover staat, dat de corrosiebestendigheid in allerlei media uitstekend is. Be-paalde vormen van inwerking zijn echter wel mogelijk. Kunststoffen zijn ruimtelijk gezien veel minder dicht dan metalen. Het is daarom voor kleine gas- en vloeistof-moleculen relatief gemakkelijk om tussen de kunststofketens naar binnen te dringen. Wanneer hierbij alleen zwelling optreedt, waarbij de afstanden tussen de ketens wat groter worden, is er sprake van een omkeerbaar verlopend fysisch proces. Bij hogere temperaturen kan het zwellen echter leiden tot het verbreken van bindingen tussen de ketens, hetgeen zover kan gaan dat zij geheel oplossen. Er zijn daarnaast chemische inwerkingen mogelijk die onomkeerbaar verlopen, bijvoorbeeld ketenafbraak, verandering van de binding tussen de ketens, verandering van de samenstelling van de ketens, enzovoort. Dergelijke inwerkingen zijn meestal van grotere invloed op de materiaaleigenschappen dan de zuiver fysische. Indien er sprake is van een langzame degradatie van de eigenschappen, spreekt men wel van veroudering. Dit kan bijvoorbeeld optreden door verdampen van oplosmiddel- of weekmakermoleculen die normaal in de kunststof aanwezig zijn, of door bestraling met ultraviolette straling. Ook dient rekening te worden gehouden met het mogelijk optreden van scheuren onder invloed van een mechanische belasting. Het kan gebeuren dat een kunststof die in een bepaald milieu bestendig is, toch onder invloed van een trekspanning gaat scheuren (spanningscorrosie). Dit is een fysisch-chemische inwerking, waarbij door de trekspanning en de invloed van het milieu de bindingskrachten tussen de ketens zover worden verlaagd dat scheurvorming en -voortplanting kunnen plaats vinden. Bij de groep thermoplasten, dat zijn kunststoffen waarvan de sterkte met toenemende temperatuur sterk afneemt, zijn die met een uitsluitend uit koolstofatomen bestaande hoofdketen gevoeliger voor deze vorm van spanningscorrosie dan wanneer de hoofdketen uit verschillende elementen is opgebouwd.

Voorkomen en bestrijden van elektrochemische corrosie
136
Nog beter bestand is de groep thermoharders, dat zijn kunststoffen waar de verharding juist plaats vindt door verwarming. Bij versterkte kunststoffen dient te worden gelet op grensvlakverschijnselen tussen de kunststofmoleculen en het wapeningsmateriaal, omdat aan die grensvlakken fouten aanwezig kunnen zijn die aanleiding geven tot scheurvorming.
8.4.6.2 Beton
Beton is een kunstmatig soort steen, gevormd uit een mengsel van cement, water en een vulstof, bijvoorbeeld grind. De voor aantasting gevoelige component is de cement. De bestanddelen hiervan zijn kalk (CaO), aluminiumoxide (Al2O3 ) en kiezelzuur (SiO2 x H2O). Bij de uitharding van beton vormen zich hydratatieproducten, die bepalend zijn voor de sterkte van het beton, en calcium hydroxide (Ca(OH)2) kristallen (circa 20vol %). Beton is daardoor basisch waardoor de pH hoog is: circa 12.5. Verlaging van de pH kan optreden door inwerking van kooldioxide (CO2) op de kalkkristallen, waarbij kalksteen (CaCO3) ontstaat (carbonatatie). Ook zwaveldioxide (SO2) kan een dergelijk effect hebben. Voor alle vormen van aantasting is beton beter bestendig naarmate het een dichtere structuur bezit. Deze wordt vooral bepaald door de soort en de hoeveelheid cement en de verhouding water/cement, die bij aanmaak wordt toegepast. Ook vulstoffen (grind) worden voor dit doel toegevoegd omdat zij voor een verdere verdichting van de structuur zorgen. Verder worden wel oppervlaktebehandelingen uitgevoerd of beschermende lagen aangebracht. Bij gewapend beton is het wapeningsstaal onder gunstige condities passief, dankzij de hoge pH. Het wapeningsstaal zal echter worden aangetast, indien in de gecarbo-natiseerde zone die veel minder basisch is, water en/of chloride tot op het staaloppervlak kunnen doordringen. De roest die dan ontstaat, heeft een circa 9× zo groot volume als het staal waaruit het is ontstaan. Dit heeft tot gevolg, dat het beton van het staal wordt gedrukt en gaat scheuren. Behalve de reeds genoemde beschermingsmaatregelen zijn in dit verband belangrijk: − het vermijden van chloriden bij de aanmaak van beton, in het bijzonder door
geen chloridehoudende hardingversnellers te gebruiken; − een voldoende dikke afdichtende laag beton op het wapeningsstaal aan de
buitenkant; − in bijzondere gevallen een voorbehandeling van het betonstaal, zoals thermisch
verzinken of toepassen van een andere deklaag; − een juiste nabehandeling van het beton om scheurvorming aan het oppervlak te
voorkomen.
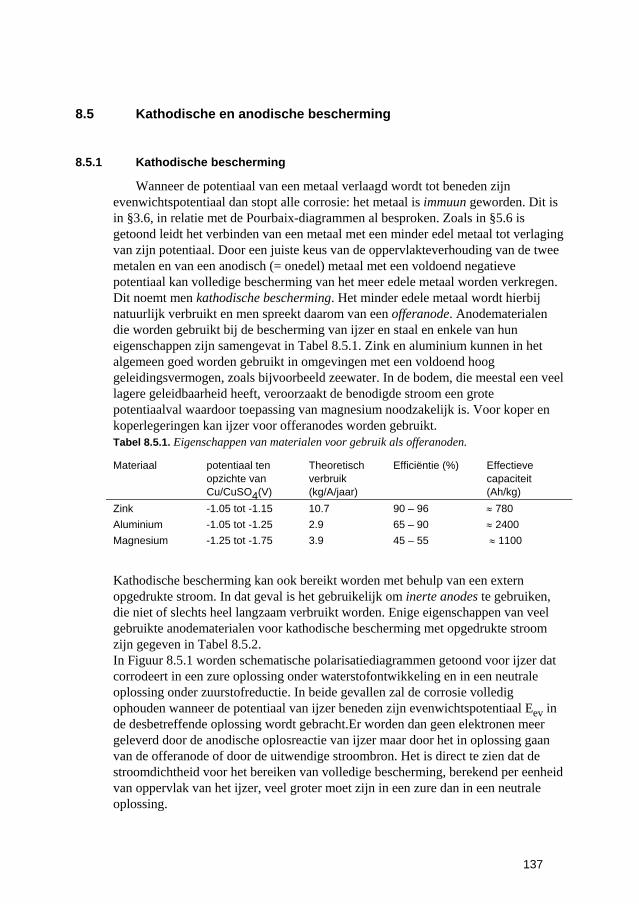
137
8.5 Kathodische en anodische bescherming
8.5.1 Kathodische bescherming
Wanneer de potentiaal van een metaal verlaagd wordt tot beneden zijn evenwichtspotentiaal dan stopt alle corrosie: het metaal is immuun geworden. Dit is in §3.6, in relatie met de Pourbaix-diagrammen al besproken. Zoals in §5.6 is getoond leidt het verbinden van een metaal met een minder edel metaal tot verlaging van zijn potentiaal. Door een juiste keus van de oppervlakteverhouding van de twee metalen en van een anodisch (= onedel) metaal met een voldoend negatieve potentiaal kan volledige bescherming van het meer edele metaal worden verkregen. Dit noemt men kathodische bescherming. Het minder edele metaal wordt hierbij natuurlijk verbruikt en men spreekt daarom van een offeranode. Anodematerialen die worden gebruikt bij de bescherming van ijzer en staal en enkele van hun eigenschappen zijn samengevat in Tabel 8.5.1. Zink en aluminium kunnen in het algemeen goed worden gebruikt in omgevingen met een voldoend hoog geleidingsvermogen, zoals bijvoorbeeld zeewater. In de bodem, die meestal een veel lagere geleidbaarheid heeft, veroorzaakt de benodigde stroom een grote potentiaalval waardoor toepassing van magnesium noodzakelijk is. Voor koper en koperlegeringen kan ijzer voor offeranodes worden gebruikt. Tabel 8.5.1. Eigenschappen van materialen voor gebruik als offeranoden.
Materiaal potentiaal ten opzichte van Cu/CuSO4(V)
Theoretisch verbruik (kg/A/jaar)
Efficiëntie (%) Effectieve capaciteit (Ah/kg)
Zink -1.05 tot -1.15 10.7 90 – 96 ≈ 780 Aluminium -1.05 tot -1.25 2.9 65 – 90 ≈ 2400 Magnesium -1.25 tot -1.75 3.9 45 – 55 ≈ 1100
Kathodische bescherming kan ook bereikt worden met behulp van een extern opgedrukte stroom. In dat geval is het gebruikelijk om inerte anodes te gebruiken, die niet of slechts heel langzaam verbruikt worden. Enige eigenschappen van veel gebruikte anodematerialen voor kathodische bescherming met opgedrukte stroom zijn gegeven in Tabel 8.5.2. In Figuur 8.5.1 worden schematische polarisatiediagrammen getoond voor ijzer dat corrodeert in een zure oplossing onder waterstofontwikkeling en in een neutrale oplossing onder zuurstofreductie. In beide gevallen zal de corrosie volledig ophouden wanneer de potentiaal van ijzer beneden zijn evenwichtspotentiaal Eev in de desbetreffende oplossing wordt gebracht.Er worden dan geen elektronen meer geleverd door de anodische oplosreactie van ijzer maar door het in oplossing gaan van de offeranode of door de uitwendige stroombron. Het is direct te zien dat de stroomdichtheid voor het bereiken van volledige bescherming, berekend per eenheid van oppervlak van het ijzer, veel groter moet zijn in een zure dan in een neutrale oplossing.
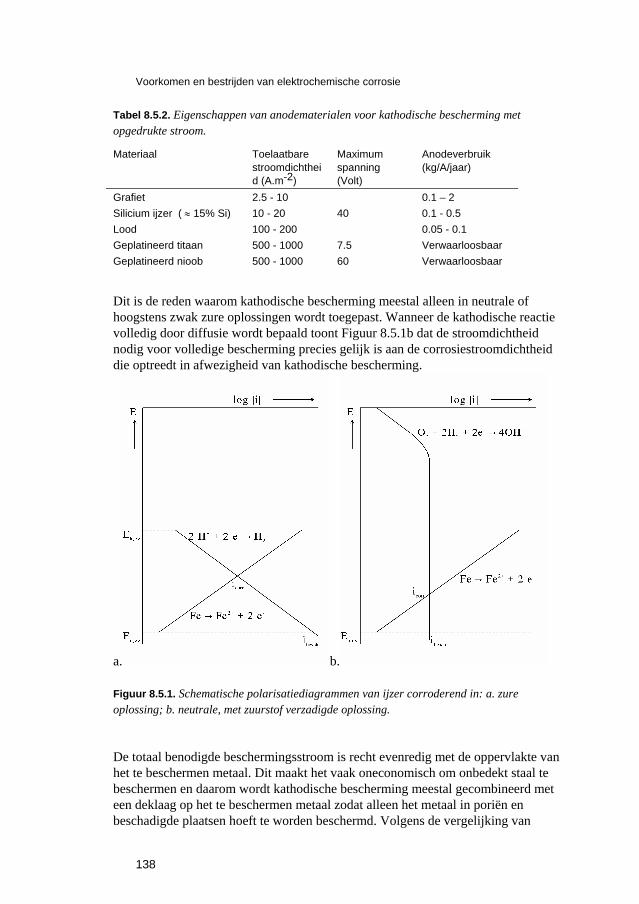
Voorkomen en bestrijden van elektrochemische corrosie
138
Tabel 8.5.2. Eigenschappen van anodematerialen voor kathodische bescherming met opgedrukte stroom.
Materiaal Toelaatbare stroomdichtheid (A.m-2)
Maximum spanning (Volt)
Anodeverbruik (kg/A/jaar)
Grafiet 2.5 - 10 0.1 – 2 Silicium ijzer ( ≈ 15% Si) 10 - 20 40 0.1 - 0.5 Lood 100 - 200 0.05 - 0.1 Geplatineerd titaan 500 - 1000 7.5 Verwaarloosbaar Geplatineerd nioob 500 - 1000 60 Verwaarloosbaar
Dit is de reden waarom kathodische bescherming meestal alleen in neutrale of hoogstens zwak zure oplossingen wordt toegepast. Wanneer de kathodische reactie volledig door diffusie wordt bepaald toont Figuur 8.5.1b dat de stroomdichtheid nodig voor volledige bescherming precies gelijk is aan de corrosiestroomdichtheid die optreedt in afwezigheid van kathodische bescherming.
a. b.
Figuur 8.5.1. Schematische polarisatiediagrammen van ijzer corroderend in: a. zure oplossing; b. neutrale, met zuurstof verzadigde oplossing.
De totaal benodigde beschermingsstroom is recht evenredig met de oppervlakte van het te beschermen metaal. Dit maakt het vaak oneconomisch om onbedekt staal te beschermen en daarom wordt kathodische bescherming meestal gecombineerd met een deklaag op het te beschermen metaal zodat alleen het metaal in poriën en beschadigde plaatsen hoeft te worden beschermd. Volgens de vergelijking van

139
Nernst zou in een omgeving die volledig vrij is van ijzer een oneindige potentiaal nodig zijn om kathodische bescherming te bereiken. In de praktijk zal in het begin altijd enige corrosie optreden met als gevolg een lage, maar eindige concentratie van ijzerionen in het milieu. Hierdoor lijkt het erop dat het eigenlijk onmogelijk zou zijn om een bruikbaar criterium te vinden wanneer voldoende kathodische bescherming is verkregen omdat de Eev afhangt van de feitelijke metaalionenconcentratie. Maar, samen met de kinetische beschouwing in samenhang met Figuur 8.5.1, is een benaderd, praktisch criterium dat, wanneer de metaalionenconcentratie beneden de 10-5 tot 10-7 mol.l-1 wordt gehouden de corrosie verwaarloosbaar is omdat in dat geval het wegdiffunderen van de metaalionen ook te verwaarlozen is. Uit de vergelijking van Nernst volgt dat de beschermingspotentiaal dan ongeveer 0.18 V beneden de standaardpotentiaal moet liggen. Voor ijzer is dit – 0.62 V ten opzichte van de standaard waterstofelektrode, dan wel – 0.84 V ten opzichte van de Ag/AgCl-elektrode of – 0.94 V ten opzichte van de Cu/CuSO4-elektrode die beide in de praktijk veel worden toegepast omdat ze veel gemakkelijker te hanteren zijn dan de waterstofelektrode. In de praktijk blijkt dit criterium iets te streng zijn: in de meeste gevallen is –0.75 V ten opzichte van de Ag/AgCl-elektrode of – 0.85 V ten opzichte van de Cu/CuSO4-elektrode voldoende. Alleen in aanwezigheid van sulfaatreducerende bacteriën moet het strengere criterium wèl worden toegepast. Tabel 8.5.3 geeft de benodigde stroomdichtheid voor bescherming van staal onder verschillende omstandigheden.
Tabel 8.5.3. Stroomdichtheden (gebaseerd op het totale oppervlak) nodig voor kathodische bescherming van staal
Object Omgeving Deklaag Leeftijd Typische stroomdichtheid (mA.m-2)
Staal zoet water Geen - 40 - 70 Staal zoet water Goed - 0.1 - 1 Staal stilstaand
zeewater Geen voor vorming
van afzetting 500 - 1000
Staal stilstaand zeewater
Geen na vorming van afzetting
50 - 100
Staal stromend zeewater
Geen na vorming van afzetting
200 - 300
Staal zeewater Goed - 1 - 10 Pijpleiding bodem Asfalt circa 10 jaar 0.2 - 10 Pijpleiding bodem Plastic - 0.001 - 0.1
In de praktijk wordt kathodische bescherming zeer veel toegepast. Voorbeelden van op deze manier beschermde objecten zijn: ondergrondse tanks en pijpleidingen, koelwatersystemen, waterkasten van condensors, verdampers, schepen, haven- en offshore-installaties, enzovoort.

Voorkomen en bestrijden van elektrochemische corrosie
140
a. b.
Figuur 8.5.2. Praktische voorbeelden van kathodische bescherming: a. inwendige bescher-ming van warmwatertank met offeranode; b. uitwendige bescherming van ondergrondse tank of leiding met opgedrukte stroom (Met toestemming overgenomen uit [2]). In Figuur 8.5.2 is een schematische tekening gegeven van twee praktische gevallen van kathodische bescherming.
Figuur 8.5.3. Onderdeel van offshore boorplatform met een aluminium offeranode (Foto beschikbaar gesteld door VanderVelde Protection, Zoetermeer).
In Figuur 8.5.3 wordt een onderdeel van een offshore boorplatform getoond waarop een aluminium offeranode is aangebracht. Figuur 8.5.4a toont een titanium anode voor kathodische bescherming met opgedrukte stroom. Het kunststofscherm waarop de anode is geplaatst is bedoeld om plaatselijke overbescherming te voorkomen. Door de kleinere weerstand zou de stroom naar het te beschermen object dichtbij de anode zo groot kunnen worden dat waterstofontwikkeling op gaat treden. Wanneer kathodische bescherming en deklagen gecombineerd worden toegepast, wat zoals hierboven reeds vermeld vaak gebeurt, kan dit bovendien leiden tot beschadiging van de deklaag.

141
Figuur 8.5.4b toont een foto van een Ag/AgCl-elektrode voor potentiaalcontrole bij kathodische bescherming zoals die in de praktijk, speciaal bij installaties van kathodische bescherming met opgedrukte stroom wordt gebruikt. De keuze tussen kathodische bescherming met offeranodes dan wel met opgedrukte stroom wordt vooral bepaald door economische factoren samenhangende met noodzakelijke investering, onderhouds- en vervangingskosten, productieverlies bij vervanging van anodes, enzovoort. Verder is de mogelijkheid van automatische con-trole en bewaking bij toepassen van een opgedrukte stroom vaak een belangrijke overweging. Het hier gegeven overzicht van kathodische bescherming is betrekkelijk beknopt en voor een uitvoeriger behandeling van theorie en toepassing van kathodische bescherming wordt naar de literatuur verwezen [12, 13, 14, 15, 16].
a b
Figuur 8.5.4. a. Titaananode met iridium mengoxidecoating op kunststofscherm voor opgedrukte stroom; b. Ag/AgCl-elektrode voor potentiaalcontrole bij kathodische bescherming (Foto's beschikbaar gesteld door VanderVelde Protection, Zoetermeer).
8.5.2 Anodische bescherming
In §5.7 is bij de bespreking van passiviteit gewezen op de mogelijkheid van anodische bescherming [12, 17] door een metaal door potentiaalverhoging in de passieve toestand te brengen. Anodische bescherming is, in tegenstelling tot kathodische, niet zonder gevaar omdat door verkeerde toepassing de corrosie juist kan worden versneld. Anodische bescherming kan dan ook alleen toegepast worden

Voorkomen en bestrijden van elektrochemische corrosie
142
wanneer men een goed inzicht heeft zowel in het beschermingsmechanisme als in de specifieke omstandigheden waar deze eventueel moet worden toegepast. Tot nog toe wordt anodische bescherming nog slechts op bescheiden schaal toegepast. Voorbeelden zijn: staal in geconcentreerd zwavelzuur en nitraatoplossingen (kunstmest) en roestvast staal in verdund salpeterzuur. Er zijn echter veel meer potentiële toepassingen die zeker het overwegen waard zijn.
8.6 Corrosiebestrijding met deklagen
8.6.1 Algemeen
In veel gevallen kan geen materiaal worden gevonden dat aan alle hieraan door constructie en toepassing gestelde eisen tegelijk kan voldoen. Heel vaak is het dan wèl mogelijk een materiaal te bedekken met een beschermende laag waardoor het contact tussen het eigenlijke constructiemateriaal en de omgeving wordt verbroken, zodat geen (of vrijwel geen) corrosie meer op kan treden. De toepassing van beschermende deklagen is dan ook één van de meest gebruikte methoden om corrosie te bestrijden [18, 19, 20] en een overzicht over de omvang van het gebruik is gegeven in bijlage 11.14. In die bijlage zijn ook (internet)adressen gegeven van organisaties die zich hiermee bezighouden en waarbij aanvullende gegevens kunnen worden verkregen. Een eerste vereiste wanneer deklagen worden toegepast is dat het te bedekken oppervlak goed en volledig kan worden gereinigd, want de kwaliteit en duurzaamheid van iedere deklaag staat of valt met de zorgvuldige voorbereiding van het materiaaloppervlak. Voor de vormgeving betekent dit in de eerste plaats dat alle te bedekken oppervlakken goed toegankelijk moeten zijn, zowel voor de reiniging als voor de bedekking zelf. In een aantal gevallen betekent dit dat de deklaag al moet worden aangebracht voordat samenbouw van de onderdelen, waaruit een constructie bestaat, geschiedt. Daarbij moet er dan natuurlijk wèl voor worden gezorgd dat de al aangebrachte deklaag niet beschadigd wordt bij de montage. Een apart geval wordt gevormd door deklagen die door dompelen worden aangebracht. Indien deze zowel inwendig als uitwendig moeten worden aangebracht geeft dit speciale eisen die aan de vormgeving moeten worden gesteld hetgeen in §8.7 verder wordt besproken. De verschillende in de praktijk toegepaste deklagen kunnen in drie hoofdgroepen worden verdeeld: de metallische, de anorganische-niet-metallische en de organische deklagen. Voor het ontwerp is van belang te bedenken hoe deze deklagen zich in de loop van de tijd gedragen en in hoeverre reparatie en/of onderhoud mogelijk is. Zo is bijvoorbeeld bij metallische deklagen, die meestal òf uit de smelt òf elektrolytisch worden aangebracht, ter plekke reparatie of onderhoud vrijwel nooit mogelijk. Wanneer voorzien wordt dat herstel van bijvoorbeeld beschadigde plaatsen toch zou moeten gebeuren dient de constructie dusdanig te zijn dat de betreffende onderdelen

143
zonder al te veel moeite of kosten kunnen worden gedemonteerd. Deze kunnen dan fabrieksmatig worden voorzien van een nieuwe deklaag.
Figuur 8.6.1. Goede en slechte toegankelijkheid voor schilderen (Met toestemming overgenomen uit [1]).
Bij deklagen die wèl ter plaatse kunnen worden onderhouden of gerepareerd, zoals de meeste verfsoorten, moet natuurlijk, zoals hierboven ook al genoemd is, de constructie zodanig zijn dat daarvoor het gehele oppervlak goed toegankelijk is. Dit laatste geldt zowel voor het bij het onderhoud nodige reinigen en voorbehandelen van het oppervlak, als voor het eigenlijke aanbrengen van de laag. Figuur 8.6.1 geeft hiervoor enkele voorbeelden, waarbij òf gezorgd moet worden voor voldoende afstand tussen de constructiedelen òf spleten volledig moeten worden vermeden. Wanneer een ventilatiekoker moet worden aangebracht, bijvoorbeeld op een schip, dan is de constructie van Figuur 8.6.2a ongeschikt omdat daarbij noch de wanden, noch de koker voor onderhoud bereikbaar zijn. In zo'n geval is de constructie waarbij de ventilatiekoker integraal in de wanden is opgenomen, zoals weergegeven in Figuur 8.6.2b veel beter.

Voorkomen en bestrijden van elektrochemische corrosie
144
Figuur 8.6.2. Ventilatiekoker in ruimte met metalen wanden.
8.6.2 Voorbehandeling
De kans dat goede deklagen worden verkregen hangt in hoge mate af van de voorbehandeling van het te bedekken oppervlak. Zou bijvoorbeeld niet van een goede hechting sprake zijn door een onzorgvuldige voorbehandeling, dan kan een beschadiging van de laag de inleiding zijn van ernstige corrosie ten gevolge van aantasting onder de aangebrachte huid, waardoor deze door de sterk volumineuze corrosieproducten wordt weggedrukt. Een achtergebleven laagje vocht kan zelfs onder een zo goed mogelijk gesloten huid de corrosie gewoon doen doorgaan, zodat na enige tijd ook in dit geval de huid zal afspringen en de corrosie ongehinderd zijn gang kan gaan. Om roest-, wals- of giethuiden te verwijderen, kan men stralen met staalgrit, schuren, slijpen, borstelen of beitsen in sterk zuur. Grote oppervlakken, zoals scheepshuiden, kan men van oxiden ontdoen door vlamstralen. De oxiden springen ten gevolge van de verhitting van het oppervlak af door het verschil in uitzetting tussen oxide en metaal. Vet en olie kan men met behulp van organische oplosmiddelen of met alkalische ontvettingsbaden verwijderen, maar worden natuurlijk bij vlamstralen ook verwijderd door verbranding. Na drogen dient men het schoon gemaakte oppervlak niet meer met de hand of zelfs maar een vinger aan te raken, daar de op de menselijke huid aanwezige stoffen veelal aanleiding geven tot een soms in eerste instantie niet zichtbare, maar daarom niet minder ernstige aantasting, waardoor de aan te brengen laag binnen korte tijd volledig wordt ondermijnd wegens slechte hechting! Ook moet de bedekkingslaag zo snel mogelijk na de voorbehandeling aangebracht worden, omdat anders, bijvoorbeeld door een regenbui of ten gevolge van een hogere relatieve vochtigheid en/of de afzetting van stofdeeltjes op het schone oppervlak, alle inspanning teniet wordt gedaan.
8.6.3 Metallische deklagen
Hierbij kunnen we twee gevallen onderscheiden: − de deklaag is edeler dan het te beschermen materiaal; − de deklaag is onedeler dan het te beschermen materiaal.

145
In het eerste geval zijn poriën in de laag uit corrosieoogpunt gevaarlijk bij volledige bevochtiging: we krijgen dan contactcorrosie met een klein anodisch oppervlak en een zeer groot kathodisch oppervlak. Voorbeelden hiervan zijn tin op ijzer en koper op ijzer. In het tweede geval zijn poriën of beschadigingen ongevaarlijk: het grote onedele oppervlak wordt slechts weinig versneld aangetast. Het onderliggende metaal wordt hierbij kathodisch beschermd door de deklaag. Het belangrijkste voorbeeld hiervan is zink op ijzer (staal). Voor het aanbrengen van de deklaag bestaat een aantal methoden waarvan de belangrijkste zijn: a. "cladding" of bekleden. Hierbij wordt een plaat van het beschermende metaal
aangebracht op een plaat van het te beschermen metaal. Een voorbeeld hiervan is "clad steel", dat bestaat uit on- of laaggelegeerd staal met daarop aangebracht een betrekkelijke dunne laag roestvast staal (of soms monel, een legering bestaande uit 70% nikkel en 30% koper). "Cladden" krijgt pas zin als het te vervangen materiaal een bepaalde dikte overschrijdt. Deze grens ligt bij staal bij ongeveer 6 mm. Bij dunnere platen is het goedkoper om massief roestvast staal te gebruiken, dan om een dunne plaat ongelegeerd staal te nemen en deze te bedekken met een dun laagje roestvast staal. Verwant hieraan zijn de zogenaamde duplexpijpen die bestaan uit twee in elkaar gewalste dunwandige pijpen die vooral in warmtewisselaars worden toegepast waar geen materiaal gevonden kan worden dat tegen zowel het "inwendige" als tegen het "uitwendige" milieu bestand is. Voorbeelden van dergelijke combinaties zijn: messing-staal, koper-roestvast staal, monel-roestvast staal, enzovoort. Een nadeel is de hoge prijs, zodat ze alleen in zeer bepaalde omstandigheden worden toegepast.
b. "Hot dipping" of dompelen. Deze techniek wordt vrijwel alleen gebruikt voor het aanbrengen van zink (thermisch verzinken, zie voor een uitvoerige bespreking [21, 22]) of tin op ongelegeerd staal. De laag wordt aangebracht door het staal onder te dompelen in een bad van gesmolten zink of tin. Dit kan zowel continu van band of plaat, als discontinu voor losse onderdelen gebeuren. Vooral bij discontinu thermisch verzinken is het mogelijk om in één keer grote constructiedelen, zoals stalen spanten, trappen, enzovoort in hun geheel in- en uitwendig te bedekken. (Bij heel grote afmetingen is vaak een tweezijdige onderdompeling, de zogenaamde "double-dip" mogelijk).
c. Galvanisch bedekken. Hierbij wordt de deklaag aangebracht door elektrolytisch neerslaan uit een oplossing van een zout van het metaal. Dit proces wordt onder meer toegepast om metalen te bedekken met koper, nikkel, zink, chroom, cadmium, zilver, goud, rhodium, enzovoort. Blik, dat is met tin bedekt staal, wordt tegenwoordig meer vervaardigd door galvanische bedekking dan door "hot dipping" omdat galvanisch bedekken gevolgd door een warmtebehandeling leidt tot een dunner, gelijkmatiger en gladder tinlaagje.
d. Opspuiten. Hierbij wordt een draad van het beschermende metaal in een vlam gebracht. Het gesmolten metaal wordt druppelsgewijs door de vlam meegenomen en nog vloeibaar op het te beschermen metaal neergeslagen. Voor

Voorkomen en bestrijden van elektrochemische corrosie
146
een goede hechting is een schoon, maar vooral ook ruw oppervlak nodig. Deze methode is vooral belangrijk, omdat het hiermee mogelijk is in constructies plaatselijk beschermende lagen aan te brengen. Opspuiten is mogelijk met metalen die niet al te hoge smeltpunten hebben en wordt daarom onder andere toegepast bij aluminium en zink. In dat geval dient men met een ondermaat zuurstof te werken om oxidatie van het op te spuiten aluminium, respectievelijk zink te voorkomen. Dit is verder ook een mogelijkheid om beschadigde of gecorrodeerde metallische deklagen ter plaatse te repareren.
In het bijzonder voor de bescherming van ongelegeerd staal worden metallische deklagen veel toegepast. De meest gebruikte zijn tin, zink, cadmium en nikkel. Blik wordt voornamelijk gebruikt als verpakkingsmateriaal voor levensmiddelen, genotmiddelen, verven en lakken, olie, enzovoort. Zink en cadmium worden speciaal gebruikt voor bescherming tegen atmosferische corrosie. Een belangrijk voordeel van cadmium boven zink is dat het heel goed soldeerbaar is, vandaar dat het veel wordt gebruikt in elektronische apparatuur. Nadelen van cadmium zijn echter de giftigheid en de hoge prijs. Zink wordt in grote hoeveelheden gebruikt ter bescherming van kleine en grote staalconstructies zoals kranen, pijpen, bruggen, wapeningsstaal, hoogspanningsmasten, enzovoort. Een eerste voorbeeld van een verzinkte constructie, namelijk de staalconstructie van een fabriekshal, wordt getoond in Figuur 8.6.3.
Figuur 8.6.3. Een in aanbouw zijnde fabriekshal met een volledig verzinkte constructie (foto ter beschikking gesteld door de Stichting Doelmatig Verzinken).

147
Figuur 8.6.4. Thermisch verzinkte trap en leuningen van galerijwoningen (foto ter beschikking gesteld door de Stichting Doelmatig Verzinken).
Figuur 8.6.5. Balconplaten en hekken in thermisch verzinkte uitvoering (foto ter beschikking gesteld door de Stichting Doelmatig Verzinken).
Een tweede voorbeeld betreft trappen en leuningen van galerijwoningen, zoals te zien in Figuur 8.6.4. Het is hierbij van belang te bedenken dat de beschermingsduur direct samenhangt met de dikte van de zinklaag. Op kleinere onderdelen, bijvoorbeeld bouten en moeren wordt zink dikwijls galvanisch aangebracht in geringe laagdikte, tussen 5 en 12 μm . De levensduur daarvan in een agressieve atmosfeer is meestal niet meer dan 2 tot 3 jaar, in tegenstelling tot 25 tot 30 jaar

Voorkomen en bestrijden van elektrochemische corrosie
148
onder gelijke omstandigheden voor thermisch verzinkt staal met een zinklaagdikte van 60 tot 80 μm . Voor bevestiging van buitenconstructies moeten dus ook thermisch verzinkte bouten en moeren worden gebruikt om corrosie van de bevestigingen te voorkomen. Een voorbeeld van balconplaten en hekken aan een groot flatgebouw worden getoond in Figuur 8.6.5. Voor andere voorbeelden en verdere bespreking van eigenschappen en toepassingen van verzinkt staal zie ook referentie 22. Nikkellagen worden veel gebruikt ter bescherming van constructieonderdelen vooral tegen door lucht en water veroorzaakte corrosie. Nikkel is nogal duur en daarom wordt vaak eerst een laag koper aangebracht en daarna pas nikkel. Omdat nikkel in de atmosfeer, speciaal ten gevolge van de aanwezigheid van sporen zwavel-verbindingen, vaak geel tot lichtbruin wordt, brengt men meestal op het nikkel nog een dunne laag chroom aan. Aldus "verchroomt" men onderdelen van meubels, fietsen en (vooral vroeger) bumpers van auto's. Aangezien de koper, nikkel en chroom bevattende laag edel is ten opzichte van staal wordt de kwaliteit daarvan in eerste instantie bepaald door het aantal poriën dat erin voorkomt, en omdat het aantal daarvan meestal afneemt bij toenemende dikte, dus ook door de dikte.
8.6.4 Anorganische niet-metallische deklagen
Ook hier kunnen we twee typen onderscheiden: a. glas en email b. conversielagen. a. Glas en email zijn beide silicaten die in gesmolten of verweekte toestand op een
metaal, in de meeste gevallen staal, worden aangebracht. Zij zijn zeer goed bestand tegen bijna alle chemicaliën, met uitzondering van met name fluoriden en sterk alkalische oplossingen. Zij zijn ook erg hard, glad en weinig poreus, hetgeen vooral van belang is in verband met de hygiëne. Deze lagen zijn echter nogal bros en reparatie van beschadigingen is vrijwel onmogelijk. Desondanks worden deze deklagen op tamelijk ruime schaal toegepast in de voedingsmiddelen- en geneesmiddelenindustrie, in de huishouding en in de medische sector, waarbij met name ook hun goede bestendigheid tegen hoge temperaturen (kookgerei en sterilisatie) een grote rol speelt. Voorbeelden van toepassingen zijn: huishoudelijke machines en apparaten, baden en douchekuipen, wandpanelen, benzinepompen, wegwijzers en verkeerstekens, uitlaten en geluidsdempers, enzovoort. Een integraal geëmailleerd uitlaatspruitstuk dat bestand moet zijn tegen hoge

149
temperatuur wordt getoond in Figuur 8.6.6.
Figuur 8.6.6. Geëmailleerd uitlaatspruitstuk (foto ter beschikking gesteld door Ferro(Holland) B.V. Rotterdam).
Paneelplaten gebruikt voor het lichteiland Goeree worden getoond in Figuur 8.6.7. Hier is het vooral de blijvende goede zichtbaarheid door het behoud van kleur en glans die van doorslaggevend belang zijn.
Figuur 8.6.7. Lichtplatform Goeree met geëmailleerde platen die na meer dan 30 jaar nog volledig schoon zijn (foto ter beschikking gesteld door Ferro(Holland) B.V. Rotterdam)

Voorkomen en bestrijden van elektrochemische corrosie
150
Figuur 8.6.8. Zonnepanelen op woonhuizen (foto ter beschikking gesteld door Ferro(Holland) B.V. Rotterdam)
Ook bij het gebruik van zonne-energie wordt veel email toegepast, onder andere voor panelen (zie bijvoorbeeld Figuur 8.6.8), voor leidingwerk, 1 warmteopslagvaten, enzovoort.
b. Conversielagen. Deze ontstaan door de omzetting van een metaal aan het oppervlak in één of andere onoplosbare verbinding zoals fosfaat, oxide, enzovoort. Fosfateren is een zeer vaak toegepaste techniek, waarbij het staal in fosforzuur wordt gedompeld. Hierdoor wordt een dun, onoplosbaar laagje ijzerfosfaat gevormd, dat op het oppervlak achterblijft na het uit het bad halen van het staal dat een goede tijdelijke bescherming biedt tegen atmosferische corrosie. Nog belangrijker is echter dat de structuur van dit laagje zodanig is dat verflagen er beter op hechten dan op niet behandeld staal en dat bij beschadigde plekken minder snel onderroest wordt gevormd. Een ander type conversielaag wordt gevormd door het zogenaamde anodiseer-, ook wel eloxeerproces. Dit is vooral bekend voor aluminium en titaan. Op het metaal wordt daarbij een oxidelaag aangebracht door het anodisch te maken in een elektrochemische cel. Er wordt hierbij een dikke, honingraatachtige oxidestructuur gevormd bovenop een geheel gesloten dunne huid. Veel van die poriën kunnen worden gesloten door behandeling met heet water of stoom ("sealen" van de laag). Deze lagen geven een goede bescherming tegen atmosferische corrosie. Zij zijn bovendien tamelijk hard en worden daarom minder gemakkelijk beschadigd dan de van nature aanwezige, maar veel dunnere oxidelaag. Men kan in de poriën een kleurstof aanbrengen alvorens ze te sluiten. Dit doet men door dompeling in een kleurstofoplossing. Het resultaat treft men aan in vele keukens: de gekleurde deksels van aluminium pannen hebben op deze manier hun opvallende uiterlijk gekregen.
151
8.6.5 Organische deklagen
De belangrijkste organische deklaag is verf. Dit wordt meestal toegepast ter bescherming tegen atmosferische corrosie. Het is een heterogeen mengsel bestaande uit een vaste stof, het pigment, gesuspendeerd in een organische vloeistof: de drager of binder. Na aanbrengen op het te beschermen oppervlak moet de verflaag hard worden om de ondergrond blijvend te beschermen. Hiervoor bestaat een drietal mechanismen: a: Reactie van de verf met zuurstof uit de lucht. De zuurstof moet hierbij in de laag
diffunderen, hetgeen wil zeggen dat de laag een voldoende doorlaatbaarheid voor gassen en dus een zekere porositeit moet hebben. Een klassiek voorbeeld is lijnolieverf. Ter versnelling van de droging wordt vaak nog een oxidatiekatalysator (siccatief) toegevoegd.
b: Verdamping van het oplosmiddel. Ook in dit geval is een zekere porositeit nodig om het oplosmiddel te kunnen afstaan.
c: Harding door inwendige reactie. Hier is sprake van een polymerisatiereactie tussen twee componenten (de zogenaamde tweecomponenten verf) die kort voor het aanbrengen worden gemengd.
Meestal is sprake van een combinatie van twee van de drie genoemde mechanismen.
Tabel 8.6.1. Invloed van voorbehandeling op duurzaamheid van verfsystemen (Sheffield, Engeland)
Oppervlaktebehandeling 2 lagen loodmenie + 2 lagen ijzeroxide verf
2 lagen loodmenie
Onbeschadigde walshuid 8.2 jaar 3.0 jaar Verweerd en gestaalborsteld 2.3 1.2 Gebeitst 9.5 4.6 Gestaalstraald 10.4 6.3
Wat algemeen geldt, is ook van groot belang in het geval van verven: de wijze van voorbehandeling van het metaaloppervlak dient zeer grondig en zorgvuldig te zijn! In Tabel 8.6.1 zijn enkele gegevens samengevat die het belang tonen van de voorbehandeling van een te schilderen oppervlak. Duidelijk is dat men meestal beter een slecht verfsysteem op een goed voorbehandeld oppervlak kan aanbrengen dan omgekeerd! De voorbehandeling en het aanbrengen van de verf bepalen meestal het grootste deel van de kosten. Zo bedragen de kosten voor het ontroesten van staal ongeveer 40% van de totale kosten en kan het aanbrengen van de verf wel vijf maal duurder zijn dan de verf zelf. Bezuiniging op het verfsysteem is dus onverstandig, temeer omdat bij gebruik van een duurder systeem het aantal onderhoudsbeurten gedurende de standtijd kleiner is, waardoor op de totale kosten aanzienlijk kan worden bespaard. Om dezelfde reden wordt dikwijls gekozen voor gecombineerde bescherming: in het bijzonder verzinken plus schilderen, hetgeen een duplex-systeem wordt genoemd. Ook hiervan zijn de investeringskosten aanzienlijk hoger dan van een enkelvoudig

Voorkomen en bestrijden van elektrochemische corrosie
152
systeem. De onderhoudsperioden worden echter zoveel langer en de kosten van onderhoud zoveel lager, omdat voor de zinklaag slechts weinig of geen roestvorming optreedt in eventuele beschadigingen, dat dit op de duur toch voordeliger is (vergelijk de grafiek in Figuur 8.2.2). De beschermingsduur is namelijk minstens twee maal zo lang als de som van beide methoden afzonderlijk. Globaal geldt ook nog dat, hoe agressiever de omgeving is, des te eerder de gecombineerde bescherming economisch verantwoord is. Voor een uitvoerige bespreking van uitvoering en toepassing van duplex-systemen zie het boek van van Eijnsbergen [23]. Gewoonlijk bestaat een verfsysteem uit meerdere lagen die meestal verschillende pigmenten bevatten. Een meerlaagsysteem is om diverse redenen te prefereren boven één waarbij slechts een enkele verflaag wordt aangebracht: 1. er is dan minder kans op doorlopende poriën; 2. dikke lagen drogen vaak onvoldoende, hetgeen in het bijzonder geldt voor de
hierboven genoemde hardingsmechanismen a. en b.; 3. het is vaak onmogelijk om in één keer de gewenste dikte aan te brengen omdat
de nog vloeibare verf dan van het oppervlak afstroomt. Voor de eerste verflaag kiest men vaak een pigment dat ook een actieve corrosiebeschermende werking heeft. Bekend zijn loodmenie en zinkchromaat, die beide een passiverende werking hebben, en metallisch zink dat kathodische bescherming geeft. Dit betekent dat bij gebruik hiervan in beschadigde plaatsen minder roestvorming optreedt, waardoor de totale levensduur van het verfsysteem aanzienlijk wordt verlengd. Op verdere details van de beschermende werking van deze componenten gaan wij hier niet in (zie hiervoor de literatuur [24]).
Figuur 8.6.9. Plaatvormige pigmentdeeltjes in een verflaag.
Een speciale functie hebben pigmenten zoals ijzerglimmer en aluminium die in de vorm van dunne plaatjes in de verf aanwezig zijn. Deze rangschikken zich in de

153
verflaag ongeveer parallel aan het oppervlak, zoals schematisch aangegeven in Figuur 8.6.1. Omdat de polymere binder altijd een zekere doorlaatbaarheid voor water heeft kan onder de verflaag, wanneer nog resten agressieve bestanddelen op het metaaloppervlak zijn achtergebleven, toch corrosie gaan optreden. Door de in de figuur aangegeven rangschikking van de pigmentdeeltjes wordt deze waterdoorlaatbaarheid en daardoor ook de kans op corrosie sterk verminderd.
8.6.6 Tijdelijke corrosiebestrijdingsmiddelen
In veel gevallen moeten gefabriceerde onderdelen of volledige apparaten, motoren, enzovoort getransporteerd of opgeslagen worden alvorens in gebruik gesteld te worden. In het bijzonder bewerkte oppervlakken die essentieel zijn voor de goede werking, zoals assen, lagers, schroefdraad, en dergelijke meer, mogen tijdens opslag of transport niet corroderen. Van groot belang is dat een tijdelijk corrosiebestrijdingsmiddel gemakkelijk te verwijderen is bij de ingebruikstelling van het onderdeel of apparaat. Alle tijdelijke corrosiebestrijdingsmiddelen zijn organische verbindingen die vaak kunnen worden verwijderd door afspoelen met een geschikt oplosmiddel. In sommige gevallen is geen aparte behandeling nodig namelijk wanneer het tijdelijke corrosiebestrijdingsmiddel oplost in smeerolie. Deze kunnen bijvoorbeeld worden gebruikt voor de inwendige bescherming van verbrandingsmotoren die dan na opslag of transport zonder speciale maatregelen ineens in gebruik kunnen worden genomen. Tijdelijke corrosiebestrijdingsmiddelen bestaan in zeer verschillende consistenties. Aan de ene kant zijn er relatief dun vloeibare oliën die door spuiten of met de kwast kunnen worden aangebracht. Aan de andere kant zijn er vaste, vette tijdelijke corrosiebestrijdingsmiddelen die soms als zodanig kunnen worden aangebracht, maar vaker met de kwast of door spuiten worden opgebracht na oplossen in een oplosmiddel dat na het opbrengen verdampt. Tenslotte zijn er relatief harde, wasachtige vast tijdelijke corrosiebestrijdingsmiddelen die kunnen worden aangebracht door dompelen in de gesmolten stof. Bij het aanbrengen van tijdelijke corrosiebestrijdingsmiddelen is de oppervlaktevoorbehandeling soms niet zo kritisch als bij de toepassing van permanente deklagen. Één reden hiervoor is dat meestal alleen bewerkte oppervlakken daarmee worden behandeld zodat roest, walshuid en oxidelagen meestal afwezig zijn. Maar aan de andere kant zorgt schoon- en droogmaken van het oppervlak voor een betere en langer durende bescherming, in het bijzonder door het verwijderen van agressieve stoffen van het oppervlak. In veel gevallen worden onderdelen die met tijdelijke corrosiebestrijdingsmiddelen worden beschermd ook verpakt. Een belangrijke functie van de verpakking is het voorkomen van beschadiging van de onderdelen zelf en van het tijdelijke corrosiebestrijdingsmiddel en het buitensluiten van vocht en andere agressieve bestanddelen uit de omgeving. Maar dan moet er wel op worden gelet dat poreuze verpakkingsmaterialen, zoals papier, niet vochtig worden want daardoor kan ernstige corrosie worden veroorzaakt.

Voorkomen en bestrijden van elektrochemische corrosie
154
Een speciale groep van tijdelijke corrosiebestrijdingsmiddelen zijn de dampfase inhibitoren en deze worden vaak met het verpakkingsmateriaal gecombineerd. Dit zijn vluchtige verbindingen waarmee papier geïmpregneerd kan worden en die de verpakte onderdelen tegen corrosie beschermen. Door hun lage dampspanning en lage verdampingssnelheid kunnen deze middelen alleen in gesloten ruimtes worden gebruikt. Een meer gedetailleerde beschrijving van eigenschappen en gebruik van deze tijdelijke corrosiebestrijdingsmiddelen wordt gegeven in British Standard 1133 [25]. Daarin worden ook de eisen besproken die voor verschillende toepassingen gelden alsmede het testen van deze verbindingen. Voor verdere nuttige informatie wordt naar de literatuur verwezen [26, 27, 28].
8.7 Corrosiebestrijding door goede vormgeving
Corrosiebestrijding moet op de tekentafel beginnen
Dit is een zeer uitgebreid en belangrijk gebied van corrosiebestrijding dat zeker niet in zijn geheel kan worden behandeld. We geven alleen een aantal typische voorbeelden van verschillende aard met een korte toelichting waarom de ene mogelijkheid beter is dan de andere. Voor meer voorbeelden en een meer diepgaande behandeling verwijzen we naar de boeken van Pludek [29], Parkins en Chandler [30] en naar de aan dit onderwerp gewijde hoofdstukken van referenties 10 en 11. Vaten en tanks moeten vaak op een betonnen voet of een staalconstructie worden geplaatst. Typische voorbeelden zijn beitsbaden, voorraadvaten, galvanische baden, enzovoort. Het ontwerp van Figuur 8.7.1a is in dit opzicht erg slecht. Gemorste vloeistof kan daar in de spleet tussen de bodem van het vat en de ondersteuning kruipen waar schoonmaken vrijwel onmogelijk is. Dit kan tot ernstige corrosie leiden, zowel van de bodem als van de ondersteuning. Veel beter is het ontwerp van Figuur 8.7.1b, waar de afdruipplaat het binnendringen van gemorste vloeistof , condensaat of regen verhindert.
Figuur 8.7.1. Ondersteuning van vaten (Met toestemming overgenomen uit [2]).

155
Men realiseert zich vaak niet dat corrosie alleen op kan treden indien een agressieve vloeistof (in veel gevallen een waterige oplossing) aanwezig is en dat de omvang van de corrosie vooral bepaald wordt door de tijdsduur gedurende welke het metaal met dat milieu in aanraking is. Door goede vormgeving kan dikwijls voorkomen worden dat een vloeistof te lang in een apparaat of vat blijft staan wanneer dat voor de werking van apparaat of installatie niet noodzakelijk is. Dit is bijvoorbeeld van groot belang in verband met het voorkomen van onnodige corrosie van bodems van vaten, reactoren, enzovoort. In dit opzicht is dus de con-structie van Figuur 8.7.2a verkeerd en die in b goed. Uiteraard moet bij het later aanbrengen van zo'n uitloop of bij reparatie of vervanging daarvan bij onderhoudsbeurten hieraan ook terdege aandacht worden besteed.
Figuur 8.7.2. a. Verkeerde uitloop van tank of reactie ketel; b. goede vormgeving van uitloop (Met toestemming overgenomen uit [2]).
Figuur 8.7.3. Foute en goede constructie van versterkingsribben (boven) en steunbalken.

Voorkomen en bestrijden van elektrochemische corrosie
156
Ook voor andere constructiedelen geldt dit en enkele illustraties staan in Figuur 8.7.3. Iets dergelijks komen we tegen wanneer een deklaag moet worden aangebracht door onderdompelen in een vloeistof (galvanisch bedekken, verzinken, elektroforetisch schilderen, enzovoort). Het ontwerp moet dan vanzelfsprekend zo zijn dat volledige bevochtiging mogelijk is, dus geen ingesloten luchtbellen, en dat de constructie volledig kan leeglopen, zoals schematisch aangegeven in Figuur 8.7.4b. Ditzelfde geldt natuurlijk voor onderdelen die gebeitst of ontvet moeten worden. Voor specifieke aanwijzingen voor goede vormgeving bij thermisch verzinken zie de brochure [31].
Figuur 8.7.4. Plaatsing van gaten in constructies die in een vloeistof moeten worden ondergedompeld voor oppervlaktebehandeling of –bedekking (Met toestemming overgenomen uit [2]).
Verbindingen tussen onderdelen van een constructie zijn bekend om hun gevoeligheid voor corrosie. Met name daar komen we spleetcorrosie en contactcorrosie tegen en we besteden daarom aan dit onderwerp ook enige bijzondere aandacht. De in hoofdstuk 5 reeds besproken contactcorrosie is vooral ernstig wanneer een klein, relatief onedel oppervlak in contact staat met een groot edel oppervlak omdat dan de stroomdichtheid, die een directe maat is voor de corrosiesnelheid, zeer groot kan worden. Het omgekeerde is meestal veel minder ernstig (denk aan het gebruik van relatief edele bronzen afsluiters in overigens van het veel onedeler staal gemaakte verwarmingssystemen). In Figuur 8.7.5 wordt het gevaar van het gebruik van verkeerde klinknagels getoond: een onedele klinknagel (Figuur 8.7.5a, klein ten opzichte van de te verbinden platen) corrodeert snel, terwijl een klinknagel van hetzelfde metaal (Figuur 8.7.5b) wèl kan worden toegepast. Bij toepassing van een edeler klinknagel wordt weliswaar in theorie de corrosie van de verbonden platen vergroot, maar omdat dit effect zich over een zeer groot oppervlak uitspreidt is dat in de praktijk te verwaarlozen.

157
Figuur 8.7.5. Contactcorrosie bij geklonken verbinding, a. stalen klinknagel in koperen platen, b. stalen klinknagel in stalen platen (Met toestemming overgenomen uit [2]).
Figuur 8.7.6. Voorkomen van spleetcorrosie bij boutverbinding (Met toestemming overgenomen uit [2]).
Het belang van de oppervlakteverhouding bij de koppeling van ongelijke metalen geldt natuurlijk ook voor boutverbindingen. Een ander punt van groot belang bij dit type verbindingen is het vermijden van de ook al hiervoor besproken spleetcorrosie, bijvoorbeeld door het kiezen van enigszins vervormbare onderlegringen bij boutverbindingen waardoor eventuele spleten volledig worden gesloten. Dit is geïllustreerd in Figuur 8.7.6. Wanneer twee platen aan elkaar gelast moeten worden is de in Figuur 8.7.7a. gegeven constructie fout omdat in de spleet tussen de platen agressieve componenten achter kunnen blijven en er gevaar voor spleetcorrosie is. Het volledig sluiten van de spleet, zoals in Figuur 8.7.7b, geeft al een duidelijke verbetering, maar dan moet, ook aan de uiteinden, de lasspleet wèl volledig gesloten zijn en blijven, zodat dit in het algemeen toch niet is aan te bevelen. Het beste is aansluitend lassen van de platen zoals weergegeven in Figuur 8.7.7c.

Voorkomen en bestrijden van elektrochemische corrosie
158
Figuur 8.7.7. Schematische weergave van lassen als verbinding van twee platen (Met toestemming overgenomen uit [2]).
Van zeer groot belang is dat lokale verschillen in concentratie of temperatuur worden vermeden. Wanneer bijvoorbeeld een geconcentreerd zuur aan een reactiemengsel moet worden toegevoegd dient de inlaat van dat zuur zo te worden geplaatst dat er geen direct contact is van het geconcentreerde zuur met de wand van het reactievat. In Figuur 8.7.8a tot c zijn onjuiste constructies weergegeven waar de toegevoegde vloeistof langs de vatwand loopt, of daartegen kan spatten. Figuur 8.7.8d toont een veel betere constructie waar de vloeistof wordt toegevoegd onder het oppervlak van de al aanwezige oplossing en zover mogelijk van de wand.
Figuur 8.7.8. Plaatsing van inlaat van sterk zure oplossing in reactievat: a, b, c: fout, d: goed (Met toestemming overgenomen uit [2]).
Om analoge redenen moeten verhittingselementen niet te dicht bij een wand of bodem worden geplaatst zoals in Figuur 8.7.9a is getekend. Het gevaar is dan groot dat oververhitting optreedt of de oplossing plaatselijk gaat koken met het gevaar van
159
toenemende concentratie, met daaraan gekoppelde grotere agressiviteit, door verdamping. Een betere plaatsing is getoond in Figuur 8.7.9b waar de warmte zich goed kan verspreiden in de vloeistof en dus ook het ontstaan van concentratieverschillen veel kleiner is.
Figuur 8.7.9. Goede en foute plaatsing van verhittingselement (Met toestemming overgenomen uit [2]).
Figuur 8.7.10. Voorkomen van condensaatcorrosie door juist aangebrachte isolatie (Met toestemming overgenomen uit [2]).
Wanneer in een vochtige ruimte afkoeling optreedt bestaat altijd het gevaar voor condensvorming en daarmee dus ook voor corrosie wanneer de temperatuur daalt beneden het dauwpunt van het gas. Men spreekt in dit type gevallen dan ook van dauwpuntcorrosie of condensaatcorrosie. Vooral in slecht geventileerde, tamelijk

Voorkomen en bestrijden van elektrochemische corrosie
160
afgesloten ruimten zoals holle onderdelen (autodeuren!), bestaat hiervoor gevaar en de beste methode om dit te ondervangen is volledig afsluiten van zo'n ruimte zodat er geen vocht in kan komen. Is dat niet mogelijk dan is het beslist noodzakelijk voor een goede luchtverversing te zorgen. Afkoeling kan ook plaatselijk optreden, zoals geschetst in Figuur 8.7.10 voor een geïsoleerde pijp, waardoor warm vochtig gas stroomt, bij een ophanging die eigenlijk als koelrib werkt. In dit geval is de beste oplossing om de ophanging zelf ook te isoleren waardoor het koeleffect niet meer op kan treden. Analoge situaties kunnen ook voorkomen bij de ondersteuning met stalen profielbalken van vaten waarin zich warme, vochtige gassen bevinden. Een andere belangrijk punt betreft de stromingssnelheid die uiteraard ook door de vormgeving sterk kan worden beïnvloed. Bij kleine stromingssnelheden kunnen afzettingen van vaste stoffen, die in het milieu aanwezig zijn, leiden tot de hiervoor al genoemde "deposit-attack" die in wezen een vorm van spleetcorrosie is. Om dat te voorkomen moet, wanneer zuivering van het milieu niet mogelijk is om de afzettingen onmogelijk te maken, de stromingssnelheid groter worden gemaakt. Anderzijds kan, vooral bij te hoge stromingssnelheden, erosie-corrosie optreden. Wanneer vaste stoffen in het milieu de oorzaak zijn van de erosie-corrosie kan dit verschijnsel in principe door zuiveren worden voorkomen wanneer dat verontreinigingen betreft, maar soms is de aanwezigheid van de vaste stoffen een onderdeel van het proces. Het kan worden bestreden door het vermijden van een te sterke richtingsverandering, zoals weergegeven in Figuur 8.7.11. Daarnaast kan het kleiner maken van de stromingssnelheid helpen om erosie-corrosie te voorkomen, waarbij ook de vormgeving een grote rol speelt door het voorkomen van abrupte vernauwingen, bijvoorbeeld ten gevolge van een niet goed passende pakking, zoals schematisch aangegeven in Figuur 8.7.12.
Figuur 8.7.11. Erosie-corrosie door te sterke richtingsverandering (Met toestemming overgenomen uit [2]).

161
In wezen komt het bij het voorkomen van erosie-corrosie bijna steeds erop neer dat een loodrecht op de wand van een pijp of vat staande beweging van de vloeistof moet worden vermeden. Zo'n beweging kan enerzijds natuurlijk ontstaan door de vormgeving (zie Figuur 8.7.11), anderzijds door sterke turbulentie (zie het voorbeeld van Figuur 8.7.12). Vooral wanneer vaste deeltjes in een vloeistofstroom aanwezig zijn kan echter ook een sterke beweging langs een oppervlak leiden tot erosie-corrosie. In zo'n geval moet òf de vloeistof worden gereinigd, òf moeten slijtplaten worden aangebracht die gemakkelijk te vernieuwen zijn.
Figuur 8.7.12. Erosie-corrosie door vernauwing in vloeistofstroom (Met toestemming overgenomen uit [2]).
Waar ook goed op moeten worden gelet is de instroming van vloeistof in reactievaten, koelers, enzovoort. Zowel te sterke turbulentie langs de wand, als die langs in zo'n apparaat geplaatste onderdelen, zoals bijvoorbeeld koelbuizen, verwarmingselementen en dergelijke, moet worden voorkomen om te zorgen dat erosie-corrosie niet kan optreden. Zoals besproken in §6.10 (zie ook Figuur 6.10.4) worden in de inlaatzijde van koeler- en condensorpijpen soms inzetstukken geplaatst omdat daar juist het gevaar voor erosie-corrosie het grootst is. Deze inzetstukken kunnen òf van een meer erosie-bestendig materiaal worden gemaakt òf periodiek worden vervangen. De keuze tussen deze mogelijkheden hangt natuurlijk van economische factoren af. Twee verschijnselen die nauw verwant zijn met erosie-corrosie en in hoofdstuk 6 al werden genoemd zijn:

Voorkomen en bestrijden van elektrochemische corrosie
162
− inslag-aantasting (Engels: impingement attack), ten gevolge van gasbellen in een vloeistof die tegen een metaaloppervlak botsen, en:
− cavitatie-corrosie, waarbij dampbellen worden gevormd in de vloeistof door sterke onderdruk, die vervolgens imploderen op het metaal en daar putvormige schade veroorzaken.
De belangrijkste bestrijdingsmethoden hierbij zijn analoog aan die van erosie-corrosie, namelijk het vermijden van de sterke relatieve beweging van vloeistof en metaal.
8.8 Economische aspecten van corrosiebestrijding
Zoals in het voorgaande al verscheidene keren is aangegeven is het van groot belang om bij de keuze van de methoden van voorkomen of bestrijden van corrosie ook steeds voldoende aandacht te schenken aan de economische aspecten die daarmee zijn verbonden. Helaas leidt corrosie vrijwel altijd tot verhoogde kosten. Dit kan het gevolg zijn van de kosten van onderhoud, reparatie of vervanging van gecorrodeerde onderdelen of van de kosten van maatregelen genomen om corrosie te voorkomen of bestrijden. Er bestaat geen magische manier waarmee het mogelijk is om goedkope materialen als ongelegeerd staal of gietijzer te gebruiken zonder dat daar extra kosten door corrosie of corrosiebestrijding aan verbonden zijn. Het is dus van groot belang om een manier te vinden waarop de kosten kunnen worden geminimaliseerd Centraal hierbij staat het vergelijken van de economische merites van de verschillende methoden voor het voorkomen of bestrijden van corrosie. Er bestaan verschillende manieren om dit soort economische vergelijking uit te voeren. Hier beperken we ons tot de methode gebaseerd op de berekening van de netto contante waarde. Voor een uitvoeriger behandeling en een vergelijking met andere methoden moeten we naar de literatuur verwijzen [32, 3334]. Het basisprincipe van de netto contante waarde is het vergelijkbaar maken van inkomsten en uitgaven die op verschillende tijdstippen voorvallen. Aan inkomsten in latere perioden worden lagere waarden toegekend. Wanneer deze latere inkomsten namelijk eerder waren binnengekomen hadden zij op een bankrekening rente kunnen genereren of hadden zij misschien voor een winstgevend project kunnen worden ingezet. De huidige waarde P(n,r) van € 1 die n jaar na heden wordt ontvangen (of betaald), wordt gegeven door:€
n)r1( ƒ)r,n(P −+= (8.8.1)
Hierin is r de “prijs van kapitaal”, dat wil zeggen het door de onderneming geëiste minimale rendement op het bij corrosiebestrijding ingezette vermogen. Een tweede maat van de netto contante waarde die we voor ons doel nodig hebben is de tegenwoordige waarde A(n,r) van € 1 die gedurende n jaar ieder jaar wordt ontvangen of betaald en deze is gelijk aan:

163
{ }
r)r1(1€
)r1()r1()r1(€)r,n(An
n21
−
−−−
+−=
++++++= L
(8.8.2)
Er zijn tabellen van P(n,r) en A(n,r) gepubliceerd [32] en een korte versie daarvan wordt gegeven in bijlage 11.17. De netto contante waarde van een project is nu gelijk aan:
NCWproject = Incidentele uitgaven × P(n,r) + jaarlijks terugkerende uitgaven × A(n,r) (8.8.3)
Wanneer bijvoorbeeld uitgaven voor onderhoud niet op regelmatige tijdstippen terugkeren is het gemakkelijker om hun netto contante waardes afzonderlijk uit te rekenen en deze bij de initiële kosten van de verschillende alternatieven op te tellen. Een in vele gevallen beter alternatief is het vergelijken op basis van de equivalente kosten Ceq,i. Voor verschillende investeringen Ii met verschillende levensduren ni zijn deze gelijk aan:
)r,n(AIC iii,eq = (8.8.4)
waarbij is aangenomen dat de installatie na ni jaar wordt vervangen. In veel gevallen zijn er ook bedrijfskosten Cb,i per jaar die bij de equivalente kosten moeten worden opgeteld. Verder zijn er uitgaven voor onderhoud Mi die iedere mi jaar terugkeren. Deze leiden tot de equivalente kosten Meq,i die gelijk zijn aan:
)r,m(AMM iii,eq = (8.8.5)
De totale kosten Ctot,i per jaar worden dan gegeven door: MCCC ieq,i,bi,eqi,tot ++= (8.8.6)
We gaan hier niet dieper in op allerlei verfijningen van deze vergelijkingen samenhangend met investeringssubsidies, verschillende belasting voor investering en bedrijfskosten, restwaarde aan het eind van de levensduur, verschillen in milieubelasting, ongelijke perioden voor onderhoud, enzovoort. Vanzelfsprekend zal die investering de voorkeur verdienen waarvoor de totale jaarlijkse kosten Ctot,i of de NCW het laagste zijn. We bespreken nu de toepassing van deze principes op enkele (vereenvoudigde) praktijkgevallen. De hieronder genoemde prijzen zijn niet noodzakelijkerwijs de nu geldende en moeten uitsluitend als voorbeelden worden gezien. Het eerste voorbeeld betreft de materiaalkeuze voor een warmtewisselaar voor een geval waar uit ervaring is gebleken dat een ongelegeerd stalen warmtewisselaar een levensduur had van ongeveer 3 jaar bij een aanschaffingsprijs van € 12000. Als alternatieve materialen komen in aanmerking roestvast staal 304 (18% Cr, 8% Ni) en 316 (18% Cr, 10% Ni, 1.5% Mo). Hiervoor wordt een levensduur van 10 jaar verwacht terwijl de aanschaffingsprijs voor 304 € 39000 en voor 316 € 45000 is. De transport en installatiekosten bedragen € 4500 en zijn onafhankelijk van het gekozen materiaal. In Tabel 8.8.1 zijn de gegevens voor dit voorbeeld samengevat. Het blijkt dat bij lage kosten van kapitaal de 304 warmtewisselaar het goedkoopste is, bij hoge kosten van kapitaal de ongelegeerd stalen warmtewisselaar. Het is
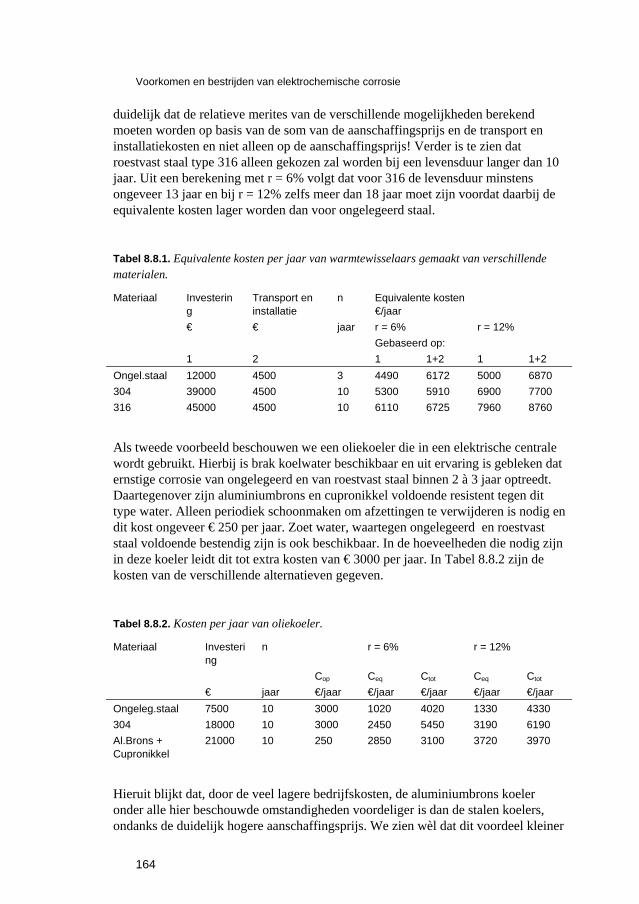
Voorkomen en bestrijden van elektrochemische corrosie
164
duidelijk dat de relatieve merites van de verschillende mogelijkheden berekend moeten worden op basis van de som van de aanschaffingsprijs en de transport en installatiekosten en niet alleen op de aanschaffingsprijs! Verder is te zien dat roestvast staal type 316 alleen gekozen zal worden bij een levensduur langer dan 10 jaar. Uit een berekening met r = 6% volgt dat voor 316 de levensduur minstens ongeveer 13 jaar en bij r = 12% zelfs meer dan 18 jaar moet zijn voordat daarbij de equivalente kosten lager worden dan voor ongelegeerd staal.
Tabel 8.8.1. Equivalente kosten per jaar van warmtewisselaars gemaakt van verschillende materialen.
Materiaal Investering
Transport en installatie
n Equivalente kosten €/jaar
€ € jaar r = 6% r = 12% Gebaseerd op: 1 2 1 1+2 1 1+2 Ongel.staal 12000 4500 3 4490 6172 5000 6870 304 39000 4500 10 5300 5910 6900 7700 316 45000 4500 10 6110 6725 7960 8760
Als tweede voorbeeld beschouwen we een oliekoeler die in een elektrische centrale wordt gebruikt. Hierbij is brak koelwater beschikbaar en uit ervaring is gebleken dat ernstige corrosie van ongelegeerd en van roestvast staal binnen 2 à 3 jaar optreedt. Daartegenover zijn aluminiumbrons en cupronikkel voldoende resistent tegen dit type water. Alleen periodiek schoonmaken om afzettingen te verwijderen is nodig en dit kost ongeveer € 250 per jaar. Zoet water, waartegen ongelegeerd en roestvast staal voldoende bestendig zijn is ook beschikbaar. In de hoeveelheden die nodig zijn in deze koeler leidt dit tot extra kosten van € 3000 per jaar. In Tabel 8.8.2 zijn de kosten van de verschillende alternatieven gegeven.
Tabel 8.8.2. Kosten per jaar van oliekoeler.
Materiaal Investering
n r = 6% r = 12%
Cop Ceq Ctot Ceq Ctot € jaar €/jaar €/jaar €/jaar €/jaar €/jaar Ongeleg.staal 7500 10 3000 1020 4020 1330 4330 304 18000 10 3000 2450 5450 3190 6190 Al.Brons + Cupronikkel
21000 10 250 2850 3100 3720 3970
Hieruit blijkt dat, door de veel lagere bedrijfskosten, de aluminiumbrons koeler onder alle hier beschouwde omstandigheden voordeliger is dan de stalen koelers, ondanks de duidelijk hogere aanschaffingsprijs. We zien wèl dat dit voordeel kleiner

165
wordt wanneer de kosten van kapitaal hoger worden. In het hier beschouwde geval wordt ongelegeerd staal voordeliger bij r groter dan ongeveer 15%. De hier gegeven voorbeelden tonen enkele van de meest belangrijke aspecten in de economische beoordeling van corrosiebestrijding en preventie. In het bijzonder be-treft dit het vinden van een balans tussen de initiële kosten (investering) en de onder-houds- en bedrijfskosten , gecombineerd met de kosten van kapitaal. De relatieve merites van de hier behandelde gevallen kunnen zeker anders worden bij verande-ringen in de kosten van kapitaal, van de arbeidskosten of van de agressiviteit van de omgeving. Ieder geval moet afzonderlijk worden beoordeeld, waarbij alle relevante omstandig-heden mee in beschouwing moeten worden genomen. Er is zeker geen universeel antwoord op de vraag welke het beste, in de betekenis van het goedkoopste, systeem van bescherming tegen corrosie is.
8.9 Opgaven
1. Voor offshore constructies die in diep water staan is kathodische bescherming met offeranoden vaak de enige mogelijkheid van corrosiebestrijding. De hiervoor te gebruiken anodes moeten groot genoeg zijn om bescherming te geven gedurende de hele verwachte levensduur omdat onderhoud of vervanging vaak niet mogelijk is. Wanneer de beschermstroomdichtheid van onbedekt staal onder gegeven omstandigheden 0.1 A.m-2 is bereken dan de hoeveelheid zink of aluminium die nodig is om een oppervlak van 100 m2 gedurende 20 jaar te beschermen.
2. Onbeschermd staal corrodeert in zeewater met een corrosiesnelheid van 25 mdd. 1. Wanneer zuurstofreductie de enige kathodische reactie is schets dan het
polarisatiediagram. Neem aan dat de concentratie van de ijzerionen 10-7 mol.l-1 is. De uitwisselingsstroomdichtheid van de anodische reactie is i0 = 10-4 A.m-2 en de helling van de Tafel-lijn is βa = 0.12 Volt.
2. Bepaal de corrosiepotentiaal onder deze omstandigheden. 3. Bereken de minimum stroomdichtheid in mA.m-2 die nodig is om het staal
volledig kathodisch te beschermen. 4. Afzetting van calciumcarbonaat ten gevolge van de kathodische
bescherming veroorzaakt een lineaire afname van de voor kathodische bescherming benodigde stroomdichtheid tot 15% van de oorspronkelijke waarde in 2 jaar, waarna die weer constant blijft. De kathodische bescherming wordt uitgevoerd met zinkanodes van 30 kg die niet vaker dan eens in de 10 jaar mogen worden vervangen. Wanneer de efficiëntie van de anodes 95% is hoeveel m2 staal kan dan met één anode worden beschermd?
3. In Figuur 8.9.1 is een doorsnede gegeven van een tank waarin een hete, chloride bevattende oplossing wordt opgeslagen. Het volume is 2 m3, de tank staat op 4 buisvormige poten van 8 cm diameter die aan de roestvast stalen bodem van de tank zijn gelast. De verticale zijde van de tank is gemaakt van ongelegeerd staal, aan de binnenkant geschilderd met een zuurbestendige verf.

Voorkomen en bestrijden van elektrochemische corrosie
166
Bespreek de essentiële fouten die in dit ontwerp en bij de materiaalkeuze zijn gemaakt. Geef een voorstel voor een verbeterde constructie en materiaalkeuze en geef de redenen voor de voorgestelde wijzigingen.
Figuur 8.9.1. Doorsnede van tank voor hete, chloridehoudende oplossing.
3. a. We beschouwen een proceskoeler waarvan het koelelement bestaat uit
buizen met een wanddikte van 1.5 mm. De wanddikte mag door corrosie niet kleiner worden dan 0.75 mm. Alleen aan de koelwaterzijde van de pijpen treedt corrosie op en voor ongelegeerd staal bedraagt deze 0.12 A.m-2: bereken de maximale levensduur (afgerond in gehele jaren) van deze pijpen. Het betreffende koelelement kost ƒ 100 per m2 warmtewisselend oppervlak. Als zo'n koelelement een oppervlak van 100 m2 heeft, wat zijn dan de equivalente kosten per jaar, bij de hiervoor berekende levensduur (wanneer men die niet heeft kunnen berekenen dan vragen aan surveillant) en kapitaalskosten van 12 % . Wanneer men de koeler maakt van roestvast stalen buizen type 304 en de corrosie daarvan is onder de gegeven omstandigheden verwaarloosbaar, wat mag dan de aanschafprijs maximaal zijn om niet duurder uit te komen dan

167
bij regelmatige vervanging van ongelegeerd staal, uitgaande van een gewenste levensduur van 20 jaar?
b. Om de corrosie te verminderen kan men ook een zogenaamde adsorptie-inhibitor toevoegen. Deze vormt een laag op het staaloppervlak waardoor de zuurstofdiffusie wordt vertraagd (aangenomen mag worden dat de corrosiesnelheid volledig wordt bepaald door diffusiepolarisatie van de zuurstofreductie reactie). Experimenteel is gebleken dat 10 mg.l-1 van een bepaalde inhibitor de corrosie met een factor 0.9 verlaagt. Dit heeft wel tegelijk als gevolg dat de warmteoverdracht per 10 mg.l-1 met een factor 0.985 omlaag gaat. Hoeveel inhibitor moet in het koelwater aanwezig zijn om de levensduur op 20 jaar te brengen en hoe groot moet men dan het warmtewisselend oppervlak maken. Wanneer de inhibitor ƒ 75 per kg kost, de inhoud van het totale systeem 20 m3 is en per dag 0.25 m3 water wordt gesuppleerd, wat worden dan de totale kosten in dit geval. Geef aan welke conclusie u uit het resultaat van deze berekening trekt.
c. Een alternatieve mogelijkheid om de corrosie te verminderen is het wegnemen van (een deel van) de in het water aanwezige zuurstof, bijvoorbeeld door met stoom te ontluchten. Wanneer de hoeveelheid lucht in het koelwater 8 mg.l-1 is hoeveel moet dit dan worden verlaagd om ook weer een levensduur van 20 jaar te bereiken. De aanschafprijs van een ontluchter die dit mogelijk maakt bedraagt ƒ 8000. Wat mag de maximale uitgave voor stoom per jaar zijn om een levensduur van 20 jaar te bereiken en niet duurder uit te komen dan bij vervanging iedere 5 jaar.
d. Tenslotte kan ook nog overwogen worden om kathodische bescherming toe te passen. Wanneer dit gedaan wordt wat is dan de totale stroom die nodig is om het gehele koelende oppervlak te beschermen? Om de stroom goed over het hele oppervlak te verdelen wil men 5 aluminium opofferingsanoden toepassen. Deze hebben een efficiency van 70 %, en men wil deze niet meer dan eens in de 2 jaar vervangen. Hoe zwaar moeten deze anodes dan zijn? Het anodemateriaal kost € 16 per kg, de anodekabel die iedere keer moet worden vervangen per stuk € 85, het de eerste keer monteren kost € 60 en het demonteren + monteren bij vervanging € 120 per anode. Wat worden voor dit geval de totale kosten?
8.10 Literatuur
1. P.J. Gellings, Corrosie en Onderhoud, in: Handboek Onderhoudsmanagement, Alphen aan de Rijn, Samsom BedrijfsInformatie, 1996, p. K3010-1 - 56.
2. P.J. Gellings en T. Fransen, Corrosiebewust ontwerpen in: Handboek Werktuigbouwkundig ontwerpen en construeren, Alphen aan de Rijn, Samsom BedrijfsInformatie, 1993, p. B4400-1 - 40.

Voorkomen en bestrijden van elektrochemische corrosie
168
3. Aanbevelingen ter voorkoming van corrosie en ketelsteenvorming in watervoerende
installaties, ISSO publicatie 13, Stichting ISSO, Rotterdam (1983). 4. Uitwendige corrosie, Aanbevelingen ter voorkoming van uitwendige corrosie van
verwarmings- en luchtbehandelingsinstallaties, ISSO publicatie 26, Stichting ISSO, Rotterdam (1990)
5. P.J. Gellings en F. IJsseling, Corrosie en Corrosiebestrijding. Materiaalkeuze en Constructieve Aspecten, NCC brochure 2, Delft, Waltman.
6. F.Walsh, G. Mills, D. Barker, and T. Oliver, Preventing corrosion by effective design and maintenance, Corrosion Management Oct/Nov 1995, p. 18 - 21.
7. A. Snel en E.D.D. During, Corrosiebestrijding door waterbehandeling, NCC brochure 5, Leiden/Delft, SMD/Waltman (1993)
8. Reviews on corrosion inhibitor science and technology, Ed. by A. Raman and P. Labine, Houston, NACE (1991)
9. Corrosion inhibition – Theory and Practice, NACE Reference 7, Ed. by R.H. Hausler, Houston, NACE (1988)
10. S.A. Bradford, Corrosion Control, New York, Van Nostrand Reinhold, 1993. 11. P.J. Gellings en F. IJsseling, Corrosie en Corrosiebestrijding. Materiaalkeuze en
Constructieve Aspecten, NCC brochure 2, Delft, Waltman. 12. A.G.C. Kobussen en B.H. Wijngaard, Kathodische en Anodische Bescherming, NCC
brochure 3, Delft, Waltman (1989) 13. Kathodische bescherming On-Shore / Buisleidingen en constructies van metaal,
Nederlandse Praktijk Richtlijn NPR 6912, Concept, Nederlands Normalisatie Instituut (1994).
14. J.H. Morgan, Cathodic protection, London, Leonard Hill Ltd. (1971). 15. W. von Baeckmann, W. Schwenk und W.Prinz, Handbuch des kathodischen
Korrosionsschutzes, neubearbeitete Auflage, Weinheim, Verlag Chemie (1989). 16. V. Ashworth and C. Googan, Cathodic protection, theory and practice, Chichester, Ellis
Horwood (1993) 17. O.L. Riggs and C.E. Locke, Anodic protection, New York/London, Plenum Press (1981) 18. T. van der Klis, Vademecum Oppervlakte Technieken Materialen, deel 1: Metalen,
Bilthoven, Vereniging Oppervlaktetechnieken van Materialen (1982). 19. T. van der Klis, Corrosiebestrijding door metallische en anorganische deklagen, NCC
brochure 4, Leiden/Delft, SMD/Waltman (1993) 20 J.H.W. de Wit, Inorganic and organic coatings, in: Corrosion mechanisms in theory and
practice, ed. by P.Marcus and J.Oudar, New York, Marcel Dekker (1995) 21. P. Maaß und P. Peißker, Handbuch Feuerverzinken¸ 2te Aufl., Leipzig-Stuttgart,
Deutscher Verlag für Grundstoffindustrie (1993) 22. Zink op staal, Sassenheim/Rotterdam, Stichting Doelmatig Verzinken/Staalbouw
Instituut (1994) 23. J.F.H. van Eijnsbergen, Duplex systems. Hot-dip galvanizing plus painting, Amsterdam,
Elsevier (1994) 24. T. van der Klis, Corrosiebestrijding door organische deklagen, NCC brochure 6,
Delft/Leiden, Waltman/SMD (1995) 25. Package Code. Part 6: Protection of metal surfaces during transport and storage. 6.1.
Cleaning and drying of metal surfaces. 6.2. Temporary protectives and their applications. British Standard 1133.
26. Packaging for handling and transporting of coated goods in the Construction industry, Guides to practice in corrosion control, no. 3, London, HMSO (1979)
27. Temporary protection, Guides to practice in corrosion control, no. 6, London, HMSO (1981)

169
28. P.D. Donovan, Protection of metals from corrosion in storage and transit, Chichester,
Ellis Horwood (1986) 29. V.R. Pludek, Design and Corrosion Control, London, MacMillan (1977) 30. R.N. Parkins and K.A. Chandler, Corrosion Control in Engineering Design, London,
HMSO (1978) 31. Constructieve wenken voor thermisch te verzinken onderdelen en constructies,
Sassenheim/Rotterdam, Stichting Doelmatig Verzinken/Stichting Staalcentrum Nederland (1981)
32. H. Bierman Jr. And S. Smidt, The capital budgeting decision: economic analysis of investmentprojects, 7th ed.New York, MacMillan (1988)
33. E.D. Verink, Economic appraisals of corrosion control measures, J.Educational Modules in Materials Science and Engineering 3 (1981) 239 - 268
34. A.M.M. Blommaert en J.M.J. Blommaert, Bedrijfseconomische analyses, 3de druk, Houten, Stenfert Kroese (1995)


9
Hoge-temperatuuroxidatie en de bestrijding daarvan


173
9.1 Inleiding
In de industriële praktijk worden steeds vaker hoge temperaturen gebruikt en daarvoor zijn verschillende redenen. De eerste is het welbekende feit dat de thermodynamische efficiency van verbrandingsmotoren, turbines en stoommachines toeneemt met een groter verschil tussen de hoogste en laagste temperatuur waarbij zo'n apparaat werkt. Verhoging van de maximum temperatuur leidt dus tot energiebesparing. Een tweede reden is dat veel chemische reacties alleen bij hoge temperatuur voldoende snel verlopen of daarbij pas mogelijk zijn, hetgeen heeft geleid tot het gebruik van steeds hogere temperaturen in de chemische industrie. Een derde reden is dat de materialen die nodig zijn voor de apparatuur waarin deze hoge temperaturen worden toegepast uiteraard bij nog hogere temperaturen moeten worden gemaakt en/of behandeld. In de overgrote meerderheid van deze gevallen wordt het metaal gebruikt in een gasvormige omgeving bij hoge temperatuur en de samenstelling van het gas is bepalend voor de aard van de reacties die op kunnen treden. In de meeste gevallen bevat het gas zuurstof zodat oxidatie van het metaal het gevolg is. Bij het kraken van zwavelhoudende zware aardoliefracties of de verbranding of vergassing van zwavelhoudende brandstoffen treedt sulfidatie op. Wanneer het gas chloorverbindingen bevat, bijvoorbeeld de verbrandingsgassen die ontstaan bij de verbranding van afval dat polyvinylchloride of andere soortgelijke kunststoffen bevat, kunnen metaalchloriden worden gevormd. Het is duidelijk dat de "gewone" elektrochemische corrosie, zoals die in de voorgaande hoofdstukken is behandeld, onder deze omstandigheden niet op kan treden omdat de gassen die hierbij voor de corrosie verantwoordelijk zijn geen elektrische stroom geleiden. Meestal worden in deze gevallen vaste corrosieproducten gevormd, alleen onder betrekkelijk weinig voorkomende omstandigheden ontstaan vloeibare of vluchtige producten. De aanwezigheid van vaste producten op het oppervlak kan natuurlijk een sterke invloed uitoefenen op de kinetiek van de aantasting, net zoals dat bij de passiviteit het geval is. Omdat de vaste producten in veel gevallen (half)geleiders zijn kunnen toch elektrochemische beschouwingen van nut zijn, zoals onder andere besproken door de Wit en Fransen [1], maar daarop wordt hier niet verder ingegaan. In dit hoofdstuk gaan we vooral in op de hoge-temperatuuroxidatie van metalen omdat, uitgaande van het aantal tonnen materiaal dat onderworpen is aan hoge-temperatuurcorrosie, die verreweg het meeste voorkomt. Daarnaast zal ook kort aandacht worden besteed aan de corrosie van metalen in andere gassen.
9.2 Thermodynamica van oxidatiereacties
Voor de reactie van nikkel met zuurstof: Ni + ½ O2 = NiO (9.2.1)

Hoge-temperatuuroxidatie en de bestrijding daarvan
174
kan de verandering van de vrije-enthalpie, op dezelfde wijze en met dezelfde notatie als besproken in hoofdstuk 2, worden geschreven als het verschil tussen de vrije-enthalpie van het product NiO min die van de reactanten Ni en O2:
NiOO21
Ni 2g μ+μ−μ−=Δ (9.2.2)
Het gebruik van een kleine letter g geeft aan dat we vorming van 1 mol NiO beschouwen, dat wil zeggen de reactie verloopt éénmaal zoals geschreven. De thermodynamische beschouwingen, gegeven in hoofdstuk 2, maken het mogelijk voor de vrije-enthalpieverandering van reactie (451H9.2.1) af te leiden:
2O0 pln
2RTgg −Δ=Δ (9.2.3)
waarin 2Op de partiële zuurstofdruk is.
Bij een gegeven temperatuur is Δg0 een constante. In evenwicht is Δg = 0 en hieruit kunnen we afleiden dat:
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ=
RTg2expp
0evO2
(9.2.4)
Wanneer de zuurstofdruk lager is dan deze waarde, die de evenwichtsdruk of dissociatiedruk wordt genoemd, zien we dat Δg > 0 hetgeen betekent dat reactie (451H9.2.1) de neiging heeft spontaan naar links te verlopen: het oxide dissocieert spontaan in metaal plus zuurstof. Wanneer de zuurstofdruk echter hoger is dan de dissociatiedruk is dat Δg < 0 en heeft het metaal juist de neiging om geoxideerd te worden.
Tabel 9.2.1. Evenwichtsdrukken van enkele oxiden bij 1273 K (= 1000 °C).
Oxide evO2
p (bar)
FexO 9.3 × 10-16 Fe3O4 8.6 × 10-15 Fe2O3 3.4 × 10-14 Cr2O3 8.7 × 10-23 NiO 4.9 × 10-11 MnO 7.2 × 10-25 Al2O3 4.1 × 10-26 TiO2 2.8 × 10-30 SiO2 1.4 × 10-28 Y2O3 2.8 × 10-43 CeO2 1.1 × 10-34
Voor NiO is de dissociatiedruk 10-74 atm bij kamertemperatuur en iets kleiner dan 10-10 atm bij 1000 °C en dit betekent dus dat de oxidatie van nikkel in lucht (
2Op =
0.2 atm) in dit hele temperatuurgebied een spontane reactie is. Gebruik makend van

175
de Δg0-waarden uit bijlage 11.6 kunnen de evO2
p -waarden van andere oxiden op
dezelfde wijze worden berekend en een aantal waarden voor belangrijke oxiden wordt gegeven in Tabel 9.2.1. We zien hieruit dat voor bijna alle metalen die in de praktijk worden gebruikt de reactie:
m M + (n/2) O2 = MmOn (9.2.5) spontaan naar rechts verloopt bij praktisch belangrijke drukken en temperaturen. Bijna de enige uitzonderingen zijn goud, waarvan bij kamertemperatuur geen stabiel oxide bestaat, zilver (Ag2O dissocieert boven circa 200 °C) en kwik (HgO dissocieert boven 270 °C). Ditzelfde geldt voor de reactie van veel metalen met stoom. Bijvoorbeeld voor de reactie:
3 Fe + 4 H2O → Fe3O4 + 4 H2 (9.2.6) kan met behulp van de thermodynamische gegevens van bijlage 11.6 worden berekend dat de evenwichtsverhouding OHH 22
pp bij 350 °C ongeveer 10 is.
Wanneer deze reactie dus in evenwicht zou zijn bij deze temperatuur zou een stalen oververhitter meer waterstof dan stoom produceren! De experimenteel waargenomen verhouding van ongeveer 10-6 wordt veroorzaakt door een kinetische barrière en wordt in ketelinstallaties soms continu gemeten als controle op eventueel optredende ernstige corrosie. Dit is één van de oudste methoden van continue corrosiebewaking. Uit het bovenstaande volgt dat het al dan niet toepasbaar zijn van metalen bij hoge temperatuur onder oxiderende omstandigheden volledig afhankelijk is van de kinetiek van de oxidatie omdat thermodynamisch gezien oxidatie altijd een spontane reactie is.
9.3 Kinetische vergelijkingen voor hoge-temperatuuroxidatie
Vanuit een praktisch gezichtspunt worden de twee meest belangrijke types hoge-temperatuuroxidatie beschreven met behulp van de parabolische en de lineaire snelheidsvergelijkingen die in Figuur 9.3.1 worden geïllustreerd. Parabolische oxidatie treedt op wanneer een ondoordringbare, hechtende oxidehuid wordt gevormd. De snelheidsvergelijking daarvan heeft de vorm:
tk)x( p2=Δ (9.3.1)
waarin Δx kan staan voor de gewichtstoename tijdens de oxidatie, de dikte van de oxidehuid of het gewichtsverlies van het metaal na verwijdering van de oxidehuid, die allemaal evenredig zijn met elkaar. In deze vergelijking is kp de parabolische snelheidsconstante. Een typisch voorbeeld van zo'n oxidehuid wordt getoond in Figuur 9.3.2. Omdat de snelheid, dat wil zeggen d(Δx)/dt in dit geval met toenemende tijd steeds kleiner wordt spreekt men vaak van beschermende kinetiek.
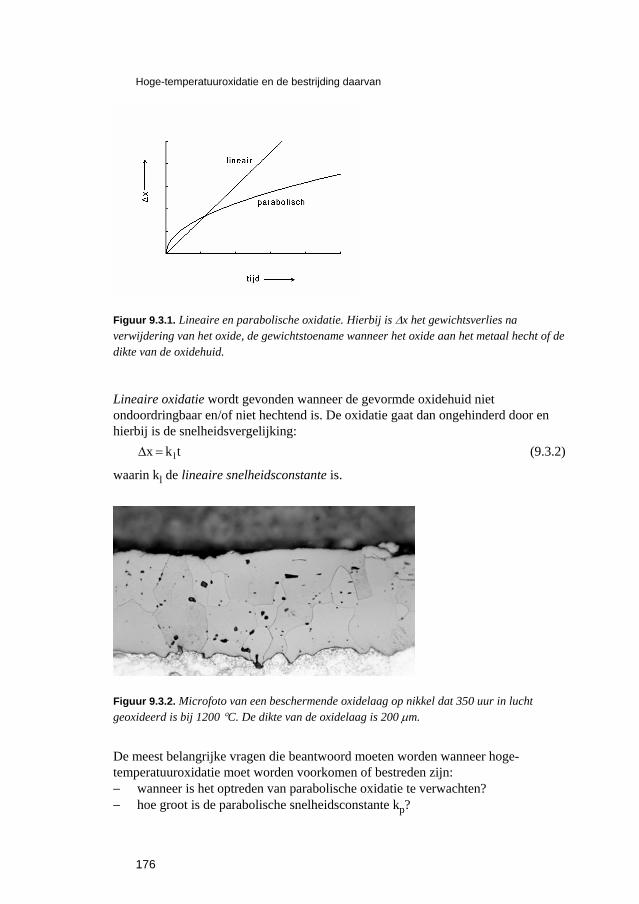
Hoge-temperatuuroxidatie en de bestrijding daarvan
176
Figuur 9.3.1. Lineaire en parabolische oxidatie. Hierbij is Δx het gewichtsverlies na verwijdering van het oxide, de gewichtstoename wanneer het oxide aan het metaal hecht of de dikte van de oxidehuid.
Lineaire oxidatie wordt gevonden wanneer de gevormde oxidehuid niet ondoordringbaar en/of niet hechtend is. De oxidatie gaat dan ongehinderd door en hierbij is de snelheidsvergelijking:
tkx l=Δ (9.3.2)
waarin kl de lineaire snelheidsconstante is.
Figuur 9.3.2. Microfoto van een beschermende oxidelaag op nikkel dat 350 uur in lucht geoxideerd is bij 1200 °C. De dikte van de oxidelaag is 200 μm.
De meest belangrijke vragen die beantwoord moeten worden wanneer hoge-temperatuuroxidatie moet worden voorkomen of bestreden zijn: − wanneer is het optreden van parabolische oxidatie te verwachten? − hoe groot is de parabolische snelheidsconstante kp?

177
Deze vragen worden in de hierna volgende paragrafen beantwoord. (Overigens zijn er nog diverse andere snelheidsvergelijkingen waargenomen en afgeleid, bijvoorbeeld "kubische oxidatie" en "logaritmische oxidatie" waarvoor we naar de literatuur verwijzen [2, 3, 4, 5]).
9.4 Parabolische oxidatie
Zoals hierboven aangegeven vinden we vaak een met de tijd afnemende snelheid van hoge-temperatuuroxidatie die beschreven wordt met een parabolische kromme en we spreken dan van parabolische oxidatie. Deze treedt op wanneer een ondoordringbare en hechtende oxidehuid op het metaal wordt gevormd. De oxidatie blijft doorgaan, zij het met een steeds afnemende snelheid, ten gevolge van diffusie door het oxide. Voor deze diffusie kunnen twee mechanismen onderscheiden worden: a. Een atoom van metaal M, met oxide MO, staat twee elektronen af onder
vorming van een M2+-ion. De metaalionen en de elektronen bewegen dan door het oxide naar het oxide-gas grensvlak. De elektronen worden door zuurstof opgenomen waarbij O2-_ionen worden gevormd. Samen met de M2+-ionen veroorzaken deze de groei van het oxide, In dit geval groeit het oxide aan de buitenkant en dit wordt schematisch getoond in Figuur 9.4.1a.
b. Een metaalatoom geeft weer twee elektronen af aan het oxide onder vorming van een M2+-ion. In dit geval bewegen de elektronen door het oxide naar het oxide-gas grensvlak waar ze weer combineren met zuurstof onder vorming van O2-_ionen. Deze diffunderen dan in omgekeerde richting door het oxide naar het oxide-metaal grensvlak en vormen daar met de M2+-ionen het oxide. Hier vindt de groei dus aan de binnenkant van het oxide plaats zoals schematisch getoond in Figuur 9.4.1b.
a. b.
Figuur 9.4.1. Tijdens hoge-temperatuuroxidatie plaatsvindende processen: a. met diffusie van metaal-ionen; b. met diffusie van zuurstofionen.

Hoge-temperatuuroxidatie en de bestrijding daarvan
178
Of er nu wel of niet een gesloten oxidehuid wordt gevormd hangt af van de zogenaamde Pilling-Bedworth verhouding (PB):
ox
m
nm
dAMmdMM
)metaalgeoxideerdvolume/()OMoxidevanvolume(PB
×××
=
= (9.4.1)
waarin MM = molaire massa van oxide, AM = atomaire massa van metaal, dm = dichtheid van metaal, dox = dichtheid van oxide. Wanneer PB < 1 dan is het oxide niet in staat het metaal te volledig bedekken waardoor het of zal scheuren of poreus zal zijn. Dit vinden we bij de alkali- en aard-alkali-metalen en ook bij een aantal lanthaniden. Geheel volgens de verwachting vertonen deze metalen dan ook lineaire oxidatie. Wanneer PB = 1 kan een gesloten laag worden gevormd zonder inwendige spanningen, maar dit zal slechts zelden worden gevonden. Voor de meeste metalen is PB > 1. Dit betekent dat een gesloten laag kan worden gevormd, maar omdat het volume van het oxide groter is dan van het metaal waaruit het is gevormd, zal in deze laag meestal een drukspanning heersen. Wanneer het mechanisme van de oxidatie is zoals geschetst in Figuur 9.4.1a zijn deze spanningen alleen aanwezig in een dunne oppervlaktelaag waar ze meestal gemakkelijk kunnen verdwijnen zodat parabolische oxidatie kan optreden. Metalen die dit type gedrag vertonen zijn bijvoorbeeld nikkel (PBNiO = 1.52), ijzer (PBFeO = 1.77), chroom (PBCr2O3
= 2.02) en koper (PBCuO = 1.68). Wanneer daarentegen het mechanisme van Figuur 9.4.1b wordt gevolgd worden grote inwendige spanningen in de oxidelaag opgebouwd die in het algemeen niet gemakkelijk vanzelf kunnen verdwijnen. De oxidelaag zal dan afspringen (Engels: "spalling") en dit kan leiden tot versnelde (Engels "break-away") oxidatie. Metalen die dit vertonen zijn bijvoorbeeld zirkoon (PBZrO2
= 1.53) en titaan (PBTiO2 = 1.95).
In deze gevallen wordt een initiële periode met parabolische oxidatie gevolgd door lineaire oxidatie. In sommige gevallen van dit type neemt de oxidatiebestendigheid toe met toenemende temperatuur. De verklaring hiervan is dat bij verhoogde temperatuur het oxide meer plastisch wordt waardoor de spanningen kunnen afnemen en de kans op scheuren wordt verminderd. Voor een diepergaande beschouwing betreffende de invloed van de morfologie van de oxidehuid op de hog-temperatuuroxidatie zie referentie 6.
9.5 Snelheid van parabolische oxidatie
Het is natuurlijk niet voldoende om in een bepaald geval alleen te weten dat parabolische oxidatie optreedt. Het is daarnaast ook nodig om te weten met welke snelheid deze dan verloopt en hoe die snelheid kan worden beïnvloed. De hiervoor algemeen geaccepteerde theorie is voorgesteld door Wagner omstreeks 1930. In het navolgende zal één voorbeeld nader worden uitgewerkt om de belangrijkste punten naar voren te brengen. Het basisidee hierbij is dat in alle kristallijne vaste stoffen defecten aanwezig zijn, dat wil zeggen afwijkingen van de ideale kristalstructuur. Bij

179
de theorie van hoge-temperatuuroxidatie speelt de defectchemie dus een belangrijke rol. We onderscheiden een aantal types defecten waarvan het eerste is de metaalvacature, dat is een plaats in het rooster waar een metaalion ontbreekt dat op die plaats in een ideaal kristal aanwezig is. Analoog kunnen er ook anionvacatures (bijvoorbeeld een zuurstofvacature) aanwezig zijn. Verder kunnen er elektrongaten aanwezig zijn, dat wil zeggen dat een elektron ontbreekt dat er in een ideaal kristal wèl zou zijn, dus bijvoorbeeld is er een Fe3+-ion aanwezig in plaats van een Fe2+-ion. Op analoge wijze kunnen ook vrije elektronen aanwezig zijn (voor een vollediger overzicht en bespreking van defecten in kristallen zie bijlage 11.16) Een belangrijk punt is dat er, in afwezigheid van elektrische velden, altijd combinaties van defecten moeten zijn zodanig dat het kristal als geheel overal elektrisch neutraal is. Als bijvoorbeeld metaalvacatures aanwezig zijn, dat wil zeggen een tekort aan positieve lading, dan moet een equivalent aantal positief geladen defecten aanwezig zijn, bijvoorbeeld elektrongaten. Deze speciale combinatie van defecten komt voor in oxiden zoals FeO, CoO, NiO, Cu2O, enzovoort. Voor de vacatureconcentratie Cvac kan worden afgeleid (zie bijlage 11.16) dat:
n/1vac 2O
pKC ′= (9.5.1)
met n = 6 voor FeO, CoO, NiO en n = 8 voor Cu2O en waarin K' een van het oxide afhankelijke constante is. Zoals besproken in §9.2 is de evenwichtsdruk ev
O2p voor een metaal in evenwicht met
zijn oxide meestal extreem laag. Aan het grensvlak metaal-metaaloxide kan aangenomen worden dat de zuurstofdruk gelijk is aan die evenwichtsdruk mits de oxidatiesnelheid niet al te hoog is. Omdat de zuurstofdruk aan de buitenzijde van de oxidelaag vrijwel altijd veel hoger is dan de evenwichtsdruk volgt uit vergelijking (9.5.1) dat een vacatureconcentratiegradiënt in de oxidehuid ontstaat. Als gevolg daarvan diffunderen de vacatures door het oxide, hetgeen overeenkomt met een flux van metaalionen in de omgekeerde richting. Volgens de tweede wet van Fick is de vacaturediffusiesnelheid evenredig met deze gradiënt, zodat de oxidatiesnelheid daar ook evenredig mee is.

Hoge-temperatuuroxidatie en de bestrijding daarvan
180
Figuur 9.5.1. Concentraties tijdens oxidatie van nikkel volgens de theorie van Wagner.
Onder de condities zoals geschetst in Figuur 9.5.1. vinden we dan:
xADC
x]CC[AD
dtxd buitenz,vacbinnenz,vacbuitenz,vac
Δ≈
Δ
−=
Δ (9.5.2)
Hierin is Δx de dikte van het oxide, A een evenredigheidsconstante, D de diffusiecoëfficiënt, Cvac,buitenz de vacatureconcentratie bij het grensvlak met het gas en Cvac,binnenz die aan het grensvlak tussen metaal en oxide.De tweede vorm van vergelijking (466H9.5.2) is in dit soort gevallen meestal een goede benadering omdat vrijwel altijd
22 OevO pp << .
Integreren van vergelijking (466H9.5.2) en invullen Δx = 0 voor t = 0 geeft dan de parabolische snelheidsvergelijking:
t.C.AD2)x( buitenz,vac2=Δ (9.5.3)
waarbij de tweede vorm van vergelijking (466H9.5.2) gebruikt is. Vergelijken met vergelijking (459H9.3.1) geeft voor de parabolische snelheidsconstante:
n/1Obuitenz,vacp 2
p'ADK2ADC2k == (9.5.4)
waarbij voor de tweede vorm gebruik is gemaakt van vergelijking (462H9.5.1). Een belangrijk gevolg van dit resultaat is dat de parabolische snelheidsconstante afneemt met dalende partiële zuurstofdruk in de atmosfeer, maar we zien ook dat deze afname veel minder dan evenredig is. Zowel K', die in feite een evenwichtsconstante is, als D nemen exponentieel toe met toenemende temperatuur. Uit vergelijking (472H9.5.4) volgt dat de oxidatiesnelheid dus op dezelfde manier toeneemt. Volledig analoge beschouwingen en conclusies zijn van toepassing op oxiden met andere types defecten, hoewel de details van de afleiding en de resultaten natuurlijk verschillen. We gaan daarop hier niet verder in en verwijzen de geïnteresseerde lezer naar bijlage 11.16 en de literatuur [2 - 5].

181
Tabel 9.5.1. Parabolische snelheidsconstanten kp (in g2 cm-4 min-1) voor nikkel-chroomlegeringen bij 1000 °C.
Gewichts% chroom Kp atmosfeer kp 0 3.8 × 10-10 O2 2.5 × 10-10 0.3 15 × 10-10 O2 + Li2O 5.8 × 10-11 1.0 28 × 10-10 3.0 36 × 10-10 10.0 5 × 10-10
Met behulp van deze theorie kan ook de invloed van legeringselementen op de oxidatiesnelheid worden verklaard. Wanneer bijvoorbeeld chroom wordt toegevoegd aan nikkel is dit in het oxide aanwezig als Cr3+-ionen. Om de elektroneutraliteit te handhaven neemt de hoeveelheid nikkelvacatures toe en het aantal elektrongaten af (zie bijlage 11.16). Daardoor stijgt de vacatureconcentratie boven de waarde gegeven door vergelijking (462H9.5.1) en dit leidt dan tot een evenredige verhoging van de oxidatiesnelheid. Tabel 9.5.1 toont dat dit in overeenstemming is met de experimentele waarneming voor niet te hoge chroomconcentratie in de legering. Wanneer Li+-ionen in het oxide worden opgenomen, hetgeen mogelijk is door de oxidatie uit te voeren in aanwezigheid van gasvormig Li2O, gebeurt het omgekeerde, zoals te zien is in Tabel 9.5.1. Bij hoge chroomgehalten gaat deze theorie niet meer op omdat chroom een veel hogere affiniteit voor zuurstof heeft dan nikkel (zie bijlage 11.6) en chroom preferentieel wordt geoxideerd bij hogere concentraties. De chroomconcentratie in het oxide is dan veel hoger dan in de legering en inplaats van een NiO-type oxide ontstaat een oxide van het type NiCr2O4. De diffusie in dat oxide is, door de geheel andere structuur, veel langzamer dan in NiO en dit verklaart de afname van de oxidatiesnelheid bij chroomgehaltes boven 5 tot 6 gewichts%. Voor preferentiële oxidatie zie ook opgave 6 van §9.9. Omdat volgens het hier besproken mechanisme bij hoge-temperatuur oxidatie sprake is van het transport van geladen deeltjes, kunnen hierop net als bij corrosie in waterige oplossing, ook elektrochemische beschouwingen worden toegepast. Voor een verdere bespreking hiervan verwijzen we naar het overzicht gegeven door de Wit en Fransen [1].
9.6 Voorkomen en bestrijden van hoge-temperatuuroxidatie
9.6.1 Algemeen
In §9.5 is afgeleid is één van de methoden om de snelheid van hoge-tempera-tuuroxidatie te verlagen het verlagen van de (partiële) zuurstofdruk. Maar om de oxidatiesnelheid met een factor 10 te verlagen moet de zuurstofdruk met een factor 104 tot 108 worden verlaagd zoals blijkt uit vergelijking (472H9.5.4) (zie ook opgave 2 in §9.9).

Hoge-temperatuuroxidatie en de bestrijding daarvan
182
In ketelinstallaties en fornuizen kan een verlaging van de zuurstofdruk soms worden bereikt door het verbrandingsproces zo te regelen dat stoichiometrische hoeveelheden brandstof en zuurstof worden gebruikt. De verbrandingsgassen zijn dan vrijwel zuurstofvrij zodat oxidatie wordt verminderd. In de praktijk is dit alleen mogelijk met gasvormige of vloeibare brandstoffen omdat bij gebruik van vaste brandstoffen de verbranding meestal ongelijkmatig is zodat daarbij inefficiënt gebruik van brandstof op zou treden en er toch restzuurstof in de verbrandingsgassen aanwezig zou zijn. Verder kan, zoals genoemd in §9.2 (zie ook opgaven 5 en 7 van §9.9), oxidatie ook worden veroorzaakt door andere gassen dan zuurstof. De meest belangrijke in dit opzicht zijn water en kooldioxide. Het eventuele optreden van oxidatie hangt daarbij natuurlijk af van het metaal en van de omstandigheden zoals temperatuur en partiële druk van het gas. Warmtebehandeling van onderdelen wordt vaak uitgevoerd in een beheerste atmosfeer om oxidatie van de onderdelen en daarmee kostbare nabehandelingen en/of verandering van afmetingen te voorkomen. Voorbeelden van dergelijke atmosferen zijn stikstof met 5% waterstof, gekraakte ammoniak en argon. Ovensolderen van metalen zoals roestvast staal of titaan wordt uitgevoerd in hoog vacuüm. Dit is echter niet voldoende om reeds aanwezige oxidehuiden, die bevochtiging met vloeibaar metaal verhinderen, te verwijderen en daarom worden fluxen gebruikt. Bij normale zuurstofdruk is heroxidatie zo snel dat solderen, zelfs in aanwezigheid van een flux niet mogelijk is. In een voldoend hoog vacuüm wordt de oxidatiesnelheid echter zo klein dat bevochtiging sneller is dan heroxidatie.
9.6.2 Materiaalkeuze
Vanzelfsprekend is het bij oxidatie van onderdelen die warmtebehandeld moeten worden niet mogelijk hoge-temperatuuroxidatie te voorkomen door een ander materiaal te kiezen: die keuze wordt door andere overwegingen bepaald. Maar voor onderdelen of constructies die continu bij hoge temperatuur worden gebruikt is het in het algemeen wèl mogelijk materialen te kiezen die onder de heersende omstandigheden beter tegen hoge-temperatuuroxidatie bestand zijn.

183
Tabel 9.6.1. Maximale bedrijfstemperatuur van verschillende legeringen zonder sterke oxidatie in lucht (metaalverlies kleiner dan circa 1 mm per jaar).
Legering Typische handelsnaam
Maximale bedrijfstemperatuur (°C)
ongelegeerd staal 500 - 550 ijzer met 1%Cr, 0.5%Mo 600 ijzer met 4-6%Cr 650 ijzer met 9%Cr 750 ijzer met 13%Cr 750 - 850 ijzer met 17%Cr 850 - 900 ijzer met 18%Cr, 8% Ni 900 ijzer met 27%Cr 1050 – 1100 ijzer met 25%Cr, 20% Ni 1150 ijzer met 25%Cr, 1% Y 1350 ijzer met 30%Cr, 5% Al, 0.5% Si megapyr 1300 ijzer met 24%Cr, 5.5% Al, 2% Co kanthal A 1300 Nikkel 900 - 950 nikkel met 20% Cr nichroom 20 1100 nikkel met 16% Cr, 7% Fe inconel 1050 nikkel met 10% Cr chromel P 1100 nikkel met 2% Al, 2% Mn, 1% Si alumel 1100 platina met 10% Rh 1400
Zoals aangetoond in §9.5 (zie ook bijlage 11.16) kan de oxidatiebestendigheid van een materiaal worden verhoogd door het toevoegen van geschikte legeringselementen. Deze kunnen één van de twee volgende effecten hebben: vermindering van de diffusie door een bepaald oxide, of de vorming van een ander, beter beschermend oxide. In feite wordt in de grote meerderheid van de gevallen van de tweede mogelijkheid gebruik gemaakt. Typische voorbeelden zijn chroom, silicium en aluminium in ijzer, chroom en aluminium in nikkel, aluminium in koper, enzovoort. Tabel 9.6.1. geeft de maximale bedrijfstemperatuur van een aantal legeringen die veel gebruikt worden bij hoge temperatuur in oxiderende atmosfeer. Het kiezen van een geschikt materiaal is in veel gevallen de enige mogelijkheid om hoge-temperatuuroxidatie effectief te bestrijden. In principe is het ook mogelijk om hoge-temperatuuroxidatie te bestrijden door het toepassen van beschermende lagen. Maar deze zijn in dit geval beperkt tot anorganische en metallische deklagen. Een voorbeeld van de tweede mogelijkheid is het aluminiseren van staal. Dit wordt uitgevoerd door het staal te bedekken met een dunne aluminiumlaag waarna deze een warmtebehandeling ondergaat waarbij een uit ijzer-aluminiumverbindingen bestaande oppervlaktelaag bestaat. Deze hebben een veel hoger smeltpunt dan zuiver aluminium (650 °C) gecombineerd met een grote bestendigheid tegen oxidatie.

Hoge-temperatuuroxidatie en de bestrijding daarvan
184
Als niet-metallische deklagen kunnen keramische lagen, email en op silicaten gebaseerde verfsoorten worden toegepast. Dit is een nogal specialistisch gebied, ook in verband met andere eisen zoals thermische schokbestendigheid, mechanische sterkte, hechting, enzovoort zodat het niet gemakkelijk is om algemene regels te geven. Voor toepassing en eigenschappen van keramische deklagen wordt verwezen naar het overzicht van Haanappel en Fransen [7].
9.6.3 Vormgeving
Hoewel de vormgeving meestal niet de mogelijkheid biedt om hoge-tempera-tuuroxidatie volledig tegen te gaan kunnen de gevolgen daarvan echter duidelijk minder ernstig worden indien de vormgeving is aangepast aan de gebruiksomstandigheden. Hoge-temperatuuroxidatieweerstand berust altijd op de aanwezigheid van een hechtende en ondoordringbare oxidehuid. Wanneer deze plaatselijk beschadigd of verzwakt wordt oxidatie versneld. Zo kunnen plaatselijke temperatuurverschillen tot voldoend hoge thermische spanningen leiden dat scheuren in het oxide ontstaan. Een voorbeeld hiervan wordt gegeven in Figuur 9.6.1. Dit betreft oververhitter pijpen die aangebracht waren in tamelijk nauwsluitende stalen steunplaten. Deze platen werkten in feite als verwarmingsribben waardoor de pijpwandtemperatuur plaatselijk veel hoger was dan op basis van de stoomtemperatuur kon worden verwacht. Op zich leidt dit al tot versnelde oxidatie. Maar bovendien veroorzaakte het temperatuurverschil ook verhoogde spanningen en daardoor scheuren in de oxidehuid. Dit leidde tot lineaire oxidatiekinetiek en daarmee tot het zeer snelle falen na slechts enkele maanden in bedrijf te zijn geweest.
a b
Figuur 9.6.1. Plaatselijk versnelde oxidatie van oververhitterpijp: a. foto van pijpen na falen; b. schematische doorsnede van de constructie.
Het is tenslotte goed nog eens te benadrukken dat oxidatieweerstand altijd afhangt van de aanwezigheid van een dichte en hechtende oxidelaag, Alles wat kan leiden tot (plaatselijk) verwijderen, beschadigen of verzwakken van die laag, zoals

185
mechanische beschadiging, plotselinge grote temperatuurverschillen (thermische schok), grote plaatselijke temperatuurverschillen, enzovoort moeten dus vermeden worden. Geen enkel hoge-temperatuur materiaal is beter dan de daarop aanwezige oxidehuid!
9.7 Hoge-temperatuurcorrosie in andere gassen
Naast hoge-temperatuuroxidatie kunnen metalen bij hoge temperatuur ook met andere gasvormige componenten reageren en de belangrijkste hiervan zijn zwavel en chloor. In Omdat zwavel onder veel omstandigheden voorkomt in gassen is sulfidatie erg belangrijk. In atmosferen waarin alleen zwavel voorkomt is de sulfidatiesnelheid in het algemeen aanzienlijk groter dan de oxidatiesnelheid bij overigens vergelijkbare omstandigheden. Enkele voorbeelden zijn gegeven in Tabel 9.7.2. Sulfidatie verloopt, net als oxidatie, vaak volgens een parabolische kinetiek en de in §488H9.5 besproken Wagner-theorie is daarop van toepassing. Dat de sulfidatiesnelheid zoveel groter is komt door de kleinere roosterenergie van sulfiden ten opzichte van die van de oxiden. In de eerste plaats is daardoor de concentratie van puntfouten veel groter en in de tweede plaats is de activeringsenergie van de diffusie daardoor veel kleiner. Tezamen leidt dit tot een veel sneller transport van defecten door de sulfidehuid en daarmee tot een veel grotere snelheid dan bij oxidatie, onder overigens vergelijkbare omstandigheden.
Tabel 9.7.1 zijn samenstellingen gegeven van enkele praktisch belangrijke gasatmosferen uitgedrukt als de partiële drukken van O2, S2 en Cl2. Omdat zwavel onder veel omstandigheden voorkomt in gassen is sulfidatie erg belangrijk. In atmosferen waarin alleen zwavel voorkomt is de sulfidatiesnelheid in het algemeen aanzienlijk groter dan de oxidatiesnelheid bij overigens vergelijkbare omstandigheden. Enkele voorbeelden zijn gegeven in Tabel 9.7.2. Sulfidatie verloopt, net als oxidatie, vaak volgens een parabolische kinetiek en de in §9.5 besproken Wagner-theorie is daarop van toepassing. Dat de sulfidatiesnelheid zoveel groter is komt door de kleinere roosterenergie van sulfiden ten opzichte van die van de oxiden. In de eerste plaats is daardoor de concentratie van puntfouten veel groter en in de tweede plaats is de activeringsenergie van de diffusie daardoor veel kleiner. Tezamen leidt dit tot een veel sneller transport van defecten door de sulfidehuid en daarmee tot een veel grotere snelheid dan bij oxidatie, onder overigens vergelijkbare omstandigheden.

Hoge-temperatuuroxidatie en de bestrijding daarvan
186
Tabel 9.7.1. Samenstellingen van enige in de praktijk voorkomende gasatmosferen waarin hoge temperatuur oxidatie, sulfidatie of chloreren op kan treden.
Gasmengsel 2Op (bar)
2Sp (bar) HClp (bar)
Oxiderend lucht 2.0 × 10-1 ··· ··· verbrandingsgas 3.0 × 10-2 ··· ··· vuilverbranding 1.4 × 10-2 2.0 × 10-31 1.0 × 10-2 Reducerend vergassing 1.3 × 10-22 ··· ··· vergassing met zwavel 1.3 × 10-22 1.4 × 10-7 ··· vergassing met zwavel en chloor 1.3 × 10-22 1.4 × 10-7 1.0 × 10-2
Tabel 9.7.2. Parabolische snelheidsconstante kpvoor oxidatie en sulfidatie van enkele metalen in O2 en S2 bij 1 atm.
Reagerend gas kp (g.cm-4s-1) Ni Co Fe Cr O2 9.1 × 10-11 1.6 × 10-9 5.5 × 10-8 4.5 × 10-12 (1000 °C ) (950 °C ) (800 °C ) (1000 °C ) S2 8.5 × 10-4 6.7 × 10-6 8.1 × 10-6 8.1 × 10-7 (620 °C ) (720 °C ) (800 °C ) (1000 °C )
Wanneer zwavel en zuurstof beide in het gas aanwezig zijn hangt het van de partiële drukken af welk van de processen zal overheersen. Bij voldoend hoge zuurstofdruk kan een beschermende oxidehuid worden gevormd, waardoor sulfidatie niet of vrijwel niet optreedt. Bij lage zuurstofdruk, dat wil zeggen onder reducerende omstandigheden, kan echter toch sulfidatie optreden. Thermodynamisch wordt dit aangegeven met zogenaamde stabiliteitsdiagrammen waarvan in Figuur 9.7.1 een eenvoudig voorbeeld is gegeven voor een enkelvoudig metaal M. Boven de lijn 2 - 1 is het sulfide stabiel ten opzichte van het metaal, rechts van de lijn 3 - 1 is het oxide stabiel ten opzichte van het metaal. Op de lijn 1 - 4 zijn sulfide en oxide met elkaar in evenwicht, in gebied I is het sulfide stabiel ten opzichte van het oxide, in gebied II juist omgekeerd.

187
Figuur 9.7.1. Stabiliteitsdiagram van enkelvoudig metaal M in atmosferen die zowel zuurstof als zwavel bevatten.
In het gebied MO + II is dus een eventueel beschermende oxidelaag mogelijk, in de gebieden MS + I zal een sulfidelaag stabiel zijn en is het gevaar voor snelle sulfidatie aanwezig. Bij legeringen moeten twee of meer diagrammen van dit type worden gesuperponeerd, maar daarop kan in dit kader niet verder worden ingegaan. Wanneer een metaal wordt blootgesteld aan een gas dat chloorverbindingen, bijvoorbeeld in de vorm van HCl of Cl2, bevat blijkt in de praktijk een volledige verandering van de kinetiek op te treden. Dit wordt veroorzaakt door het feit dat vele metaalchloriden bij hoge temperatuur vluchtig zijn. Hierdoor treedt, zelfs als toch nog een oxidehuid wordt gevormd toch een gewichtsverlies in plaats van een gewichtstoename op. Ook het oxide zal namelijk aan de gaszijde met de chloorverbindingen reageren, bijvoorbeeld:
)g(O)g(OH3)g(ClCo3)g(HCl6)s(OCo2 225
24243 ++↑→+
Hierdoor neemt de dikte van de oxidehuid af en is de gecombineerde oxidatie plus chloridevorming veel sneller dan de oxidatie alleen. Dit is weer een tamelijk gespecialiseerd gebied en we verwijzen voor een uitvoeriger behandeling naar de literatuur [8].
9.8 Dauwpuntcorrosie
Een speciale vorm van corrosie, die in bepaalde opzichten tussen hoge-temperatuurcorrosie en elektrochemische corrosie in ligt is dauwpuntcorrosie of condensaatcorrosie.. Een voorbeeld hiervan is al genoemd in §8.7 (zie Figuur 8.7.10). Deze vorm van corrosie treedt op wanneer hete gassen, in de meeste gevallen verbrandingsproducten, zover worden afgekoeld dat condensatie optreedt. In het bijzonder in gevallen waar zwaveldioxide of zwaveltrioxide in het gas aanwezig zijn, bijvoorbeeld bij de verbranding van zwavelhoudende brandstoffen,

Hoge-temperatuuroxidatie en de bestrijding daarvan
188
kan het dauwpunt, dat wil zeggen de temperatuur waarbij condensatie op begint te treden, wel bij 150 tot 200 °C liggen. Het daarbij gevormde sterk zure condensaat is zeer agressief en kan tot ernstige aantasting van on- en laaggelegeerd staal leiden. Één van de meest belangrijke methoden om dit type corrosie te voorkomen is de temperatuur van het gas zo hoog te houden dat die overal boven het dauwpunt blijft. Een typisch geval wordt geïllustreerd in Figuur 9.8.1. waarin zowel het oorspronke-lijke als het verbeterde ontwerp van een geluiddemper van een diesellocomotief worden getoond. Om de demping van het geluid te verbeteren was het oorspronke-lijke ontwerp voorzien van een dubbele wand met een geperforeerde binnenplaat. Tussen de binnen- en buitenplaten was een laag glaswol aangebracht voor extra geluiddemping. Deze laag werkte echter ook als warmte-isolatie waardoor de buitenplaat, door afkoeling aan de buitenlucht, zo koud werd dat daarop aan de binnenzijde condensatie optrad, met als gevolg ernstige corrosie. Door een dichte plaat als binnenplaat te gebruiken werd het binnendringen van de agressieve verbrandingsgassen voorkomen en werd de corrosie volledig tegengegaan.
a b
Figuur 9.8.1. Schets van doorsnede van een deel van een geluiddemper van een diesel-locomotief: a. oorspronkelijk ontwerp; b. verbeterd ontwerp.
Een tweede methode om deze vorm van corrosie te voorkomen of in ieder geval te verminderen is gebruik te maken van brandstoffen waaruit bij verbranding geen agressieve verbrandingsproducten ontstaan. Wanneer geen van deze methoden kan worden toegepast is het noodzakelijk over te gaan tot het gebruik van een ander materiaal of van een deklaag of binnenbekleding. De keuze tussen de verschillende methoden hangt natuurlijk weer af van economi-sche factoren en van de afmetingen van de installatie. Het is een heel verschil of het gaat om een industriële schoorsteen van misschien 4 à 5 meter diameter en 75 meter hoogte of het uitlaatsysteem van een auto!
9.9 Opgaven
1. Bereken de Pilling-Bedworth-verhouding voor Al2O3 op Al, MgO op Mg en Na2O op Na. Verwacht u dat deze oxiden beschermend zijn?

189
2. Bereken met welke factor de oxidatiesnelheid van nikkel verkleind wordt wanneer dat in lucht bij een druk van 0.1 mm Hg wordt geoxideerd in plaats van bij atmosferische druk. Bereken dit ook voor een druk van 10-6 mm Hg.
3. Om de evenwichtsdruk van oxiden te berekenen: 1. Leidt een vergelijking af voor Δg van de vorming van enkele metaaloxiden
(bijvoorbeeld Fe3O4, Cu2O, Cr2O3, enzovoort). Leidt uit deze vergelijking een vergelijking voor de dissociatie- of evenwichtsdruk van deze oxiden af.
2. Gebruik dit resultaat en de gegevens in bijlage 11.6 om de dissociatiedruk van een aantal oxiden te berekenen bij kamertemperatuur, 500 °C en 1000 °C.
4. Bereken de temperatuur waarbij Ag2O in evenwicht is met metallisch zilver en lucht (pO2 = 0.2 atm). Wat gebeurt er indien zilver boven deze temperatuur wordt verhit in lucht?
5. Een bespreking zoals in §8.2 is gegeven voor de oxidatie door zuurstof is ook toe te passen op oxidatie door andere oxidatiemiddelen zoals stoom: 3 Fe + 4 H2O = Fe3O4 + 4 H2. Leidt een vergelijking af voor de Δg van deze reactie en toon aan dat de neiging van ijzer om geoxideerd te worden nu bepaald wordt door de verhouding van de partiële drukken van stoom en waterstof. Gebruik de gegevens uit bijlage 11.6 om de evenwichtsverhouding H2O / H2 te berekenen van stoom in contact met ijzer bij 350 °C.
6. Wanneer in een binaire legering van de metalen M en N het oxide van M stabieler is dan dat van N is preferente oxidatie van M mogelijk. a. in welke richting zal de reactie
NO + M = N + MO dan de neiging hebben om spontaan te verlopen?
Wanneer aangenomen wordt dat de legering zich gedraagt als een ideale oplossing van de metalen in elkaar wordt de thermodynamische potentiaal μi (i = M of N) van de component in de legering gegeven door: μi = μi
0 + RT ln xi waarin xi de molfractie van i in de legering is, waarbij x1 + x2 = 1 voor een binaire legering. b. Toon aan dat voor tweewaardige metalen preferente oxidatie van M alleen
plaats vindt boven een kritische molfractie (xM)c die gegeven wordt door:
[ ]RT)gg(expA
)A1(A)x(0NO
0MO
cM
Δ−Δ=
+=
waarin 0jgΔ de vrije vormingsenthalpie van oxide j is.
c. Bereken de kritische concentratie, in gewichts%, voor preferentiële oxidatie bij 750 °C van ijzer, opgelost in nikkel, onder de aanname dat alleen FeO en NiO worden gevormd.

Hoge-temperatuuroxidatie en de bestrijding daarvan
190
7. In een bepaald type kernreactor wordt CO2 gebruikt als koelmiddel en grafiet als moderator. Dezelfde stoffen, maar dan in andere concentraties komen voor bij de vergassing van kolen en olie. a. Leidt een vergelijking af voor Δg voor de reactie
M + CO2 → MO + CO. In zo'n systeem is het zogenaamde Boudouard-evenwicht: CO2 + C = 2 CO
meestal ingesteld. Bij een druk van 60 atm betekent dit dat: bij 500 °C : pCO = 0.523 atm bij 800 °C : pCO = 17.77 atm.
b. Bereken uit deze gegevens of bij 500 °C en bij 800 °C koper (onder vorming van CuO), ijzer (onder vorming van FeO) of nikkel (onder vorming van NiO) de neiging vertonen spontaan geoxideerd te worden door het overeenkomstige CO2/CO-mengsel.
9.10 Literatuur
1. J.H.W. de Wit and T. Fransen, Corrosion studies, in: CRC Handbook of Solid State Electrochemistry, ed. by P.J. Gellings and H.J.M. Bouwmeester, Boca Raton, CRC Press (1997)
2. J.H.W. de Wit, High temperature oxidation of metals, J.Educational Modules in Materials Science and Engineering 3 (1981) 333 - 377
3. P. Kofstad, High temperature corrosion, New York, Elsevier (1988). 4. O. Kubaschewski and B.E. Hopkins, Oxidation of metals and alloys, London,
Butterworths (1962) 5. N. Birks and G.H. Meier, Introduction to high temperature oxidation of metals, London,
Edward Arnold (1983) 6. J.H.W. de Wit, The influence of metal oxide morphology on the high temperature metal
oxidation, J.Educational Modules in Materials Science and Engineering 5 (1983) 7 - 29 7. V.A.C. Haanappel en T. Fransen, Technische keramiek als bescherming tegen hoge-
temperatuurcorrosie, Klei, Glas en Keramiek 13 (nr. 6/7, 1992) 209 - 213. 8. V.A.C. Haanappel, T. Fransen and P.J. Gellings, Chlorine-induced high-temperature
corrosion: I Metals and alloys - A review, High Temperature Materials and Processes 10 (1992) 67 - 89.

10
Epiloog: hoe kunnen we corrosie voorkomen of bestrijden?


193
In §1.1 is gezegd dat de term corrosie zowel betrekking heeft op het proces van de aantasting zelf als op de schade die door de aantasting is veroorzaakt. In de verdere hoofdstukken zijn veel verschillende methoden besproken die be-schikbaar zijn om corrosie, in beide betekenissen van het woord, te voorkomen en te bestrijden. Daarbij is ook de achtergrond van die methoden aan de orde gekomen. Vanzelfsprekend komen in een bepaalde praktische situatie maar een beperkt aantal van die methoden voor toepassing in aanmerking. Om van de met dit boek verworven kennis gebruik te maken is het altijd nodig: 1. te beslissen welke methoden in een bepaald geval toepasbaar zijn; 2. die methode te kiezen die de grootste kans op succes geeft en tegelijkertijd
economisch acceptabel is. De bedoeling van dit hoofdstuk is het nog eens overzien van de verschillende moge-lijkheden die beschikbaar zijn voor het voorkomen en bestrijden van corrosie en aanwijzingen te geven langs welke weg men kan komen tot een beslissing hierover in bepaalde praktische situaties. Als hulpmiddel in dit overzicht wordt gebruik gemaakt van het ontwerpschema van Figuur 8.2.1, dat in verband met het belang daarvan in Figuur 10.1 nog eens wordt herhaald. Wanneer we er van uit gaan dat de functie en de gewenste levensduur in een bepaald geval zijn gegeven is de eerste stap het verkrijgen van een juiste interpretatie van het milieu, of de milieus, waarin de te ontwerpen apparatuur of constructie moet worden gebruikt. Omdat het gedrag van veel materialen sterk beïnvloed kan worden door componenten die in kleine hoeveelheden aanwezig zijn, bijvoorbeeld koper en koperlegeringen zijn zeer gevoelig voor zelfs kleine hoeveelheden ammoniumverbindingen, is het niet voldoende alleen een globaal idee te hebben van de samenstelling van het milieu. Verder moeten natuurlijk het temperatuurgebied waarin de apparatuur wordt gebruikt, de stromingssnelheid van de vloeistof, de eventuele aanwezigheid van gesuspendeerde vaste stoffen in de vloeistof en andere omstandigheden, zoals intermitterende of continue bevochtiging, enzovoort allemaal bekend zijn. In bepaalde gevallen, bijvoorbeeld in verbrandingsmotoren, zijn verschillende delen van de apparatuur tegelijkertijd in aanraking met verschillende milieus en/of zijn onderhevig aan verschillende omstandigheden, zodat ook deze allemaal bekend moeten zijn. De volgende stap is het verkrijgen en interpreteren van corrosiegegevens van verschillende, eventueel in aanmerking komende materialen, in het milieu of (de milieus) onder de heersende omstandigheden. Dit kan worden gedaan met de in hoofdstuk 7 besproken methoden, dat wil zeggen het gebruik van gepubliceerde gegevens indien die beschikbaar zijn of het uitvoeren van geschikte metingen, indien mogelijk in de praktijk of in het laboratorium onder praktijkomstandigheden. De resultaten hiervan worden dan gebruikt bij het gedetailleerde ontwerp van de onderhavige apparatuur. De corrosiegegevens moeten natuurlijk worden gecombineerd met de gegevens van andere eisen die hier niet worden besproken, zoals mechanische sterkte, bewerkbaarheid, enzovoort. Met nadruk moet er echter

Epiloog: hoe kan corrosie worden voorkomen of bestreden
194
op worden gewezen dat het rekening houden met corrosie een integraal onderdeel van het ontwerpproces dient te zijn.
Figuur 10.1. Algemene aspecten van het voorkomen en bestrijden van corrosie.
Zoals aangegeven in Figuur 10.1 omvat het ontwerp niet alleen de vormgeving (het meetkundige ontwerp) maar ook de materiaalkeuze, de keuze van een beschermende laag en beslissingen betreffende eventuele wijzigingen of aanpassingen van milieu en/of omstandigheden. Ook al zijn deze verschillende punten afzonderlijk in het schema getoond vertonen zij een sterke onderlinge samenhang en moeten ze in werkelijkheid als één samenhangende activiteit worden gezien, dat in de figuur met de gestippelde rechthoek is aangegeven. Tijdens het ontwerp moet de economische betekenis van de verschillende alternatieven ook in beschouwing worden genomen, bijvoorbeeld door het toepassen van de methoden die besproken zijn in §8.8. De keuze van het materiaal is in de eerste plaats gebaseerd op de gegevens verkregen met de in §7.2 besproken methoden, gebruikmakend van de algemene achtergrond die in §8.4 is gegeven. Wanneer het niet lukt om een materiaal te vinden dat bestand tegen het beschouwde milieu onder de gegeven omstandigheden moet van één of andere methode van corrosiebescherming worden toegepast zoals hieronder verder wordt besproken. In ieder geval is het noodzakelijk om, na vaststelling dat een bepaald corrosiebestendig materiaal aan alle eisen voldoet, na te gaan hoe de kosten zich verhouden tussen toepassen van een duur materiaal zonder of een goedkoper materiaal met additionele corrosiebescherming. Wanneer corrosiebescherming nodig is zijn de algemene mogelijkheden: 1. toepassen van een beschermende deklaag; 2. veranderen van milieu of omstandigheden; 3. toepassen van elektrochemische bescherming; 4. een combinatie van twee (of meer) van de bovenstaande methoden.

195
De meest belangrijke soorten beschermende lagen zijn besproken in §8.6, samen met de belangrijkste toepassingsgebieden, eigenschappen en aanbrengmethoden. Het belang van goede oppervlakte voorbehandeling en van de invloed van de vormgeving (zie ook §8.7) zijn daarbij ook benadrukt. Het is ook belangrijk om te beseffen dat sommige tijdelijke corrosiebestrijdingsmiddelen (zie §8.6.6) kunnen leiden tot zacht worden of zwellen van bepaalde verfsoorten. Ook weer een reden om reeds in het ontwerpstadium met deze en andere wisselwerkingen rekening te houden. Corrosiebestrijding door verandering van het milieu kan op verschillende manieren worden uitgevoerd zoals in het bijzonder is besproken in de hoofdstukken 5, 6 en 8. Het verlagen van de elektrolyt (= zout-)concentratie kan leiden tot verminderde aantasting ten gevolge van de verhoogde elektrische weerstand van het milieu (zie §5.5). Verhogen van de pH vermindert vaak de aantasting, behalve bij amfotere materialen, zoals aluminium. Het effect van verhoogde pH kan te danken zijn aan een verminderde bijdrage van waterstofontwikkeling aan de kathodische stroomdichtheid (§5.3) of aan het bevorderen van de vorming van een passieve laag (§5.7). Toevoeging van inhibitoren of beitsremmen (§5.3) of passivatoren (§5.7) geeft ook een vermindering van de corrosiesnelheid. Verwijderen van zuurstof vermindert de corrosiesnelheid wanneer zuurstofreductie de kathodische reactie is (§5.4) en ook bij hoge-temperatuuroxidatie (hoofdstuk 9). De tweede mogelijkheid is het veranderen van de omstandigheden. In dit opzicht is vooral de stromingssnelheid van de vloeistof een belangrijke factor. Wanneer er gevaar is voor put- of afzettingscorrosie kan de aantasting worden verminderd door de stromingssnelheid te vergroten (§6.3 en 6.4) maar als er gevaar is voor erosie- of cavitatiecorrosie (§6.10) moet die juist worden verkleind. Ook spanningen zijn in hoge mate afhankelijk van het ontwerp en moeten zo laag mogelijk worden gehouden om spanningscorrosie te voorkomen (§6.8). Elimineren of verminderen van trillingen en kleine relatieve bewegingen van onderdelen wordt toegepast om passingroest te voorkomen (§6.11). Verbeterde isolatie of toepassing van andere methode van temperatuurbeheersing kunnen het gevaar voor dauwpuntcorrosie verkleinen (§9.8). Tenslotte kan elektrochemische bescherming worden toegepast. De twee hiervoor beschikbare methoden zijn kathodische en anodische bescherming (zie §8.5 en 5.7). Kathodische bescherming wordt vaak gecombineerd met de toepassing van deklagen om de benodigde stroom te verkleinen. Wanneer men klaar is met al deze punten moet de apparatuur nog gefabriceerd worden. Zoals ook genoemd in §8.7 moet de fabricage in overeenstemming zijn met het ontwerp. Dus moeten om te beginnen volledige en duidelijke instructies aan de werkplaats worden verstrekt. Maar het betekent ook dat regelmatige inspectie van de corrosiebeschermingsaspecten nodig is, hetgeen helaas vaak over het hoofd wordt gezien. Bovendien moet niet alleen achteraf worden geïnspecteerd, ook tijdens de constructie en het aanbrengen van de beschermingsmiddelen moet regelmatig worden geïnspecteerd. Hierbij is ook terugkoppeling naar de ontwerper van groot

Epiloog: hoe kan corrosie worden voorkomen of bestreden
196
belang. Omdat vaak onverwachte problemen opdoemen die veranderingen in het ontwerp nodig maken is het vanzelfsprekend nodig om de invloed daarvan op de corrosie mede in beschouwing te nemen! Hier moet nog aan toe worden gevoegd dat ook de ervaringen die worden opgedaan tijdens gebruik en onderhoud een onderdeel van het gehele proces dienen te vormen. Ook hier is terugkoppeling naar de ontwerper van groot belang, zowel voor het oplossen van daarbij geconstateerde problemen als voor het voorkomen daarvan in toekomstige apparaten en constructies. Uit het bovenstaande volgt dat het onmogelijk is om één standaardvoorschrift te geven voor een universele oplossing van alle corrosieproblemen. Ieder praktijkgeval moet afzonderlijk en in detail worden bestudeerd en meestal is het niet mogelijk een volledige oplossing in de corrosieliteratuur te vinden. Maar wanneer alle activiteiten in verband met ontwerp, fabricage en inspectie georganiseerd worden langs de in Figuur 10.1 geschetste lijnen zoals in dit hoofdstuk besproken en daarnaast gebaseerd zijn op een begrip van corrosieprocessen en van de manieren waarop deze kunnen worden beïnvloed, is het in de meeste gevallen mogelijk een adequate oplossing te vinden. Dit maakt het dan mogelijk om de besparingen, genoemd in bijlage 11.1, inderdaad te realiseren door het voorkomen van onnodige corrosiekosten door het toepassen van bestaande kennis van het voorkomen en bestrijden van corrosie.

197
11
Bijlagen


199
11.1 Potentiële besparingen van corrosiekosten
Van de totale geschatte kosten van corrosie in Groot Brittanië in 1971, ten bedrage van 1365 miljoen pond, kan ongeveer 25% worden bespaard en de potentiële besparingen, samen met de maatregelen die nodig zijn om deze te bereiken, zijn gegeven in onderstaande tabel. (Overgenomen uit: Report of the Committee on Corrosion and Protection, Chairman T.P. Hoar, London, H.M.S.O., 1971). Industrie of organisatie Geschatte
mogelijke besparing M£
Veranderingen nodig om besparing te bereiken
Bouw en constructie 50 Meer aandacht voor keuze, specificatie en controle van aanbrengen van bescherming
Voedingsmiddelen 4 Meer aandacht voor keuze van apparatuur en methoden van bescherming tegen corrosie
Algemene techniek 35 Grotere aandacht voor gevaren van corrosie in het ontwerpstadium en tijdens fabricage
Overheidsdiensten 20 Vooral preventief door beter ontwerp en betere procedures
Maritiem 55 Verbeterd ontwerp, meer aandacht voor en beter aanbrengen van bescherming
Metaal raffinage en halffabrikaten
2 Toename van bewustheid in apparatuur en product bescherming
Olie en chemische industrie 15 Meer effectiviteit in selectie van materialen en methoden van bescherming tegen corrosie
Energie 25 Meer toepassing van kathodische bescherming en meer aandacht voor procesomstandigheden bij het ontwerp
Transport 100 Verandering van materiaal voor uitlaten en meer aandacht voor corrosie bij het ontwerp
Water 4 Meer aandacht voor corrosie en bescherming daartegen
Totaal 310

Bijlagen
200
11.2 Constanten en conversiefactoren
Getal van Avogadro 6.022 × 1023 mol-1 Gas constante R = 8.314 J.mol-1.K-1 Faraday F = 96487 C.(g.equiv)-1 = 26.8 A.h.equiv-1 2.303 RT / F = 59.2 mV Druk 1 atm = 1.013×105 N.m-2 = 1.013 bar Energie 1 J = 0.2383 cal = 1 Ws = 1.78×10-7 kWh 1 cal = 4.184 J, 1 l.atm = 101.33 J Lengte 1 inch = 2.54 cm; 1 mil = 0.001 inch Tijd 1 dag = 8.64×104 s;
1 jaar = 3.156×107 s = 8.76×103 h Hoeveelheid 1 ppm = 1 deel per miljoen (bijvoorbeeld 1 mg/kg,
1 cm3/ m3) Volume van 1 mol gas Vm = 22.414 dm3mol-1 (0 °C , 1 atm)
Vm = 24.465 dm3mol-1 (25 °C , 1 atm)

201
11.3 Conversie van eenheden voor corrosiesnelheden
In de tabellen worden de factoren gegeven om een eenheid die aan de linkerkant staat om te rekenen in één die aan de bovenkant staat, bijvoorbeeld: 1 cm.jaar-1 = 2050 mdd. Gebruikte afkortingen:
ipy = inch per jaar; mdd = mg.dm-2.dag-1 n = waardigheid van het beschouwde metaal M = atoom massa van het beschouwde metaal d = dichtheid in g.cm-3 van het beschouwde metaal
mA.cm-2 cm.jaar-1 ipy mdd g.m-2.dag-1 mA.cm-2 1 0.326M/nd 0.129M/nd 89.2M/n 8.92M/n cm.jaar-1 3.06nd/M 1 0.394 274d 27.4d Ipy 7.75nd/M 2.54 1 694d 69.4d mdd 0.0112n/M 0.00365/d 0.00144/d 1 0.1 g.m-2.dag-1 0.112n/M 0.0365/d 0.0144/d 10 1
Voor een typisch metaal zoals ijzer, koper, zink, enzovoort met M ≈ 60, d ≈ 7.5 g.cm-3 en n = 2 geeft dit de volgende tabel. Deze is voor de meeste praktische toepassingen van dit soort metalen nauwkeurig genoeg.
mA.cm-2 cm.jaar-1 ipy mdd g.m-2.dag-1 mA.cm-2 1 1.3 0.52 2676 268 cm.jaar-1 0.77 1 0.394 2050 205 Ipy 1.92 2.54 1 5200 520 Mdd 3.6×10-4 4.9×10-4 1.92×10-4 1 0.1 g.m-2.dag-1 3.6×10-3 4.9×10-3 1.92×10-3 10 1

Bijlagen
202
11.4 Periodiek systeem der elementen
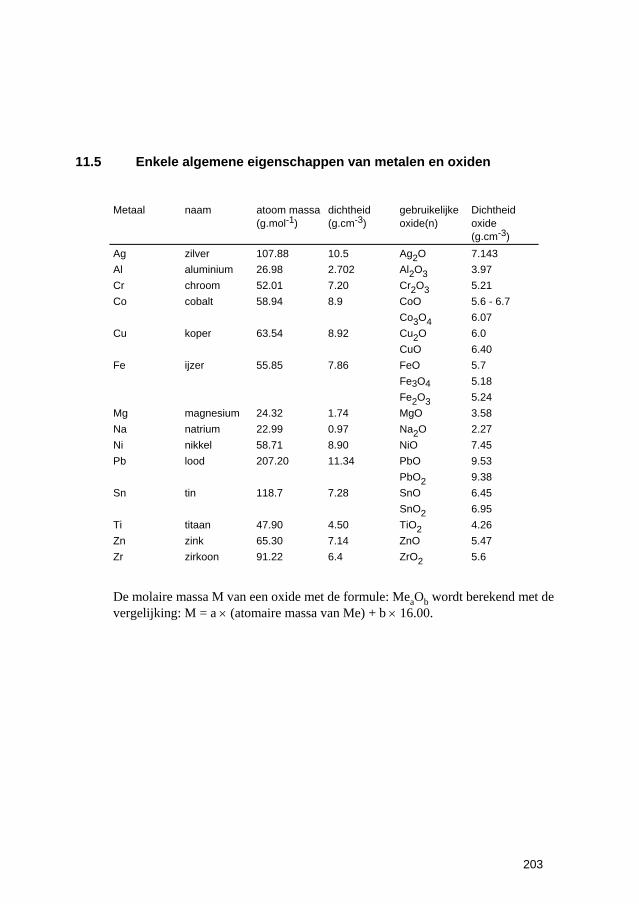
203
11.5 Enkele algemene eigenschappen van metalen en oxiden
Metaal naam atoom massa
(g.mol-1) dichtheid (g.cm-3)
gebruikelijke oxide(n)
Dichtheid oxide (g.cm-3)
Ag zilver 107.88 10.5 Ag2O 7.143 Al aluminium 26.98 2.702 Al2O3 3.97 Cr chroom 52.01 7.20 Cr2O3 5.21 Co cobalt 58.94 8.9 CoO 5.6 - 6.7 Co3O4 6.07 Cu koper 63.54 8.92 Cu2O 6.0 CuO 6.40 Fe ijzer 55.85 7.86 FeO 5.7 Fe3O4 5.18 Fe2O3 5.24 Mg magnesium 24.32 1.74 MgO 3.58 Na natrium 22.99 0.97 Na2O 2.27 Ni nikkel 58.71 8.90 NiO 7.45 Pb lood 207.20 11.34 PbO 9.53 PbO2 9.38 Sn tin 118.7 7.28 SnO 6.45 SnO2 6.95 Ti titaan 47.90 4.50 TiO2 4.26 Zn zink 65.30 7.14 ZnO 5.47 Zr zirkoon 91.22 6.4 ZrO2 5.6
De molaire massa M van een oxide met de formule: MeaOb wordt berekend met de vergelijking: M = a × (atomaire massa van Me) + b × 16.00.
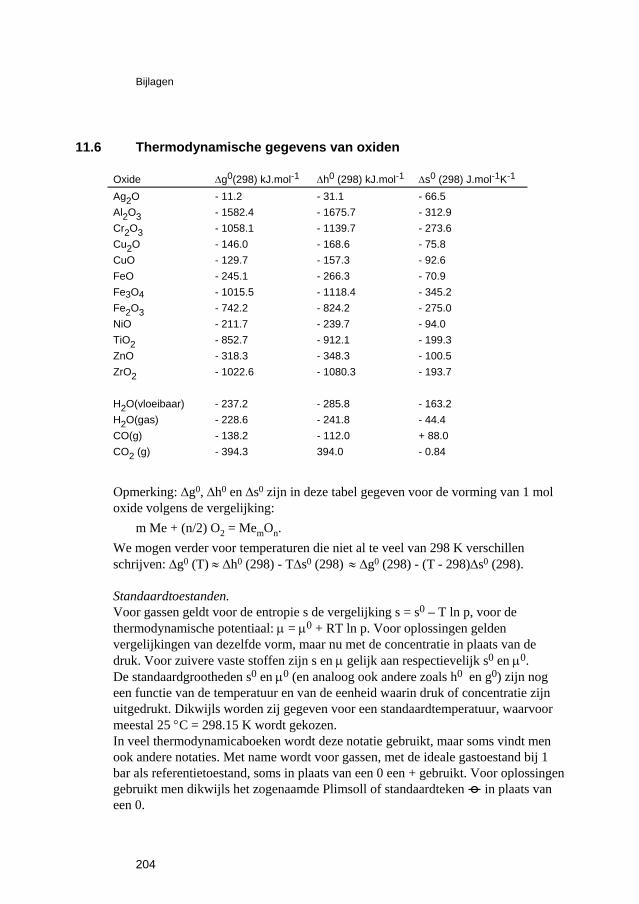
Bijlagen
204
11.6 Thermodynamische gegevens van oxiden
Oxide Δg0(298) kJ.mol-1 Δh0 (298) kJ.mol-1 Δs0 (298) J.mol-1K-1 Ag2O - 11.2 - 31.1 - 66.5 Al2O3 - 1582.4 - 1675.7 - 312.9 Cr2O3 - 1058.1 - 1139.7 - 273.6 Cu2O - 146.0 - 168.6 - 75.8 CuO - 129.7 - 157.3 - 92.6 FeO - 245.1 - 266.3 - 70.9 Fe3O4 - 1015.5 - 1118.4 - 345.2 Fe2O3 - 742.2 - 824.2 - 275.0 NiO - 211.7 - 239.7 - 94.0 TiO2 - 852.7 - 912.1 - 199.3 ZnO - 318.3 - 348.3 - 100.5 ZrO2 - 1022.6 - 1080.3 - 193.7 H2O(vloeibaar) - 237.2 - 285.8 - 163.2 H2O(gas) - 228.6 - 241.8 - 44.4 CO(g) - 138.2 - 112.0 + 88.0 CO2 (g) - 394.3 394.0 - 0.84
Opmerking: Δg0, Δh0 en Δs0 zijn in deze tabel gegeven voor de vorming van 1 mol oxide volgens de vergelijking:
m Me + (n/2) O2 = MemOn. We mogen verder voor temperaturen die niet al te veel van 298 K verschillen schrijven: Δg0 (T) ≈ Δh0 (298) - TΔs0 (298) ≈ Δg0 (298) - (T - 298)Δs0 (298). Standaardtoestanden. Voor gassen geldt voor de entropie s de vergelijking s = s0 – T ln p, voor de thermodynamische potentiaal: μ = μ0 + RT ln p. Voor oplossingen gelden vergelijkingen van dezelfde vorm, maar nu met de concentratie in plaats van de druk. Voor zuivere vaste stoffen zijn s en μ gelijk aan respectievelijk s0 en μ0. De standaardgrootheden s0 en μ0 (en analoog ook andere zoals h0 en g0) zijn nog een functie van de temperatuur en van de eenheid waarin druk of concentratie zijn uitgedrukt. Dikwijls worden zij gegeven voor een standaardtemperatuur, waarvoor meestal 25 °C = 298.15 K wordt gekozen. In veel thermodynamicaboeken wordt deze notatie gebruikt, maar soms vindt men ook andere notaties. Met name wordt voor gassen, met de ideale gastoestand bij 1 bar als referentietoestand, soms in plaats van een 0 een + gebruikt. Voor oplossingen gebruikt men dikwijls het zogenaamde Plimsoll of standaardteken in plaats van een 0.

205
11.7 Celspanningen en elektrodepotentialen
Om te komen tot een ondubbelzinnige toekenning van het teken van een celspanning en van elektrodepotentialen zijn in internationaal overleg de volgende regels vastgesteld: 1. De cel en de celreactie worden in dezelfde volgorde geschreven, dat wil zeggen
dat de metaalatomen aan dezelfde zijde van de reactievergelijking staan als de overeenkomstige elektrode in de cel: cel: Fe | Fe2+(aq) | Cu2+(aq) | Cu reactie: Fe + Cu2+ → Fe2+ + Cu.
2. Het teken van de celspanning is gelijk aan de polariteit van de rechter elektrode bij open circuit. Als E > 0 voor de cel dan is ΔG < 0 voor de celreactie en de reactie heeft de neiging spontaan naar rechts te verlopen.
3. Wanneer de waterstofelektrode als standaardelektrode wordt gebruikt dan wordt die aan de linkerkant van de cel geplaatst. Moleculaire waterstof staat dus ook aan de linkerkant in de cel reactie.
4. De standaardspanning van zo'n cel, weer genomen met de polariteit van de rechter elektrode, wordt de standaardpotentiaal van die elektrode genoemd. Dus geldt dat de standaardspanning van de cel: H2 | H+(aq) |Cu2+(aq) | Cu met de celreactie: H2 + Cu2+ → 2 H+ + Cu de standaardpotentiaal van koper is.
5. De celspanning kan worden berekend uit de evenwichtspotentialen van de samenstellende elektroden door de elektrodepotentiaal van de linker elektrode af te trekken van die van de rechter elektrode. Voor de cel: Zn | Zn2+ | Fe2+ | Fe geldt dus: Ecel = EFe - EZn.
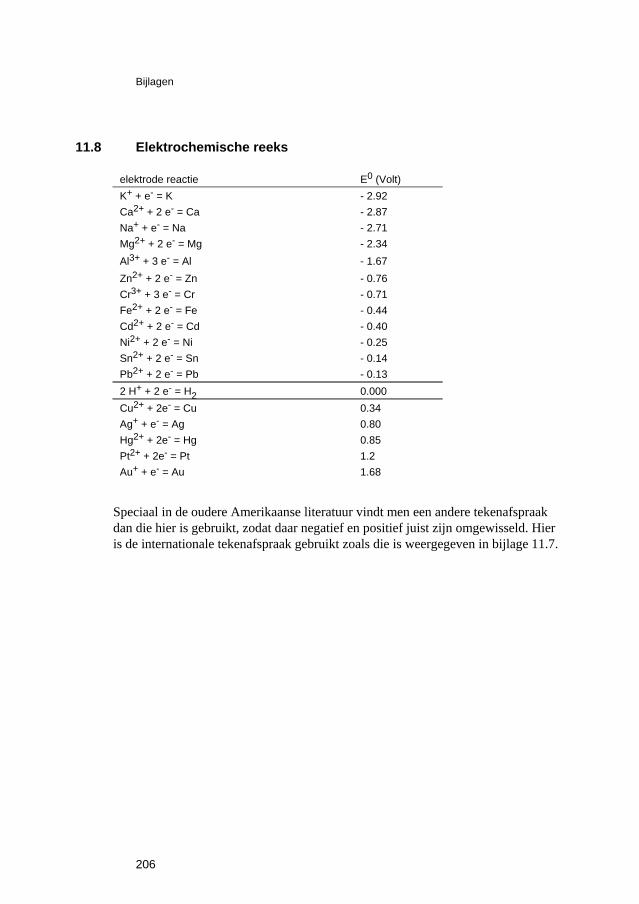
Bijlagen
206
11.8 Elektrochemische reeks
elektrode reactie E0 (Volt) K+ + e- = K - 2.92 Ca2+ + 2 e- = Ca - 2.87 Na+ + e- = Na - 2.71 Mg2+ + 2 e- = Mg - 2.34 Al3+ + 3 e- = Al - 1.67 Zn2+ + 2 e- = Zn - 0.76 Cr3+ + 3 e- = Cr - 0.71 Fe2+ + 2 e- = Fe - 0.44 Cd2+ + 2 e- = Cd - 0.40 Ni2+ + 2 e- = Ni - 0.25 Sn2+ + 2 e- = Sn - 0.14 Pb2+ + 2 e- = Pb - 0.13 2 H+ + 2 e- = H2 0.000 Cu2+ + 2e- = Cu 0.34 Ag+ + e- = Ag 0.80 Hg2+ + 2e- = Hg 0.85 Pt2+ + 2e- = Pt 1.2 Au+ + e- = Au 1.68
Speciaal in de oudere Amerikaanse literatuur vindt men een andere tekenafspraak dan die hier is gebruikt, zodat daar negatief en positief juist zijn omgewisseld. Hier is de internationale tekenafspraak gebruikt zoals die is weergegeven in bijlage 11.7.

207
11.9 Standaardpotentialen van redoxreacties
elektrode reactie Eo (Volt) Cr3++ e- = Cr2+ - 0.41 2 H+ + 2 e- = H2 0.000 Cu2+ + e- = Cu+ 0.153 Fe(CN)6
3- + e- = Fe(CN)64- 0.356
O2 + 2 H2O + 4 e- = 4 OH- 0.401 I2 + 2 e- = 2 I- 0.53 O2 + 2 H+ + 2 e- = H2O2 0.69 Fe3+ + e- = Fe2+ 0.771 Br2 + 2 e- = 2 Br- 1.06 O2 + 4 H+ + 4 e- = 2 H2O 1.230 Cl2 + 2 e- = 2 Cl- 1.360 H2O2 + 2 H+ + 2 e- = 2 H2O 1.77 F2 + 2 e- = 2 F 2.85

Bijlagen
208
11.10 Praktische galvanische reeks in belucht, neutraal zeewater
metaal potentiaal (Volt) magnesium - 1.32 zink legering Zamak Z400 - 0.94 zink - 0.78 aluminium 99.5% - 0.67 koolstof staal - 0.40 gietijzer GG 22 - 0.35 13% Cr-staal (actief) - 0.30 18% Cr-8% Ni-staal (actief) - 0.30 lood 99.9% - 0.26 messing 60-40 - 0.07 koper + 0.10 monel K + 0.12 70-30 cupronikkel + 0.34 Cr- en Cr-Ni-staalsoorten (passief) ≈ + 0.40
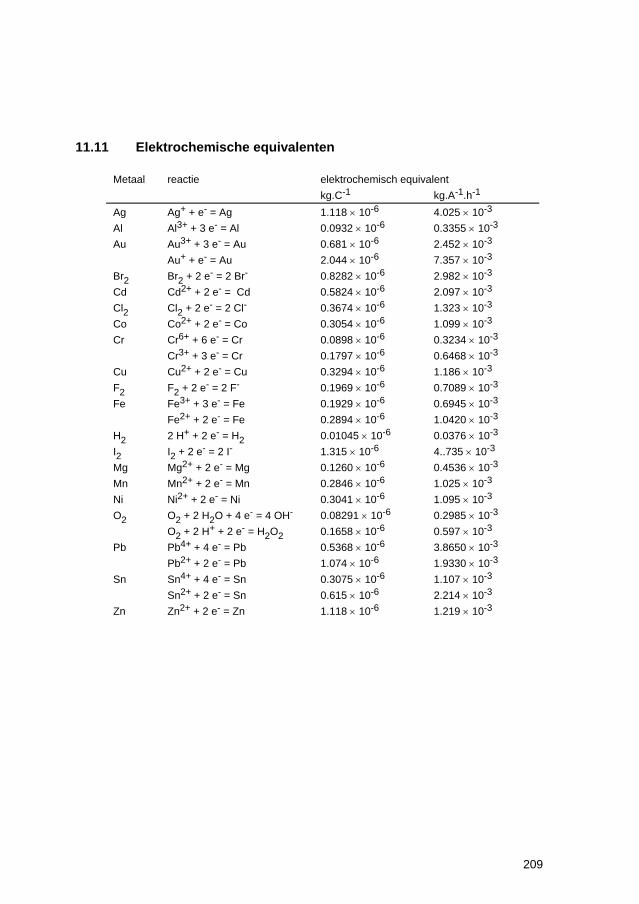
209
11.11 Elektrochemische equivalenten
Metaal reactie elektrochemisch equivalent kg.C-1 kg.A-1.h-1 Ag Ag+ + e- = Ag 1.118 × 10-6 4.025 × 10-3 Al Al3+ + 3 e- = Al 0.0932 × 10-6 0.3355 × 10-3 Au Au3+ + 3 e- = Au 0.681 × 10-6 2.452 × 10-3 Au+ + e- = Au 2.044 × 10-6 7.357 × 10-3 Br2 Br2 + 2 e- = 2 Br- 0.8282 × 10-6 2.982 × 10-3 Cd Cd2+ + 2 e- = Cd 0.5824 × 10-6 2.097 × 10-3 Cl2 Cl2 + 2 e- = 2 Cl- 0.3674 × 10-6 1.323 × 10-3 Co Co2+ + 2 e- = Co 0.3054 × 10-6 1.099 × 10-3 Cr Cr6+ + 6 e- = Cr 0.0898 × 10-6 0.3234 × 10-3 Cr3+ + 3 e- = Cr 0.1797 × 10-6 0.6468 × 10-3 Cu Cu2+ + 2 e- = Cu 0.3294 × 10-6 1.186 × 10-3 F2 F2 + 2 e- = 2 F- 0.1969 × 10-6 0.7089 × 10-3 Fe Fe3+ + 3 e- = Fe 0.1929 × 10-6 0.6945 × 10-3 Fe2+ + 2 e- = Fe 0.2894 × 10-6 1.0420 × 10-3 H2 2 H+ + 2 e- = H2 0.01045 × 10-6 0.0376 × 10-3 I2 I2 + 2 e- = 2 I- 1.315 × 10-6 4..735 × 10-3 Mg Mg2+ + 2 e- = Mg 0.1260 × 10-6 0.4536 × 10-3 Mn Mn2+ + 2 e- = Mn 0.2846 × 10-6 1.025 × 10-3 Ni Ni2+ + 2 e- = Ni 0.3041 × 10-6 1.095 × 10-3 O2 O2 + 2 H2O + 4 e- = 4 OH- 0.08291 × 10-6 0.2985 × 10-3 O2 + 2 H+ + 2 e- = H2O2 0.1658 × 10-6 0.597 × 10-3 Pb Pb4+ + 4 e- = Pb 0.5368 × 10-6 3.8650 × 10-3 Pb2+ + 2 e- = Pb 1.074 × 10-6 1.9330 × 10-3 Sn Sn4+ + 4 e- = Sn 0.3075 × 10-6 1.107 × 10-3 Sn2+ + 2 e- = Sn 0.615 × 10-6 2.214 × 10-3 Zn Zn2+ + 2 e- = Zn 1.118 × 10-6 1.219 × 10-3

Bijlagen
210
11.12 Teken van de overspanning
Voor een celreactie zoals A + B+ = A+ + B (11.12.1)
geldt volgens vergelijking (3.3.2): Δg = - nFE (11.12.2)
waarin het teken van de celspanning E is gekozen in overeenstemming met de afspraken van bijlage 11.7. Wanneer reactie (494H11.12.1) spontaan naar rechts verloopt is Δg < 0 en dit geeft, met vergelijking (495H11.12.2):
E > 0 (11.12.3) Wanneer de reactie naar rechts verloopt gaat de stroom I van elektrode B naar elektrode A, dat is van de positieve naar de negatieve elektrode, zodat:
E × I > 0 (11.12.4) Als aangenomen wordt dat de potentiaal van elektrode B niet door de stroom wordt beïnvloed, hetgeen in goede benadering mag wanneer de oppervlakte van B veel groter is dan van elektrode A, dan is de celspanning gelijk aan de overspanning van elektrode A en geldt:
ηA × I > 0 (11.12.5) Analoog vinden we dat wanneer de reactie naar links verloopt zowel het teken van Δg en E aan de ene kant als de richting van de stroom aan de andere kant beide worden omgekeerd, zodat de vergelijkingen (500H11.12.4) en (501H11.12.5) geldig blijven.
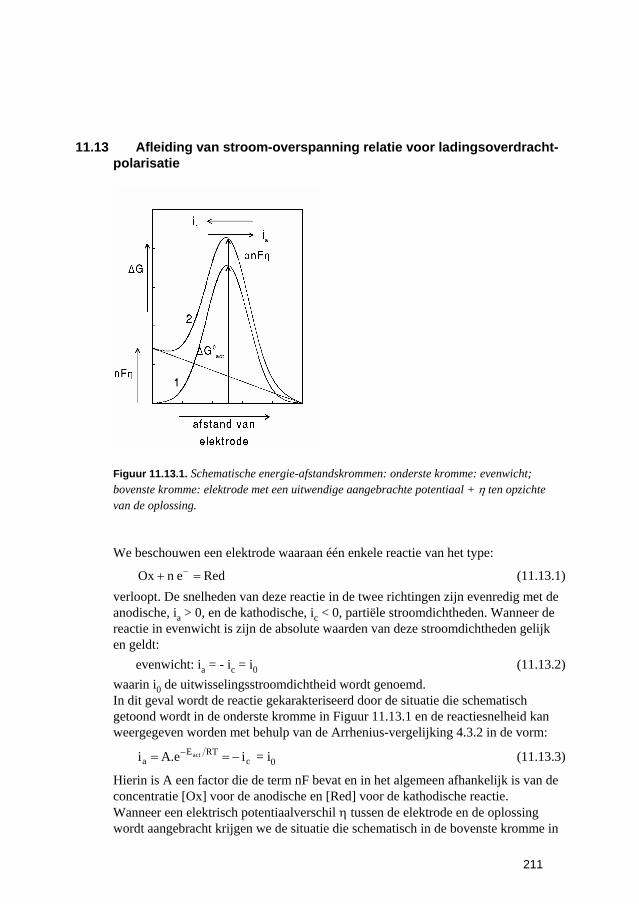
211
11.13 Afleiding van stroom-overspanning relatie voor ladingsoverdracht-polarisatie
Figuur 11.13.1. Schematische energie-afstandskrommen: onderste kromme: evenwicht; bovenste kromme: elektrode met een uitwendige aangebrachte potentiaal + η ten opzichte van de oplossing.
We beschouwen een elektrode waaraan één enkele reactie van het type:
Red en Ox =+ − (11.13.1) verloopt. De snelheden van deze reactie in de twee richtingen zijn evenredig met de anodische, ia > 0, en de kathodische, ic < 0, partiële stroomdichtheden. Wanneer de reactie in evenwicht is zijn de absolute waarden van deze stroomdichtheden gelijk en geldt:
evenwicht: ia = - ic = i0 (11.13.2) waarin i0 de uitwisselingsstroomdichtheid wordt genoemd. In dit geval wordt de reactie gekarakteriseerd door de situatie die schematisch getoond wordt in de onderste kromme in Figuur 11.13.1 en de reactiesnelheid kan weergegeven worden met behulp van de Arrhenius-vergelijking 4.3.2 in de vorm:
cRTE
a i A.e i act −== − = i0 (11.13.3)
Hierin is A een factor die de term nF bevat en in het algemeen afhankelijk is van de concentratie [Ox] voor de anodische en [Red] voor de kathodische reactie. Wanneer een elektrisch potentiaalverschil η tussen de elektrode en de oplossing wordt aangebracht krijgen we de situatie die schematisch in de bovenste kromme in

Bijlagen
212
Figuur 11.13.1 is weergegeven. Ten gevolge van de lading n die in de reactie wordt overgedragen is de (vrije) energie van de reagerende stof aan de elektrode verminderd met nFη. Als gevolg hiervan zien we in Figuur 11.13.1 dat de activeringsenergieën voor de heen- en teruggaande reactie nu niet meer gelijk zijn. Voor de anodische reactie is deze kleiner geworden en is nu gelijk aan (Eact - αnFη) waarin α de fractie van elektrische potentiaalverschil op de plaats van de top van de energiekromme is. De grootheid α wordt de overdrachtscoëfficiënt genoemd en heeft in veel gevallen een waarde dichtbij 0.5. Analoog is de activeringsenergie voor de kathodische reactie verhoogd en deze is nu (Eact + (1 - α)nFη). Zoals in de figuur aangegeven betekent dit dat de anodische stroomdichtheid is toegenomen, terwijl de absolute waarde van de kathodische stroomdichtheid is verlaagd. In overeen-stemming met de verwachting is de netto stroom nu anodisch. Met deze veranderde activeringsenergieën krijgen we voor de partiële stroomdicht-heden de vergelijkingen:
RTnF0
RT)nFE(a .eiA.ei act ηαηα−− == (11.13.4)
RTnF)1(0
RT)nF)1(E(a .eiA.ei act ηα−−ηα−+− =−= (11.13.5)
Deze komen volledig overeen met de vergelijkingen (4.3.3.a) en (4.3.3.b).

213
11.14 Gebruik van beschermende deklagen
De gegevens in deze tabel gelden voor 1994 en de bedragen zijn daarom nog in guldens opgegeven. Type deklaag Beschermd materiaal Omvang gebruik Verf (noot 1) Staal 44,000,000 m2 Aluminium 6,000,000 m2 Thermisch verzinken (noot 2) Staal 300,000,000 kg Galvanische bedekking (noot 3) Diverse 1,200,000,000 m2 Anodiseren Aluminium (noot 4) 10,000,000 m2 Noten 1. Hiervoor gebruikt: natlak 39,000,000 kg; poederlak 4,500,000 kg,
gemiddelde prijs per m2 van ƒ 10 tot ƒ 17. 2. Zinkverbruik 30,000,000 kg. 3. Totale omzet van de branche ƒ 700,000,000 4. Hiervan: kleine producten 3,000,000 m2, platen en profielen 7,000,000 m2. De totale omzet in deze industrietak bedroeg in 2005: 3.5 miljard €. Deze gegevens zijn beschikbaar gesteld door de Vereniging voor Oppervlakte-technieken van Materialen (VOM), een brancheorganisatie voor de industrie op dit gebied. Nadere inlichtingen en het adres zijn te vinden op de web-site: www.vom.nl. Binnen de VOM zijn een groot aantal andere organisaties als branche groepen verenigd en ook via de VOM bereikbaar: Stichting Email. Vereniging Industriële Spuit- en Moffelbedrijven VISEM (www.visem.nl). Vereniging van Leveranciers voor de Galvanotechniek. Vereniging voor Leveranciers van Verfspuitapparatuur. Vereniging voor Thermische Spuittechnieken VTS (www.thermisch-spuiten.nl). Nederlandse Galvano Ondernemers (NGO) - Stichting Bevordering Galvano-technische Industrie (SBG) (www.ngo-svb.nl). Stichting Anodiseren (STANOD). Stichting Doelmatig Verzinken SDV (www.sdvonline.nl). Centrum voor Onderzoek en Technisch Advies COT (www.cot-nl.com). Op alle genoemde web-sites zijn uitvoerige gegevens beschikbaar over de diverse oppervlaktebehandelingsmethoden inclusief publicaties op deze gebieden die een waardevolle aanvulling vormen op het in dit boek behandelde.
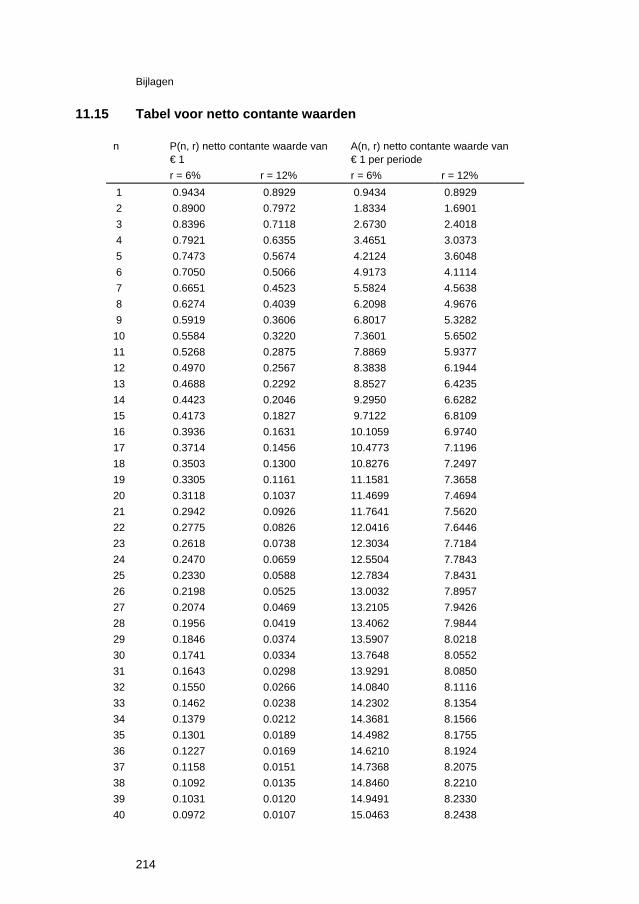
Bijlagen
214
11.15 Tabel voor netto contante waarden
n P(n, r) netto contante waarde van € 1
A(n, r) netto contante waarde van € 1 per periode
r = 6% r = 12% r = 6% r = 12% 1 0.9434 0.8929 0.9434 0.8929 2 0.8900 0.7972 1.8334 1.6901 3 0.8396 0.7118 2.6730 2.4018 4 0.7921 0.6355 3.4651 3.0373 5 0.7473 0.5674 4.2124 3.6048 6 0.7050 0.5066 4.9173 4.1114 7 0.6651 0.4523 5.5824 4.5638 8 0.6274 0.4039 6.2098 4.9676 9 0.5919 0.3606 6.8017 5.3282 10 0.5584 0.3220 7.3601 5.6502 11 0.5268 0.2875 7.8869 5.9377 12 0.4970 0.2567 8.3838 6.1944 13 0.4688 0.2292 8.8527 6.4235 14 0.4423 0.2046 9.2950 6.6282 15 0.4173 0.1827 9.7122 6.8109 16 0.3936 0.1631 10.1059 6.9740 17 0.3714 0.1456 10.4773 7.1196 18 0.3503 0.1300 10.8276 7.2497 19 0.3305 0.1161 11.1581 7.3658 20 0.3118 0.1037 11.4699 7.4694 21 0.2942 0.0926 11.7641 7.5620 22 0.2775 0.0826 12.0416 7.6446 23 0.2618 0.0738 12.3034 7.7184 24 0.2470 0.0659 12.5504 7.7843 25 0.2330 0.0588 12.7834 7.8431 26 0.2198 0.0525 13.0032 7.8957 27 0.2074 0.0469 13.2105 7.9426 28 0.1956 0.0419 13.4062 7.9844 29 0.1846 0.0374 13.5907 8.0218 30 0.1741 0.0334 13.7648 8.0552 31 0.1643 0.0298 13.9291 8.0850 32 0.1550 0.0266 14.0840 8.1116 33 0.1462 0.0238 14.2302 8.1354 34 0.1379 0.0212 14.3681 8.1566 35 0.1301 0.0189 14.4982 8.1755 36 0.1227 0.0169 14.6210 8.1924 37 0.1158 0.0151 14.7368 8.2075 38 0.1092 0.0135 14.8460 8.2210 39 0.1031 0.0120 14.9491 8.2330 40 0.0972 0.0107 15.0463 8.2438
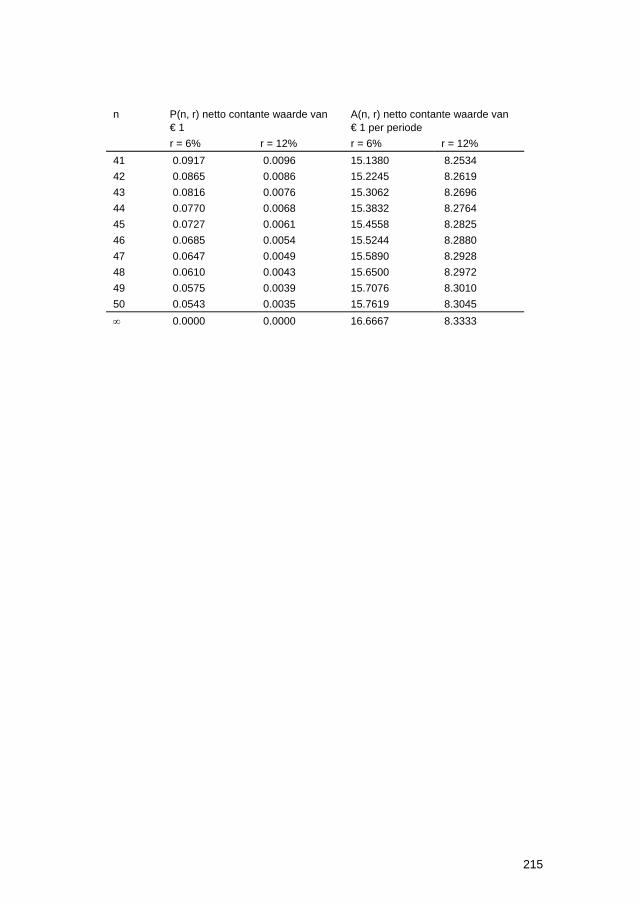
215
n P(n, r) netto contante waarde van € 1
A(n, r) netto contante waarde van € 1 per periode
r = 6% r = 12% r = 6% r = 12% 41 0.0917 0.0096 15.1380 8.2534 42 0.0865 0.0086 15.2245 8.2619 43 0.0816 0.0076 15.3062 8.2696 44 0.0770 0.0068 15.3832 8.2764 45 0.0727 0.0061 15.4558 8.2825 46 0.0685 0.0054 15.5244 8.2880 47 0.0647 0.0049 15.5890 8.2928 48 0.0610 0.0043 15.6500 8.2972 49 0.0575 0.0039 15.7076 8.3010 50 0.0543 0.0035 15.7619 8.3045 ∞ 0.0000 0.0000 16.6667 8.3333

Bijlagen
216
11.16 Elementen van defectchemie
Thermodynamisch kan worden aangetoond dat ieder kristal bij alle temperaturen boven het absolute nulpunt een eindig aantal puntdefecten bevat. Voor metalen zijn de eenvoudigste typen puntdefecten de vacature, dat wil zeggen een roosterplaats waar het metaalatoom ontbreekt en een interstitiëel, dat is een metaalatoom op een plaats die leeg is in het ideale kristal. De bedoelde afleiding toont dat de defectcon-centratie x, indien deze niet al te hoog is, gegeven wordt door:
RT/Eex −= (11.16.1)
Hierin is E de energie die nodig is om het defect te maken. Wanneer bijvoorbeeld E = 40 kJ.mol-1, dan is x bij 300 K ongeveer 10-7 en bij 1000 K ongeveer 10-2. In ionogene kristallen, zoals metaaloxiden, zijn afzonderlijke defecten niet mogelijk omdat het kristal als geheel steeds elektrisch neutraal moet blijven. In stoichiome-trische kristallen zijn de eenvoudigste typen puntfouten: − een paar vacatures of Schottky-defect zoals geïllustreerd in Figuur 11.16.1a − een interstitieel ion plus een vacature of Frenkel-defect, schematisch getoond in
Figuur 11.16.1b
Figuur 11.16.1. Puntfouten in stoichiometrische kristallen: a: Schottky-defect; b. Frenkel-defect.

217
a.
O2- Ni2+ O2- Ni3+ O2- Ni2+ Ni3+ O2- O2- Ni2+ O2- O2- Ni2+ O2- Ni2+ O2- Ni3+ Ni2+ O2- Ni2+ O2- Ni2+ O2- O2- O2- Ni3+ O2- Ni2+
b.
O2- Zn2+ O2- Zn2+ O2- Zn2+ e' Zn2+ Zn2+ O2- Zn2+ O2- Zn2+ O2- e' e' O2- Zn2+ O2- Zn2+ O2- Zn2+ Zn2+ O2- Zn2+ O2- Zn2+ O2- Zn2+ e' O2- Zn2+ O2- Zn2+ O2- Zn2+
c.
Zr4+ O2- Zr3+ O2- Zr3 O2- Ni3+ O2- Zr4+ O2- Zr4+ O2- Zr4+ O2- Ni2+ O2- Zr3+ O2- Zr4+ O2- Zr3+ O2-
Figuur 11.16.2. Puntdefecten in niet-stoichiometrische kristallen: a. NiO: metaal tekort; b. ZnO: metaal overschot; c. ZrO2: zuurstoftekort.
Van groot praktisch belang is niet-stoichiometrie in oxiden omdat die vaak een beslissende invloed heeft op de oxidatiekinetiek. Drie veel voorkomende voorbeel-den worden schematisch getoond in Figuur 11.16.2. Als eerste nikkeloxide, NiO, dat een nikkel tekort heeft in de vorm van Ni2+-vacatures en daarom wel geschreven wordt als Ni1-xO. Om het daardoor veroorzaakte tekort aan positieve lading te com-

Bijlagen
218
penseren hebben een equivalent aantal Ni2+-ionen een elektron verloren en zijn in Ni3+-ionen omgezet. De nikkelvacature wordt met het symbool NiV ′′ waarin het dubbele accent een effectieve lading 2 aangeeft. De Ni3+-ionen zijn in wezen elek-trongaten die aangegeven worden met het symbool h· waarin de punt een effectieve lading van plus 1 aangeeft. De effectieve ladingen worden gedefinieerd ten opzichte van het ideale rooster, zonder defecten, als referentie. Het tweede geval is dat van zinkoxide, ZnO, met interstitiële zink ionen ( ••
iZn ) en vrije elektronen (e'). Als derde is zirkoonoxide, ZrO2, getoond waarin zuurstof-vacatures ( ••
OV ) en vrije elektronen (e') bevat. ZnO en ZrO2 hebben allebei een metaal overschot, echter van verschillende aard die kunnen worden aangegeven als Zn1+xO en ZrO2-y. In Tabel 11.16.1 is een overzicht gegeven van verschillende soorten defecten en van de symbolen die gebruikt worden om ze aan te geven. Met deze symbolen kunnen we, precies als met de symbolen voor de chemische elemen-ten, reactievergelijkingen schrijven voor het ontstaan en verdwijnen van defecten.
Tabel 11.16.1. Types en symbolen van defecten volgens Kröger en Vink∗
Defect symbool opmerkingen lege M plaats (= M-vacature) MV ′′ MX, met tweewaardige ionen
M op eigen roosterplaats MM als voorbeeld genomen
lege X plaats (=X-vacature) ••XV • = effectief positieve lading
X op eigen roosterplaats XX ' = effectief negatieve lading
interstitiëel M-ion ••iM
interstitiëel X-ion iX ′′
Elektron e' elektron gat h· driewaardig ion N op M-plaats •
MN
eenwaardig ion A op M-plaats MA′
Wanneer we bijvoorbeeld nikkeloxide beschouwen, dat in contact staat met zuurstof, kunnen we de reactievergelijking van de defectvorming schrijven als:
ONi221 Oh2V)g(O ++′′= • (11.16.2)
Met deze vergelijking geven we het proces aan, dat ook schematisch is afgebeeld in Figuur 11.16.3, waarin één zuurstofatoom twee elektronen opneemt waardoor twee elektrongaten worden gevormd en het zelf een O2- wordt dat in het rooster wordt op-genomen. Er zou dan ook een nikkel-ion kunnen worden opgenomen hetgeen wil zeggen dat een nikkelvacature wordt gevormd. De evenwichtsconstante van ver-gelijking (517H11.16.2) kan geschreven worden als:
∗ F.A. Kröger en H.J. Vink, Solid State Physics, 3 (1956) 307.

219
[ ][ ] [ ]2/1
O
O2
Ni
2p
OhVK
•′′= (11.16.3)
of, omdat OO constant is:
[ ][ ]2/1
O
2Ni
2p
hVK
•′′= (11.16.4)
Figuur 11.16.3. Vormingsreactie van nikkelvacatures en elektrongaten in nikkeloxide.
Om de concentraties te kunnen berekenen is een tweede vergelijking nodig omdat vergelijking (521H11.16.4) nog twee onbekenden bevat. Deze tweede vergelijking is de elektroneutraliteitsvoorwaarde:
[ ] [ ]•=′′ hV2 Ni (11.16.5)
Deze voorwaarde drukt uit dat om de elektrische neutraliteit te behouden voor iedere nikkelvacature twee elektrongaten moeten worden gevormd, dat wil zeggen dat het aantal (en dus ook de concentratie) van de elektrongaten tweemaal zo hoog is als van de nikkelvacatures. Substitutie van vergelijking (523H11.16.5) in vergelijking (521H11.16.4) geeft, na enige omvorming:

Bijlagen
220
[ ] 61
2
31
ONi p4KV ×⎟⎠⎞
⎜⎝⎛=′′ (11.16.6)
Soortgelijke beschouwingen kunnen gegeven worden voor andere oxiden. Voor de andere twee voorbeelden van Figuur 11.16.2 zijn dan de reactievergelijkingen:
ZnOi221 ZnOe2Zn)g(O:ZnO +=′++ •• (11.16.7)
OO221
2 Oe2V)g(O:ZrO =′++ •• (11.16.8)
Omdat niet stoichiometrische oxiden meestal halfgeleiders zijn noemt men oxiden met elektrongaten p-type oxiden en oxiden met overmaat elektronen n-type oxiden. In de n-type oxiden zijn dan naast de elektronen interstitiële metaalionen (ZnO) of zuurstofvacatures (ZrO2) aanwezig. Legeringselementen aanwezig hebben een duidelijke invloed op de defectconcen-traties wanneer hun waardigheid verschilt van die van het moedermateriaal. Wanneer bijvoorbeeld Cr3+-ionen ingebouwd worden in NiO bezetten zij een nik-kelpositie en kunnen dus geschreven worden als •
NiCr . Dit betekent dat de elektro-neutraliteitsvoorwaarde (523H11.16.5) moet worden veranderd en nu luidt:
[ ] [ ] [ ]•• +=′′ NiNi CrhV2 (11.16.9)
Wanneer we deze vergelijking combineren met vergelijking (521H11.16.4) dan is direct te zien dat een toename in de chroomconcentratie leidt tot een toename van de nikkelvacatureconcentratie. Omdat, zoals in hoofdstuk 8 is besproken, de oxidatiesnelheid toeneemt met de vacatureconcentratie zal toevoegen van chroom in eerste instantie dus ook tot een toename van de oxidatiesnelheid leiden (zie verder §8.5). Analoog, wanneer lithium wordt ingebouwd in NiO, dan gebeurt dat als NiiL ′ zodat de elektroneutraliteitsvoorwaarde dan wordt:
[ ] [ ] [ ]•=′+′′ hiLV2 NiNi (11.16.10)
Dit betekent dus dat nu de nikkelvacatureconcentratie wordt verlaagd, hetgeen leidt tot een verlaging van de oxidatiesnelheid. Volledig analoge effecten kunnen natuurlijk ook worden afgeleid voor ZnO, ZrO2 en andere oxiden.
11.16.1 Opgaven
1. Toon aan dat de oxidatiesnelheid van zink toeneemt wanneer het gelegeerd wordt met een eenwaardig metaal, zoals lithium, en afneemt bij legeren met een driewaardig metaal, zoals aluminium.
2. Toon aan dat de concentratie van zuurstofvacatures in ZrO2 evenredig is met 6
1
2Op− .

221
11.17 Corrosiepublicaties van het Nederlands Corrosie Centrum
Het Nederlands Corrosie Centrum (NCC, Postbus 190, 2700 AD Zoetermeer, Tel: 079-353 14 11, Fax: 079-353 13 65, E-mail: [email protected]) heeft een grote reeks publicaties uitgegeven op vele gebieden van corrosie en corrosiebestrijding, die een uitstekende aanvulling kunnen vormen op dit boek. Deze publicaties kunnen bij het NCC worden besteld, de prijzen hiervan zijn vermeld in de lijst op de web-site van het NCC: http://www.corrosiecentrum.nl waar ook verdere actuele gegevens en activiteiten van het NCC kunnen worden gevonden. .
De aan het eind van de lijst genoemde video is speciaal erg geschikt om te worden vertoond als onderdeel en toelichting van cursussen op corrosiegebied en bij lessen of colleges gebaseerd op dit boek.
Corrosiepublicaties 1. "Corrosiebeproevingsmethoden" (fotokopieën)
(Voordrachten gehouden tijdens de Vakantieleergang Corrosie, georganiseerd door de TH-Delft op 5 en 6 januari 1967)
2. "Corrosie en oxidatie bij hoge temperatuur" (fotokopieën)
(Voordrachten gehouden tijdens de Vakantieleergang Corrosie, georganiseerd door de TH-Twente op 9 en 10 januari 1969).
3. "Structuur en corrosiewerende eigenschappen van metallieke deklagen op
metalen" (Voordrachten gehouden tijdens de Vakantieleergang Corrosie, georganiseerd door de TH-Eindhoven op 8 en 9 januari 1970).
4. "Vijf voordrachten op corrosiegebied"
(Voordrachten gehouden tijdens bijeenkomsten van de Bond van Materialenkennis op 29 januari 1969 te Apeldoorn en op 27 november 1969 te Nijmegen).
5. "Spanningscorrosie"
(Voordrachten gehouden tijdens de Vakantieleergang Corrosie, georganiseerd door de TH-Delft op 6 en 7 januari 1972).
6. "Het beschermen van constructiestaal, in het bijzonder in de atmosfeer"
Voordrachten gehouden tijdens de Praktische Corrosiedag, georganiseerd door het NCC in samenwerking met de Stichting Centrum Bouwen in Staal en de Stichting Verftoepassing op 26 november 1971 te Utrecht).

Bijlagen
222
7. "Kathodische bescherming" (Voordrachten gehouden tijdens de Studiedag van het NIRIA op 2 maart 1972 te Utrecht).
8. "Corrosie door luchtverontreiniging"
(Rapport van de NCC-werkgroep "Invloed van Luchtverontreiniging op de atmosferische corrosie van materialen").
9. "Enkele voordrachten op corrosiegebied"
(5 voordrachten gehouden tijdens bijeenkomsten van de Bond van Materialenkennis in 1970 en 1971).
10. "Middelen voor tijdelijke corrosiewering"
(Kritisch overzicht van bestaande standaardvoorschriften, samengesteld door de NCC-werkgroep "Voorschriften").
11. "Richtlijnen betreffende de voorbehandeling van constructiestaal; mechanische
reiniging" (overgenomen uit publicatie nr. 201 van het Staalcentrum Nederland, aan welke uitgave tevens hebben meegewerkt: Stichting Bouwen in Staal, Stichting Verftoepassing en NCC).
12. "Maritieme Corrosie" (fotokopieën)
(Voordrachten gehouden tijdens de Leergang Maritieme Corrosie, georganiseerd door het corrosielaboratorium van het Koninklijk Instituut voor de Marine in samenwerking met de Stichting Materiaalonderzoek in de Zee en de Tussenafdeling der Metaalkunde van de TH-Delft, op 4 t/m 7 juni 1973 te Delft).
13. "Corrosie in warmtewisselaars, in het bijzonder koelsystemen en condensors"
(Voordrachten gehouden tijdens de Praktische Corrosiedag, georganiseerd door het NCC in samenwerking met de Tussenafdeling der Metaalkunde van de TH-Delft, op 19 november 1973 te Delft).
14. "Kathodische bescherming" (fotokopieën)
(Voordrachten gehouden tijdens de Vakantieleergang Corrosie, georganiseerd door de TH-Twente op 3 en 4 april 1975).
15. "Meetmethoden bij corrosieonderzoek en corrosiebestrijding"
(Voordrachten gehouden tijdens de Leergang Corrosie, georganiseerd door de TH-Eindhoven op 6 en 7 januari 1977).
16. "Corrosie en Corrosiebestrijding bij weg- en railtransport"
(Voordrachten gehouden tijdens de Praktische Corrosiedag, TH-Eindhoven, 10 juni 1977)

223
17. "De invloed van het milieu op de scheurvorming in mechanische belaste constructiedelen" (Voordrachten gehouden tijdens de Vakantieleergang Corrosie, georganiseerd door de TH-Delft op 9 en 10 april 1979).
18. "Corrosie en corrosiebestrijding bij Elektriciteitsproductie"
(Voordrachten gehouden tijdens de Corrosiedag georganiseerd door de NV KEMA onder auspiciën van het NCC op 14 september 1982 te Arnhem).
19. "Toepassing van elektrochemische meetmethoden bij corrosie-onderzoek"
(Voordrachten gehouden tijdens de Corrosiedag 21 november 1984). 20. Syllabus van de contactdag "Sector Bescherming Buisleidingsystemen SBB"
op 23 mei 1985. 21. Syllabus van de contactdag "Sector Bescherming Buisleidingsystemen SBB" op
11 juni 1986. 22. Syllabus van de corrosiedag "Sector Metaal en Metaalbescherming" op 17
oktober 1986. 23. Syllabus van de najaarsvergadering "Werkgroep Spanningscorrosie" op 6
oktober 1987. 24. Syllabus van de corrosiedag "Sector Metaal en Metaalbescherming" op 17
september 1987. 25. Syllabus van de contactdag "Sector Bescherming Buisleidingsystemen SBB" op
21 oktober 1988 (fotokopieën). 26. Syllabus van de corrosiedag "Systematische corrosiebeheersing van
staalconstructies "Sector Metaal en Metaalbescherming" op 17 november 1988. 27. Syllabus van de contactdag "Kwaliteitszorg in de Praktijk" "Sector
Bescherming Buisleidingsystemen" op 14 juni 1990. 28. Syllabus van de corrosiedag "Hechting en Hechtingsproblemen" "Sector Metaal
en Metaalbescherming" op 31 oktober 1990. 29. Syllabus van de corrosiedag "Sector Energie" en "Sector Procestechniek,
Apparatenbouw en Engineering" op 22 november 1990. 30. Syllabus van een studiedag "Corrosiebeheersing met elektrochemische
meetmethoden" door Werkgroep Elektrochemische Meetmethoden op 7 april 1992.

Bijlagen
224
31. Syllabus van de contactdag "Corrosiepreventie offshore pijpleidingen" van
"Sector Bescherming Buisleidingsystemen SBB" op 16 juni 1992. 32. Syllabus van de corrosiedag "Praktische corrosiebestrijding van
staalconstructies" van "Sector Metaal en Metaalbescherming SMM" op 18 juni 1993.
33. Seminar "Corrosion by seawater" op 8 december 1992. 34. Syllabus van de corrosiedag "Onderhoud en materiaalkeuze" van "Sector
Energie SE" en "Sector Procestechniek, Apparatenbouw en Engineering SPAE" op 10 december 1992.
35. Syllabus van de contactdag "Levensduurbeheersing in de transportsector van
ontwerp tot en met hergebruik" van "Sector Transport ST i.o" op 13 mei 1993. 36. Syllabus van het 1e Nederlandse Koelwater Symposium op 9 juni 1993. 37. Syllabus van het symposium "Verwerking en gebruik van roestvast staal" op 8
juni 1994. 38. Syllabus van de contactdag van de NCC-Sector Transport "Lichtgewicht en
levensduur" op 3 mei 1994 (fotokopieën). 39. Syllabus van de contactdag van de NCC-Sector Bescherming
Buisleidingsystemen (SBB) op 15 september 1994. 40. Syllabus van de contactdag van de NCC-Sector Maritiem SM/SMOZ op 5
oktober 1994. 41. Syllabus van het symposium "Corrosie, fouling en onderhoud van warmte-
wisselaars", sub-thema: Problemen en oplossingen; een initiatief van de NCC-Sector Procesindustrie, Energie en Engineering (SPEE), in samenwerking met Novem BV, gehouden op 2 november 1994 in de Jaarbeurs, Utrecht.
42. Syllabus van het symposium "Filiforme corrosie" van de NCC-Sector Metaal en
Metaalbescherming (SMM) in samenwerking met het Aluminium Centrum op 3 november 1994 in de Jaarbeurs, Utrecht.
43. Syllabus van het Watersymposium 1995 d.d. 17 en 18 mei in 'het Turfschip' in
Breda op 17 mei en 18 mei 1995.

225
44. Bundel lezingen van de contactdag van de NCC-Sector Metaal en Metaalbescherming (SMM), tezamen met het Staalbouwkundig Genootschap (SG), op 14 juni 1995 in De Efteling in Kaatsheuvel.
45. Syllabus van het CorrosieCongres'95 met als thema 'Praktische beheersing van
corrosie in bouw en industrie' van 31 oktober t/m 2 november 1995 in de Jaarbeurs in Utrecht.
46. Syllabus van de kennisoverdrachtbijeenkomst van het IOP
Oppervlaktetechnologie "Innovatie in oppervlaktetechnologie (vanuit bestaande kennis)" op 12 september 1996 in de Jaarbeurs in Utrecht, georganiseerd in opdracht van Senter.
47. Syllabus van het Watersymposium '97 d.d. 15 april 1997 in het Turfschip in
Breda Thema: Technieken en problematieken bij (her)gebruik van water; ook uw kunt hierop kosten besparen
48. PMP Researchdag van 06-11-97. Thema: Van onderzoek naar praktijk 49. Syllabus van de contactdag “RVS, uw juiste keuze” op 14 mei 1998. 50. Syllabus van het “Watersymposium ‘99” € 93,75 / *€ 75,00 51. Syllabus van de contactdag “Vezelversterkte Kunststof Sluisdeuren” € 47,00 /
*€ 37,50 52. Syllabus Symposium Spanningscorrosie d.d. 25 mei 2000. € 47,00 / *€ 37,50 53. Praktijk richtlijn voor inspectie en onderhoud van (ophang)constructies,
bevestigingsmiddelen en voorzieningen in overdekte zwembaden € 20,00 Praktische Corrosie Handleidingen 1. Algemene inleiding ƒ 23,50 prof.dr. P.J. Gellings, prof.dr. F.P. IJsseling 2. Materiaalkeuze en constructieve aspecten ƒ 23,50 prof.dr. P.J. Gellings, prof.dr. F.P. IJsseling 3. Kathodische en anodische bescherming ƒ 23,50 drs. A.G.C. Kobussen, ing. B.H. Wijngaard 4. Corrosiebestrijding door metallische en anorganische deklagen ƒ 23,50 T. van der Klis 5. Corrosiebestrijding door waterbehandeling ƒ 27,00 A. Snel, E.D.D. During 6. Corrosiebestrijding door organische deklagen ƒ 32,50

Bijlagen
226
T. van der Klis 8. Corrosie van wapening en andere metalen in beton ƒ 32,50 dr. R.B. Polder, prof.dr. J.M.J.M. Bijen Video, samengesteld uit vijf bestaande video's: Corrosie en corrosiebestrijding: − Corrosion in action; − Corrosion under insulation; − Ontwerp en corrosiebestrijding; − Corrosiebestrijding door verandering van omgeving; − Corrosiebestrijding door deklagen Prijs: ƒ 75,--

227
11.18 Internationale organisaties op het gebied van Corrosie en Corrosiebestrijding.
Er zijn twee belangrijke internationale organisaties op het gebied van corrosie en corrosiebestrijdding die veel en nuttige publicaties uitgeven en ook actief zijn op het gebied van voorlichting en onderwijs op dit gebied. In de eerste plaats is dat de EUROPEAN FEDERATION OF CORROSION
die een groot aantal publicaties op corrosiegebied heeft uitgegeven. De meest recente lijst hiervan is te vinden op het internet: http://www.efcweb.org/List_of_Publications.html. Op de website van de EFC: http://www.efcweb.org/ zijn ook veel andere gegevens over de activiteiten van de EFC te vinden. De publicaties van de EFC kunnen worden besteld via de hierboven genoemde website van de EFC of bij: Maney Publishing Subscription Department Hudson Road Leeds LS9 7DL UK Tel: +44 (0) 113 113 249 7481 Fax: +44 (0) 113 248 6983 E-mail: [email protected]. Daarnaast is belangrijk de NACE = National Association of Corrosion Engineers. Oorspronkelijk was dit een Amerikaanse organisatie maar inmiddels is deze ook duidelijk internationaal gericht. Deze organisatie geeft onder andere de tijdschriften: "Corrosion" en "Materials Protection" uit die van groot belang zijn voor iedereen die in de praktijk met corrosie en corrosiebestrijding te maken heeft en die van de nieuwste ontwikkelingen op de hoogte wil blijven. Voor meer gegevens over de actuele activiteiten en publicaties is de NACE-website de beste ingang: http://www.nace.org. Daar zijn de publicaties en cursus-activiteiten te vinden en wordt de mogelijkheid geboden deze te bestellen respectievelijk daarvoor in te schrijven.

Bijlagen
228
11.19 Literatuurlijst
Chemie P.W. Atkins and J.A. Beran, General Chemistry, New York, W.H.Freeman & Co.Ltd, (1992) P.W. Atkins and Julio de Paulo, Physical Chemistry, 7th ed. Oxford, Oxford University Press
(2001) D.F. Shriver and P.W. Atkins, Inorganic Chemistry, Oxford, Oxford University Press (2006) Algemene boeken: M.G. Fontana, Corrosion Engineering, 3rd ed. New York, McGrawHill (1986) J.T.N. Atkinson and H. Van Droffelaar, Corrosion and its Control. An Introduction to the
Subject. 2nd ec. Houston, NACE (1994) S.A. Bradford, Corrosion Control, Canada, Casti Publishing Inc. (2001) K.R. Trethewey and J. Chamberlain, Corrosion for students of science and engineering,
Upper Saddle Hill NJ, Prentice Hall (1996) M. Kowaka, Metal corrosion damage and protection technology, Allerton Press (1990) H.H. Uhlig, Corrosion and corrosion control – An introduction to corrosion science and
engineering, 3rd ed., NewYork, John Wiley (1985) Thermodynamica O. Kubaschewski, C.B. Alcock and P.J. Spencer, Materials Thermochemistry, London,
Butterworth-Heinemann Ltd. (1993) C.H.P. Lupis, Chemical thermodynamics of materials, New York/Amsterdam, North Holland
(1983) Electrochemie en elektrochemische corrosie A.J. Bard and L.R. Faulkner, Electrochemical Methods, Fundamentals and Applications, New
York, John Wiley, (1980) H. Kaesche, Metallic corrosion, 2nd ed., Houston, NACE (1985) D.L. Piron, The electrochemistry of corrosion, Houston, NACE (1991) M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions, Houston, NACE
(1974) M. Pourbaix, Lectures on electrochemical corrosion, 3rd English ed., Houston, NACE (1995) Electrochemical Impedance – Analysis and interpretation, ed. by J.R. Scully, D.C. Silverman,
and W.M. Koenig, Philadelphia, ASTM (1990) Electrochemical Techniques for Corrosion, ed. R. Baboian, Houston, NACE (1977) Corrosiegegevens en praktijkgevallen E.D.D. During, Corrosion Atlas. A collection of illustrated case histories. 3rd expanded and
revised edition, Amsterdam, Elsevier (1997) C.P. Dillon, Forms of corrosion – recognition and prevention, NACE Handbook 1, Houston,
NACE (1982) Corrosion Data Survey – Metals Section, ed. by .L. Graver, 6th ed., Houston, NACE (1985) ASM Handbook, Vol 13: Corrosion, Materials Park, Ohio, ASM International (1987) Dechema Werkstoff Tabelle, (losbladig, wordt regelmatig aangevuld en verbeterd), Frankfurt
am M., Dechema

229
P.A. Schweitzer, Corrosion Resistance Tables, 4th ed., 3 volumes, New York, Marcel Dekker (1995)
Corrosiemetingen en standaardisatie Handbook on corrosion testing and evaluation, ed. by W.H. Ailor, New York, John Wiley
(1971) Techniques for corrosion measuremen, ed. by A. Bronson, G. Warren, Houston, NACE
(1992) R. Baboian, Corrosion tests and standards – Application and interpretation, Philadelphia,
ASTM (1995) Application of accelerated corrosien tests to service life prediction of materials, ed. by G.A.
Cragnolino and N. Sridhar, Philadelphia, ASTM (1994) F. Mansfeld, The polarization technique for measuring corrosion currents, in: Adv.Corrosion
Science and Technology, vol. VI, New York, Plenum Press (1976) NACE Book of Standards, Houston, NACE. Ontwerp en corrosiebestrijding V.R. Pludek, Design and Corrosion Control, London, MacMillan (1977) R.N. Parkins and K.A. Chandler, Corrosion Control in Engineering Design, London, HMSO
(1978) Kathodische en anodische bescherming J.H. Morgan, Cathodic protection, Houston, NACE (1987) W. von Baeckmann, W. Schwenk und W.Prinz, Handbuch des kathodischen
Korrosionsschutzes, neubearbeitete Auflage, Weinheim, Verlag Chemie (1989) W. von Baeckmann, W. Schwenk and W.Prinz, Handbook of cathodic corrosion protection –
Theory and practice of electrochemical protection processes, 3rd ed., Houston, Gulf Publishing Company (1997)
V. Ashworth and C.J.L.Booker, Cathodic protection, theory and practice, Chichester, Ellis Horwood (1986)
V. Ashworth and C. Googan, Cathodic protection,a practical guide, Chichester, Ellis Horwood (1993)
M.E. Parker and E.G. Peattie, Pipeline corrosion and cathodic protection, 4th ed., Houston, Gulf Publishing (2001)
O.L. Riggs and C.E. Locke, Anodic protection, New York/London, Plenum Press (1981) Materialen A.J. Sedriks, Corrosion of stainless steels, 2nd ed., New York, John Wiley (1996) Carbon and alloy steels, ASM Specialty Handbook, ed. by J.R. Davis, Materials Park, Ohio,
ASM International (1995) F.C. Porter, Corrosion of zinc and zinc-alloys, New York, Marcel Dekker (1994) F.W. Gibson, Corrosion, concrete and chlorides – Steel corrosion in concrete: Causes and
restraints, ACI (1987) M. Szeliga, Corrosion in prestressed concrete: pipes, piles and decks, Houston, NACE
(1995) R.A. McCauley, Corrosion of ceramics, New York, Marcel Dekker (1995) P.A. Scweitzer, Corrosion resistance of elastomers, New York, Marcel Dekker (1990)

Bijlagen
230
Deklagen T. van der Klis, Vademecum Oppervlakte Technieken Materialen, deel 1: Metalen, Bilthoven,
Vereniging Oppervlaktetechnieken van Materialen (1982) J.F.H. van Eijnsbergen, Duplex systems. Hot-dip galvanizing plus painting, Amsterdam,
Elsevier (1994) P.D. Donovan, Protection of metals from corrosion in storage and transit, Chichester, Ellis
Horwood (1986) I.Suzuki, Corrosion-resistant coatings technology, New York, Marcel Dekker (1989) Corrosion control by organic coatings, ed. by H. Leidheiser, Jr., Houston, NACE (1981) Hoge-temperatuuroxidatie P. Kofstad, High temperature corrosion, New York, Elsevier (1988). O. Kubaschewski and B.E. Hopkins, Oxidation of metals and alloys, London, Butterworths
(1962) N. Birks and G.H. Meier, Introduction to high temperature oxidation of metals, London,
Edward Arnold (1983) J.H.W. de Wit and T. Fransen, Corrosion studies, in CRC Handbook of Solid State
Electrochemistry, ed. by P.J. Gellings and H.J.M. Bouwmeester, Boca Raton, CRC Press (1997)
Economische beschouwingen H. Bierman Jr. And S. Smidt, The capital budgeting decision: economic analysis of
investmentprojects, 7th ed.New York, MacMillan (1988) E.D. Verink, Economic appraisals of corrosion control measures, J.Educational Modules in
Materials Science and Engineering 3 (1981) 239 - 268 A.M.M. Blommaert en J.M.J. Blommaert, Bedrijfseconomische analyses, 3de druk, Houten,
Stenfert Kroese (1995) A.B. Dorsman, Vlottend financieel management, analyse en planning, 3de druk, Deventer,
Kluwer Bedrijfswetenschappen (1996) Speciale onderwerpen H. McArthur, Corrosion prediction and prevention in motor vehicles, Chichester, Ellis
Horwood (1988) K. Barton, Protection against atmospheric corrosion, New York, John Wiley (1976) S.W. Borenstein, Microbiologically influenced corrosion handbook, Cambridge, Woodhead
Publishing Ltd. (1994) S.C. Dexter, Biologically induced corrosion, Houston, NACE (1986) G. Kobrin, A practical manual of microbiologically induced corrosion, Houston, NACE
(1993) H.L. Logan, The stress corrosion of metals, New York, John Wiley (1966). R.B. Waterhouse, Fretting corrosion, Oxford, Pergamon Press (1972) Dewpoint corrosion, ed. by D.R. Holmes, Chichester, Ellis Horwood (1985) Corrosion in the water and waste water industries, ed. by M. Szeliga, Houston, NACE (1996) P. McIntyre and A.D. Mercer, Corrosion and related aspects of materials for potable water
supplies, London, The Institute of Materials (1993) A.V. Levey, Solid particle ersion and erosion-corrosion of materials, Materials Park, Ohio,
ASM International (1995)

231
11.20 Oplossingen van opgaven
Opgaven §1.6 Opgave 2 Het metaaloppervlak dat in contact is met de vloeistof is 1.5 × π × 1.0 + ¼ × π × 1.02 = 5.5 m2, en het volume van het reactiemengsel is 1.5 × ¼ × π × 1.02 = 1.178 m3. Een corrosiesnelheid van 0.1 mm.jaar-1 = 0.01 cm.jaar-1 en volgens de eerste tabel van bijlage 11.3 is dit gelijk aan 0.01 × 27.4 × 8.92 = 2.44 g.m-2.dag-1, waarbij de dichtheid uit bijlage 11.5 is genomen. Hieruit volgt dat in 8 uur over het hele oppervlak 2.44 × 5.5 / 3 = 4.48 g koper in oplossing gaat. Met het hierboven berekende volume geeft dit een concentratie van 4.48 / 1/178 = 3.8 ppm Opgave 3 Inwendig oppervlak blikje = π × 15 × 10 + 2 × ¼ × π × 102 = 628.3 cm2 Volume blikje = ¼ × π × 102 × 15 = 1178 cm3. Maximaal 5 ppm = 5 mg/1000 cm3 dus in totaal mag in 3 maanden maximaal 5.89 mg oplossen, dat is in een jaar 4 × 5.89 = 23.56 mg dSn = 7.28 g.cm-3 dus maximaal mag (28.56 × 10-3)/7.28 cm3 tin per jaar oplossen, dat is per cm2 maximaal (28.56 × 10-3)/(7.28 × 628.3) = 5.15 × 10-6 cm/jaar ofwel 5.15 × 10-5 mm/jaar. De laagdikte is 5 μm = 5 × 10-3 mm. De levensduur is dus (5 × 10-3)/(5.15 × 10-5) = 97 jaar! De beperking ligt dus in de hoeveelheid tin die in oplossing gaat en niet in de levensduur Opgaven §2.7 Opgave 1 p.ΔV is bij p = 1 atm = 1.013 × 105 N.m-2, ΔV (per mol) = 22.4 × 10-3 m3/mol, dus: p.ΔV = 2269 N.m-2.m3.mol-1 = 2.269 kJ.mol-1. In de meeste gevallen is dit kleiner dan 1 à 2% van de in bijlage 11.6 gegeven Δh0-waarden Opgave 2 Als de concentratie m wordt uitgedrukt in g.l-1 hebben we [A] = m / M mol.l-1 waarbij M de molaire massa van A is, zodat:
µA = µA0 + RT ln(m / M) = (µA
0 - RT ln M) + RT ln m = µA,m0 + RT ln m
Als de concentratie w gewichts% is dan geldt [A] = 10 × w / M mol.l-1 waarbij M de molaire massa van A is, dus:
µA = µA0 + RT ln(10 × w / M) = (µA
0 + RT ln(10 / M)) + RT ln w = µA,w0 + RT ln w

Bijlagen
232
Opgave 3
22+2
(aq)+HFe(s)
2+2+
H20
)g(H0
(aq)Fe00
)g(H(aq)Fe(aq)HFe(s)
plnRT]Feln[RT]Hln[RT2) 2 - -(
2 - -= g
++−μ+μ+μμ=
μ+μ+μμΔ
++
dus:
0)g(H
0(aq)Fe
0002
H2
02
+2(aq)+HFe(s)
2 2 - -=gmet]H[
p]Fe[lnRT2g= g μ+μ+μμΔ
+−ΔΔ
+
en de evenwichtsconstante is: 2
H2
]H[
p]Fe[K 2
+=
+
Opgave 4 Snelheid bij 300 K is r1, bij 310 K r2, r2 = 2 × r1 en ri = RTactEe . Dus is:
⎟⎠⎞
⎜⎝⎛
×===∴
==
⎟⎠⎞
⎜⎝⎛ −
×−×−
31030010
314.8E
)2ln(zodate2rr
erener
act3101
3001
314.8actE
1
2
)310314.8(actE2
)300314.8(actE1
dit geeft dan:
1act mol.kJ6.53
10310300)2ln(314.8E −=
×××=
Opgaven §3.9 Opgave 1 Volgens bijlage 11.7 regel 1 hoort bij de reactie Cd2+ + Fe → Cd + Fe2+ de cel: Fe | Fe2+ | Cd2+ | Cd en de celspanning daarvan is volgens regel 5: Ecel = ECd – EFe. Opgave 2 De reactie die kan verlopen is Ag+(aq)+Fe2+(aq)=Ag(s)+Fe3+(aq). Uit bijlage 11.8 volgt voor de deelreacties: EAg/Ag+ = 0.8 + 0.059 log[Ag+] en EFe3+/Fe2+ = 0.771 + 0.059 log[Fe3+]/[Fe2+]. Evenwicht als EAg/Ag+ = EFe3+/Fe2+ dus als: 0.8 + 0.059 × – 6 = 0.771 + 0.059 log[Fe3+]/[Fe2+]. Hieruit volgt dat: log[Fe3+]/[Fe2+] = 0.029/0.059 – 6 = -5.508 dus: [Fe3+]/[Fe2+]evenw = 3.1 × 10-6. Wanneer [Fe3+]/[Fe2+] = 1 dan is het quotiënt 1 > 3.1 × 10-6. De reactie loopt dan spontaan naar links: zilver lost op, Fe3+ wordt gereduceerd en reageert weg. Idem als [Fe3+]/[Fe2+] = 10–7 alleen wordt het evenwicht dan veel sneller bereikt. Wanneer [Ag+] = 1 mol.l–1 dan wordt log[Fe3+]/[Fe2+] = 0.029/0.059 = 0.49 en [Fe3+]/[Fe2+]evenw = 3.1, In dat geval wordt Fe2+ geoxideerd en zilver slaat neer.

233
Opgave 3 Bij pH = 7 is EH/H2 = 0 + 0.059/2 × log[H+]2 = – 0.059 × pH = – 0.413 V. Dus als [Mn+] = 1mol.l–1 dan spontaan oplossen wanneer EM/Mn+ < – 0.413 V en dat is van K tot en met Fe. als [Mn+] = 10–6 mol.l–1 dan spontaan oplossen wanneer EM/Mn+ – (6 × 0.059)/n < – 0.413 V en dat is van K tot en met Ni. (Ni: –.25 – 0.177 = – 0.427V, Sn: – 0.14 – 0.177 = – 0.317V) pH = 12 dan EH/H2 = – 12 × 0.059 = – 0.708V. als [Mn+] = 1 mol.l–1 en [Mn+] = 10-6 mol. –1 dan spontaan oplossen wanneer
EM/Mn+ < – 0.708 V en dat is van K tot en met Cr. Opgave 4 Volgens vergelijking 3.6.2 is EO2/H2O = 1.228 – 0.059pH +(0.059/4) logpO2.
PH Ei Ei (Volt) 0 1.228 – 0.01 1.218 7 1.228–0.059×7–0.01 0.805 14 1.228–0.059×14–0.01 0.392 [Mn+] = 1mol.l–1 dan corrosie mogelijk wanneer EM/Mn+ < Ei 0 K → Pt 7 K → Ag 14 K → Cu [Mn+] = 10-6 mol.l-1 dan corrosie mogelijk wanneer EM/Mn+ - (0.059 × 6 / n) = EM/Mn+ - 0.177 < Ei 0 K → Pt 7 K → Hg 14 K → Cu Opgave 5 Reactie: Au+ + e– = Au(s) met als potentiaal: E = E0 + 0.059 log [Au+]. Complexvormingsreactie: Au+ + 2 CN– = Au(CN)2
– met evenwichtsconstante: Kcompl = [Au(CN)2
–]/[Au+][CN–]2 = 3 ×1038. Massabalans: [Au+] + [Au(CN)2–] =
10–6. Met [Au+] = x geldt volgt uit de vergelijking voor K: (10–6 – x) = (0.1)2 × 3 × 1038 → x = 3.33 ×10–43. Invullen in de vergelijking van de potentiaal geeft dan: E = –0.826 V: dit betekent dat in aanwezigheid van cyanide goud zich onedel gedraagt met een evenwichtspotentiaal lager dan die van ijzer bij [Fe2+] = 10–6 mol.l–1. Opgave 6 Het oplosbaarheidsproduct van CuS is: Ksol = [Cu2+][S2–] = 10–44 (mol2.l–2). [S2–] = 1 mg.l–1 = 10–3/32 = 3.125×10–5 mol.l–1 [Cu2+] = Ksol/[S2–] = 10–44/3.125×10–5 = 3.2×10–40 mol.l–1. Voor een koperelektrode geldt: Eev = E0 + 0.0295 log [Cu2+] = 0.34 + 0.0295 log(3.2×10–40) = –0.825 V. Dit betekent dat in aanwezigheid van sulfide koper zich onedel gedraagt met een evenwichtspotentiaal lager dan die van ijzer bij [Fe2+] = 10–6 mol.l–1.

Bijlagen
234
Opgave 7 Waterstofontwikkelingsreactie: 2 H+ + 2 e– = H2(g) met als evenwichtspotentiaal: Eev(H+/H2) = –0.059 pH – 0.0295 log p(H2). De maximum Cu2+-concentratie hebben we als evenwicht heerst dus als Eev(Cu/Cu2+) = Eev(H+/H2). 4. Bij p(H2) = 1 atm en pH = 0.1 hebben we dan: 0.34 + 0.0295 log[Cu2+] = –
0.059 × 0.1, dus log[Cu2+] = –0.34 / 0.0295 – 2 × 0.1 = –11.725 dus [Cu2+] = 1.19 × 10–7, maal 63.54×103 geeft dit 1.19×10–7 ppm.
5. Bij p(H2) = 10–8 atm en pH = 0.1 krijgen we: 0.34 + 0.0295 log[Cu2+] = –0.059 × 0.1 + 0.0295 × 8, dus log[Cu2+] = –0.34 / 0.0295 – 2 × 0.1 + 8 = –3.725 dus [Cu2+] = 1.88 × 10–4, maal 63.54×103 geeft dit 11.9 ppm.
Opgave 8 1. zonder O2 hebben we Cd + 2 H2O → Cd2+ + 2 OH– + H2
of: Cd + 2 H+ → Cd2+ + H2 met O2 : 2 Cd + 2 H2O + O2 → 2 Cd2+ + 4 OH– of: 2 Cd + 4 H+ + O2 → 2 Cd2+ + 2 H2O
2. A. 2 H+ + 2 e– → H2, het Cd is immuun en is kathode B: Cd → Cd2+ + 2 e– ; 2 H+ + 2 e– → H2, het Cd is een meervoudige elektrode en dus tegelijk anode en kathode C: Cd → Cd2+ + 2 e–, het cadmium is anode
3. Dan wordt het cadmium bedekt door een gesloten laag Cd(OH)2, dit wordt passiviteit genoemd.
Opgaven §4.6 Opgave 1 1 mA gedurende 24 h is 10–3 × 24 × 3600 As = C. De reactie is Fe → Fe2+ + 2 e–. Dus geldt: aantal mol × molaire massa = (10–3 × 24 × 3600)/(2 × 96487) × 55.85 = 0.025 g. (met getal uit bijlage 11.11 voor elektrochemisch equivalent: 1.042 × 10+3 kg.A–1.h–1 krijgen we 10–3 × 24 × 1.042 × 10+3 ×10–3 = 0.025 g) Opgave 2 Aantal mol zink × molaire massa = (10 × 10–3 ×5 × 3600 C)/(2 × 96487) × 65.3 = 0.061 g. Volume zink = 0.061 / 71.4 (g.cm–3, bijlage 11.5) = 8.53 ×10–3 cm3 en dit geeft een dikte van (8.53 / 15) ×10–3 ×104 = 5.7 μm. Opgave 3 Reactie: 2 H+ + 2 e– → H2, hiervoor geldt dan: (5 × 10–3 × 15 × 3600 C)/(2 × 96487 C/mol) × 22447 (cm3mol–1) = 31.37 cm3. Opgave 4 Reactie: O2 + 4 e– + 2 H2O → 4 OH–, hiervoor krijgen we: (2 × 10–3 × 48 × 3600 C)/(4 × 96487 C/mol) × 22447 (cm3mol–1) = 20.14 cm3.
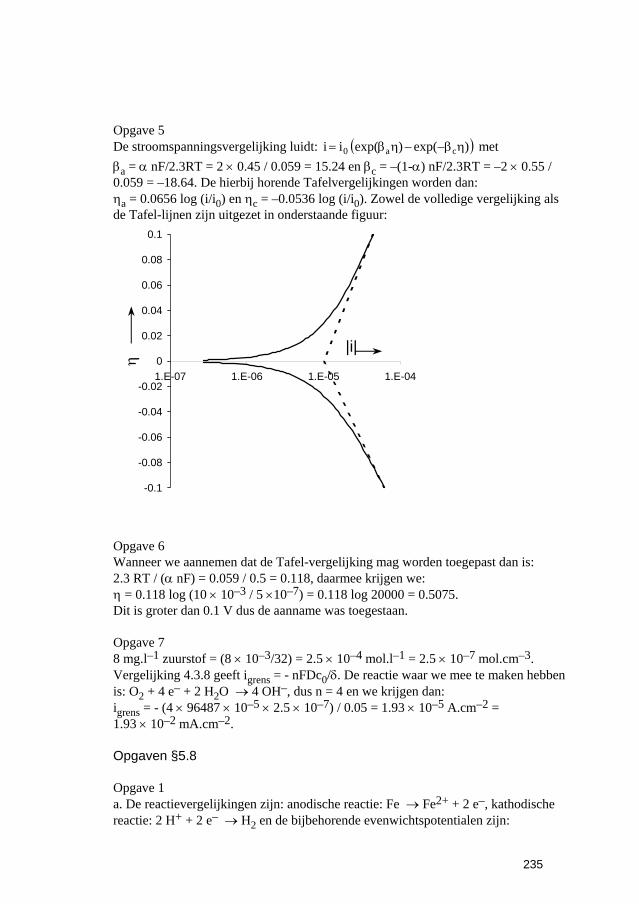
235
Opgave 5 De stroomspanningsvergelijking luidt: ( ))exp()exp(ii ca0 ηβ−−ηβ= met βa = α nF/2.3RT = 2 × 0.45 / 0.059 = 15.24 en βc = –(1-α) nF/2.3RT = –2 × 0.55 / 0.059 = –18.64. De hierbij horende Tafelvergelijkingen worden dan: ηa = 0.0656 log (i/i0) en ηc = –0.0536 log (i/i0). Zowel de volledige vergelijking als de Tafel-lijnen zijn uitgezet in onderstaande figuur:
-0.1
-0.08
-0.06
-0.04
-0.02
0
0.02
0.04
0.06
0.08
0.1
1.E-07 1.E-06 1.E-05 1.E-04
|i|
η
Opgave 6 Wanneer we aannemen dat de Tafel-vergelijking mag worden toegepast dan is: 2.3 RT / (α nF) = 0.059 / 0.5 = 0.118, daarmee krijgen we: η = 0.118 log (10 × 10–3 / 5 ×10–7) = 0.118 log 20000 = 0.5075. Dit is groter dan 0.1 V dus de aanname was toegestaan. Opgave 7 8 mg.l–1 zuurstof = (8 × 10–3/32) = 2.5 × 10–4 mol.l–1 = 2.5 × 10–7 mol.cm–3. Vergelijking 4.3.8 geeft igrens = - nFDc0/δ. De reactie waar we mee te maken hebben is: O2 + 4 e– + 2 H2O → 4 OH–, dus n = 4 en we krijgen dan: igrens = - (4 × 96487 × 10–5 × 2.5 × 10–7) / 0.05 = 1.93 × 10–5 A.cm–2 = 1.93 × 10–2 mA.cm–2. Opgaven §5.8 Opgave 1 a. De reactievergelijkingen zijn: anodische reactie: Fe → Fe2+ + 2 e–, kathodische reactie: 2 H+ + 2 e– → H2 en de bijbehorende evenwichtspotentialen zijn:

Bijlagen
236
V5875.052059.044.0)Fe/Fe(E
V118.0pH.059.00.0)H/H(E
2ev
2ev
−=−×+−=
−=−=
+
+
(1)
b. De polarisatielijnen worden gegeven door de Tafel-vergelijkingen (4.3.6):
96.0ilog.12.05875.0E)a(EE
7.0ilog.1.0118.0E)c(EE
aaevaa
ccevcc
+=+=−=η
−−=+=−=η (2)
De hiermee berekende lijnen zijn in onderstaande figuur getekend.
-0.6
-0.5
-0.4
-0.3
-0.2
-0.1-9 -8 -7 -6 -5 -4 -3
log | i |
E 0.0388V
log icorr =-4.502
log icorr =-5.411
-0.277V
-0.168V
{
Fe / Fe2+
H+ / H2
Eev(H2) =-0.118V
Eev(Fe) =-0.5875V
Het snijpunt kan grafisch uit de figuur worden bepaald of, nauwkeuriger, worden berekend door in de Tafelvergelijkingen (2) in te vullen: Ea = Ec = Ecorr en |ia| = |ic| = icorr, hetgeen, door combinatie van de twee Tafelvergelijkingen, geeft:
V277.0E7.0)ilog(.1.0)c(EEcm.A1088.3i66.1)ilog(.22.0)c(E)a(E
corrcorrevcorr
26corrcorrevev
−=→−−=×=→−−=− −−
(3)
Dit is ook in de figuur aangegeven. Als er een weerstand in het systeem is moeten we figuur 5.5.1 toepassen en dan wordt het potentiaalverschil tussen de elektroden dus met de Ohmse-spanningsval ΔE = i × R verminderd. Als R = 0.1 Ω dan wordt dit 0.1 × 3.88 × 10-6 = 3.88 × 10-7 Volt, hetgeen volledig verwaarloosbaar is ten opzichte van het potentiaalverschil van 0.4695 tussen de evenwichtspotentialen en de corrosiestroomdichtheid blijft dus gelijk aan de hierboven berekende waarde. Wanneer R = 10000 Ω wordt ΔE = 0.0388 Volt. In de figuur wordt dan het punt gezocht waar dit juist de afstand tussen de polarisatielijnen is, dat is de dunne, verticale lijn. Dit geeft een i = 2.5 × 10-6 A.cm-2 en zou dus tot een kleinere ΔE

237
leiden. Een waarde tussen de oorspronkelijke en nu gevonden stroomdichtheid geeft na 1 of 2 keer proberen een nieuwe waarden van ongeveer 3 × 10-6 A.cm-2. Nauwkeuriger kunnen we dit berekenen met de volgende vergelijking, die we krijgen door de term ΔE = i × R toe te voegen aan de eerste vergelijking (3) voor icorr:
66.1)ilog(.22.0i100004695.0iR)c(E)a(EE)c(E)a(E
corrcorr
correvevevev
−−=×+−=×+−=Δ+−
en deze kan òf door proberen òf met behulp van een programmeerbare calculator worden opgelost, met als resultaat dat in dit geval de corrosiestroomdichtheid gelijk is aan 2.87 × 10-6 A.cm-2. c. Dit is te berekenen door te nemen i'0 = 10–5 A.cm–2 (1% van het oppervlak!) in de vergelijking: η(H2) = E – Eev(H2) = E + 0.118 = –0.1 log(i/10–5). Dus, analoog aan eerste vergelijking (3): -0.5875 + 0.118= – 0.22 log i – 0.12 × 8 – 0.1 × 5 en dit geeft log i = -4.5023 (i = 3.146 × 10–5 A.cm–2), dus meer dan 10 maal zo groot als zonder de onzuiverheid. Ecorr wordt in dit geval – 0.168 V. Opgave 2 Kathodische reactie: ηc = E – Eev,c = – βc log |i/i0c| Anodische reactie: ηa = E – Eev,a = βa log |i/i0a| In stationaire toestand is E = Ecorr, invullen in de vergelijkingen en aftrekken geeft: – Eev,c + Eev,a = – (βc + βa) log i + βc log i0c + βa log i0a en hieruit volgt:
)i(log)i(logEE
ilog a0ca
ac0
ca
c
ca
a,evc,evcorr β+β
β+
β+ββ
+β+β
−= .
Opgave 3 De snelheidsbepalende stap voor corrosie in neutrale, beluchte oplossing is zuurstof-reductie die in alle gevallen over het hele oppervlak van de proefstukken met gelijke snelheid verloopt omdat deze reactie volledige diffusiepolarisatie vertoont. De totale kathodische stroom is dus in alle gevallen gelijk, zodat ook de totale anodi-sche stroom in alle gevallen gelijk moet zijn, zodat ook de gewichtsverandering in alle gevallen gelijk is. Omdat de corrosiesnelheid op het onbedekte deel evenredig is met de stroomdichtheid wordt deze groter wanneer het onbedekte deel kleiner wordt. De penetratie (= dikteafname) van het onbedekte deel neemt dus toe met toenemen-de bedekking van het oppervlak. Opgave 4 Uit tabel 5.7.1 zien we dat bij pH = 8.4 de kritische passiveringsstroomdichtheid van ijzer 10–5 A.cm–2 is. De kathodische reductie stroomdichtheid berekend in opgave 4.6.7 is 1.93 × 10–5 A.cm–2, dus iets hoger dan de kritische passiveringsstroom-dichtheid. Bij een pH iets beneden 8.4 wordt ijzer dus spontaan gepassiveerd. Opgave 5 Antwoorden hangen af van de gekozen gemeenschappelijke eenheid!

Bijlagen
238
Opgave 6 In zuur corrodeert ijzer volgens de reacties: anodische reactie: Fe → Fe2+ + 2 e–, kathodische reactie: 2 H+ + 2 e– → H2. a. er wordt waterstof gevormd en de totale reactie is Fe + H+ → Fe2+ + H2. Het oppervlak van de proefplaatjes is 2 × 100 × 50 + 2 × 100 × 2 + 2 × 50 × 2 = 10600 mm2 = 106 cm2. 256.7 ml H2 per 10 min = 1.146 × 10–2 mol/10 min = 1.65 mol/dag 37.4 ml H2 per 60 min = 1.670 × 10–3 mol/60 min = 4.01 × 10–2 mol/dag 1 mol Fe per dag = 55.85 g/dag = 55.85/7.86 =7.11 cm3/dag. b. zonder beitsrem wordt de aantasting (1.65 × 7.11) *10 / 106 = 1.11 mm/dag.
Met de gegevens uit bijlage 11.3 en 1 mm/dag = 36.5 cm/jaar = 36.5 × 3.06 × nd / M = 31.4 mA.cm–2 = 0.0314 A.cm–2 krijgen we: 1.11 × 0.0314 = 3.49 × 10–2 A.cm–2. met beitsrem wordt de aantasting (4.01× 10–2 × 7.11) *10 / 106 = 0.027 mm/dag. Net als hiervoor krijgen we: 0.027 × 0.0314 = 8.49 × 10–4 A.cm–2.
Bij pH = 1 is Eev(H2) = –0.059 V en bij 10 g Fe/liter is Eev(Fe) = - 0.44 + 0.059/2 log 10/55.85 = – 0.462 V c. Volgens de Tafelvergelijking geldt:
Ecorr = – 0.462 + 0.1 log (icorr / 10–5) (anodische reactie) Ecorr = – 0.059 – 0.1 log (icorr /i0c) (kathodische reactie) Aftrekken en omwerken geeft dan: log i0c = 0.97 + d log icorr. Invullen van de onder b gevonden waarden geeft dan: zonder beitsrem: log i0c = 0.97 + 2 log 3.49 × 10–2 = - 1.944 zodat i0c = 1.13 ×10-2 A.cm–2 met beitsrem: log i0c = 0.97 + 2 log 8.49 × 10–2 = - 5.172 zodat nu i0c = 6.73 ×10–2 A.cm–2 In onderstaande figuur is het bijbehorende polarisatiediagram getekend.
-0.5-0.45
-0.4-0.35
-0.3-0.25
-0.2-0.15
-0.1-0.05
0-7 -6 -5 -4 -3 -2 -1 0
log | i |
E
-0.462 V
-0.059 V
log icorr :-3.071
log icorr :-1.457
log i0c :-5.172
log i0c :-1.944

239
Opgaven §7.4 Opgave 1 Rdraad = (80 × 10–6 × 20)/(0.25 × π × 0.052) = 8.1487 × 10–1 Ω. 24 uur = 1/365 jaar, 0.25 mm.jaar–1 = 0.25 / 365 mm/24 h. Dikteafname = 2 × 0.25 / 365 = 1.37 × 10–3 mm = 1.37 × 10–4 cm. R2 = (80 × 10–6 × 20)/(0.25 × π × (0.05 – 1.37 × 10–4)2) = 8.13935 × 10–1 Ω. Dus ΔR = 4.48 × 10–3 Ω, ΔR/R = 5.5 × 10–3 = 0.55%. Opgave 2 In feite alleen kwantitatief voor algemene corrosie. Weerstandsmethode eventueel ook voor put- of interkristallijne corrosie, maar dan alleen kwalitatieve aanduiding van mate van penetratie. Opgave 3 Het water is gedurende 4 s in contact met het koper. In die 4 s is per 0.5 m3 dus 1.5 × 500 = 750 mg = 0.75 g koper opgenomen. Dit is gebeurd vanaf een oppervlak van 70 m2. Per dag is dus 0.75 × 15 × 60 × 24 / 70 = 231 g koper opgenomen, corrosiesnelheid = 231 g.m–2d–1 = 231 × 0.0365 × 10/ 8.92 = 9.5 mm.jaar–1 (formule uit bijlage 11.3, dichtheid koper uit bijlage 11.5). Opgaven §8.9 Opgave 1 Voor de bescherming van 100 m2 is nodig 0.1 × 100 = 10 A.Uit tabel 8.5.1 volgt: Zn: nodig 10.7 / 0.93 = 11.5 kg per A.jaar bij 93% efficiëntie, Al: nodig 2.9 / 0.65 = 4.46 kg per A.jaar bij 65% efficiëntie. Dus: Zn: nodig 10 × 20 × 11.5 = 2300 kg, Al: nodig 10 × 20 × 4.46 = 892 kg. Opgave 2 1. Corrosiesnelheid: 25 mdd = 90 × 10–4 mA.cm–2 = 90 mA.m–2 en dus moet de
grensstroomdichtheid van zuurstofreductie hieraan gelijk zijn (log icorr = 1.95). Eev(Fe) = – 0.44 + 0.0295 × –7 = – 0.647 V. Zie figuur voor polarisatiediagram.
-0.7
-0.5
-0.3
-0.1
0.1
0.3
0.5
0.7
0.9
-2 -1 0 1 2 3
log | i |E
- 0.647 V
- 0.292 V

Bijlagen
240
2. Ecorr = – 0.647 + 0.12 × log(90 / 10–1) = – 0.292 V 3. De minimale beschermstroomdichtheid is gelijk aan de corrosiestroomdichtheid
(zie §8.5.1) en is dus 90 mA.m-2 = 0.09 A.m-2. 4. Volgens Tabel 8.5.1 levert wordt 10.7 kg per A.jaar zink verbruikt. Een anode
van 30 kg levert dus bij 95% efficiëntie 30 × (0.95 / 10.7) = 2.664 A.jaar. Nodig
is: ⎟⎠⎞
⎜⎝⎛ ××+
×+× 09.015.08
209.015.009.02 = 0.2115 A.jaar.m–2. Er kan dus
2.664 / 0.2115 = 12.6 m2 met 1 anode worden beschermd. Opgave 3 Slechte punten: − Bedekken van onedel metaal in contact met edeler metaal is gevaarlijk: in
poriën of beschadigde plaatsen is een klein onedel oppervlak in contact met een groot edel oppervlak → snelle corrosie.
− Ondersteunen van het vat met 4 dunne poten geeft grote spanningsconcentratie bij de bevestigingsplaats van de poten en voor roestvast staal in contact met hete chloride-oplossing leidt dit zeker tot spanningscorrosie.
− Roestvast staal is niet erg resistent tegen hete chloride-oplossing en zeer gevoelig voor putcorrosie.
− Las tussen roestvast en ongelegeerd staal kan leiden tot verhoogde koolstof-concentratie in de las en/of het roestvaste staal en daardoor interkristallijne corrosie veroorzaken.
Verbeteringen: − Maak tank in zijn geheel van één materiaal. Bij voorkeur koperlegering of
(eventueel) roestvast staal met verhoogd molybdeengehalte (bijvoorbeeld 316) dat beter bestendig is tegen put- en spanningscorrosie dan 304.
− Ondersteun de tank met een rondom daaraan gelaste ring zodat de belasting gelijkmatig is verdeeld en minder hoog is.
Opgave 4 a. Uit de bovenste tabel van bijlage 11.3. volgt dat de corrosiesnelheid dan 0.12 ×
1000 × 1.158 / 10000 = 0.014 cm.jaar-1 = 0.14 mm.jaar-1 is. De levensduur is dan 0.75 / 0.14 = 5.4, dus afgerond 5 jaar. Met de onderste tabel vinden we 0.12 × 1000 × 1.3 × 10 / 10000 = 0.156 mm.jaar-1, dus ook 5 jaar als levensduur. 1 koelelement van 100 m2 kost € 10000, equivalente kosten C1 = 10000 / 3.6048 = € 2775 per jaar. De maximale prijs van 304 moet dan zodanig zijn dat Pmax / 7.4694 ≤ 2775, dus Pmax ≤ € 20727.
b. De corrosiesnelheid moet met een factor 4 worden verlaagd. Volgens ver-gelijking 4.3.8. betekent dit dat de diffusiecoëfficiënt ook met een factor 4 omlaag moet. Dus als x maal 10 mg.l-1 moet worden toegevoegd dan geldt: 0.9x = 0.25 zodat x = 13.2 of zekerheidshalve naar boven afgerond is 14 × 10 mg.l-1 = 140 mg.l-1 nodig. De prijs van de eerste hoeveelheid is dus 20000 × 0.14 × 75 / 1000 = € 210, hetgeen 210 / 7.4694 = € 28 bijdraagt aan de equivalente kosten. De bedrijfs-

241
kosten zijn 0.25 × 0.14 × 365 × 75 = € 958 per jaar. Het benodigde oppervlak is nu een factor 1 / 0.98514 = 1.236, afgerond 1.25 maal zo groot. Dit betekent dat de investering nu € 12500 wordt en de equivalente kosten dus 12500 / 7.4694 = € 1673 per jaar. De totale kosten worden dan 28 + 1673 + 958 = € 2659. Dit is iets lager dan de totale kosten bij vervanging iedere 5 jaar, dus verdient dit de voorkeur (bovendien dan geen productie-onderbreking nodig voor de vervanging).
c. De corrosiesnelheid moet ook hier een factor 4 omlaag, hetgeen volgens vergelijking 4.3.8. ook betekent dat de O2 -concentratie met diezelfde factor omlaag moet: deze mag dus hoogstens nog 2 mg.l-1 zijn. De totale aanschafprijs wordt nu € 18000, dus de totale equivalente kosten 18000 / 7.4694 = € 2409. De kosten voor het gebruik van stoom mogen dus niet hoger zijn dan € (2775 - 2409) = € 366 per jaar.
d. Gezien het gegeven van diffusiepolarisatie moet de beschermingsstroom-dichtheid gelijk zijn aan de corrosiestroomdichtheid: 0.12 A.m-2, de totale beschermstroom wordt dus 100 × 0.12 = 12 A. Volgens tabel 8.5.1. is theoretisch nodig 2.9 kg per A.jaar, dus 12 × 2.9 = 34.8 kg. De efficiency is 70 % dus in werkelijkheid is nodig 34.8 / 0.7 = 50 kg, dat wil zeggen 10 kg per anode. De totale investering wordt € 10000 (koeler) + 5 × € (10 × 16 + 85 + 60) = € 11525. De equivalente kosten worden dan 11525 / 7.4694 + 1825 / 1.69 = € 2623.
Opgaven §9.9 Opgave 1 Pilling-Bedworth verhouding = PB = (mol.massa oxide × dichtheid metaal) / (m × mol.massa metaal × dichtheid oxide), het oxide is MmOn. Al2O3: PB = ((2 × 26.98 + 3 × 16) × 2.702) / 2 × 26.98 × 3.97 = 1.286 → beschermend; MgO: PB = ((24.32 + 16) × 1.74) / 24.32 × 3.58 = 0.806 → niet beschermend; Na2O: PB = ((2 × 22.99 + 16) × 0.97) / 2 × 22.99 × 2.27 = 0.576 → niet beschermend. Opgave 2 Volgens vergelijking (9.5.4) is kp evenredig met n/1
O2p en volgens bijlage 11.16 is
voor NiO: n = 1/6. Hieruit vinden we:
kp(0.1 mm Hg) =
61
2.0760
1.02.0
⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜
⎝
⎛ ×= 0.226 × kp(1 atm)

Bijlagen
242
kp(10–6 mm Hg) =
61
6
2.0760
102.0
⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜
⎝
⎛×
−
= 0.0331 × kp(1 atm)
Opgave 3 De algemene reactievergelijking is: m M (s) + n/2 O2(g) → MmOn(s) en hiervoor geldt: Δg = – m μM – n/2 μO2 + μMmOn = (– m μ0
M – n/2 μ0O2 + μ0
MmOn) – n/2RT ln (pO2) = Δg0 – n/2 RT ln (pO2). Evenwichtsdruk wanneer Δg = 0, dus als Δg0 = n/2 RT ln (pO2) en hieruit krijgen
we: ⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ=
RTgexpp
2n
0evO 2
Kamertemperatuur = 25 °C = 298 K, 500 °C = 773 K, 1000 °C = 1273 K. We gebruiken de formule (zie bijlage 11.6: Δg0(T) = Δg0(298) – (T – 298)Δs0(298) In onderstaande tabel zijn de benodigde thermodynamische gegevens samengevat.
Oxide Δg0(298) Δs0(298) Δg0(773) Δg0(1273) Fe3O4 – 1015.5 – 345.2 – 851.5 – 576.1 Cu2O – 146.0 – 75.8 – 110.0 – 72.1 Cr2O3 – 1058.2 – 273.6 – 928.1 – 791.3 Al2O3 – 1582.4 – 312.9 – 1433.8 – 1277.3 In de volgende tabel staan de hiermee berekende evenwichtsdrukken in atm.
Oxide evO2
p (298) evO2
p (773) evO2
p (1273)
Fe3O4 1.0 ×10–52 1.7 ×10–29 1.5 ×10–12 Cu2O 6.6 ×10–52 1.36 ×10–15 1.2 ×10–6 Cr2O3 2.2 ×10–124 1.54 ×10–42 2.3 ×10–22 Al2O3 1.2 ×10–185 2.57 ×10–55 1.2 ×10–30 Opgave 4 De oxidatiereactie is: Ag (s) + ½ O2 → Ag2O en hiervoor geldt: Δg = Δg0 + ½RT ln
2Op = Δh0 –TΔs0 + ½RT ln 2Op en in evenwicht is Δg = 0.
2Op = 0.2 atm onder normale omstandigheden.
Invullen: (– 31100 + 66.5 T) + (8.314 × T / 2) ln (0.2) = 0, uitwerken geeft dan:

243
T = 5.66
2609.1314.8
31100
+×
= 425 K = 152 °C.
Opgave 5 Reactie: 3 Fe (s) + 4 H2O(g) → Fe3O4(s) + 4 H2(g) waarvoor: Δg = – 3 μFe – 4 μH2O + μFe3O4 + 4 μH2 = (– 3 μ0
Fe – 4 μ0H2O + μ0
Fe3O4 + 4 μ0H2) + 4RT ln (pH2/pH2O)
= Δg0 + 4RT ln (pH2/pH2O). In evenwicht (bij constante p en T) is Δg = 0 en dat geeft:
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ−=⎟
⎟⎠
⎞⎜⎜⎝
⎛
RT4gexp
pp 0
evOH
H
2
2
350 °C = 623 K Δg0 = – 1118400 + 623 * 345.2 ← Fe3O4 + 4 × 241800 – 4 × 623 × 44.4 ← H2O
= – 46875 J.mol-1 en dit geeft: evOH
H
2
2
pp
⎟⎟⎠
⎞⎜⎜⎝
⎛= 9.56. In evenwicht zou dus wanneer
stoom in contact met ijzer komt de partiële waterstofdruk ongeveer 10 maal zo groot worden als de partiële waterdruk. In (zuivere) stoom zou ijzer dus altijd moeten oxideren! Opgave 6 a. Oxide van M stabieler dan van N: de reactie NO + M = N + MO wil naar rechts verlopen. Dan geldt: Δg = – μNO – μM + μMO + μN = (– μ0
NO – μ0M + μ0
MO + μ0N) + RT ln (xN/xM)
= Δg0 + RT ln (xN/xM) Er heerst evenwicht wanneer Δg = 0 en daaruit volgt:
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ−Δ=⎟⎟
⎠
⎞⎜⎜⎝
⎛− RT
ggexp
x1x 0
NO0MO
evM
M
Met x/(1-x) = A krijgen we na omvormen x = a/(1+A), wat bewezen moest worden. b. Bij 750 °C = 1023 K krijgen we: Δg0(FeO) = – 266300 + 70.9 × 1023 = – 193769 Δg0(NiO) = – 239700 + 94.0 × 1023 = – 143538 Aftrekken geeft Δg0(FeO) - Δg0(NiO) = – 50231.3 en A = 2.724 × 10–3. De kritische concentratie xc = A/(1+A)= 2.72 × 10–3 = 0.26 gewichts%

Bijlagen
244
Opgave 7 Voor de reactie M + CO2 → MO + CO geldt: Δg = (– μ0
M – μ0CO2 + μ0
MO + μ0CO) + RT ln (pCO/pCO2) =
Δg0 + RT ln (pCO/pCO2) zodat: ⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ−=⎟
⎟⎠
⎞⎜⎜⎝
⎛
RTgexp
pp 0
evCO
CO
2
RT ln (pCO/pCO2) = – 30420 bij 500 °C = 773 K en – 7720 bij 800 °C = 1073 K. Δg0 = Δg 0(MO) – Δg0(CO2) + Δg0(CO) Δg0(CO) – Δg0(CO2) = – 112000 + 394000 – 773 × 88 – 773 × 0.84 = 213330 bij 773 K en Δg0(CO) – Δg0(CO2) = 186670 bij 1073 K. 500°C = 773 K: Δg0(CuO) = – 85720, Δg0(FeO) = – 211490, Δg0(NiO) = – 167040 800°C = 1073 K: Δg0(CuO) = – 57940, Δg0(FeO) = – 190224, Δg0(NiO) = –138838 500°C: Δg(CuO) = – 85720 + 213330 – 30420 = 97190 → geen oxidatie 500°C: Δg(FeO) = – 211490 + 213330 – 30420 = – 28580 → oxidatie 500°C: Δg(NiO) = – 167040 + 213330 – 30420 = 15870 → geen oxidatie 800°C: Δg(CuO) = – 57940 + 186670 – 7720 = 121010 → geen oxidatie 800°C: Δg(FeO) = – 190224 + 186670 – 7720 = – 11274 → oxidatie 800°C: Δg(NiO) = – 138338 + 186670 – 7720 = 40612 → geen oxidatie Opgaven §11.16 Opgave 1 Hierbij geldt de reactievergelijking
ZnOi221 ZnOe2Zn)g(O +=′++ ••
en de evenwichtsconstante hiervan is:
2iO
2iO
ZnO
]e][Zn[p1
]e][Zn[p]Zn][O[
K2
1
2
21
2′
=′
=••••
met de elektroneutraliteitsvoorwaarde:
]e[]Zn[2 i ′=•• . Bij aanwezigheid van lithium wordt dit: ]iL[]e[]Zn[2 Zni ′+′=•• . Samen met de evenwichtsconstante geeft dit dat de interstitiële zinkionencon-centratie, en daarmee de oxidatiesnelheid, toeneemt. In aanwezigheid van aluminium wordt de elektroneutraliteitsvoorwaarde:
]e[]Zn[2]Al[ iZn ′=+ ••• . Samen met de evenwichtsconstante geeft dit dat de interstitiële zinkionenconcentratie, en daarmee de oxidatiesnelheid, afneemt. Opgave 2 Hiervoor hebben we de reactievergelijking:
OO221 Oe2V)g(O =′++ •• (11.16.11)
met de evenwichtsconstante: 2
OO2
OO
O
]e][V[p1
]e][V[p]O[
K2
1
2
21
2′
=′
=••••
en de elektroneutraliteitsvoorwaarde: ]e[]V[2 O ′=•• .

245
Invullen hiervan geeft dan: 6
1
2
21
221
2
OOO3
O3OO
p]V[:ofp]V[:omvormenna]V[p4
1K −••−••••
∝∝=
wat bewezen moest worden.


247
Index
A aantasting
algemene, 83 inslag-, 105, 163 plaatselijke, 83
actief-passief element, 76 activeringsenergie, 21, 212 activeringspolarisatie, 48 afzettingscorrosie, 91 Ag/AgCl-elektrode, 32, 140 alfa-ijzerstructuur, 14 algemene aantasting, 83 algemene corrosie, 83 aluminiseren, 183 aluminium en aluminiumlegeringen, 135 aluminiumbrons, 135 aluminiummessing, 134 amfoteer, 21, 90 anionvacature, 179 anode, 29, 39
inerte, 138 offer, 138
anodische bescherming, 79, 142 anodische reactie, 28 anodische stroomdichtheid, 212 anodiseren, 151 anorganische deklagen, 149 Arrhenius-vergelijking, 21, 211 atmosfeer
industriële, 86 landelijke, 85 maritieme, 86 stedelijke, 86
atmosferische corrosie, 84, 147 atoombouw, 13 atoommassa, 13 atoomnummer, 13 austenietstructuur, 14 austenitisch roestvast staal, 133 Avogadro
getal van, 13
B bacteriën
aërobe, 93 anaërobe, 93 ijzer- en mangaan, 93 slijmvormende, 93 sulfaat-reducerende, 93 zuurvormende, 93 zwavel-oxiderende, 93
bedrijfskosten, 164 beitsrem, 68, 128 beluchting
differentiële, 91 bescherming
anodische, 79, 142 kathodische, 37, 138
beschermingspotentiaal, 140 bestrijding hoge temperatuur corrosie
materiaalkeuze, 183 vormgeving, 184
beton, 137 bimetallische corrosie, 73 binder, 152 binding
covalente, 14 ionogene, 15 metallische, 15
biofilm, 93 blik, 146 brons, 134
C calomel-elektrode, 32 carbideprecipitatie, 98 cavitatie-corrosie, 105, 163 celspanning, 29, 205 chemische potentiaal, 18 chemische thermodynamica, 17 chromaten, 78 chroomverarming, 98 condensaatcorrosie, 160, 187

Bijlagen
248
contactcorrosie, 73 oppervlakteverhouding, 73
contante waarde netto, 163
continue corrosiebewaking, 118 corrosie
algemene, 83 atmosferische, 84, 147 bimetallische, 73 cavitatie, 105, 163 condensaat, 160, 187 contact, 73 dauwpunt, 160, 187 erosie-, 105 galvanische, 73 interkristallijne, 96 microbiologisch beïnvloede, 93 microbiologisch geïnduceerde, 93 plaatselijke, 83 spannings, 4, 99, 114, 121, 130, 133
corrosiebestendigheid, 6 corrosiebewaking
continue, 118 corrosiekosten, 4 corrosiepotentiaal, 63 corrosieproeven
versnelde, 116 corrosiestroomdichtheid, 63 corrosievermoeiing, 99 corrosion monitoring, 118 covalente binding, 14 Cu/CuSO4-elektrode, 32, 140 cupronikkel, 134
D dampfase inhibitoren, 155 dauwpunt, 188 dauwpuntcorrosie, 160, 187 defectchemie, 179, 216 defecten, 178 definitie van corrosie, 3 deklagen, 143
anorganische, 149 metallische, 145 niet-metallische, 149
organische, 152 diagram
E-pH-, 34 Evans, 47 polarisatie, 47 potentiaal-pH, 34 Pourbaix, 34 stabiliteits, 186 stroom-spanning, 47 Tafel, 49
differentiële beluchting, 91 diffusiegrenslaag, 51 diffusiemodel van Nernst, 51 diffusiepolarisatie, 55 dissociatie, 16 dissociatiedruk, 174 dissociatieproduct, 20 drager, 152 drijvende kracht, 17 duplex roestvast staal, 133 duplex-systeem, 152
E economische aspekten, 163 edel metaal, 27, 32, 65, 73, 95, 137, 145 eenheidscel, 14 effectieve lading, 218 elektrochemisch
equivalent, 45 elektrochemische
cel, 28, 46, 151 corrosie, 45 kinetiek, 46 metingen, 118 reeks, 32, 39, 40, 47
elektrochemische impedantie spectroscopie, 118
elektrode, 28 Ag/AgCl, 32, 140 calomel-, 32 Cu/CuSO4, 32, 140 standaard, 30, 205 waterstof-, 31, 205
elektrodepotentiaal, 30, 205 elektrolytweerstand, 70

249
elektroneutraliteitsvoorwaarde, 219 elektrongat, 179, 218 element, 13 eloxeren, 151 enthalpie, 16
molaire, 18 entropie, 17
molaire, 18 equivalent
elektrochemisch, 45 equivalente kosten, 164 erosie-corrosie, 105 Evans-diagram, 47 evenwichtsconstante, 19 evenwichtsdruk, 174
F Faraday
wet van, 45 ferritisch roestvast staal, 131 Fick
wet van, 51, 179 fosfateren, 109, 151 Frenkel-defect, 216 fretting corrosion, 108
G galvanisch verzinken, 148 galvanische
praktische, reeks, 40, 208 galvanische corrosie, 73 gamma-ijzerstructuur, 14 getal van Avogadro, 13 Gibbs vrije energie, 17 grensstroomdichtheid, 52, 55 groei
put, 88 scheur, 100
H half-cel, 30 halfgeleiders, 220 hoge-temperatuur oxidatie, 175
HSLA-staal, 87 hydrazine, 70
I ideale gassen, 18 immuniteit, 37, 39 immuun, 37, 39, 137 impedantie spectroscopie
elektrochemische, 118 impingement attack, 105 inerte anodes, 138 inhibitoren, 67, 68, 103, 109, 115, 128
dampfase, 155 gemengde werking, 67 kathodische werking, 67
initiatie put, 88 scheur, 100
inslag-aantasting, 105, 163 interkristallijne corrosie, 96 interkristallijne scheuren, 99 interstitiëel, 216 ionen, 15 ionogene binding, 15
K kathode, 29, 39 kathodische bescherming, 37, 75, 138
polarisatiediagram, 138 kathodische reactie, 29 kathodische stroomdichtheid, 212 kernlading, 13 ketelsteen, 96 kinetiek
beschermende, 175 elektrochemische, 46 kubische, 177 lineaire, 176 logaritmische, 177 parabolische, 175
koper en koperlegeringen, 134 kosten
bedrijfs-, 164 corrosie, 4, 199

Bijlagen
250
directe, 4 equivalente, 164 indirecte, 5 onderhouds-, 164
kristalrooster, 14 kritische passiveringsstroomdichtheid, 54 kubische kinetiek, 177 kubische oxidatie, 177 kubische structuur
ruimtelijk gecentreerd, 14 vlakken gecentreerd, 14
kunststof spanningscorrosie, 136
kunststoffen, 136
L lading
effectieve, 218 ladingsoverdrachtpolarisatie, 54 lasbederf, 98 lineaire
kinetiek, 176 oxidatie, 176
lineaire snelheidsconstante, 176 logaritmische
kinetiek, 177 oxidatie, 177
M marinemessing, 134 martensitisch roestvast staal, 130 materiaalkeuze, 6, 129 meervoudige elektrode, 55, 61 mengpotentiaal, 63 messing, 134 metaal
edel, 27, 32, 65, 73, 95, 137, 145 onedel, 32, 63, 65, 73, 74, 95, 137, 146
metaalvacature, 179 metallische binding, 15 metallische deklagen, 145 microbiologisch beïnvloede corrosie, 93 microbiologisch geïnduceerde corrosie,
93
mol, 13 monitoring
corrosion, 118
N natriumchloride structuur, 15 Nernst-vergelijking, 30, 32, 39 netto contante waarde, 163 niet-metallische deklagen, 149 niet-stoichiometrie, 217 nikkel en nikkellegeringen, 135 nikkelvacature, 220 nitrieten, 78 n-type
oxide, 220
O offeranode, 138 onderhoudskosten, 164 onedel, 40 onedel metaal, 32, 63, 65, 73, 74, 95, 137,
146 ongelegeerd staal, 129 ontluchten, 70, 127 ontwerpschema, 125, 193 ontzinking, 95
laagvormige, 95 propvormige, 95
ontzouting, 72 oplosbaarheidsproduct, 34 oppervlakteverhouding, 73 organische deklagen, 152 overbescherming, 104, 135, 141 overdrachtscoëfficiënt, 49, 212 oxidatie, 28, 173
kubische, 177 lineaire, 175 logaritmische, 177 parabolische, 175 preferentiele, 181
oxidatieslijtage, 108 oxide
n-type, 220 p-type, 220

251
P parabolische
kinetiek, 175, 185 oxidatie, 177, 178 snelheidsconstante, 175, 180 snelheidsvergelijking, 180
partiële druk, 18 passief, 55 passief-actief-element, 89 passieve stroomdichtheid, 54 passingroest, 108 passivatoren, 78, 90, 128 passiveringspotentiaal, 54 passiviteit, 37, 40, 53, 75 periodiek systeem, 13, 202 pH, 20 pigment, 152, 153 Pilling-Bedworth verhouding, 178 plaatselijke aantasting, 83 plaatselijke corrosie, 83 polarisatie
activerings-, 48 diffusie-, 48 kromme, 49 ladingsoverdracht-, 48 weerstand, 49, 50, 113, 117, 119
polarisatiediagram, 47 kathodische bescherming, 138
potentiaal beschermings, 140 passiverings, 54 transpassieve, 54
potentiaal-pH diagram, 34 potentiaalval, 70, 71, 138 Pourbaix-diagram, 34 praktische galvanische reeks, 40, 208 precipitatie hardend roestvast staal, 134 preferentiele oxidatie, 181 p-type
oxide, 220 puntdefect, 216 putcorrosie, 88
kathodische bescherming, 90 putgroei, 88 putinitiatie, 88
putvormige corrosie, 88
R reactie
aflopende, 16 anodische, 28 evenwichts-, 16 kathodische, 29
reactiesnelheid, 21 redox-koppel, 33 redox-reactie, 33 reductie, 28 roest, 84 roestvast staal, 130
austenitisch, 133 duplex, 133 ferritisch, 131 martensitisch, 130 precipitatie hardend, 134
S scheuren
interkristallijne, 99 transkristallijne, 99
scheurgroei, 100 scheurinitiatie, 100 scheurvoortplantingssnelheid, 101 Schottky-defect, 216 season cracking, 102 seizoenziekte, 102 selektief oplossen, 95 sensitiseren, 98 siccatief, 152 snelheidsconstante, 21
lineaire, 176 parabolische, 175, 180
spanningscorrosie, 4, 99, 114, 121, 130, 133 kunststof, 136
spleetcorrosie, 91 staal
HSLA, 87 ongelegeerd, 129 roestvast, 77, 130

Bijlagen
252
weerbestendig, 129 weerbestendig, 87
stabiliteitsdiagram, 186 standaardelektrode, 30, 205 standaardpotentiaal, 32, 39, 205 stationaire stroomdichtheid, 63 sterke zuren, 20 stoichiometrische verbranding, 182 stroomdichtheid
anodische, 212 corrosie, 63 grens, 52 kathodische, 212 kritische passiverings-, 54 passieve, 54 stationaire, 63 uitwisseling-, 49, 50, 54, 64, 66, 76,
78, 211 stroom-spanningsdiagram, 47 structuur
austeniet-, 14 ferriet-, 14 NaCl, 15
sulfidatie, 173, 185 sulfidatiesnelheid, 185 sulfiet, 70
T Tafel-diagram, 49 Tafel-vergelijking, 49 thermisch verzinken, 137, 146, 157 thermodynamische potentiaal, 18 tijdelijke corrosiebeschermingsmiddelen,
86, 109, 154 transkristallijne, 99 transpassieve potentiaal, 54 tribochemische reacties, 108 tweecomponenten verf, 152
U uitwisselingsstroomdichtheid, 49, 50, 54,
64, 66, 76, 78, 211
V vacature, 216
anion, 179 metaal, 179 nikkel, 220 zuurstof, 179, 220
verbinding, 13 verbranding
stoichiometrische, 182 verf, 152
tweecomponenten, 152 versnelde corrosieproeven, 116 verzinken
galvanisch, 148 thermisch, 137, 146, 157
vormgeving, 184 vrije enthalpie, 17, 174
W waterbehandeling, 127 waterstofelektrode, 31, 205 weerbestendig staal, 87, 129 weerbestendige staalsoorten, 87 weld-decay, 98 wet
van Faraday, 45 van Fick, 51, 179
Z zelfionisatie, 20 zuursterkte, 20 zuurstofbinders, 70 zuurstofcorrosie, 68 zuurstofvacature, 179, 220 zuurstofverwijdering, 70 zwakke zuren, 20


















