
Tijdschr. voor Geneeskunde, 62, nr. 4, 2006 · 283 Samenvatting De klinische benadering van...
13
283 Samenvatting De klinische benadering van congenitale luchtwegmalformaties is over de laatste jaren voor een belangrijk deel verschoven van post- naar prenataal. Dit overzichtsartikel wil een illustratie zijn van de positieve impact van deze prenatale diagnostiek op het postnataal inter- nistisch en chirurgisch beleid. Voor de gynae- coloog-echografist doen deze aangeboren luchtwegmalformaties zich meestal voor als uni- of bilaterale hyperdense longen – de zg. „bright fetal lungs”. Bij bilaterale hyperdensi- teit gaat het meestal om een „congenital high airway obstruction syndrome” (CHAOS) of een bilaterale congenitale cystische adenoma- toïde malformatie (CCAM). De prognose van deze zeldzame bilaterale letsels is veelal infaust. De prognose van unilaterale congenitale lucht- wegmalformaties is daarentegen globaal goed tot zeer goed te noemen na postnatale correc- tie. Terughoudendheid met zwangerschapson- derbreking is daarom een nuttige boodschap. Het is belangrijk te onderlijnen dat in-utero- regressie van dergelijke unilaterale hyperdense long eerder de regel is dan de uitzondering. Deze „vanishing lesions” moeten postnataal nochtans precies nagezien worden. De pasgeborene met een congenitale lucht- wegmalformatie is vaak asymptomatisch bij geboorte. Prenatale diagnostiek zal daarom een electieve postnatale aanpak bevorderen. Postnatale (meestal niet-urgente) chirurgie is de regel bij zowel CCAM, longsekwester, bronchogene cyste als bij de meeste gevallen van congenitaal lobair emfyseem (CLE). Indien asymptomatisch, wordt electieve heel- kunde aangeraden tussen 3 en 6 maanden. Conserverende segmentectomie leidt tot een grotere kans op recidief. Lobectomie is daarom de aangewezen ingreep. Bij asymp- tomatisch CLE kan een conservatief beleid overwogen worden, hoewel dit inhoudt dat de einddiagnostiek later onjuist kan blijken. Tijdschr. voor Geneeskunde, 62, nr. 4, 2006 PERINATALE AANPAK BIJ CONGENITALE LUCHTWEGMALFORMATIES 1 P. VANHAESEBROUCK 2, 7 , P. DEFOORT 3 , K. SMETS 2 , H. VERMEERSCH 4 , V. MEERSSCHAUT 5 , L. GOOSSENS 2 , S. VAN DAELE 6 , F. DE BAETS 6 , M. TEMMERMAN 3 1 „Perinatale Club N° 4” 19 oktober 2004: postgraduaat onderwijs georganiseerd door het perinataal centrum van het Universitair Ziekenhuis te Gent. 2 Vakgroep Pediatrie-Genetica, Dienst Neonatale Intensieve Zorgen, 3 Vakgroep Uro-Gynaecologie, Vrouwenkliniek, 4 Vakgroep Neus-, Keel- en Oorheelkunde, Dienst Hoofd- en Halschirurgie, 5 Vakgroep Radiologie, Dienst Radiologie en Medische Beeldvorming, 6 Vakgroep Pediatrie-Genetica, Dienst Pediatrie, Afdeling Kinderpneumologie, Universitair Ziekenhuis Gent. 7 Correspondentieadres: prof. dr. P. Vanhaesebrouck, Dienst Neonatale Intensieve Zorgen, Behandelingsblok 1B1, Uni- versitair Ziekenhuis Gent, De Pintelaan 185, 9000 Gent; e-mail: [email protected] Inleiding De nomenclatuur van congenitale longziek- ten staat nog ter discussie (1). Zoektermen zoals „congenital cystic lung diseases”, „congenital lungbud anomalies”, „bronchopulmonary fore- gut malformations” en „congenital pulmonary airway malformations” dekken eenzelfde lading. Wij zullen ons in dit overzicht beperken tot de meest gebruikelijke benaming „congenitale luchtwegmalformatie”. De pathologisch-anato- mische beschrijving van de letsels wordt hier grotendeels buiten beschouwing gelaten. Ons doel is praktijkgericht, uitgaand van een foetale waarneming. De eerste invalshoek is eenvoudig: gaat het om bilaterale hyperdense longen of is slechts één long hyperdens? De lokalisatie, de homogeniciteit en het kleurendopplerpatroon van het aangeboren letsel zullen in een later sta- dium vaak richtinggevend zijn voor een meer precieze foetale diagnostiek. Bilaterale „bright lung” Het „congenital high airway obstruction syndrome” (CHAOS) is een zeldzame entiteit
Transcript of Tijdschr. voor Geneeskunde, 62, nr. 4, 2006 · 283 Samenvatting De klinische benadering van...

283
Samenvatting
De klinische benadering van congenitaleluchtwegmalformaties is over de laatste jarenvoor een belangrijk deel verschoven van post-naar prenataal. Dit overzichtsartikel wil eenillustratie zijn van de positieve impact van dezeprenatale diagnostiek op het postnataal inter-nistisch en chirurgisch beleid. Voor de gynae-coloog-echografist doen deze aangeborenluchtwegmalformaties zich meestal voor alsuni- of bilaterale hyperdense longen – de zg.„bright fetal lungs”. Bij bilaterale hyperdensi-teit gaat het meestal om een „congenital highairway obstruction syndrome” (CHAOS) ofeen bilaterale congenitale cystische adenoma-toïde malformatie (CCAM). De prognose vandeze zeldzame bilaterale letsels is veelal infaust.De prognose van unilaterale congenitale lucht-wegmalformaties is daarentegen globaal goedtot zeer goed te noemen na postnatale correc-tie. Terughoudendheid met zwangerschapson-derbreking is daarom een nuttige boodschap.Het is belangrijk te onderlijnen dat in-utero-regressie van dergelijke unilaterale hyperdenselong eerder de regel is dan de uitzondering.Deze „vanishing lesions” moeten postnataalnochtans precies nagezien worden.
De pasgeborene met een congenitale lucht-wegmalformatie is vaak asymptomatisch bijgeboorte. Prenatale diagnostiek zal daaromeen electieve postnatale aanpak bevorderen.Postnatale (meestal niet-urgente) chirurgie isde regel bij zowel CCAM, longsekwester,bronchogene cyste als bij de meeste gevallenvan congenitaal lobair emfyseem (CLE).Indien asymptomatisch, wordt electieve heel-kunde aangeraden tussen 3 en 6 maanden.Conserverende segmentectomie leidt tot eengrotere kans op recidief. Lobectomie isdaarom de aangewezen ingreep. Bij asymp-tomatisch CLE kan een conservatief beleidoverwogen worden, hoewel dit inhoudt dat deeinddiagnostiek later onjuist kan blijken.
Tijdschr. voor Geneeskunde, 62, nr. 4, 2006
PERINATALE AANPAK BIJ CONGENITALE LUCHTWEGMALFORMATIES 1
P. VANHAESEBROUCK 2, 7, P. DEFOORT 3, K. SMETS 2, H. VERMEERSCH 4,V. MEERSSCHAUT 5, L. GOOSSENS 2, S. VAN DAELE 6, F. DE BAETS 6, M. TEMMERMAN 3
1 „Perinatale Club N° 4” 19 oktober 2004: postgraduaatonderwijs georganiseerd door het perinataal centrum vanhet Universitair Ziekenhuis te Gent.
2 Vakgroep Pediatrie-Genetica, Dienst Neonatale IntensieveZorgen,
3 Vakgroep Uro-Gynaecologie, Vrouwenkliniek,4 Vakgroep Neus-, Keel- en Oorheelkunde, Dienst Hoofd-
en Halschirurgie,5 Vakgroep Radiologie, Dienst Radiologie en Medische
Beeldvorming,6 Vakgroep Pediatrie-Genetica, Dienst Pediatrie, Afdeling
Kinderpneumologie, Universitair Ziekenhuis Gent.7 Correspondentieadres: prof. dr. P. Vanhaesebrouck, Dienst
Neonatale Intensieve Zorgen, Behandelingsblok 1B1, Uni-versitair Ziekenhuis Gent, De Pintelaan 185, 9000 Gent;e-mail: [email protected]
Inleiding
De nomenclatuur van congenitale longziek-ten staat nog ter discussie (1). Zoektermen zoals„congenital cystic lung diseases”, „congenitallungbud anomalies”, „bronchopulmonary fore-gut malformations” en „congenital pulmonaryairway malformations” dekken eenzelfde lading.Wij zullen ons in dit overzicht beperken tot demeest gebruikelijke benaming „congenitaleluchtwegmalformatie”. De pathologisch-anato-mische beschrijving van de letsels wordt hiergrotendeels buiten beschouwing gelaten. Onsdoel is praktijkgericht, uitgaand van een foetalewaarneming. De eerste invalshoek is eenvoudig:gaat het om bilaterale hyperdense longen of isslechts één long hyperdens? De lokalisatie, dehomogeniciteit en het kleurendopplerpatroonvan het aangeboren letsel zullen in een later sta-dium vaak richtinggevend zijn voor een meerprecieze foetale diagnostiek.
Bilaterale „bright lung”
Het „congenital high airway obstructionsyndrome” (CHAOS) is een zeldzame entiteit
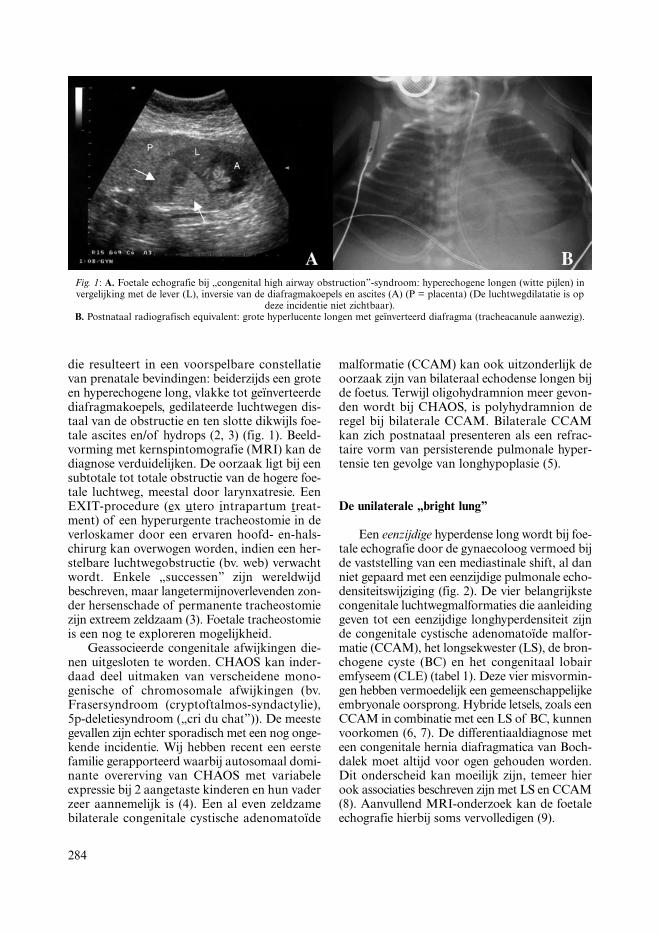
284
Fig. 1: A. Foetale echografie bij „congenital high airway obstruction”-syndroom: hyperechogene longen (witte pijlen) invergelijking met de lever (L), inversie van de diafragmakoepels en ascites (A) (P = placenta) (De luchtwegdilatatie is op
deze incidentie niet zichtbaar).B. Postnataal radiografisch equivalent: grote hyperlucente longen met geïnverteerd diafragma (tracheacanule aanwezig).
die resulteert in een voorspelbare constellatievan prenatale bevindingen: beiderzijds een groteen hyperechogene long, vlakke tot geïnverteerdediafragmakoepels, gedilateerde luchtwegen dis-taal van de obstructie en ten slotte dikwijls foe-tale ascites en/of hydrops (2, 3) (fig. 1). Beeld-vorming met kernspintomografie (MRI) kan dediagnose verduidelijken. De oorzaak ligt bij eensubtotale tot totale obstructie van de hogere foe-tale luchtweg, meestal door larynxatresie. EenEXIT-procedure (ex utero intrapartum treat-ment) of een hyperurgente tracheostomie in deverloskamer door een ervaren hoofd- en-hals-chirurg kan overwogen worden, indien een her-stelbare luchtwegobstructie (bv. web) verwachtwordt. Enkele „successen” zijn wereldwijdbeschreven, maar langetermijnoverlevenden zon-der hersenschade of permanente tracheostomiezijn extreem zeldzaam (3). Foetale tracheostomieis een nog te exploreren mogelijkheid.
Geassocieerde congenitale afwijkingen die-nen uitgesloten te worden. CHAOS kan inder-daad deel uitmaken van verscheidene mono-genische of chromosomale afwijkingen (bv.Frasersyndroom (cryptoftalmos-syndactylie),5p-deletiesyndroom („cri du chat”)). De meestegevallen zijn echter sporadisch met een nog onge-kende incidentie. Wij hebben recent een eerstefamilie gerapporteerd waarbij autosomaal domi-nante overerving van CHAOS met variabeleexpressie bij 2 aangetaste kinderen en hun vaderzeer aannemelijk is (4). Een al even zeldzamebilaterale congenitale cystische adenomatoïde
malformatie (CCAM) kan ook uitzonderlijk deoorzaak zijn van bilateraal echodense longen bijde foetus. Terwijl oligohydramnion meer gevon-den wordt bij CHAOS, is polyhydramnion deregel bij bilaterale CCAM. Bilaterale CCAMkan zich postnataal presenteren als een refrac-taire vorm van persisterende pulmonale hyper-tensie ten gevolge van longhypoplasie (5).
De unilaterale „bright lung”
Een eenzijdige hyperdense long wordt bij foe-tale echografie door de gynaecoloog vermoed bijde vaststelling van een mediastinale shift, al danniet gepaard met een eenzijdige pulmonale echo-densiteitswijziging (fig. 2). De vier belangrijkstecongenitale luchtwegmalformaties die aanleidinggeven tot een eenzijdige longhyperdensiteit zijnde congenitale cystische adenomatoïde malfor-matie (CCAM), het longsekwester (LS), de bron-chogene cyste (BC) en het congenitaal lobairemfyseem (CLE) (tabel 1). Deze vier misvormin-gen hebben vermoedelijk een gemeenschappelijkeembryonale oorsprong. Hybride letsels, zoals eenCCAM in combinatie met een LS of BC, kunnenvoorkomen (6, 7). De differentiaaldiagnose meteen congenitale hernia diafragmatica van Boch-dalek moet altijd voor ogen gehouden worden.Dit onderscheid kan moeilijk zijn, temeer hierook associaties beschreven zijn met LS en CCAM(8). Aanvullend MRI-onderzoek kan de foetaleechografie hierbij soms vervolledigen (9).
285
Fig. 2: Echogram van de foetus waarbij de mediastinaleshifting (H = hart) het merkteken is voor een unilaterale
hyperechogene long (= L) (congenitale cystischeadenomatoïde malformatie type 3).
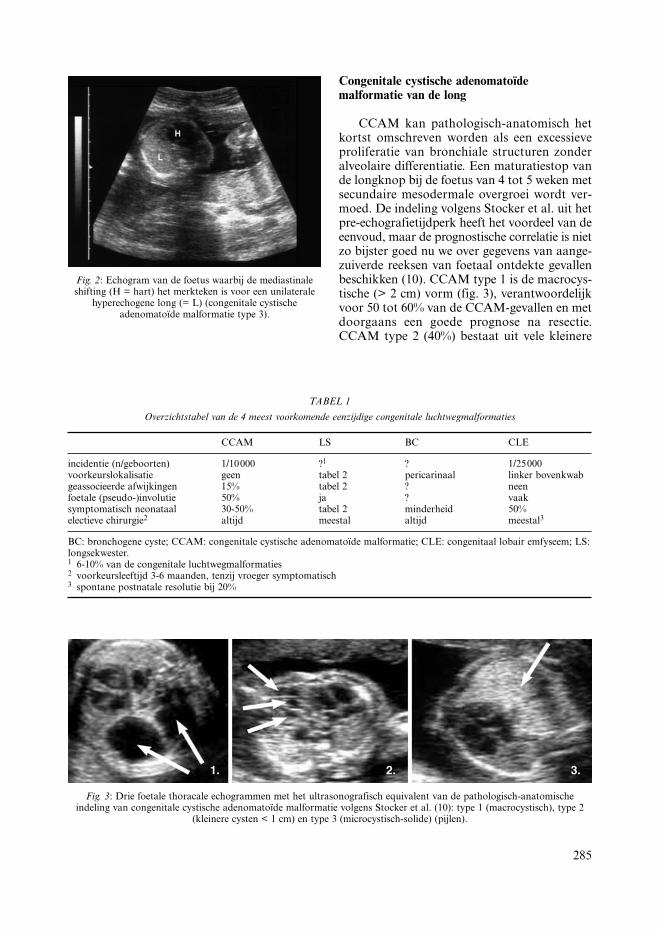
Congenitale cystische adenomatoïdemalformatie van de long
CCAM kan pathologisch-anatomisch hetkortst omschreven worden als een excessieveproliferatie van bronchiale structuren zonderalveolaire differentiatie. Een maturatiestop vande longknop bij de foetus van 4 tot 5 weken metsecundaire mesodermale overgroei wordt ver-moed. De indeling volgens Stocker et al. uit hetpre-echografietijdperk heeft het voordeel van deeenvoud, maar de prognostische correlatie is nietzo bijster goed nu we over gegevens van aange-zuiverde reeksen van foetaal ontdekte gevallenbeschikken (10). CCAM type 1 is de macrocys-tische (> 2 cm) vorm (fig. 3), verantwoordelijkvoor 50 tot 60% van de CCAM-gevallen en metdoorgaans een goede prognose na resectie.CCAM type 2 (40%) bestaat uit vele kleinere
TABEL 1
Overzichtstabel van de 4 meest voorkomende eenzijdige congenitale luchtwegmalformaties
CCAM LS BC CLE
incidentie (n/geboorten) 1/10000 ?1 ? 1/25000voorkeurslokalisatie geen tabel 2 pericarinaal linker bovenkwabgeassocieerde afwijkingen 15% tabel 2 ? neenfoetale (pseudo-)involutie 50% ja ? vaaksymptomatisch neonataal 30-50% tabel 2 minderheid 50%electieve chirurgie2 altijd meestal altijd meestal3
BC: bronchogene cyste; CCAM: congenitale cystische adenomatoïde malformatie; CLE: congenitaal lobair emfyseem; LS:longsekwester.1 6-10% van de congenitale luchtwegmalformaties2 voorkeursleeftijd 3-6 maanden, tenzij vroeger symptomatisch3 spontane postnatale resolutie bij 20%
Fig. 3: Drie foetale thoracale echogrammen met het ultrasonografisch equivalent van de pathologisch-anatomischeindeling van congenitale cystische adenomatoïde malformatie volgens Stocker et al. (10): type 1 (macrocystisch), type 2
(kleinere cysten < 1 cm) en type 3 (microcystisch-solide) (pijlen).

286
cysten (< 1 cm). De prognose is vooral afhan-kelijk van de geassocieerde afwijkingen (bij 8tot 15%). CCAM type 3 (10%) doet zich voorals een microcystisch-solide massa en heeft deslechtste prognose (vaak letale longhypoplasie).
In tegenstelling met de vroegere (pre-ultra-sonografische) reeksen kan nu globaal gesteldworden dat de prognose van de meeste foetaleluchtwegmalformaties, inclusief CCAM, goedtot zeer goed is met een sterfte die nu onder10% ligt (11-17). Naast het type zijn ook degraad van mediastinale shifting, de aanwezig-heid van polyhydramnion en de uitgebreidheidvan de longaantasting bepalend voor de uitein-delijke prognose. De belangrijkste sterfte bijCCAM (2/3) is op dit ogenblik het gevolg vanzwangerschapsonderbreking.
CCAM komt voor bij 1/10000 geboorten. Bij15% worden andere afwijkingen gevonden.CCAM kent geen voorkeurslongkwab. Het risicovan het ontwikkelen van hydrops wordt zeer wis-selend ingeschat van 10 tot 40%. Het klinischspectrum gaat neonataal van vaak asymptoma-tisch (50-70%) tot ernstige ademhalingsinsuffi-ciëntie of zelfs refractaire pulmonale hypertensie(door longhypoplasie). Bij de meer voorkomendemanifestatie na de neonatale periode staan recur-rente longinfecties op de voorgrond.
Nagenoeg de helft van de foetaal vastgesteldeCCAM’s regresseert of verdwijnt echografischvóór de term wordt bereikt, typisch rond de zwan-gerschapsleeftijd van 30-32 weken (18). Postna-taal zal de röntgenopname van de thorax even-eens normaal zijn. Dit belet niet dat in de meestegevallen van „vanishing” CCAM wel degelijk nogafwijkingen zullen gevonden worden bij het ver-richten van een spiraal-CT-scan. Gezien het risicovan latere verwikkelingen (recurrente infectie,maligniteit, air trapping,...), is het aangewezen ditpostnataal onderzoek altijd te verrichten, ook bijde prenataal geïnvolueerde gevallen. De ontwik-keling van ascites en vooral van hydrops bijCCAM wordt meestal als zeer omineus gezien methoge foetale/neonatale sterfte. Recent is echter eenreeks gevallen beschreven waar de hydrops en/of deascites spontaan in utero regresseerden, gevolgddoor een voldragen geboorte met succesvollelobectomie postnataal. In Brazilië, waar zwanger-schapsonderbreking illegaal is, blijkt bij 20% vande gevallen deze regressie plaats te vinden (19-21).
De perinatale aanpak van CCAM zal vooralgestuurd worden door de eventuele geassocieerdeernstige afwijkingen en het al of niet ontwikke-
len van hydrops (22, 23). De meeste moeders meteen CCAM-foetus zullen – zo geen zwanger-schapsonderbreking werd uitgevoerd – de termbereiken en een asymptomatisch kind baren. Bijhydropische omvorming zal (door sommigen) eenpartus praematurus overwogen worden zo dezwangerschapsleeftijd van 32 weken is bereikt.Thoraco-amniotische shunting in utero werdreeds met succes toegepast bij extreem pretermenmet CCAM en hydrops en/of pleura-effusies (23).In het licht van een reële kans op spontane regres-sie van de hydrops kan een afwachtende „no fetalintervention”-houding verdedigd worden.
Postnataal worden bij CCAM zeer variabeleradiologische longbeelden gevonden, soms metniveaubeelden (fig. 4). Spiraal-CT zal ook hier hetonderzoek bij uitstek zijn om de letsels preopera-tief goed te omschrijven. Postnataal wordt eenonmiddellijke lobectomie aanbevolen indien deneonatus respiratoire symptomen vertoont (24-27). Preoperatieve transthoracale drainage kanlevensreddend zijn bij airtrapping en acute over-expansie (28). Hoogfrequente oscillatieventilatieen/of selectieve intubatie van de contralateralebronchus kunnen hierbij noodzakelijk zijn. Seg-mentectomie wordt afgeraden gelet op de groterekans op recidief. Indien de pasgeborene symp-toomloos is en blijft, wordt algemeen een electievelobectomie aanbevolen rond de leeftijd van 3 tot6 maanden. De argumenten voor deze electieveingreep bij asymptomatische CCAM zijn veel-vuldig. Acute airtrapping met (spannings)pneu-mothorax tot gevolg is een reëel risico. Recurrenteinfecties bemoeilijken de chirurgie. Vooral vroegemaligne degeneratie bij conservatief behandeldeCCAM wordt vaker beschreven (29). 8% van dezeldzame longkankers bij kinderen (pleuropul-monaal blastoom, rhabdomyosarcoom, bron-choalveolair carcinoom) is geassocieerd met eer-der vastgestelde aangeboren cystischelongafwijkingen. De compensatoire longgroeiwordt bij extrapolatie vanuit dierexperimenteelonderzoek als effectiever beschouwd vóór de leef-tijd van 2 jaar. Een zuigeling herstelt vlugger vaneen thoracotomie dan een ouder kind, met korterehospitalisatieduur tot gevolg. Een postnataal vast-gestelde CCAM involueert nooit volledig. De pre-natale resolutie is vaak „vals” omdat postnataalalleen een radiografie van de thorax werd verricht.En ten slotte wordt ook geargumenteerd datCCAM „ooit wel eens” symptomatisch wordt,zelfs op hoge leeftijd op een ogenblik van alge-meen verminderde weerstand (27).
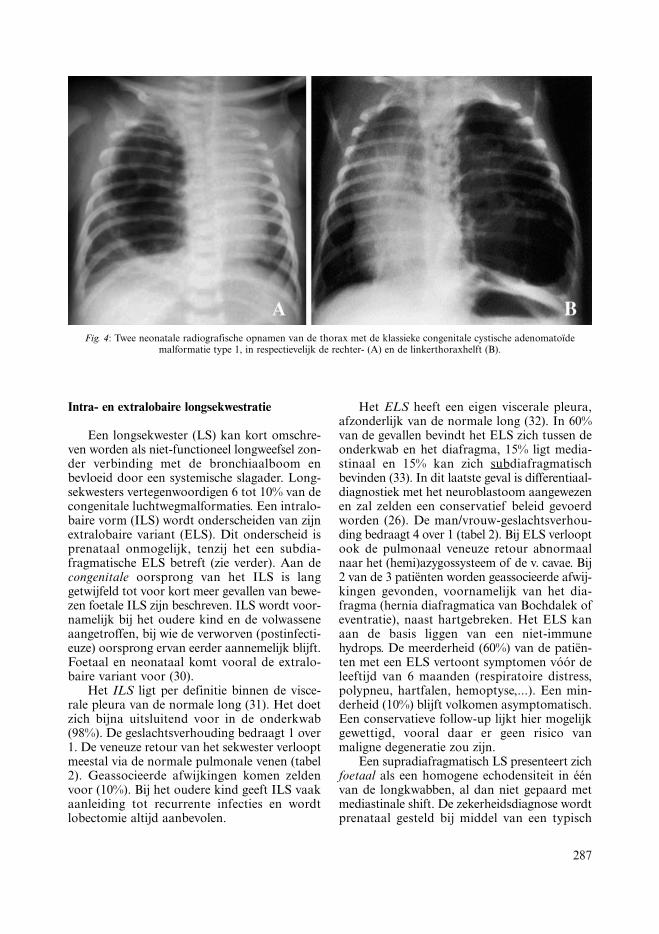
287
Fig. 4: Twee neonatale radiografische opnamen van de thorax met de klassieke congenitale cystische adenomatoïdemalformatie type 1, in respectievelijk de rechter- (A) en de linkerthoraxhelft (B).
Intra- en extralobaire longsekwestratie
Een longsekwester (LS) kan kort omschre-ven worden als niet-functioneel longweefsel zon-der verbinding met de bronchiaalboom enbevloeid door een systemische slagader. Long-sekwesters vertegenwoordigen 6 tot 10% van decongenitale luchtwegmalformaties. Een intralo-baire vorm (ILS) wordt onderscheiden van zijnextralobaire variant (ELS). Dit onderscheid isprenataal onmogelijk, tenzij het een subdia-fragmatische ELS betreft (zie verder). Aan decongenitale oorsprong van het ILS is langgetwijfeld tot voor kort meer gevallen van bewe-zen foetale ILS zijn beschreven. ILS wordt voor-namelijk bij het oudere kind en de volwasseneaangetroffen, bij wie de verworven (postinfecti-euze) oorsprong ervan eerder aannemelijk blijft.Foetaal en neonataal komt vooral de extralo-baire variant voor (30).
Het ILS ligt per definitie binnen de visce-rale pleura van de normale long (31). Het doetzich bijna uitsluitend voor in de onderkwab(98%). De geslachtsverhouding bedraagt 1 over1. De veneuze retour van het sekwester verlooptmeestal via de normale pulmonale venen (tabel2). Geassocieerde afwijkingen komen zeldenvoor (10%). Bij het oudere kind geeft ILS vaakaanleiding tot recurrente infecties en wordtlobectomie altijd aanbevolen.
Het ELS heeft een eigen viscerale pleura,afzonderlijk van de normale long (32). In 60%van de gevallen bevindt het ELS zich tussen deonderkwab en het diafragma, 15% ligt media-stinaal en 15% kan zich subdiafragmatischbevinden (33). In dit laatste geval is differentiaal-diagnostiek met het neuroblastoom aangewezenen zal zelden een conservatief beleid gevoerdworden (26). De man/vrouw-geslachtsverhou-ding bedraagt 4 over 1 (tabel 2). Bij ELS verlooptook de pulmonaal veneuze retour abnormaalnaar het (hemi)azygossysteem of de v. cavae. Bij2 van de 3 patiënten worden geassocieerde afwij-kingen gevonden, voornamelijk van het dia-fragma (hernia diafragmatica van Bochdalek ofeventratie), naast hartgebreken. Het ELS kanaan de basis liggen van een niet-immunehydrops. De meerderheid (60%) van de patiën-ten met een ELS vertoont symptomen vóór deleeftijd van 6 maanden (respiratoire distress,polypneu, hartfalen, hemoptyse,...). Een min-derheid (10%) blijft volkomen asymptomatisch.Een conservatieve follow-up lijkt hier mogelijkgewettigd, vooral daar er geen risico vanmaligne degeneratie zou zijn.
Een supradiafragmatisch LS presenteert zichfoetaal als een homogene echodensiteit in éénvan de longkwabben, al dan niet gepaard metmediastinale shift. De zekerheidsdiagnose wordtprenataal gesteld bij middel van een typisch

288
TABEL 2
Differentiërende kenmerken voor de intralobaire en extralobaire variant van het longsekwester
ILS ELS
voorkeurslokalisatie 98% onderkwab 60% tussen onderkwab endiafragma, 15% mediastinaal en
15% subdiafragmatisch
geslachtsverhouding (M/V) 1/1 4/1
laterisatie 55% links 65% links
voorkomen in de viscerale pleura van de long eigen viscerale pleura
arteriële bevloeiing 70% thoracale aorta, 80% thoracale of abdominale20% abdominale aorta aorta
veneuze afvoer normaal via pulmonale venen meestal abnormale veneuze retour(95%) via (hemi)azygos of vv. cavae
geassocieerde afwijkingen 10% 65% (HDB, hartgebrek)
kliniek zeldzaam onder 2 jaar tenzij 60% symptomen bij < 6 maandenprenatale diagnose; hoog (RDS, voedingsproblemen,
infectierisico bij het oudere kind: hartfalen, hydrops); 10%altijd resectie asymptomatisch
ELS = extralobair longsekwester; HDB = hernia diafragmatica van Bochdalek; ILS = intralobair longsekwester; RDS = respi-ratoir distress syndroom.
Fig. 5: Echogram van de foetus met een uitgesproken mediastinale shift en een hyperechogene linkerlong (zie pijlen)(H = hart). B. Het kleurendopplerpatroon toont de systemische bevloeiing (V) vanuit de aorta (AO) als pathognomonisch
teken voor een longsekwester.
kleurendopplerflowpatroon (fig. 5). Een afzon-derlijke arteriële bevloeiing (meestal rechtstreeksvanuit de aorta) binnen de homogene echoden-siteit is inderdaad pathognomonisch voor eenLS. Het LS (zowel extra- als intralobair) kaneveneens intra-uterien involueren of zelfs „ver-dwijnen”. Een postnataal verrichte CT-scan zalvaak opnieuw aantonen dat het LS nog steedsaanwezig is.
Het postnataal radiografisch beeld van eenLS kan zeer discreet zijn. Een driehoekige basaledensiteit kan in de context van recurrente long-infecties de handtekening zijn van een ILS (fig.6). CT-scan met contrast of MRI-angiogramzullen de diagnose bevestigen. Invasieve aorto-grafie voor de aanduiding van de supra- oftransdiafragmatische arteriële bevloeiing behoorttot het verleden. Wanneer – zij het ongewoon –
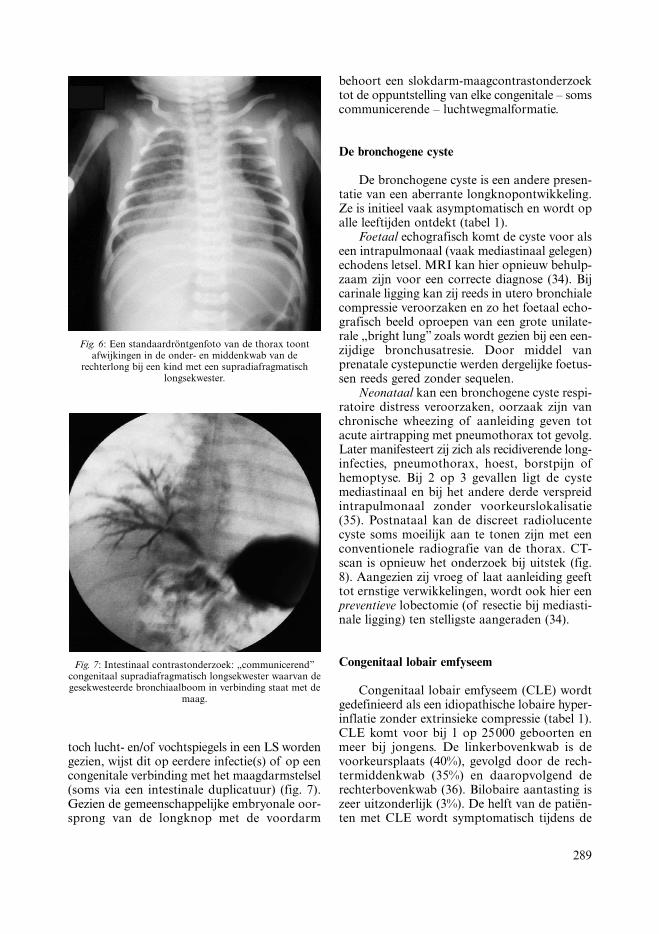
289
Fig. 6: Een standaardröntgenfoto van de thorax toontafwijkingen in de onder- en middenkwab van de
rechterlong bij een kind met een supradiafragmatischlongsekwester.
Fig. 7: Intestinaal contrastonderzoek: „communicerend”congenitaal supradiafragmatisch longsekwester waarvan degesekwesteerde bronchiaalboom in verbinding staat met de
maag.
toch lucht- en/of vochtspiegels in een LS wordengezien, wijst dit op eerdere infectie(s) of op eencongenitale verbinding met het maagdarmstelsel(soms via een intestinale duplicatuur) (fig. 7).Gezien de gemeenschappelijke embryonale oor-sprong van de longknop met de voordarm
behoort een slokdarm-maagcontrastonderzoektot de oppuntstelling van elke congenitale – somscommunicerende – luchtwegmalformatie.
De bronchogene cyste
De bronchogene cyste is een andere presen-tatie van een aberrante longknopontwikkeling.Ze is initieel vaak asymptomatisch en wordt opalle leeftijden ontdekt (tabel 1).
Foetaal echografisch komt de cyste voor alseen intrapulmonaal (vaak mediastinaal gelegen)echodens letsel. MRI kan hier opnieuw behulp-zaam zijn voor een correcte diagnose (34). Bijcarinale ligging kan zij reeds in utero bronchialecompressie veroorzaken en zo het foetaal echo-grafisch beeld oproepen van een grote unilate-rale „bright lung” zoals wordt gezien bij een een-zijdige bronchusatresie. Door middel vanprenatale cystepunctie werden dergelijke foetus-sen reeds gered zonder sequelen.
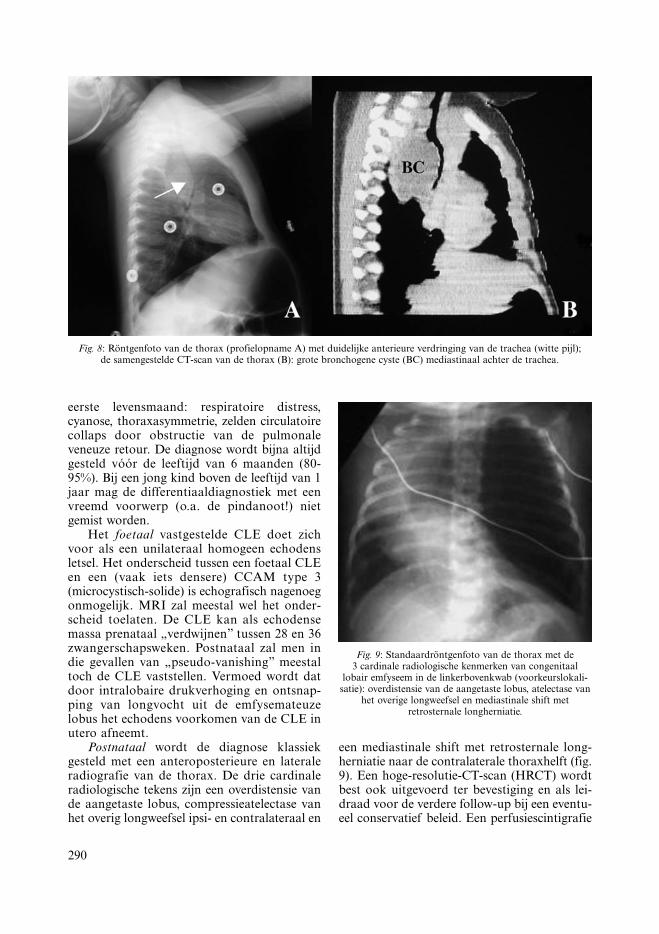
Neonataal kan een bronchogene cyste respi-ratoire distress veroorzaken, oorzaak zijn vanchronische wheezing of aanleiding geven totacute airtrapping met pneumothorax tot gevolg.Later manifesteert zij zich als recidiverende long-infecties, pneumothorax, hoest, borstpijn ofhemoptyse. Bij 2 op 3 gevallen ligt de cystemediastinaal en bij het andere derde verspreidintrapulmonaal zonder voorkeurslokalisatie(35). Postnataal kan de discreet radiolucentecyste soms moeilijk aan te tonen zijn met eenconventionele radiografie van de thorax. CT-scan is opnieuw het onderzoek bij uitstek (fig.8). Aangezien zij vroeg of laat aanleiding geefttot ernstige verwikkelingen, wordt ook hier eenpreventieve lobectomie (of resectie bij mediasti-nale ligging) ten stelligste aangeraden (34).
Congenitaal lobair emfyseem
Congenitaal lobair emfyseem (CLE) wordtgedefinieerd als een idiopathische lobaire hyper-inflatie zonder extrinsieke compressie (tabel 1).CLE komt voor bij 1 op 25000 geboorten enmeer bij jongens. De linkerbovenkwab is devoorkeursplaats (40%), gevolgd door de rech-termiddenkwab (35%) en daaropvolgend derechterbovenkwab (36). Bilobaire aantasting iszeer uitzonderlijk (3%). De helft van de patiën-ten met CLE wordt symptomatisch tijdens de
290
Fig. 8: Röntgenfoto van de thorax (profielopname A) met duidelijke anterieure verdringing van de trachea (witte pijl);de samengestelde CT-scan van de thorax (B): grote bronchogene cyste (BC) mediastinaal achter de trachea.
Fig. 9: Standaardröntgenfoto van de thorax met de3 cardinale radiologische kenmerken van congenitaal
lobair emfyseem in de linkerbovenkwab (voorkeurslokali-satie): overdistensie van de aangetaste lobus, atelectase van
het overige longweefsel en mediastinale shift metretrosternale longherniatie.
eerste levensmaand: respiratoire distress,cyanose, thoraxasymmetrie, zelden circulatoirecollaps door obstructie van de pulmonaleveneuze retour. De diagnose wordt bijna altijdgesteld vóór de leeftijd van 6 maanden (80-95%). Bij een jong kind boven de leeftijd van 1jaar mag de differentiaaldiagnostiek met eenvreemd voorwerp (o.a. de pindanoot!) nietgemist worden.
Het foetaal vastgestelde CLE doet zichvoor als een unilateraal homogeen echodensletsel. Het onderscheid tussen een foetaal CLEen een (vaak iets densere) CCAM type 3(microcystisch-solide) is echografisch nagenoegonmogelijk. MRI zal meestal wel het onder-scheid toelaten. De CLE kan als echodensemassa prenataal „verdwijnen” tussen 28 en 36zwangerschapsweken. Postnataal zal men indie gevallen van „pseudo-vanishing” meestaltoch de CLE vaststellen. Vermoed wordt datdoor intralobaire drukverhoging en ontsnap-ping van longvocht uit de emfysemateuzelobus het echodens voorkomen van de CLE inutero afneemt.
Postnataal wordt de diagnose klassiekgesteld met een anteroposterieure en lateraleradiografie van de thorax. De drie cardinaleradiologische tekens zijn een overdistensie vande aangetaste lobus, compressieatelectase vanhet overig longweefsel ipsi- en contralateraal en
een mediastinale shift met retrosternale long-herniatie naar de contralaterale thoraxhelft (fig.9). Een hoge-resolutie-CT-scan (HRCT) wordtbest ook uitgevoerd ter bevestiging en als lei-draad voor de verdere follow-up bij een eventu-eel conservatief beleid. Een perfusiescintigrafie

291
met aanduiding van hypoperfusie in de emfyse-mateuze lobus is niet obligaat. Dit onderzoekkan toch differentiërend zijn indien er twijfelbestaat over de diagnose van CLE en bijvoor-beeld een hypoplasie van de contralaterale longde werkelijke diagnose kan zijn zoals bij een sci-mitarsyndroom (d.i. een rechterlonghypoplasiemet abnormaal veneuze retour van de rechter-long naar de v. cava inferior en dextropositievan het hart). Spontane postnatale resolutie vanCLE is zowel radiografisch als bij seriële CT-scan goed beschreven en zou bij ruim 1 patiëntop 5 een conservatieve houding toelaten (37).
Een op wetenschappelijke evidentie gestoeldantwoord voor de behandeling van CLE is nogniet voorhanden. Lobectomie is de algemeen aan-vaarde optie bij symptomatische CLE. Bij eenconservatief beleid mag niet vergeten worden dateen (beperkte) differentiaaldiagnose openblijft,aangezien de ultieme zekerheidsdiagnose van CLEeen pathologisch-anatomische diagnose is. Menmag bijvoorbeeld een CCAM type 1 niet missen.Ook een verworven obstructief emfyseem door eentumor of een vreemd voorwerp, een onderliggendecarinale bronchogene cyste met airtrapping of eencongenitale unilobaire pulmonale lymfangiëcta-sie (CUPL) kunnen een CLE precies nabootsen.Bronchoscopie – zo nodig geacht – moet in deoperatiezaal worden verricht met mogelijkheid toturgente thoracotomie. Acute overinflatie kanimmers leiden tot circulatoire collaps.
Twee curiosa
Een eenzijdige foetale bright lung zal meestalonder te brengen zijn onder 1 van de 4 boven-vermelde aangeboren luchtwegmalformaties.
Daarnaast werden recent nog enkele rariteitenonder hun foetale verschijningsvorm beschre-ven, zoals de congenitale unilobaire pulmonalelymfangiëctasie (CUPL), de linker pulmonalearteriële „sling” (LPAS), de enterogene cyste, debronchiale atresie (± mucokèle) (38), het media-stinaal cystisch teratoom en het (vanishing)neuroblastoom. De twee eerst vermelde curiosaworden hier kort besproken omdat het pasbeschreven entiteiten zijn die ook reeds prena-taal gediagnosticeerd of vermoed kunnen wor-den en uiteindelijk een goede postnatale prog-nose kennen bij tijdige perinatale aanpak.
Congenitale unilobaire pulmonale lymfangiëctasieCongenitale unilobaire pulmonale lymfan-
giëctasie (CUPL) kan er perfect uitzien als CLE,zowel radiologisch als bij CT-scan (39). Ook hieris de linkerbovenkwab de voorkeurslokalisatie.In tegenstelling met de vaak zeer infauste prog-nose bij de diffuse vorm van congenitale pul-monale lymfangiëctasie heeft deze meer zeld-zame unilobaire vorm een goede prognose. Hetnatuurlijk beloop van de ziekte is echter nogonvoldoende bekend. Lobectomie dringt zichop bij symptomen. Post hoc zal een lobectomievoor CLE pathologisch-anatomisch vaak eenCUPL blijken te zijn. Het pathologisch-anato-misch substraat kenmerkt zich door sterk gedi-lateerde peribronchovasculaire, interlobulaire ensubpleurale lymfevaten (fig. 10). De opvallendehyperlucentie op de röntgenfoto en de CT-scanwordt verklaard doordat lymfevaten bij CPLeerder met lucht gevuld zijn dan met lymfe.
„Left pulmonary artery sling”De „left pulmonary artery sling” (LPAS) is
een zeldzame vaatanomalie die respiratoire
Fig. 10: Röntgenfoto (A) en CT-scan (B) van de thorax: hyperinflatie (pijlen) van de linkerbovenkwab (LBK) metuitgesproken mediastinale shift (H). Histopathologisch onderzoek (≈ 25) met peribronchiale, septale en pleurale
lymfangiëctasie (L) en septaal oedeem (C). (Met toestemming van de uitgever ref. 39.)

292
symptomen (distress, wheezing) kan veroorza-ken omdat de linker a. pulmonalis abnormaalontspringt dorsaal van de rechter a. pulmonalisen vandaar achteraan rechts van de trachealoopt om tussen trachea en slokdarm de lin-kerhilus te bereiken. Dit aberrant verloop leidttot compressie van de carina. LPAS is vaakgeassocieerd met tracheobronchiale afwijkin-gen en hartgebreken. De mortaliteit ligt rond50%. LPAS werd recent prenataal gediagnosti-ceerd bij een foetus van 20 weken met als ken-merkend echografisch teken een unilateralebright lung met mediastinale shift (40). MRIvan de foetus op 36 zwangerschapswekentoonde een grote, uniform hyperdense rechter-long en een kleine linkerlong. De baby werd àterme geboren en vertoonde milde respiratoirelast. Een postnatale CT-scan toonde een groterechterlong, mediastinale shift en compressievan de carina. Echocardiografisch kon de linkera. pulmonalis niet aangetoond worden. Bij bron-choscopie werd supracarinaal een pulserendevernauwing gezien. Via MRI kon de abnormaalverlopende linker a. pulmonalis aangetoondworden met duidelijke distorsie van de carinaen compressie van de rechterstambronchus(fig. 11). Chirurgie met reïmplantatie van deaberrante linkerlongslagader was succesvol.Deze patiëntencasus illustreert bijzonder mooide sterke relatie tussen een precieze prenatalediagnostiek leidend tot een optimale postnataleaanpak.
Besluit
Bij de ultrasonografische evaluatie van eenzieke foetale long wordt het snel duidelijk datwat prenataal door de gynaecoloog-echografistals een echodense long of „bright lung” wordtgezien, voor de kinderarts radiologisch meestalals een „hyperlucente” long voorkomt. Wat voorde ene eerder een massa lijkt, is voor de anderevaak een zwart gat. Bij bilaterale bright lung is dedifferentiaal diagnostiek eenvoudig: „congenitalhigh airway obstruction”-syndroom (CHAOS)of zeer uitzonderlijk een bilaterale congenitalecystische adenomatoïde malformatie (CCAM).De hoeveelheid vruchtwater kan helpen ter dif-ferentiatie. De prognose is globaal infaust. He-roïsche EXIT-procedures of een hyperurgenteneonatale tracheotomie zijn bij CHAOS te over-wegen, indien geen bijkomende afwijkingen
worden gevonden en indien een redelijke kansbestaat dat de luchtwegen ad integrum kunnenhersteld worden.
Bij unilaterale bright lung dient prenataalaan de volgende punten aandacht geschonkente worden. De prognose van unilaterale conge-nitale luchtwegmalformaties mag globaal goedtot zeer goed genoemd worden met een mortali-teit die de 10% niet meer overstijgt. Terughou-dendheid bij overwegingen tot zwangerschaps-onderbreking is daarom een eerste belangrijkeboodschap. De ontwikkeling van een bijko-mende niet-immune hydrops bij unilateralehyperdense long is vaak – maar niet altijd – eenzeer ongunstig teken. Spontane regressie van dehydrops is meerdere malen beschreven. Ditdient wellicht in balans gebracht te worden metoverwegingen zoals in-utero-exerese bij geval-len van unilaterale, met hydrops verwikkeldehyperdense long. Ten derde is het belangrijk teonderlijnen dat in-utero-regressie van een uni-laterale hyperdense long eerder de regel is enniet de uitzondering. Deze „vanishing lesions”moeten ten stelligste postnataal nagezien
Fig. 11: Driedimensionaal reconstructiebeeld van tracheaen bronchiaalboom: distortie van de carina (pijl) als gevolgvan de aberrant verlopende linker pulmonaalarterie bij de„left pulmonary artery sling” (LPAS). (Met toestemming
van de uitgever ref. 40.)

293
Fig. 12: Voorstel tot een differentiaaldiagnostische beslissingsboom bij foetale hyperdense longen op basis van delateraliteit, het vruchtwatercompartiment, de homogeniciteit en het kleurendopplerpatroon.
(CCAM: congenitale cystische adenomatoïde malformatie; CHAOS: „congenital high airway obstruction”-syndroom;CLE: congenitaal lobair emfyseem; LS: longsekwester.)
worden, niet enkel met een conventionelethoraxradiografie, maar ook met spiraal-CT-scan. Differentiaaldiagnostiek tussen CCAM,intra-/extralobaire sekwestratie (ILS/ELS),bronchogene cyste (BC), congenitaal lobairemfyseem (CLE) en de andere curiosa is voorde gynaecoloog-echografist vaak onmogelijk.Deze kan zich toch laten helpen door de late-raliteit van de letsels, de homogeniciteit van dehyperdense massa (cystisch-gemengd of homo-geen hyperdens) en ten slotte door het kleuren-doppleronderzoek voor de diagnose van eenlongsekwester (LS) tegenover een CLE ofCCAM type 3 (fig. 12).
De pasgeborene is vaak asymptomatisch.De prenatale diagnostiek zal een adequatepostnatale aanpak sterk bevorderen. Postna-tale (meestal niet-urgente) chirurgie is de regelbij zowel CCAM, LS, BC als CLE. Zo nietstelt men de patiënt bloot aan verscheideneernstige verwikkelingen op elke leeftijd. Indienasymptomatisch, wordt „preventieve” chirur-gie aangeraden tussen 3 en 6 maanden voor-aleer recurrente longinfecties de operatiebemoeilijken. Lobectomie is de juiste ingreep.Conserverende segmentectomie gaat gepaardmet een grotere kans op recidief. Bij asympto-matische CLE kan een conservatief beleidoverwogen worden op gevaar af dat later vast-gesteld wordt dat een onjuiste einddiagnosegesteld werd.
Abstract
Perinatal diagnosis and management ofcongenital pulmonary airway malformations
This review on congenital pulmonary airwaymalformations intends to illustrate the positiveimpact of prenatal diagnosis on their postnatalmanagement. For the gynaecologist these fetalmalformations present as uni- or bilateral hyper-dense or so-called „bright” lungs on fetal ultra-sound. The underlying cause for bilateral hyper-dense fetal lungs is mostly related to a CongenitalHigh Airway Obstruction Syndrome (CHAOS)and exceptionally to a bilateral form of Congeni-tal Cystic Adenomatoid Malformation (CCAM).
Outcome is globally poor, although specifictherapeutic procedures may be considered forCHAOS if a reasonable chance for integralrepair of the airways exists. On the other hand,overall outcome for most of the unilateral typesof congenital pulmonary airway malformationshould be considered as fairly good to excellent.Therefore reticence upon termination of preg-nancy is a major message. It is also importantto underscore that in utero regression of theseunilateral bright lungs is rather the rule and notthe exception. Still, these „vanishing lesions”should be thoroughly evaluated after birth.
The newborn infant with congenital pulmo-nary airway malformation is often asymptomatic

294
at birth. For this reason prenatal diagnosis willpromote an elective and timely postnatalmanagement. Postnatal surgery – mostly noturgent – is the rule for CCAM, as well as forpulmonary sequestration, bronchogenic cystand for most cases of congenital lobar emphy-sema (CLE). When the baby is symptom-free,elective surgery is generally advised between 3
and 6 months of age. Conservative segmentec-tomy should be avoided because of a higherrisk of recurrence. Therefore lobectomy is thesuitable intervention. In case of asymptomaticCLE a conservative policy can be considered,although this holds the objection that in laterlife final diagnosis can be proven to be incor-rect.
LITERATUUR
1. BUSH A. Congenital lung disease: a plea for clear think-ing and clear nomenclature. Pediatr Pulmonol 2001;32: 328-337.
2. LIM FY, CROMBLEHOLME TM, HEDRICK HL, et al. Con-genital high airway obstruction syndrome: natural his-tory and management. J Pediatr Surg 2003; 38: 940-945.
3. KANAMORI Y, KITANO Y, HASHIZUME K, et al. A caseof laryngeal atresia (congenital high airway obstructionsyndrome) with chromosome 5p deletion syndrome res-cued by ex utero intrapartum treatment. J Pediatr Surg2004; 39: E25-E28.
4. VANHAESEBROUCK P, DE COEN K, DEFOORT P, et al.Probable autosomal dominant transmission of prena-tally diagnosed CHAOS. Proceedings of the 7th WorldCongress of Perinatal Medicine 2005; Sept 21-24; Zagreb.
5. BANERJEA MC, WIRBELAUER J, ADAM P, TRUSEN A,MARX A, SPEER C. Bilateral cystic adenomatoid mal-formation type III – a rare differential diagnosis of pul-monary hypertension in neonates. J Perinat Med 2002;30: 429-436.
6. CONRAN RM, STOCKER JT. Extralobar sequestrationwith frequently associated congenital cystic adenoma-toid malformation, type 2: report of 50 cases. PediatrDev Pathol 1999; 2: 454-463.
7. MCLEAN SE, PFEIFER JD, SIEGEL MJ, et al. Congenitalcystic adenomatoid malformation connected to anextralobar pulmonary sequestration in the contralateralchest: common origin? Pediatr Surg 2004; 39: el3-17.
8. MCMANUS DT, O’HARA MD. Extralobar sequestrationand type II congenital cystic adenomatoid malforma-tion in an infant with congenital diaphragmatic hernia.Pediatr Pathol Lab Med 1996; 16: 637-642.
9. BREYSEM L, BOSMANS H, DYMARKOWSKI S, et al. Thevalue of fast MR imaging as an adjunct to ultrasoundin prenatal diagnosis. Eur Radiol 2003; 13: 1538-1548.
10. STOCKER JT, MADEWELL JE, DRAKE RM. Congenitalcystic adenomatoid malformation of the lung. Classifi-cation and morphologic spectrum. Hum Pathol 1977;8: 155-171.
11. EVRARD V, CEULEMANS J, COOSEMANS W, et al. Con-genital parenchymatous malformations of the lung.World J Surg 1999; 23: 1123-1132.
12. LACY DE, SHAW NJ, PILLING DW, WALKINSHAW S. Out-come of congenital lung abnormalities detected antena-tally. Acta Paediatr 1999; 88: 454-458.
13. GORNALL AS, BUDD JLS, DRAPER ES, KONJE JC, KUR-INCZUK JJ. Congenital cystic adenomatoid malforma-tion: accuracy of prenatal diagnosis, prevalence and out-come in a general population. Prenat Diagn 2003; 23:997-1002.
14. PUMBERGER W, HORMANN M, DEUTINGER J,BERNASCHEK G, BISTRICKY E, HORCHER E. Longitudi-nal observation of antenatally detected congenital lungmalformations (CLM): natural history, clinical outcomeand long-term follow-up. Eur J Cardio-thoracic Surg2003; 24: 703-711.
15. DAVENPORT M, WARNE SA, CACCIAGUERRA S, PATEL S,GREENOUGH A, NICOLAIDES K. Current outcome ofantenatally diagnosed cystic lung disease. J Ped Surg2004; 39: 549-556.
16. HSIEH CC, CHAO AS, CHANG YL, KUO DM, HSIEH TT,HUNG HT. Outcome of congenital cystic adenomatoidmalformation of the lung after antenatal diagnosis. IntJ Gynaecol Obstet 2005; 89: 99-102.
17. KIM YT, KIM JS, PARK JD, KANG CH, SUNG SW,KIM JH. Treatment of congenital cystic adenomatoidmalformation – does resection in the early postnatalperiod increase surgical risk? Eur J Cardiothorac Surg2005; 27: 658-661.
18. ROGGIN KK, BREUER CK, CARR SR, et al. The unpre-dictable character of congenital cystic lung lesions.J Pediatr Surg 2000; 35: 801-805.
19. TSAO K, HAWGOOD S, VU L, et al. Resolution ofhydrops fetalis in congenital cystic adenomatoid mal-formation after prenatal steroid therapy. J Pediatr Surg2003; 38: 508-510.
20. DIAMOND IR, WALES PW, SMITH SD, FECTEAU A. Sur-vival after CCAM associated with ascites: a report of acase and review of the literature. J Pediatr Surg 2003;38: E1-E3.
21. TSAO K, VU L, LEE H, et al. Characteristics of con-genital cystic adenomatoid malformations associatedwith nonimmune hydrops and outcome. J Perinatal 2002;22: 622-627.
22. TSAO K, ALBANESE CT, HARRISON MR. Prenatal ther-apy for thoracic and mediastinal lesions. World J Surg2003; 27: 77-83.
23. WILSON RD, BAXTER JK, JOHNSON MP, et al. Thora-coamniotic shunts: fetal treatment of pleural effusionsand congenital cystic adenomatoid malformations. FetalDiagn Ther 2004; 19: 413-420.
24. SAUVAT F, MICHEL JL, BENACHI A, EMOND S, REVIL-LON Y. Management of asymptomatic neonatal cystic ade-nomatoid malformations. J Pediatr Surg 2003; 38: 548-552.
25. BLAU H, BARAK A, KARMAZYN B, et al. Postnatal man-agement of resolving fetal lung lesions. Pediatrics 2002;109: 105-108.
26. LABERGE JM, BRATU I, FLAGEOLE H. The managementof asymptomatic congenital lung malformations. PaedResp Rev 2004; 5 Suppl A: S305-S312.

295
27. KHOSA JK, LEONG SL, BORZI PA. Congenital cystic ade-nomatoid malformation of the lung: indication and tim-ing of surgery. Pediatr Surg Int 2004; 20: 505-508.
28. ALLEGAERT K, PROESMANS M, NAULAERS G, MOER-MAN P, LERUT T, DEVLIEGER H. Neonatal transthoracicpuncture in a case of congenital cystic adenomatoid mal-formation of the lung. J Pediatr Surg 2002; 37: 1495-1497.
29. GRANATA C, GAMBINI C, BALDUCCI T, et al. Bronchi-oloalveolar carcinoma arising in congenital cystic ade-nomatoid malformation in a child: a case report andreview on malignancies originating in congenital cysticadenomatoid malformation. Pediatr Pulmonol 1998;25: 62-66.
30. CORBETT HJ, HUMPHREY GM. Pulmonary sequestra-tion. Paediatr Respir Rev 2004; 5: 59-68.
31. CHEN JS, WALFORD N, YAN YL, ONG CL, YEO GS.Foetal intralobar lung sequestration: antenatal diagnosisand management. Singapore Med J 2003; 44: 630-634.
32. LAURIN S, HAGERSTRAND I. Intralobar bronchopul-monary sequestration in the newborn: a congenital mal-formation. Pediatr Radiol 1999; 29: 174-178.
33. CHOWDHURY M, SAMUEL M, RAMSAY A, CONSTANTI-NOU J, MCHUGH K, PIERRO A. Spontaneous postnatalinvolution of intraabdominal pulmonary sequestration.J Pediatr Surg 2004; 39: 1273-1275.
34. VAAST P, HOUFFLIN-DEBARGE V, DUBOS JP, et al. Lesmalformations pulmonaires: du foetus a l’adulte, quelleprise en charge? Diagnostic et pronostic en antenatal.Arch Pediatr 2004; 11: 518-519.
35. DE BAETS F, VAN DAELE S, SCHELSTRAETE P,HAERYNCK F, VERMASSEN F, BROERS C. Asphyxiatingtracheal bronchogenic cyst. Pediatr Pulmonol 2004; 38:488-490.
36. OLUTOYE OO, BEVERLY G. COLEMAN BG, ANNE M,HUBBARD AM, ADZICK NS. Prenatal diagnosis andmanagement of congenital lobar emphysema. J PediatrSurg 2000; 35: 792-795.
37. OZCELIK U, GOCMEN A, KIPER N, DOGRU D, DILBER E,YALCIN EG. Congenital lobar emphysema: evaluationand long-term follow-up of thirty cases at a single cen-ter. Pediatr Pulmonol 2003; 35: 384-391.
38. KAMATA S, SAWAI T, USUI N, et al. Case of congenitalbronchial atresia detected by fetal ultrasound. PediatrPulmonol 2003; 35: 227-229.
39. RETTWITZ-VOLK W, SCHLOSSER R, AHRENS P, HOR-LIN A. Congenital unilobar pulmonary lymphangiecta-sis. Pediatr Pulmonol 1999; 27: 290-292.
40. SEMPLE MG, BRICKER L, SHAW BN, PILLING DW. Leftpulmonary artery sling presenting as unilateral echogeniclung on 20-week detailed antenatal ultrasound exami-nation. Pediatr Radiol 2003; 33: 567-569.


















