
ARTSENBROCHURE - MPN Stichting · 2017-01-25 · Elke patholoog kan de drie typen ET van elkaar...
40
ARTSENBROCHURE M y e l o p r o l i f e r a t i v e D i s o r d e r s (MPD) Essentiële Trombocytemie (ET) Polycythaemia Vera (PV) Chronische Idiopathische Myelofibrose (CIMF) MPD Stichting
Transcript of ARTSENBROCHURE - MPN Stichting · 2017-01-25 · Elke patholoog kan de drie typen ET van elkaar...

ARTSENBROCHURE
M y e l o p r o l i f e r a t i v e D i s o r d e r s
(MPD)
Essentiële Trombocytemie (ET)
Polycythaemia Vera (PV)
Chronische Idiopathische Myelofibrose (CIMF)
MPD Stichting


MPD Artsenbrochure 2006
Deze artsenbrochure is samengesteld door de MPD Stichtingin samenwerking met
Prof. Dr. J.J. MichielsGoedhardt Instituut Rotterdam en Universitair Ziekenhuis Antwerpen
en
Prof. Dr. H.C. SchoutenUniversitair Medisch Centrum Maastricht
Copyright © 2006 MPD StichtingNiets uit deze brochure mag worden verveelvoudigd, opgeslagen in een geautomatiseerd gege-vensbestand of openbaar gemaakt, in enige vorm of op enige wijze, elektronisch, mechanisch, door fotokopieën, opnamen of op enig andere manier, zonder voorafgaande schriftelijke toe-stemming van de MPD Stichting.
Disclaimer:De inhoud van deze brochure is met uiterste zorgvuldigheid en in nauwe samenwerking met specialisten samengesteld. De MPD Stichting is in geen geval aansprakelijk voor schade die direct of indirect voortvloeit uit het juist of onjuist gebruiken van informatie die in deze brochure staat.
�Artsenbrochure
MPD StichtingGrondelsloot 612724 BT Zoetermeer

Inhoud
Voorwoord 3
Inleiding 4
�. Essentiële Trombocytemie (ET) 6 1.1. Wat is ET? 6 1.2. Diagnose ET 6 1.3. Klachten en symptomen ET 10 1.4. Behandeling ET 13
2. Polycythaemia Vera (PV) 18 2.1. Wat is PV? 18 2.2. Diagnose PV 18 2.3. Klachten en symptomen PV 19 2.4. Behandeling PV 21
3. Myelofibrose (MF) 26 3.1. Wat is MF? 26 3.2. Diagnose MF 27 3.3. Klachten en symptomen MF 29 3.4. Behandeling MF 30
4. Referenties 34
5. Informatie over de MPD Stichting 36
december 2006
MPD stichting2

Voorwoord
Dit is een brochure voor artsen, waarin de drie beenmergziekten Essentiële Trombocytemie (ET), Polycythaemia Vera (PV) en Myelofibrose (MF) behandeld worden.Bij alle drie de ziekten komt aan bod wat het is, welke diagnosemethoden/-criteria er gebruikt worden, welke klachten en symptomen erbij horen en welke behandeling hierbij past.
Hoewel elke ziekte apart behandeld wordt, wordt soms naar eerder gegeven informatie in de brochure verwezen, omdat ET, PV en MF als MPD-ziekten veel overlap vertonen.
Deze brochure geeft ook inzicht in de herkenning en diagnose van vroege fasen van de ziekten die geassocieerd zijn met trombocytemie. Hierdoor kunnen de vroege fase van PV en MF beter onderscheiden worden van de echte ET. Een meer adequate behandeling kan hierop afgestemd worden.
Deze brochure is tot stand gekomen door medewerking van meerdere specialisten.
Onze dank gaat uit naar zowel Prof. Dr. J.J. Michiels (Goedhardt Insituut Rotterdam en Universitair Ziekenhuis Antwerpen) als naar Prof. Dr. H.C. Schouten (Universitair Medisch Centrum, Maastricht).• Prof. Michiels voor het ter beschikking stellen van zijn wetenschappelijke kennis met betrek-
king tot diagnose, classificatie en behandeling van de MPDs ET, PV en MF.• Prof. Schouten voor zijn informatie over stamceltransplantatie.
Verder gaat onze dank uit naar de pathologen: • Dr. K.H. Lam (Erasmus Medisch Centrum, Rotterdam) • Prof. H. de Raeve (Universitair Ziekenhuis Antwerpen)Zij hebben in belangrijke mate bijgedragen tot het opstellen van de beenmergcriteria voor de diagnose en classificatie van de myeloproliferatieve ziekten. Ook zijn we hen zeer erkentelijk voor het beschikbaar stellen van foto- en beeldmateriaal.
We zijn de overheid erkentelijk voor de verleende subsidie via Stichting Fonds PGO. Hierdoor kunnen we deze brochure uitgeven.
3Artsenbrochure

Inleiding
MPD algemeen
MPD is een verzamelnaam voor ziekten in het beenmerg: Myeloproliferative Diseases. Het beenmerg bestaat uit kernhoudende bloedaanmakende stamcellen, die zichzelf als een onuit-puttelijke bron telkens vernieuwen en uitgroeien tot de drie voor het leven noodzakelijke cellen in het bloed:
• de trombocyten
Zij spelen een belangrijke rol in de bloedstolling. Bij een tekort aan trombocyten (< 20 x 109/l) treden bloedingsverschijnselen op zoals puntvormige huidbloedingen, blauwe plekken, bloe-dingen na verwondingen, neusbloedingen en slijmvliesbloedingen van het maagdarmkanaal.
• de erytrocyten
Zij bevatten rood gekleurde hemoglobine (Hb), dat het vermogen heeft om zuurstof te ver-voeren van de longen naar alle weefselcellen van het lichaam. Een tekort aan erytrocyten of hemoglobine staat bekend als anemie en gaat gepaard met bleek zien en vermoeidheid.
• de leukocyten
Zij zorgen voor de afweer van zich binnendringende micro-organismen.
Bloed bestaat uit: trombocyten 150-350 x 109/l, erytrocyten 4.5-5.8 x 1012/l, leukocyten 4-12 x 109/l en plasma.
Een myeloproliferatieve ziekte wordt gekenmerkt door proliferatie in het beenmerg van één of meer van de drie bloedaanmakende cellijnen:
1. de megakaryopoiese, voor de aanmaak van trombocyten 2. de erytropoiese, voor de aanmaak van erytrocyten 3. de myelopoiese, voor de aanmaak van leukocyten
Een myeloproliferatieve ziekte openbaart zich dan ook in drie aan elkaar verwante, maar te onderscheiden Myeloproliferative Diseases (MPD)1,2,3:
�. Essentiële Trombocytemie2. Polycythaemia Vera3. Myelofibrose
MPD stichting4

5Artsenbrochure
Essentiële Trombocytemie
�. Essentiële Trombocytemie
�.� Wat is Essentiële Trombocytemie (ET)?ET is één van de myeloproliferatieve ziekten, waarbij er een proliferatie in het myelum aanwezig is, die zorgt voor teveel trombocyten. Hierdoor kunnen in meer of mindere mate stoornissen in de bloedcirculatie ontstaan, die kunnen leiden tot stolselvorming. Echter bij een sterk verhoogd aantal trombocyten kunnen ook bloedingen voorkomen. ET wordt genoemd naar het woekerende bloedceltype, dat de meest opvallende veroorzaker is: de trombocyten. ET is een zeldzame bloedziekte en komt volgens schatting voor bij ongeveer 1,5 tot 2,5 per 100.000 mensen. Hoewel ET meer voorkomt bij oudere patiënten, kunnen ook jongere patiën-ten deze ziekte ontwikkelen. De gemiddelde leeftijd bij de diagnosestelling is ongeveer 60 jaar. Ongeveer 20% van de patiënten is jonger dan 40 jaar.
�.2 Diagnose ETVoor het stellen van de diagnose ET moet het volgende gedaan worden:• Zorgvuldige registratie van klachten en symptomen • Bloedafname uit de ader in de elleboogholte voor gericht laboratoriumonderzoek• Beenmergpunctie onder plaatselijke verdoving uit de bekkenkam • Een beenmergbiopsie uit de bekkenkam
Ook moet worden uitgesloten dat bijvoorbeeld een infectie, kanker of een andere ziekte de trombocytose veroorzaakt. In dit laatste geval spreken we van secundaire of reactieve trombo-cytose. Hierbij moet de onderliggende aandoening behandeld worden. Is de ziekte verdwenen, dan verdwijnt ook de trombocytose. Is er ondanks uitgebreid onderzoek geen onderliggende ziekte aantoonbaar, dan spreekt men van primaire of Essentiële Trombocytemie.
Tabel �. Kenmerkende laboratoriumbevindingen van de 3 verschillende typen ET3-9 ET 3 Typen Type � Type 2 Type 3a Type 3bDiagnose ET PVSG Echte ET Vroege PV CIMF-0 CIMF-�Trombocyten > 400-1500 > 400-1500 > 400-1500 > 400-1500 > 400-1500Hemoglobine N N N/↑ N N/↓Hematocriet < 0.50 < 0.45 < 0.50 < 0.45 < 0.40Leukocyten N N N N N/↑LAF-score N/↑ N ↑ ↑/N ↑/NEEC -/+ - + +/- +/-Serum EPO N/↓ N N/↓ N NJAK2V617F -/+ -/+ + +/- +/-
N = normaal, - = afwezig, + = aanwezig, ↑ = toegenomen, ↓ = verminderd
Essentiële trombocytemie (ET) vastgesteld volgens de wereldwijd toegepaste en gangbare criteria van de Polycythemia Vera Study Group (PVSG)� kan op basis van de WHO been-mergcriteria4,5,6 worden onderverdeeld in 3 typen (tabel 2 en 3):• De echte essentiële trombocytemie (ET type �) met typische kenmerken van ET in het peri-
fere bloed en beenmerg.
MPD stichting6
• ET type 2 als uiting van een vroege PV met kenmerken van ET in het perifere bloed en kenmerken van PV in het beenmerg.
• ET als uiting van een prefibrotische of vroegfibrotische fase van chronische idiopathische myelofibrose (CIMF-0 = ET type 3A of CIMF-1 = ET type 3B) met typische kenmerken van CIMF in het beenmerg.
Tabel 2. Kenmerkende bevindingen in een beenmergbiopsie, die karakteristiek zijn voor elk van de 3 typen ET6–9 (tabel 3 en figuur �).ET 3 Typen Type � Type 2 Type 3a Type 3bDiagnose ET PVSG Echte ET ET- PV ET- CIMF-0 ET- CIMF-�Trombocyten > 400-1500 > 400-1500 > 400-1500 > 400-1500 > 400-1500Megakaryocyten: groot/clusters groot/clusters groot/clusters groot/clusters groot/clusters Uitrijping N N N onrijp onrijpBeenmerg: Celrijkdom N/↑ N ↑/↑↑ ↑/↑↑ ↑/↑↑ Erythropoiese N/↑ N ↑ N/↓ N/↓ Granulopoiese N/↑ N N/↑ ↑/↑↑ ↑/↑↑Myelofibrose - - - - -Miltgrootte op echo (- = N, + = < 15 cm) -/+ -(+) -(+) -/+ -/+
N = normaal, - = afwezig, + = aanwezig, ↑ = toegenomen, ↓ = verminderd
Elke patholoog kan de drie typen ET van elkaar onderscheiden (tabel 3) in een goed beoordeelbare beenmergbiopsie (�,5 cm, figuur �) bij diagnose voordat een behandeling is ingesteld.
De echte ET is gekenmerkt door toename van grote in clusters gelegen normale, rijpe mega-karyocyten in een normaal celrijk beenmerg (figuur �a). De echte ET gaat niet over in MF. ET als uiting van een vroege PV is gekenmerkt door toename van grote in clusters gelegen kleine en grote megakaryocyten in een te celrijk beenmerg door toename van de erytropoiese (figuur �b) met als gevolg een verlaagd serum EPO. ET als uiting van CIMF-0 is gekenmerkt door toename van in clusters gelegen afwijkende, onvoldoende uitgerijpte, grote megakaryocyten met plompe onrijpe kernen (cloud-like) in een veel te celrijk beenmerg door toename van de granulopoiese met relatieve onderdrukking van de erythropoiese (figuur �c). Bij CIMF-1 is er geringe toename van reticuline fibrose, vaak ook geringe splenomegalie en geringe toename van erythroblasten in het perifere bloed. De milt is niet of slechts in geringe mate vergroot bij de echte ET en ET als uiting van een vroege PV. De milt is meestal licht tot duidelijk vergroot (< 15 cm) bij ET als uiting van CIMF-0 of CIMF-1.Het onderscheid tussen de 3 typen ET in het beenmerg is niet altijd even duidelijk en dient gezien te worden als een spectrum met overgangsvormen. De levensverwachting van de echte ET is normaal, van ET als uiting van vroege PV normaal tot bijna normaal, maar enigszins tot duidelijk verkort bij ET als uiting van CIMF-0 en CIMF-1 (tabel 4). De ET als uiting van prefi-brotische of vroegfibrotische CIMF toont in de loop van vele jaren (5 tot 15) progressie naar klassieke myelofibrose (CIMF-2) en CIMF-3, welke bepalend zijn voor de verminderde levens-verwachting (tabel 4).
�Artsenbrochure
ET

Tabel 3. Perifeer bloed- en beenmergcriteria voor de diagnose essentiële trombocytemie (ET type 1), vroege polycythaemia vera (PV), ET type 2, en trombocytemie als uiting van prefibro-tische chronische idiopathische myelofibrose (CIMF-0), ET type 3
Perifeer bloedcriteria voor ET WHO beenmergcriteria voor de diagnose ET, PV en CIMF-0
Diagnose ET: A� plus WHO ET beenmerg
Diagnose vroege PV, ET type 2 : A�/A2/A3/A4/A5 plus WHO PV beenmerg
Diagnose CIMF-0, ET type 3: A�/A� plus WHO CIMF-0 beenmerg
MPD stichting�
Aantal trombocyten groter dan 400 x 109/l; graad I 400-1000 x 109/l en graad II meer dan 1000 x 109/l. Aanwezigheid van grote trombocyten in een perifeer bloeduitstrijkpreparaat
JAK2V617F mutatie (PCR test)
Laag serum EPO bij vroege PV
Spontane groei van endogene erythroide colonies (EEC).
Hemoglobine (Hb) normaal of licht ver-hoogd: Hb (man < 11.0 mmol/l of < 17.5 g/dl, vrouw < 10 mmol/l of < 16 g/dl) en Ht (man < 0.51, vrouw < 0.48).
LAF-score normaal of verhoogd bij nor-male BSE and toegenomen “mean platelet volume” (MPV)
Milt licht tot duidelijk vergroot op de echo
Geen onderliggende oorzaak voor reactieve thrombocytose (RT).
Afwezigheid traandruppel erytrocyten en geen leukoerythroblastose
Niet voorafgegaan door of aanwezig zijn van CML, IMF of MDS en afwezigheid van het Philadelphia chromosoom (bcr/abl).
ET type �Normale celrijkdom (< 60%). Proliferatie, toename en clustering van vergrote (reuze) megakaryocyten met gehyperlobuleerde kernen en normale uitrijping van het cyto-plasma (figuur �a). Geen proliferatie en normale uitrijping van de granulopoiese of erytropoiese. Geen of lichte toename van reticuline vezels
Vroege PV, ET type 2Toegenomen celrijkdom (60%-100%) als gevolg van trilineaire myeloproliferatie mettoegenomen erytropoiese, granulopoiese en megakaryopoiese (panmyelosis). Proliferatie en clustering of kleine tot reuze (pleomor-phe) megakaryocyten (figuur �b).Geen tekenen van ontstekingreactie (plas-macytosis, cellular debris). Geen of lichte toename van reticuline vezels
Prefibrotic CIMF-0, ET type 3Toegenomen celrijkdom door bilineaire megakaryocytaire en granulocytaire mye-loproliferatie met relatieve reductie van erythroide precursors. Abnormale clustering en sterke toename van atypische matig tot sterk vergrote (reuze) megakaryocyten met plomp gelobuleerde (cloud-like) kernen en duidelijke uitrijpingsdefecten van kern en cytoplasma (figuur �c). Geen of lichte toename van reticuline vezels
A�
A2
A3
A4
A5
A6
A�
A�
A9
A�0
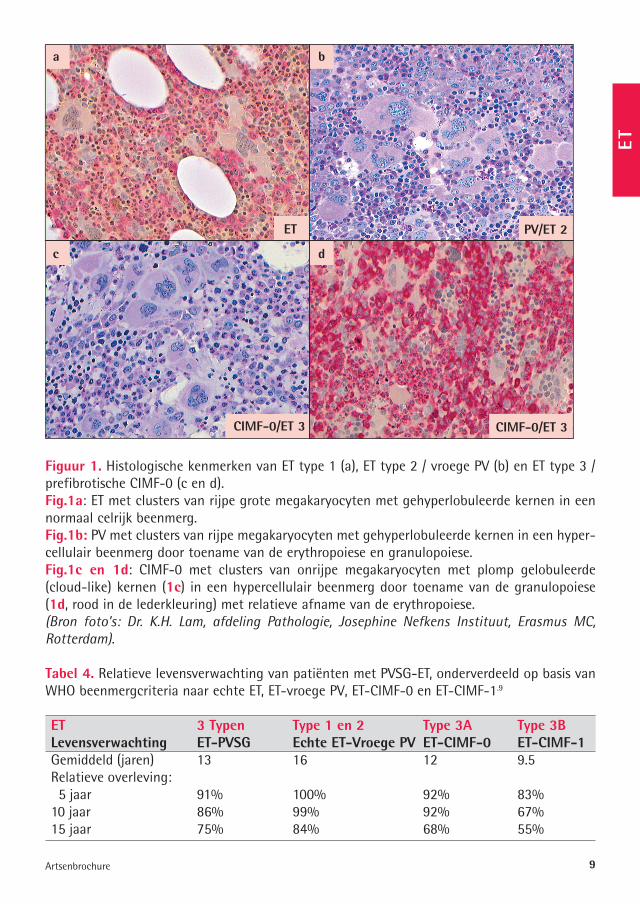
Figuur �. Histologische kenmerken van ET type 1 (a), ET type 2 / vroege PV (b) en ET type 3 / prefibrotische CIMF-0 (c en d). Fig.�a: ET met clusters van rijpe grote megakaryocyten met gehyperlobuleerde kernen in een normaal celrijk beenmerg.Fig.�b: PV met clusters van rijpe megakaryocyten met gehyperlobuleerde kernen in een hyper-cellulair beenmerg door toename van de erythropoiese en granulopoiese.Fig.�c en �d: CIMF-0 met clusters van onrijpe megakaryocyten met plomp gelobuleerde (cloud-like) kernen (�c) in een hypercellulair beenmerg door toename van de granulopoiese (�d, rood in de lederkleuring) met relatieve afname van de erythropoiese. (Bron foto’s: Dr. K.H. Lam, afdeling Pathologie, Josephine Nefkens Instituut, Erasmus MC, Rotterdam).
Tabel 4. Relatieve levensverwachting van patiënten met PVSG-ET, onderverdeeld op basis van WHO beenmergcriteria naar echte ET, ET-vroege PV, ET-CIMF-0 en ET-CIMF-1.9
ET 3 Typen Type � en 2 Type 3A Type 3BLevensverwachting ET-PVSG Echte ET-Vroege PV ET-CIMF-0 ET-CIMF-�Gemiddeld (jaren) 13 16 12 9.5Relatieve overleving: 5 jaar 91% 100% 92% 83%10 jaar 86% 99% 92% 67%15 jaar 75% 84% 68% 55%
9Artsenbrochure
ET
a b
dc
ET
CIMF-0/ET 3
PV/ET 2
CIMF-0/ET 3

JAK2V6��F mutatie als oorzaak van trilineaire MPDDe JAK2V617F mutatie kan worden aangemerkt als de oorzaak van trilineaire MPD met het ach-tereenvolgend optreden van ET, PV en MF10,11. Als de JAK2V617F positief is bij ET is er sprake van heterozygote JAK2 mutatie. ET als uiting van een vroege PV en de echte PV zijn positief voor de JAK2V617F mutatie en zijn gekenmerkt door toename van de megakaryopoiese en erytropoiese in het beenmerg. De minderheid van de echte ET (ongeveer 25%) en de helft van de ET als uiting van CIMF-0 of CIMF-1 zijn positief voor de JAK2 V617F mutatie.
�.3 Klachten en symptomen van ETDe klinische symptomen alsook de behandeling van de 3 verschillende varianten ET zijn gelijk 8,12. De 3 varianten hebben de kenmerken van ET in het perifere bloed, maar verschillen in de morfologie en celrijkdom van het beenmerg in een beenmergbiopt. Aanvankelijk heeft twee derde van de ET patiënten typische symptomen van bloedplaatjes afhankelijke microvasculaire circulatiestoornissen. Bij een derde van de ET patiënten wordt ET vaak bij toeval ontdekt door een routineonderzoek. Vermoeidheid en migraineachtige hoofdpijn zijn vaak de eerste klach-ten van ET waarmee men naar de dokter gaat. Migraineachtige hoofdpijn is typisch voor ET. Vermoeidheid komt meer voor bij CIMF-0 en CIMF-1, die gepaard gaat met geringe miltver-groting (splenomegalie). Bij een bloedplaatjesaantal van 400 - 1000 x 109/l heb je meer kans op circulatiestoornissen en trombose in de microcirculatie (figuur 2)12,13. Dit ziektebeeld is beschreven als “erythromelalgic trombotic trombocytemia” (ETT, figuur 2). Doorbloedingsstoornissen in de microcirculatie van de hersenen, ogen, hart, handen, vingers, voeten en tenen geven ernstige klachten. Enerzijds kan er spontaan stolselvorming optreden. ET patiënten hebben een overmaat aan bloedplaatjes èn hun bloedplaatjes hebben een grotere kleefkracht dan bij gezonde mensen. Is het bloed-plaatjesaantal > 1000 x 109/l, dan neemt de kans op bloedingen toe (figuur 2). Anderzijds kunnen er ook bloedingen optreden, wanneer een sterk verhoogd aantal bloedplaatjes van meer dan 1000 x109/l aanleiding geeft tot een verworven tekort van de von Willebrand factor ristocetine cofactor activiteit (vWF:RCof, figuur 2) en vWF collageenbinding (vWF:CB, figuur 2), waardoor een functiestoornis van de bloedplaatjes optreedt (figuur 2)13. ET met trombo-cyten van meer dan 1000 tot 1500 x 109/l gaat vaak gepaard met spontane huid-, mond- en slijmvliesbloedingen en vaak ook bloedingen van de tractus digestivus als gevolg van een ver-worven von Willebrand syndroom. Dit ziektebeeld is bekend en beschreven als hemorrhagische trombocytemie (hemorrhagic thrombocythemia: HT, figuur 2).13
MPD stichting�0

Figuur 2
Microvasculaire circulatiestoornissen en ET type �, 2 en 3 Symptomen met betrekking tot samenklontering van de bloedplaatjes: Circulatiestoornissen in de microcirculatie door voorbijgaande bloedplaatjespropvorming of blijvende bloedplaatjestrombose kunnen allerlei klachten en symptomen veroorzaken. Afhankelijk van de plaats in het lichaam waar deze microvasculaire stoornissen optreden, kun-nen zich de volgende symptomen/klachten voordoen:
Handen en voeten• Tintelende sensaties, prikkelingen, pijn bij vastpakken van voorwerpen en gevoeligheid in de
voorvoetzool, handpalm en één of meer vingers of tenen.• Erythromelalgie (brandende pijn in voetzolen, tenen, handpalmen en/of vingers). Dit gaat
regelmatig samen met rode huidverkleuring en zwelling (ontsteking). Het ontstaat door
��Artsenbrochure
ET

samenklontering van de trombocyten. Erythromelalgie is meestal met een eenmalige oplaaddosis aspirine van 300 tot 500 mg gevolgd door een onderhoudsdosis van �0 mg per dag adequaat te behandelen.
• Cyanose van voeten, tenen, handen en/of vingers. Dit komt door een zuurstoftekort in de kleine bloedvaten.
Hersenen• TIA’s. Dit zijn kortdurende aanvallen van enkele seconden tot enkele minuten van moeilijk pra-
ten, schrijven, lopen, woordvindingsproblemen, vertraagd maar intact herinneringsvermogen, voorbijgaand geheugen- of bewustzijnsverlies, plotselinge neiging tot vallen of zwalken of plotseling vallen met geheugenverlies al dan niet gevolgd door vaak migraineachtige hoofd-pijn. Migraineachtige aanvallen gevolgd door hoofdpijn en soms ook misselijkheid en braken. Plotselinge aanvallen van voorbijgaande verlamming van een arm of been, voorbijgaande halfzijdige verlamming, of korte tijd niet kunnen spreken of lopen kunnen zich ook voor-doen.
• Atypische TIA’s: moeite met praten, lopen of schrijven, woordvindingsproblemen en des-oriëntatie. TIA’s en atypische TIA’s bij ET dienen behandeld te worden met een eenmalige oplaaddosis aspirine van 300 tot 500 mg gevolgd door een onderhoudsdosis van 80 tot 100 mg per dag.
• Concentratiestoornissen en verminderd of slecht geheugen• Hoofdpijn, meestal migraineachtig, vaak eenzijdig en wisselend gelokaliseerd• Drukkend gevoel op of in het hoofd• Oorsuizen• Duizeligheid• Herseninfarct/beroerte
Ogen• Kortdurende voorbijgaande visuele stoornissen, zoals lichtflikkeringen voor de ogen, gele
vlekken zien en/of (gedeeltelijke) uitval van het gezichtsvermogen. Deze symptomen wor-den dan vaak, maar niet altijd gevolgd door migraineachtige hoofdpijn
• Het zien van misvormde voorwerpen
Hart• Angina pectoris • Hartinfarct
Huid• Rode, pijnlijke, gezwollen plekken in de huid van bovenarm of bovenbenen. Dit wordt
meestal geduid als oppervlakkige aderontstekingen, maar is in feite erythromelalgie van de huid, die verdwijnt met behandeling van aspirine.
Inwendige buikorganen• Onverklaarbare pijnaanvallen in de rug of buik. Dit wordt veroorzaakt door stoornissen in
de bloedcirculatie in de nieren, bijnieren of andere inwendige buikorganen
MPD stichting�2

�.4 Behandeling ET type �, 2 en 3ET is een goedaardige bloedziekte met een normale levensverwachting voor de echte ET en vroege PV mits de ziekte goed behandeld wordt. De behandeling van ET type 1, 2 en 3 is gericht op het voorkomen van trombose en bloedingen. De behandeling van ET is per patiënt verschillend. De behandelend arts of specialist, die een totaaloverzicht van de patiënt en zijn persoonlijke situatie heeft, zal daarop zijn behandeling afstemmen.
In het algemeen kun je het volgende zeggen:
Bloedplaatjes met waarden van 400 – 1500• ET patiënten zonder symptomen: geen medicijnen, maar wel nauwkeurige controle en lage
dosis aspirine kan worden overwogen of aanbevolen.• ET patiënten met symptomen zoals circulatiestoornissen, erythromelalgie, voorbijgaande
TIA’s en visuele stoornissen: oplaaddosis van 300 tot 500 mg gevolgd door lage dosis aspi-rine. � maal daags �0 mg is meestal voldoende. Ascal is ook een mogelijkheid, ascal is een calciumpreparaat van aspirine dat maagwandbescherming geeft. 100 mg Ascal komt over-een met 80 mg aspirine van Bayer. Er is bij Bayer tegenwoordig ook een maagwandsparende aspirine verkrijgbaar. Deze is voorzien van een coating, zodat de inhoud pas in de darmen vrijkomt. Dit kan een oplossing zijn als de maag geen aspirine verdraagt.
• ET patiënten met trombotische complicaties of bloedingsverschijnselen in het verleden zoals hart- of herseninfarct, maag- / darmbloedingen, hersenbloeding, trombose (hoog risico ET, (tabel 5): naast het gebruik van een lage dosis aspirine ook het toedienen van een genees-middel, waarmee de productie van bloedplaatjes wordt afgeremd. Hierbij valt te denken aan interferon of anagrelide als eerste keus en Hydrea (chemo) als tweede keus�4,�5,�6.
• Een trombocytenaantal boven 1000 x 109/l kan gepaard gaan met een verworven von Willebrand syndroom met als gevolg bloedingsverschijnselen. Bij symptomen van ver-hoogde bloedingsneiging dient men het aantal bloedplaatjes terug te brengen tot beneden 1000 x 109/l met voortzetting van lage dosis aspirine (figuur 2). Hierbij valt te denken aan interferon of anagrelide als eerste keus en Hydrea (chemo) als tweede keus.�4
Het is van belang om naast het trombocytenaantal ook naar het Ht te kijken, daar dit bij som-mige mensen ook verhoogd is en een reden kan zijn voor klachten. Een aderlating kan dan zinvol zijn.
Hemorrhagische trombocytemieTrombocytemie met bloedplaatjes met waarden van > 1000 tot 1500 x 109/l gaat gepaard met een hoge kans op bloedingen. Symptomen met betrekking tot verhoogde bloedingsneiging:- Blauwe plekken- Neus- of tandvleesbloedingen- Maag-/darmbloedingen of asymptomatisch bloedverlies uit de tractus digestivus met als
gevolg ijzergebrek zonder dat de ET patiënt daar iets van merkt- Nabloedingen bij operaties en verwondingen- Hersenbloeding (zeldzaam)
�3Artsenbrochure
ET

Geen trombose Geen bloedingen
“Circulatiestoornissen in kleine circulatie”:voeten, handen,hersenen, hart, huid
Trombose, TIA,beroerte, hartinfarct, bloedingen, diabetes, hoge bloeddruk, hoogcholesterol, aderverkalking
Onafhankelijk van symptomen
Leeftijd < 65 jaar 18 tot > 80 jaar 18 tot > 80 jaar 18 tot > 80 jaar
Lage dosis aspirinegewenst Afwachten
Lage dosis aspirine*Indien bijwerkingen
Aspirine plus trombocyten verlagen tot normaal (< 400)
Trombocyten altijd verlagen** tot < 1000 en indien nodig tot normaal plus voortzetting van aspirine
Tabel 5. De behandeling van ET type 1, 2 en 3 is afhankelijk van trombocytenaantal, leeftijd en het risico op trombose en bloedingen. De meeste ET patiënten hebben klachten of verschijnse-len van circulatiestoornissen en komen in aanmerking voor een lage dosis aspirine.
Interferon en anagrelide hebben bij ET duidelijk de voorkeur, zeker voor jongere patiënten onder de 60 tot 65 jaar, omdat bij gebruik van deze middelen geen leukemieverhogend effect is vastgesteld14,16. Interferon kan helaas wel forse bijwerkingen hebben, zoals een grieperig gevoel, koorts enz. Deze bijwerkingen worden na verloop van tijd vaak minder. Er zijn ook bepaalde tips, die de last van de bijwerkingen kunnen doen verminderen, zoals bijvoorbeeld een half uur voor het spuiten een paracetamol nemen en altijd ’s avonds spuiten. Het is ook belangrijk dat men bij aanvang van het interferongebruik met een lage dosis begint en deze geleidelijk opvoert tot de gewenste dosering, zodat het lichaam langzaam aan het interferon kan wennen.
MPD stichting�4
*Bij bloedplaatjesaantal tussen 1000 en 1500 x 109/l is de kans dat aspirine bloedingen uitlokt redelijk groot en lijkt het verstandig om bij symptomen van verhoogde bloedingsneiging het aantal bloedplaatjes terug te brengen tot beneden 1000 x 109/l met voortzetting van lage dosis aspirine
** Voor snelle trombocytendaling is Hydrea de eerste keus. Na normalisering van het trombocytenaantal kan eventueel op een ander middel, dat de beenmergactiviteit remt, worden overgegaan.
Leeftijd > 60 jaar plus afwezigheid van trombose, bloedingen, hoge bloeddruk, diabetes,hoog cholesterol en aderverkalking
Trombocyten verlagen:Leeftijd < 65 jaar: anagrelide of interferon zijn eerste keusLeeftijd > 65 jaar: keuze uit anagrelide, interferon of Hydrea• plaatjes < 1000: lage dosis aspirine • plaatjes > 1000: lage dosis aspirine en trombocyten verlagen tot < 1000
Laag risico Laag risico Hoog risico Hoog risico Trombocyten Trombocyten Trombocyten Trombocyten400 – �500 x �09/l 400 – �500 x �09/l 400 – �500 x �09/l > �500 x �09/l

PEG-interferon (Pegasys of PegIntron) geeft minder bijwerkingen dan het oude interferon (IntronA of Roferon) en hoeft maar één keer per week gespoten te worden. Bij Pegasys blijkt dat het interferon langer en gelijkmatiger in het bloed aanwezig is dan bij PegIntron. Patiënten ervaren met Pegasys minder bijwerkingen dan met PegIntron23. Er zijn ook patiënten, die interferon gebruiken en totaal geen last van bijwerkingen hebben. Interferon kan de toestand van het beenmerg verbeteren en de ziekte soms stabiliseren of terugbrengen naar een eerdere fase. Anagrelide heeft een remmende invloed op de aanmaak van de trombocyten, maar laat de andere cellijnen zoals de rode bloedcellen en de witte bloedcellen ongemoeid. Anagrelide is een goed middel, wanneer alleen de trombocyten verhoogd zijn en de andere cellijnen niet. Anagrelide kan forse bijwerkingen hebben. Patiënten met hartproblemen en/of hoge bloeddruk moeten eerst onderzocht worden of zij dit middel kunnen verdragen. Hydrea (chemo) wordt qua bijwerkingen vaak goed verdragen, maar kan op langere termijn > 10 - 15 jaar een verhoogde kans hebben om leukemie te ontwikkelen.Hydrea is een goede eerste keus en kan zonder veel bezwaar worden ingezet bij patiënten, die ouder zijn dan 65 jaar. Hydrea is een goede tweede keus voor ET patiënten, die jonger zijn dan 65 jaar, die de bijwerkingen van interferon en anagrelide niet kunnen verdragen.
Radioactief fosfor oftewel 32P komt niet meer in aanmerking om het aantal bloedplaatjes te verlagen bij ET. Het gebruik van radioactief fosfor is verouderd en niet wenselijk vanwege de duidelijk verhoogde kans op leukemie.
�5Artsenbrochure
ET

MPD stichting�6
��Artsenbrochure
ET
PV
MF
Polycythaemia Vera

2. POLYCYTHAEMIA VERA
2.� Wat is Polycythaemia Vera (PV)?PV is één van de myeloproliferatieve ziekten, waarbij er een proliferatie in het myelum aanwe-zig is, die zorgt voor teveel erytrocyten. De verhoogde productie van rode bloedcellen leidt tot een te hoog hematocriet (Ht) en hemoglobinegehalte (Hb). Het bloed wordt dik en stroperig, zodat het minder makkelijk door de kleine bloedvaten kan stromen. Dit kan bij PV leiden tot ernstige vaatziekten als gevolg van trombose. Twee derde van de PV patiënten heeft een ver-hoogd aantal trombocyten van meer dan 400 x 109/l (trombocytemie). De leukocyten kunnen ook verhoogd zijn, maar dat hoeft niet. De ziekte wordt heel toepasselijk Polycythaemia Vera genoemd, wat betekent: veel bloed in aderen.Ziekte van Vaquez-Osler, primaire polyglobulie en primaire erytrocytose zijn synoniemen van deze ziekte. Ook gebruikt men de naam primaire polycytemie voor deze ziekte, maar deze bena-ming kan alleen gebruikt worden als alleen de rode bloedcellen verhoogd zijn.PV is een zeldzame bloedziekte en komt volgens schatting voor bij 1,5 tot 3 per 100.000 men-sen. Hoewel PV meer voorkomt bij oudere patiënten, kunnen ook jongere patiënten deze ziekte ontwikkelen. De gemiddelde leeftijd bij de diagnosestelling is ongeveer 55 tot 60 jaar.
2.2 DiagnoseVoor het stellen van de diagnose PV2,3,4 moet het volgende gedaan worden:• Zorgvuldige registratie van klachten en symptomen • Bloedafname uit de ader in de elleboogholte voor gericht laboratoriumonderzoek• Beenmergpunctie onder plaatselijke verdoving uit de bekkenkam • Een beenmergbiopsie uit de bekkenkam
Soms vindt ook een bloedvolumeonderzoek plaats. Hierbij worden de erytrocyten radioactief gemerkt om zo het totale volume aan erytrocyten te meten. Er wordt ook radioactief albu-mine gebruikt om het plasmavolume te meten. Het bloedvolume (erytrocyten plus plasmavo-lume) is verhoogd bij PV en bij secundaire erytrocytose. Bloedvolumeonderzoek is veel minder nauwkeurig om PV vast te stellen en niet geschikt om de vroege fase van PV vast te stellen. Bloedvolumeonderzoek is eigenlijk niet nodig als een beenmergbiopsie is uitgevoerd en beoor-deeld (tabel 6).
MPD stichting��

Tabel 6. Perifeer bloed- en beenmergcriteria voor de diagnose Polycythaemia Vera (PV)
A� Verhoogd hematocriet man > 0.51, vrouw > 0.48 Erythrocyten > 6 x 1012/l
B� Trombocyten > 400 x 109/l
A2 Aanwezigheid van de JAK2 V617F mutatie B2 Leukocyten > 12 x 109/l
A3 Aanwezigheid van EEC en/of laag serum EPO
B3 Miltvergroting op de echo van de buik (lengtediameter van de milt > 12 cm)
A4 Trilineaire proliferatie van de erythropoiese, granulopoiese en megakaryopoiese in een beenmergbiopt passend bij PV volgens WHO criteria
Diagnose PV: A2 en A3 of A4 plus B1 is verenigbaar met vroege PV, stadium 0 A1, A2 en A3 of A4 en geen van B is vroege PV, stadium 1 A1, A2, A3 of A4 plus één van B is klassieke PV, stadium 2 of 3
Eerst moet echter worden uitgesloten dat een andere ziekte de veroorzaker van de PV is. Een te laag zuurstofgehalte in het bloed zet het beenmerg aan tot vorming van meer rode bloedcellen. Dit kan voorkomen bij patiënten met een chronische hart- of longziekte, mensen die veel roken en mensen die op grote hoogte wonen. In deze gevallen spreken we van secundaire polycyte-mie. In zeldzame gevallen kunnen bij lever- en niercysten of nier- en hersentumoren verhoogde erytropoëtinespiegels voorkomen, die ook secundaire polycytemie veroorzaken. Hierbij moet de onderliggende aandoening behandeld worden en dan verdwijnt de PV.
De oorzaak van PV was onbekend en kan sinds 2005 worden toegeschreven aan de verworven JAK2V617F mutatie bij 95% van de PV patiënten. Bij vroege en klassieke PV zijn als gevolg van de toegenomen erythropoiese in het beenmerg de erytropoëtinespiegels verlaagd.
2.3 Klachten en Symptomen van PVTwee derde van de PV patiënten heeft aanvankelijk ook trombocytemie, die gepaard gaat met circulatiestoornissen in de kleine bloedvaten. Vaak gaat trombocytemie, met meestal niet her-kende circulatiestoornissen, enkele tot vele jaren vooraf aan de PV. Lage dosis aspirine geneest en voorkomt het optreden van de circulatiestoornissen in de kleine bloedvaten. PV patiënten hebben bovendien typische klachten en symptomen die een uiting zijn van poly-cytemie. Het te dikke en te stroperige bloed kan ernstige stoornissen in de bloedcirculatie tot gevolg hebben. Er kunnen soms ook bloedingen optreden als gevolg van een stolselvormings-stoornis door te weinig bloedplasma en te veel en te dik bloed.
De symptomen met betrekking tot circulatiestoornissen in de kleine bloedvaten als gevolg van een te hoog bloedplaatjesaantal (trombocytemie) zijn precies dezelfde microcirculatie-stoornissen als beschreven voor ET. (zie blz. �� en �2 ET)
�9Artsenbrochure
PV

Typische klachten, symptomen en ernstige trombose bij PV Deze zijn het gevolg van polycytemie:
A) Algemene klachten De volgende algemene klachten/symptomen komen voort uit een overvulde bloedcircula-
tie: vermoeidheid, lusteloosheid, hoofdpijn, drukkend gevoel in het hoofd, kortademig bij inspanning, concentratiestoornissen en sterk verminderde levenslust, werklust en -vermo-gen zijn meer algemene kenmerken. Een PV patiënt voelt zich mat, suf, loom, traag, slap en futloos.
B) Tekenen van versterkte bloedaanmaak en overvulde bloedcirculatie: Verkleuringen • een pletorisch (overmatige roodverkleuring) gelaat, rode handen/vingers en voeten/
tenen. • blauw/rood verkleuring (acrocyanose) van neuspunt/gelaat en van de handen en voeten
treedt op bij toename van het Ht tot boven 0.60 door relatieve zuurstofonderverzadi-ging van de rode bloedcellen. Het bloed is veel te dik en stroperig, zodat niet alle rode bloedcellen voldoende zuurstof tot zich nemen in de longen.
• Bloeddoorlopen ogen is een versterkte vaattekening van het wit van de oogbol en zeer karakteristiek voor PV.
• Jicht veroorzaakt door een te hoog urinezuurgehalte. Allopurinol is een prima middel om de te hoge urinezuurspiegel te verlagen
C) Ernstige soms levensbedreigende trombose in de grote bloedvaten door te dik en te stro-perig bloed door een verhoogd hematocriet. De stroperigheid van het te dikke bloed door toename van de Ht verergeren de microvasculaire circulatiestoornissen tot macrovasculaire afsluitingen of trombose van de hersen-, hart- en beenarteriën met als gevolg:
• TIA’s • CVA • angina pectoris en hartinfarct • pijnlijke zwerende tenen • soms plotseling weefselversterf met mogelijk gevolg een amputatie van een teen of
vinger • claudicatio intermittens door afsluiting van een beenader in de lies • sterk verhoogde kans op aderontsteking en een trombosebeen soms gecompliceerd door
longembolie • Budd-Chiari syndroom, Vena Porta trombose of miltadertrombose
D) Verhoogd urinezuur met kans op jicht en nierstenen
E) Jeuk, vooral na het douchen Dit is zeer typisch voor PV en moeilijk te behandelen, maar reageert vaak goed op behan-
deling met interferon.
MPD stichting20

F) Nabloeden na ingrepen, operaties en verwondingen. De bloedingsneiging bij PV is het gevolg van een stolselvormingsstoornis door te weinig bloedplasma en te veel en te dik bloed. Deze neiging tot bloeden verdwijnt vaak na correctie van het Ht.
G) Lichte miltvergroting bij PV geeft meestal geen klachten.
H) PV kan na vele jaren overgaan in myelofibrose met myeloide metaplasie met symptomen zoals bij de primaire myelofibrose met myeloide metaplasie. De klachten bestaan uit bezwa-ren van een te grote milt, pijn, gauw een vol gevoel na eten, vermagering, nachtzweet, vermoeidheid etc. De frequentie van myelofibrose is 15% na 10 jaar en 40% na 16 jaar.
I) De frequentie van leukemie bij Hydrea behandelde PV patiënten is ongeveer 10% na 15 jaar.
Symptomen met betrekking tot verhoogde bloedingsneiging bij onbehandelde PV:• Soms blauwe plekken, neus- of tandvleesbloedingen • Maag-/darmbloedingen• Vaak nabloedingen bij operaties en verwondingen • Hersenbloeding (zeldzaam)
2.4 Behandeling PVDe behandeling van PV is per patiënt verschillend. De behandelend arts of specialist, die een totaaloverzicht van de patiënt en zijn persoonlijke situatie heeft, zal daarop zijn behandeling afstemmen. De eerstelijnsbehandeling van PV is aderlaten met streefwaarden van de hemato-criet tussen 0.40 en 0.44 en lage dosis aspirine 80 mg per dag, omdat bij deze combinatie de kans op microvasculaire en macrovasculaire complicaties erg gering is8,12. Het is van het hoogste belang dat de hematocriet niet hoger en altijd lager is dan 0.44 bij mannen en lager dan 0.42 bij vrouwen. Dit om uit de gevarenzone te blijven met betrekking tot vaatcomplicaties!!!
In het algemeen kun je het volgende zeggen:
Bij hematocriet < 0.44 bij mannen en < 0.42 bij vrouwen en normaal Hb• PV patiënten zonder symptomen: geen medicijnen, maar wel nauwkeurige controle.• PV patiënten met symptomen zoals circulatiestoornissen, erythromelalgie, voorbijgaande
TIA’s en visuele stoornissen: lage dosis aspirine (1 maal daags 80 mg is meestal voldoende). Ascal is ook een mogelijkheid. Ascal is een calciumpreparaat van aspirine dat maagwand-bescherming geeft. Er is bij Bayer tegenwoordig ook een maagwandsparende aspirine ver-krijgbaar. Deze is voorzien van een coating, zodat de inhoud pas in de darmen vrijkomt. Dit kan een oplossing zijn als de maag geen aspirine verdraagt. Lage dosis aspirine voorkomt het optreden van circulatiestoornissen in de kleine bloedvaten en kan bij het stijgen van het aantal trombocyten tot boven 1000 à 1250 x 109/l bloedingsverschijnselen uitlokken.
• PV patiënten met complicaties in het verleden zoals hart- of herseninfarct, maag- / darm-bloedingen, hersenbloeding, trombose etc : toedienen van een geneesmiddel, waarmee de productie van de bloedcellen wordt afgeremd
2�Artsenbrochure
PV

Tabel �. Eerste keus behandeling van de 5 stadia van polycythaemia vera:8
Stadium Vroege PV � 2 3 4 type ET 2 PV PV PV PV met MFHemoglobine mmol/l < 10.3 > 10.3 (↑↑) ↑↑ ↑↑ variabelErytrocyten x 1012/l < 6 > 6 (↑↑) ↑↑ ↑↑ ↑Hematocriet 0.45 - < 0.50 0.50 - > 0.60 0.50 - > 0.60 0.50 - > 0.60 ↑Trombocyten x 109/l > 400 - < 1000 < 400 > 400 - <1000 > 1000 variabelLeukocyten x10 9/l N N N / > 12 > 20 variabelJAK2V617F + + +/++ +/++ ++Serum EPO ↓ ↓/↓↓ ↓↓ ↓↓ ↓/↓↓Lengte milt (echo) N - < 15 cm < 15 cm < 15 cm > 15 à 18 cm > 20 cmBeenmerg: MF geen geen geen/gering gering/matig sterk
Behandeling�2,��,�� Aderlaten ja Ht < 0.45 ja Ht < 0.45 ja Ht < 0.45 ja zonodig ja zonodigLage dosis aspirine ja ja ja ja jaInterferon < 50 jaar neen neen neen/ja? ja jaHydrea > 65 jaar neen neen neen ja ja
50 tot 65 jaar: keuze uit1ste keus interferon neen neen neen/ja? ja ja2de keus Hydrea neen neen neen ja ja
N = normaal, - = afwezig, + = aanwezig, ↑ = verhoogd, ↑↑ = sterk verhoogd↓ = verlaagd, < = minder dan, > = meer dan
Met aderlaten worden de PV stadia 1 en 2 teruggebracht naar het vroege PV stadium (tabel �) en is de levensverwachting op de lange termijn normaal zolang er geen progressie naar mye-lofibrose met splenomegalie optreedt. Bij handhaving van de hematocriet rond 0.45 en lage dosis aspirine is de kans op trombotische complicaties gering, maar niet tot nul gereduceerd. In de USA geldt het advies om een hematocrietwaarde van < 0.45 voor mannen en < 0.42 voor vrouwen na te streven, hetgeen niet altijd haalbaar en nodig is.
Bij hematocriet > 0.44 bij mannen en > 0.42 bij vrouwen en/of te hoog Hb (> 9.0 mmol/l)• flebotomie en aspirine. Na het aderlaten kan aanvankelijk het trombocytenaantal omhoog-
schieten. Dit moet wel in de gaten gehouden worden, omdat hun aantal in verband met complicatiegevaar niet boven de 1000 mag komen. Een bijeffect van het aderlaten is dat het ijzergehalte afneemt en vaak te laag is. Het MCV wordt dan kleiner. Het ijzer mag niet worden aangevuld, omdat dit de frequentie van het aderlaten weer doet toenemen. Een laag ijzerniveau zorgt ervoor dat het beenmerg moeilijker rode bloedcellen kan aanmaken en dat is precies de bedoeling. De wat verhoogde kans op klachten van onwel zijn en/of duizeligheid direct na het aderlaten, kan worden tegengegaan door meteen na of tijdens het aderlaten een fysiologische zoutoplossing per infuus toe te dienen. De hoeveelheid fysiologische zoutoplossing dient overeen te komen met de hoeveelheid afgetapt bloed.
MPD stichting22

• Voor de behandeling van PV is het nadeel van aderlaten met lage dosis aspirine dat de beenmergwoekerende activiteit van de PV onverminderd doorgaat. Dit uit zich in een stijging van het aantal trombocyten en/of leukocyten, toename van de myelofibrose en miltvergroting. Ook nemen de klachten van jeuk, vermoeidheid en onwelbevinden toe.
Bij een dergelijke voortgang van de ziekte is er een duidelijke reden om te behandelen met middelen, die de activiteit van het beenmerg remmen.Hierbij valt te denken aan interferon en Hydrea. Na 5 jaar komt ongeveer 40% en na 10 jaar komt ongeveer 90% van de PV patiënten in aanmerking voor behandeling met interferon of Hydrea.
De belangrijkste redenen om van aderlaten over te gaan tot behandeling met interferon of Hydrea zijn:• als frequent aderlaten tot meer dan 10 keer per jaar onvoldoende effect heeft om normale
waarden voor de hematocriet (< 0.45) te bereiken (mannen ≤ 0.44, vrouwen < 0.42).• aantal bloedplaatjes boven de 1000 x 109/l• ernstige jeuk na douchen. Deze reageert vaak goed op interferon, maar niet of veel minder
op Hydrea.• snelle miltvergroting van meer dan 2 cm per jaar vanaf een lengtediameter van > 15 cm• symptomen van nachtzweet, vermagering en sterk vergrote milt (> 20 cm)• leukocytose, leukocyten > 20 x 109/l• fibrose in het beenmerg• complicaties met trombose in hersenen, hart of beenvaten• Budd-Chiari syndroom, Vena Porta trombose of miltadertrombose
Interferon is bij behandeling van PV de eerste keus17,18. Dit heeft bij PV duidelijk de voorkeur, zeker voor jongere patiënten onder de 60 tot 65 jaar, omdat bij gebruik van deze middelen geen leukemieverhogend effect is vastgesteld. Interferon kan de toestand van bloed en been-merg verbeteren en de ziekte soms stabiliseren of terugbrengen naar een eerdere fase met correctie van het verhoogd aantal trombocyten en leukocyten.Interferon kan helaas wel forse bijwerkingen hebben, zoals een grieperig gevoel, koorts enz. Deze bijwerkingen worden na verloop van tijd vaak minder. Er zijn ook bepaalde tips, die de last van de bijwerkingen kunnen doen verminderen, zoals bijvoorbeeld een half uur voor het spuiten een paracetamol nemen en altijd ’s avonds spuiten. Het is ook belangrijk dat men bij aanvang van het interferongebruik met een lage dosis begint en deze geleidelijk opvoert tot de gewenste dosering, zodat het lichaam langzaam aan de interferon kan wennen. PEG-interferon (Pegasys of PegIntron) geeft minder bijwerkingen dan het oude interferon (IntronA of Roferon) en hoeft maar één keer per week gespoten te worden. Bij Pegasys blijkt dat het interferon langer en gelijkmatiger in het bloed aanwezig is dan bij PegIntron. Patiënten ervaren met Pegasys minder bijwerkingen dan met PegIntron23. Er zijn ook patiënten, die interferon gebruiken en totaal geen last van bijwerkingen hebben. Interferon kan de toestand van het beenmerg verbeteren en de ziekte soms stabiliseren of terugbrengen naar een eerdere fase.
23Artsenbrochure
PV

Hydrea is een goede eerste keus en kan zonder veel bezwaar worden ingezet bij patiënten, die ouder zijn dan 65 jaar. Hydrea is een goede tweede keus voor ET patiënten, die jonger zijn dan 65 jaar, die de bijwerkingen van interferon niet kunnen verdragen.Hydrea wordt qua bijwerkingen vaak goed verdragen, maar kan op langere termijn > 10 – 15 jaar een verhoogde kans hebben om leukemie te ontwikkelen.
Langdurige behandeling met Hydrea kan de volgende klachten geven: • droge huid en acne• maagpijn en diarree• (pijnlijke) zweren in de mond en op de huid. De zweren in de mond en aan de benen ver-
dwijnen pas als de Hydrea wordt gestopt
Het is nog geen uitgemaakte zaak welke behandeling beter of geschikter is voor bepaalde groepen PV patiënten in de leeftijd tussen 50 en 65 jaar, die met interferon of die met Hydrea. Hierover wordt nog verschillend gedacht.
Radioactief fosfor oftewel 32P komt niet meer in aanmerking om de activiteit van het beenmerg af te remmen bij PV. Het gebruik van radioactief fosfor is verouderd en niet wenselijk vanwege de duidelijk verhoogde kans op leukemie.
Redenen om PV te behandelen met PEG-interferon23, Hydrea of anagrelide:
1. Er zijn aanwijzingen om te overwegen PV stadium 2 maar zeer zeker stadium 3 te gaan behandelen met lage dosis PEG-interferon ter correctie van de trombocyten, leukocyten en reductie van een vergrote milt plus aanvullende aderlating. Deze strategie zou progressie van de PV binnen de perken kunnen houden, vertragen of voorkomen.
2. Sterk verhoogd aantal bloedplaatjes van meer dan 1000 x 109/l dient te worden behandeld met anagrelide, PEG-interferon of Hydrea.
3. Bijwerkingen van aspirine zoals maagdarmbloeding of gastritis is een reden om het aantal trombocyten terug te brengen tot normaal met anagrelide, PEG-interferon of Hydrea.
4. Ernstige trombose en bloedingen (hoog risico trombocytemie, tabel 5) tijdens lage dosis aspirine is een absolute indicatie om het aantal trombocyten terug te brengen tot normaal met anagrelide, PEG-interferon of Hydrea.
5. Bij toename miltvergroting van meer dan 2 cm per jaar, en miltlengte van > 15 cm en leeftijd jonger dan 65 jaar komt PEG-interferon als eerste en Hydrea als tweede keus in aanmerking.
6. Symptomen van een sterk vergrote milt > 18 cm (PEG-interferon en/of Hydrea).7. PV gerelateerde ernstige klachten van jeuk, nachtzweet, moeheid enz. (PEG-interferon en/of
Hydrea).8. Leukocyten van > 20 à 25 x 109/l gaat vaak gepaard met splenomegalie en/of trombocytemie
(PEG-interferon en/of Hydrea).9. Onrijpe witte en rode bloedcellen in het bloed (leuko-erytroblastose) en tekenen van mye-
loide metaplasie en myelofibrose. (PEG-interferon en/of Hydrea zijn dan geïndiceerd, maar vaak weinig of niet meer effectief).
MPD stichting24
25Artsenbrochure
ET
PV
MF
Myelofibrose

3. MYELOFIBROSE
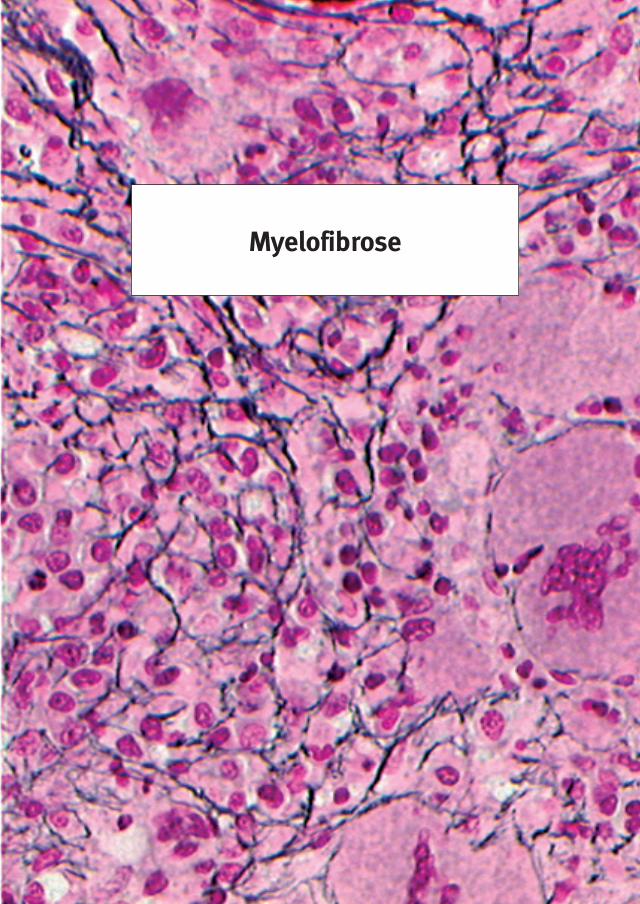
3.� Wat is Myelofibrose (MF)?Myelofibrose is één van de myeloproliferatieve ziekten waarbij een proliferatie in het beenmerg aanwezig is, waardoor er fibrose in het beenmerg ontstaat. De oorzaak van myelofibrose is onbekend. Daarom noemt men het ook wel primaire myelofibrose of chronische idiopatische myelofibrose (CIMF). De naam myelofibrose (MF) is eigenlijk niet juist, omdat MF het gevolg is van myeloide metaplasie van onbekende oorsprong. Myeloide metaplasie betekent leuko-erytroblastose in het perifere bloed met ombouw van het normale beenmerg door woekering van de witte bloedcelaanmaak en abnormale bloedplaatjesaanmaak in het beenmerg, die de rode bloedcelaanmaak verdringen. De benaming Agnogene Myeloide Metaplasie (AMM), die in Amerika wordt gebruikt, geeft niet de aard van de ziekte weer. AMM treedt op als complicatie van PV (post-PV myelofibrose), van ET (post-ET myelofibrose), of is idiopatisch (idiopatische MF). De WHO staat het gebruik van de benamingen AMM, MF, IMF, CIMF toe, maar geeft al dan niet ten onrechte de voorkeur aan de benaming CIMF. In de literatuur worden MF, IMF, CIMF en AMM door elkaar gebruikt voor één en dezelfde chronische myeloproliferatieve ziekte.Ondanks de niet adequate benaming hanteren we in deze brochure de benamingen MF en CIMF.Myelofibrose (MF) is een sterke woekering in het beenmerg met gevolgen voor de aanmaak van de leukocyten en de trombocyten. In de beginfase van MF is er meestal een sterke trom-bocytemie zonder fibrose in het beenmerg. Deze beginfase van MF is pas recent ontdekt en wordt dan ook wel prefibrotische CIMF genoemd. ET als uiting van prefibrotische of vroegfi-brotische CIMF (CIMF-0 of CIMF-�, figuur �c) is recent ontdekt als een variant van ET, die verschilt van de echte ET en van ET als uiting van een vroege PV. De beginnende en vroege stadia van MF, CIMF-0 (type 3A ET) en CIMF-1 (type 3B ET, tabel 2), gaan langzaam over in de welbekende klassieke myelofibrose (CIMF-2 en CIMF-3), waarbij de bloedaanmaak in het beenmerg langzamerhand wordt vervangen door bindweefsel en waarbij de milt langzaam sterk in grootte toeneemt. De klassieke MF gaat gepaard met bloedarmoede, een vergrote milt (tabel � en 9) doordat de bloedaanmaak zich deels naar de milt heeft verplaatst. Dit gaat gepaard met een verhoogd LDH, leuko-erytroblastose en traandruppelcellen (zie tabel � en 9). De traandruppelcellen met hun afwijkende vorm zijn zeer kenmerkend voor de klas-sieke en de gevorderde stadia van MF. De myelofibrose is langzaam progressief en verdringt de bloedaanmaak in het beenmerg wat leidt tot anemie en tot een tekort aan bloedplaatjes. De langzaam progressieve miltvergroting draagt bij tot verdere anemie en aan trombocytopenie door stapeling van rode bloedcellen en bloedplaatjes in de sterk vergrote milt. De klassieke myelofibrose is een zeldzame bloedziekte en komt volgens schatting voor bij ongeveer 0.5 tot 2,5 per 100.000 mensen. Hoewel MF meer voorkomt bij oudere patiënten, kunnen ook jongere patiënten deze ziekte ontwikkelen. De gemiddelde leeftijd bij de diagnosestelling is ongeveer 65 jaar. Ongeveer 20% tot 25% van de patiënten is jonger dan 55 jaar en 10% van de patiënten is jonger dan 45 jaar.
MPD stichting26

3.2 Diagnose MF en CIMFVoor het stellen van de diagnose MF moet het volgende gedaan worden:• zorgvuldige registratie van klachten en symptomen• bloedafname uit de ader in de elleboogholte voor gericht laboratoriumonderzoek• beenmergpunctie onder plaatselijke verdoving uit de bekkenkam• een beenmergbiopsie uit de bekkenkamDe vroege kenmerken van een beginnende myelofibrose kunnen lijken op de ziekte Essentiële Trombocytemie (ET). Een juiste beoordeling van het beenmergbiopt aan de hand van recent ontwikkelde nieuwe criteria kan ervoor zorgen dat een echte ET goed onderscheiden kan wor-den van een beginnende MF (tabel �).
Kenmerkend voor de klassieke MF is dat bij een beenmergpunctie de beenmergcellen moeilijk of niet op te zuigen zijn met een spuit. Bij deze zogenaamde “dry tap” zitten de beenmergcel-len vast in het toegenomen bindweefsel van het beenmerg. Vaak wordt een echo of scan van de buik gemaakt om te kijken of de lever en milt vergroot zijn.
Tabel �. Beenmerg en perifeer bloed criteria voor de diagnose klassieke chronische idiopathi-sche myelofibrose CIMF 1, 2 en 3.
A. Beenmerg criteria
1. Proliferatie van de granulopoiese en megakaryopiese met reductie van de erythropoiese en de aanwezigheid van dichte clusters afwijkende grote onrijpe megakaryocyten met onrijpe dysmorfe “cloud-like” compacte kernen volgens WHO criteria.
2. Myelofibrosis (MF) graad 1, 2 of 3 volgens de criteria van Thiele.
B. perifeer bloed criteria
1. Miltvergroting (splenomegalie) op de echo of bij palpatie
2. Anisopoikilocytosis met traandruppel erythrocyten
3. Aanwezigheid van jonge myeloide cellen in de leukocytendifferentiatie
4. Aanwezigheid van erythroblasten in een bloeduitstrijkpreparaat
5. Aanwezigheid van CD34-positieve cellen in het bloed
6. Anemie, hemoglobine kleiner dan 12 g/dl (7.5 mmo/l)
Diagnose klassieke CIMF-1, 2 of 3:A1 en A2 plus twee van B wanneer miltvergroting of anemie aanwezig isA 1 en A2 plus 3 van B wanneer miltvergroting of anemie afwezig is.
2�Artsenbrochure
MF
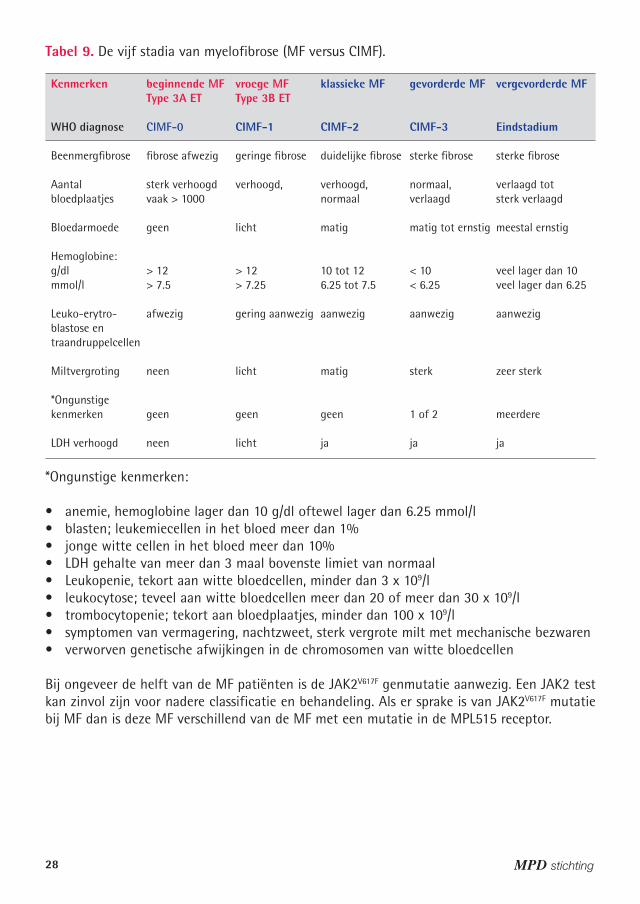
Tabel 9. De vijf stadia van myelofibrose (MF versus CIMF).
Kenmerken beginnende MF vroege MF klassieke MF gevorderde MF vergevorderde MF Type 3A ET Type 3B ET
WHO diagnose CIMF-0 CIMF-� CIMF-2 CIMF-3 Eindstadium
Beenmergfibrose fibrose afwezig geringe fibrose duidelijke fibrose sterke fibrose sterke fibrose
Aantal sterk verhoogd verhoogd, verhoogd, normaal, verlaagd totbloedplaatjes vaak > 1000 normaal verlaagd sterk verlaagd
Bloedarmoede geen licht matig matig tot ernstig meestal ernstig
Hemoglobine: g/dl > 12 > 12 10 tot 12 < 10 veel lager dan 10mmol/l > 7.5 > 7.25 6.25 tot 7.5 < 6.25 veel lager dan 6.25 Leuko-erytro- afwezig gering aanwezig aanwezig aanwezig aanwezigblastose entraandruppelcellen Miltvergroting neen licht matig sterk zeer sterk
*Ongunstige kenmerken geen geen geen 1 of 2 meerdere
LDH verhoogd neen licht ja ja ja
*Ongunstige kenmerken:
• anemie, hemoglobine lager dan 10 g/dl oftewel lager dan 6.25 mmol/l• blasten; leukemiecellen in het bloed meer dan 1%• jonge witte cellen in het bloed meer dan 10%• LDH gehalte van meer dan 3 maal bovenste limiet van normaal• Leukopenie, tekort aan witte bloedcellen, minder dan 3 x 109/l• leukocytose; teveel aan witte bloedcellen meer dan 20 of meer dan 30 x 109/l• trombocytopenie; tekort aan bloedplaatjes, minder dan 100 x 109/l • symptomen van vermagering, nachtzweet, sterk vergrote milt met mechanische bezwaren• verworven genetische afwijkingen in de chromosomen van witte bloedcellen
Bij ongeveer de helft van de MF patiënten is de JAK2V617F genmutatie aanwezig. Een JAK2 test kan zinvol zijn voor nadere classificatie en behandeling. Als er sprake is van JAK2V617F mutatie bij MF dan is deze MF verschillend van de MF met een mutatie in de MPL515 receptor.
MPD stichting2�

3.3 Klachten en symptomen van CIMFPatiënten in de beginfase van ET als uiting van CIMF-0 of CIMF-1 hebben meestal typische symptomen als beschreven bij ET. De beginfase van MF (CIMF-0 en CIMF-1) gaat meestal samen met uitgesproken trombocytemie met trombocyten van 400 tot 1500 x 109/l (zie ET type 3A en ET type 3B, tabel 2). De bloedplaatjes van ET patiënten met een myeloproliferatieve ziekte hebben vaak een grotere kleefkracht, waardoor sneller samenklontering van bloedplaatjes ontstaat dan bij gezonde mensen. Hierdoor kunnen circulatiestoornissen door het hoge aantal trombocyten in de kleine bloedvaten optreden en allerlei algemene klachten en symptomen veroorzaken zoals dat het geval is bij de echte Essentiële Trombocytemie (ET). Bij een bloed-plaatjesaantal van 400-1000 x 109/l heb je meer kans op circulatiestoornissen en trombose in de kleine bloedvaten. Is het bloedplaatjesaantal > 1000 x 109/l, dan neemt de kans op bloe-dingen toe.
Symptomen met betrekking tot samenklontering van de bloedplaatjes bij ET als uiting van een vroege fase van MF (CIMF-0 en CIMF-�) zijn dezelfde microvasculaire circulatie-stoornissen en bloedingen als beschreven bij de echte ET (type 3A en 3B ET). Zie blz. �0, �� en �2. Bij de klassieke en gevorderde MF (CIMF-2 en CIMF-3) worden de klachten en symptomen bepaald door de mate van bloedarmoede en de mate van milt-vergroting.
Typische symptomen van een zich verder ontwikkelende MF (CIMF-2 en CIMF-3) zijn:• Bleekheid en vermoeidheid door anemie• Bloedingsverschijnselen door trombocytopenie en vaak ook nog trombocytopathie• Sterke splenomegalie
Deze vergrote milt veroorzaakt op zijn beurt weer: - gauw vol gevoel na het eten door druk op de maag - drukkend en opgeblazen gevoel in de bovenbuik - gewichtsverlies, vermagering, vermoeidheid en nachtzweten en een koortsig gevoel - diarree - oedeem in de voeten en rond de enkels• Soms vergrote lever, maar leververgroting treedt vaak op na verwijdering van een sterk
vergrote milt• Budd-Chiari syndroom, Vena Porta trombose of miltadertrombose
Deze afwijkingen geven op hun beurt weer de volgende symptomen of klachten: - portale hypertensie - ascites in de buikholte - (bloedende) oesofagusvarices• Myelosclerose • Osteosclerose komt voor bij 10% van de patiënten• Jicht en nierstenen veroorzaakt door een te hoog urinezuurgehalte. Allopurinol is een prima
middel om de te hoge urinezuurspiegel te verlagen.• Buikpijn door maagzweer• Verhoogde kans op infecties met lichte koorts • Soms vergrote lymfeklieren • Miltinfarct• Pijn in de botten
29Artsenbrochure
MF

3.4 Behandeling van klassieke en gevorderde MFDe behandeling van klassieke en gevorderde MF is per patiënt verschillend19,20. Aanvankelijk is er vaak een afwachtend beleid. Men houdt de patiënt goed in de gaten door regelmatige bloedcontroles uit te voeren. De behandelende specialist, die een volledig overzicht van de patiënt en zijn persoonlijke situatie heeft, zal daarop zijn behandeling afstemmen afhankelijk van het stadium van de MF.Behandeling van een beginnende MF en een zich ontwikkelende klassieke MF: Hydrea, inter-feron en soms ook anagrelide komen in aanmerking als de MF zich nog bevindt in de vroege celrijke stadia van MF (wanneer geen of zeer geringe fibrose aanwezig is) en waarbij alleen de bloedplaatjes sterk aan het woekeren zijn, de witte bloedcellen niet of slechts gering zijn toe-genomen en waarbij de rode bloedcelaanmaak nog niet in de verdrukking is gekomen (zie tabel 9 en �0). Bij een te hoog aantal leukocyten en/of trombocyten is het wenselijk medicijnen te nemen om te proberen progressie en complicaties te vermijden. Interferon en Hydrea kunnen de trombocytemie bestrijden, miltvergroting tegengaan of de miltgrootte verkleinen.Interferon werkt het best in een vroeg stadium van myelofibrose. Interferon verbetert de com-municatie tussen de cellen onderling en reguleert de bloedwaarden. Bovendien heeft interferon vaak een positief effect op het immuunsysteem en kan interferon het ziekteproces vertragen, stoppen of zelfs terugdringen tot een eerder stadium van myelofibrose in het beenmerg. Interferon kan helaas wel forse bijwerkingen hebben, zoals een grieperig gevoel, koorts enz. De bijwerkingen van interferon treden op bij 30% van de patiënten. Ongeveer 20% van de patiën-ten staakt interferon wegens ernstige bijwerkingen. Deze bijwerkingen worden na verloop van tijd vaak minder. Er zijn ook bepaalde tips, die de last van de bijwerkingen kunnen doen ver-minderen, zoals bijvoorbeeld een half uur voor het spuiten een paracetamol nemen en altijd ’s avonds spuiten. Het is ook belangrijk dat men bij aanvang van het interferongebruik met een lage dosis begint en deze geleidelijk opvoert tot de gewenste dosering, zodat het lichaam langzaam aan de interferon kan wennen.
PEG-interferon (Pegasys of PegIntron) geeft minder bijwerkingen dan het oude interferon (IntronA of Roferon) en hoeft maar één keer per week gespoten te worden. Bij Pegasys blijkt dat het interferon langer en gelijkmatiger in het bloed aanwezig is dan bij PegIntron. Patiënten ervaren met Pegasys minder bijwerkingen dan met PegIntron23. Er zijn ook patiënten, die inter-feron gebruiken en totaal geen last van bijwerkingen hebben. Hydrea is een goede tweede keus als de bijwerkingen van interferon niet goed verdragen worden.Hydrea is een effectief geneesmiddel om de overproductie aan witte bloedcellen en/of bloed-plaatjes te bestrijden bij vroege en klassieke MF. Ook de miltgrootte kan door het gebruik van Hydrea verkleind worden. Hydrea wordt qua bijwerkingen meestal goed verdragen, maar kan op langere termijn van meer dan 10 jaar bijdragen tot een verhoogde kans op leukemie. Hydrea kan zonder veel bezwaar als eerste keus worden ingezet bij MF patiënten, die ouder zijn dan 65 jaar. Interferon draagt niet bij tot een verhoogde kans op leukemie.Het is nog geen uitgemaakte zaak welke van de twee behandelingen, Hydrea of interferon, beter of geschikter zijn voor het vroege en celrijke stadium van MF patiënten, omdat studies nog ontbreken. De meest belangrijke vraag is of behandeling van de vroege fase van MF met PEG-interferon (of anagrelide) effectief is en mogelijk gepaard gaat met uitstel van het optreden van myelofibrose, met minder leukemie en dus mogelijk wat langer overleven in vergelijking tot een behandeling met Hydrea. Interferon heeft geen effect in het gevorderde stadium van de klassieke myelofibrose met bloedarmoede, tekort aan bloedplaatjes en uitgesproken miltvergroting.
MPD stichting30

Chlorambucil en radioactief fosfor oftewel 32P zijn verouderde middelen en niet meer gewenst vanwege de sterk verhoogde kans op leukemie.
Behandeling van bloedarmoede bij MFBij behandeling van anemie kan men ook gebruikmaken van:• De combinatietherapie van thalidomide en prednison. Deze kan een (beginnende) bloed-
armoede tegengaan en de milt verkleinen21. Bij diverse mensen, die al bloedtransfusieaf-hankelijk waren, zijn door deze combinatietherapie de bloedwaarden zo gaan stijgen dat bloedtransfusies niet meer nodig zijn.
• Lenalidomide. Recent zijn gunstige resultaten beschreven van lenalidomide (Revlimid) bij een subset van patiënten met gevorderde myelofibrose22.
• Androgenen (oxymethalon, danazol) en corticosteroïden (prednison) • EPO (procrit of epogen) om de bloedaanmaak van rode bloedcellen te stimuleren• Bloedtransfusie wordt gegeven, wanneer de Hb-waarden onacceptabel laag worden. Er zijn
verschillende transfusies mogelijk. Men kan bijvoorbeeld ook bloedtransfusie geven met enkel trombocyten voor de bestrijding van bloedingsverschijnselen wanneer het trombocy-tenaantal te laag is.
• Foliumzuur kan ook voorgeschreven worden om de productie van de erytrocyten te stimu-leren
Splenomegalie en MFBehandeling van een (te) grote milt:• Bestraling van een sterk vergrote milt bij MF is geen optie, omdat dit meestal weinig en
kortdurend effectief is. Het kan splenectomie niet voorkomen en de kans op complicaties bij een latere miltverwijdering worden verhoogd.
• Een sterk vergrote milt, die niet meer reageert op Hydrea of interferon, gaat gepaard met bloedarmoede, ernstige klachten van vermoeidheid, vermagering, nachtzweet en met ern-stige bezwaren van druk op de maag. Het verwijderen van de milt kan een bloedtransfusie-behoeftige anemie en bloedingen door trombocytopenie corrigeren en heft de zware last van een sterk vergrote milt op19.
• Een sterk vergrote milt moet op tijd en goed gepland verwijderd worden. Bij sommige mensen is het noodzakelijk dat de milt wordt weggenomen, maar durft men een operatie niet aan vanwege het bloedingsgevaar of ander operatierisico. In dit geval is meestal te lang gewacht om de sterk vergrote milt te verwijderen. Alleen dan is bestraling van de milt een optie.
• Bij de gevorderde myelofibrose heerst de opvatting dat behalve in het beenmerg ook in de milt rode bloedcellen worden gevormd. Zo lang er geen sprake is van bloedtransfusie is het raadzaam om na te gaan of het echt noodzakelijk is dat een sterk vergrote milt ver-wijderd moet worden. Als het beenmerg door de myelofibrose op den duur steeds minder rode bloedcellen kan produceren, dan kun je de rode bloedcellen, die de milt mogelijk nog produceert, extra hard nodig hebben. Uiteindelijk overheerst het nadeel en dient een sterk vergrote milt chirurgisch verwijderd te worden.
• De milt speelt een rol in het immuunsysteem met betrekking tot het bestrijden van infecties. Enkele maanden voor de miltverwijdering is vaccinatie tegen pneumococcen (pneumovax) aan te raden. Als de milt is weggenomen, is het noodzakelijk dat men elk jaar een griepprik krijgt en om de 3 tot 5 jaar een pneumovax. Soms is het ook nodig dat men antibiotica gebruikt.
3�Artsenbrochure
MF

Redenen om een sterk vergrote milt te verwijderen zijn�9:• Symptomen van een sterk vergrote milt, waarbij behandeling met interferon of Hydrea niet
meer helpt• Ernstige klachten en bezwaren van een sterk vergrote milt• Transfusienoodzakelijke bloedarmoede of een ernstige trombocytopenie• Toenemende klachten van een sterk vergrote milt met vermoeidheid, kortademigheid (zon-
der inspanning), nachtzweet en vermagering• De kans van overleven of overlijden rond en na de miltverwijdering is respectievelijk 91%
en 9%.• Na de miltverwijdering is de kans op sterke toename van het aantal trombocyten 22%, op
leververgroting 16% en acute leukemie 16%.• De overleving van MF patiënten na een geslaagde miltverwijdering is vele maanden tot
meerdere jaren.
Tabel �0. Klinische score voor het schatten van de overlevingskans bij klassieke MF met teke-nen van leuko-erythroblastose in het perifere bloed
Hemoglobine mmol/l
Leukocyten x �09/l cytogenetische afwijkingen
gemiddelde levensduur
Lille score > 6.25 4 – 20 - 7.5 jaar> 6.25 4 – 20 normaal 9.3 jaar> 6.25 4 – 20 abnormaal 4.2 jaar> 6.25 < 4 of > 20 - 2.2 jaar< 6.25 4 – 20 - 2.2 jaar< 6.25 < 4 en/of > 20 - 1.1 jaar
Sheffield scoreLeeftijd < 68 jaar > 6.25 - normaal 15 jaar
> 6.25 - abnormaal 6 jaar< 6.25 - normaal 4.5 jaar< 6.25 - abnormaal 1.8 jaar
Hemoglobine mmol/l
Leukocyten x �09/l Trombocyten x �09/l gemiddelde levensduur
Keulen scoreLeeftijd < 70 jaar > 6.25 4 - 20 > 300 > 10 jaar
< 6.25 4 - 20 < 300 3.7 jaar> 6.25 > 20 < 300 3.7 jaar
Myelofibrose is te genezen door een succesvolle beenmergtransplantatie met gebruik van been-merg van een gezonde donor (meestal broer of zus) met een kans op overlijden ten gevolge van complicaties van ongeveer 20 - 40% in het eerste jaar. Na 5 jaar overleeft een groot deel van de overige patiënten met een klassieke MF. Een nieuwere vorm van stamceltransplantatie, die min-der intensief is en daardoor met minder complicaties gepaard gaat, is veelbelovend. Gezien de kans op complicaties is een zorgvuldige selectie van patiënten noodzakelijk. Patiënten met een vroege MF, met geen of slechts geringe bloedarmoede, miltvergroting en leuko-erytroblastose,
MPD stichting32

hebben een levensverwachting van meer dan 10 jaar. Zij komen vooralsnog niet in aanmerking voor beenmergtransplantatie. Allogene beenmerg-/stamceltransplantatie bij patiënten met een klassieke MF en een gemiddelde overlevingskans van rond de 5 jaar of korter (zie tabel 10), met name als ze jonger zijn dan 50 – 55 jaar, is een optie indien een geschikte donor aanwezig is.
Het ondergaan van een Allogene beenmerg-/stamceltransplantatie vergt een zorgvuldige afwe-ging. De risico’s van een beenmergtransplantatie moeten afgewogen worden tegen de nog te verwachten levensduur. De klinische scores voor het schatten van de overlevingskans bij MF in de tabel worden gebruikt om na te gaan of de risico’s van een beenmergtransplantatie in verhouding staan met de nog te verwachten levensduur.
33Artsenbrochure
MF

4. Referenties
1. Murphy S, Iland H, Rosenthal D, et al. Essential thrombocythemia: An interim report from the Polycythemia Vera Study Group. Semin Hematol 1986;23:177–182.
2. Berlin NI. Diagnosis and classification of the polycythemias. Sem Hematol 1975;12:339-351.
3. WHO classification of the chronic myeloproliferative diseases (CMPD) polycythemia vera, chronic idiopathic myelofibrosis, essential thrombocythemia and CMPD unclassifiable. In: Jaffe S, Harris NL, Stein H et al, editors. WHO Classification of Tumours. Tumours of Haematopoiesis and Lymphoid Tissues. Lyon. IARC 2001 pp 31-42.
4. Michiels JJ, Thiele J Clinical and pathological criteria for the diagnosis of essential throm-bocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metapla-sia). Int J Hematol 2002;76:133-145.
5. Thiele J, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: a comparative study of two classifications systems (PVSG-WHO) on 839 patients. Ann Hematol 2003;82:148-152.
6. Michiels JJ, De Raeve H, Berneman Z,, Van Bockstaele D, Hebeda K, Lam K, Schroyens W. The 2001 World Health Organization (WHO) and updated European clinical and patholo-gical (ECP) criteria for the diagnosis, classification and staging of the Ph1-chromosome negative chronic myeloproliferative disorders (MPD). Sem Thromb Hemostas 2006;32:307-340.
7. Thiele J, Kvasnicka HM. A critical reappraisal of the WHO classification of the chronic myeloproliferative disorders. Leukemia and Lymphoma 2006;47:381-396.
8. Michiels JJ, Kvasnicka HM, Thiele J. Doctor’s Brochure Myeloproliferative Disorders: MPDs, Essential Thrombocythemia: ET, Polycythemia Vera: PV, Chronic Idiopathic Myelofibrosis: CIMF. MPD Stichting NL; MPD Foundation NL, www.mpd-stichting.nl
9. Kvasnicka HM, Thiele J. The impact of clinicopathological studies on staging and survival in ET, PV and IMF. Sem Thromb Hemostas 2006;32:362-371.
10. Villeval JL, James C, Pisani DF, Casadevall N, Vainchenker W. New insights into the patho-genesis of JAK2V617F-positive myeloproliferative disorders and consequences for the management of patients. Sem Thromb Hemostas 2006;32:341-351.
11. Lippert E, Boissinot M, Kralovics R, Girodon F, Dobo I, Praloran V, Boiret-Dupre, Skoda RC, Hermouet S. The JAK2V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood, May 25, 2006 PubMed Ahead of publication.
12. Michiels JJ, Berneman Z, Van Bockstaele D, Van Der Planken M, De Raeve H, Schroyens W. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Sem Thromb Hemostas 2006;32:174-207.
13. Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet HHDM. The paradox of platelet activation and impaired function: platelet-von Wilebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestatons in essential thrombocythemia and polycythemia vera. Sem Thromb Hemostas 2006;32:589-604
MPD stichting34

14. Schwarz J, Pytlik R, Doubek M et al. Analysis of risk factors: the rationale of the guidelines of the Czech Hematological Society for diagnosis and treatment of chronic myeloprolife-rative disorders with thrombocythemia. Sem Thromb Hemostas 2006;32:231-245.
15. Birgegard G. Anagrelide treatment in myeloproliferative disorders. Sem Thromb Hemostas 2006;32:260-266.
16. Langer C, Lengfelder E, Thiele J, Kvasnicka HM, Pahl H, Beneke H, Schauer S, Gisslinger H, Griesshammer M. Pegylated interferon for the treatment of high risk essential thrombo-cythemia: results of a phase 2 study. Haematologica 2005;90:1333-1338.
17. Silver T. Long-term effects of the treatment of polycythemia vera with recombinant inter-feron-alpha. Cancer 2006;107:451-458.
18. Kiladjian JJ, Cassinat B, Turlure P et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon-alpha-2a. Blood 2006;108:2037-2040.
19. Tefferi A. Myelofibrosis with myeloid metaplasia. New Eng J Med 2000;342;1255-1265.20. Tefferi A,Mesa RA, Nagorney DM, Schroeder G, Silverstein MN. Splenectomy in myelofi-
brosis with myeloid metaplasia: a single-insitution experience with 223 patients. Blood 2000;95:2226-2253
21. Elliot MA, Mesa R, Li CY, Hook CC, Ansell SM, Levitt RM et al. Thalidomide treatment in myelofibrosis with myeloid metaplasia. Br J Haematol 2002;117:288-296.
22. Tefferi A, Cortes J, Verstovsek S et al. Lenalidomide (Revlimid) therapy in myelofibrosis with myeloid metaplasia. Blood 2006;108:1158-1164.
23. Alfonso Quintás-Cardama, Hagop M,. Kantarjian, Francis Giles, Srdan Verstovsek. Pegylated Interferon Therapy for Patients with Philadelphia Chromosome-Negative Myeloproliferative Disorders. Sem Thromb Hemostas 2006;32:409-416.
35Artsenbrochure

5. Informatie over de MPD Stichting
Wat doet ze?De MPD Stichting legt zich toe op een drietal zaken:• Informatieverstrekking (aan patiënten, medici en andere hulpverlenende instanties)• Lotgenotencontact• BelangenbehartigingDe Medische Adviesraad neemt een belangrijke plaats in binnen de MPD Stichting.Vooraanstaande specialisten, die hierin zitting hebben, ondersteunen het werk en voorzien zowel het bestuur als de leden van informatie.
Hoe kan ze haar werk doen?Het werk voor de MPD Stichting wordt door vrijwilligers gedaan. Om alle MPD patiënten te kunnen bereiken en zowel patiënten als artsen van goede informa-tie te kunnen voorzien is veel geld nodig. Hoewel de MPD Stichting subsidie van de overheid ontvangt, is ze ook afhankelijk van donateursbijdragen.
Wat kunt u doen?Wilt u zo goed mogelijk op de hoogte gehouden worden van de laatste ontwikkelingen in diagnose en behandeling van de ziekten ET, PV en MF? Word dan donateur!Donateurs ontvangen drie keer per jaar gratis het nieuwsblad “Pur Sang”. Ze worden ook geïn-formeerd over de contactdagen en andere activiteiten, die de MPD Stichting ontwikkelt.
Hoe kun u zich aanmelden als donateur?Vul via www.mpd-stichting.nl het aanmeldingsformulier in en verstuur het per e-mail naar de MPD Stichting.Maak de donateursbijdrage van minimaal € 25.00 per kalenderjaar over naar giro 959��5� t.n.v. MPD Stichting te Zoetermeer onder vermelding van: “aanmelding donateur en uw naam en adres”.
Pas als uw aanmeldingsformulier per post binnen is èn het te betalen bedrag gestort is op bovenstaande girorekening is uw aanmelding compleet.
Voor meer informatie:
MPD StichtingGrondelsloot 61, 2724 BT ZoetermeerTel. 079-3432423
MPD stichting36


Correspondentieadres:Secretariaat MPD Stichting
Grondelsloot 612724 BT Zoetermeer
Informatietelefoon:079 - 3432423
InformatieWebsite: www.mpd-stichting.nlE-mail: [email protected]
Giro: 9598857
Lay
out
en d
ruk:
Dru
kker
ij va
n Re
e B.
V.


















