
Ontwikkeling van een HPLC-MS methode voor de bepaling van ...
77
Vakgroep Organische Chemie Onderzoeksgroep Scheidingstechnieken Ontwikkeling van een HPLC-MS methode voor de bepaling van ceramiden in de huid Proefschrift voorgelegd tot het behalen van de graad van Master in de Chemie door Barbara DHOOP Academiejaar 2009-2010 Promotor: prof. dr. P. Sandra Copromotor: dr. F. Lynen Begeleider: dr. B. Bicalho
Transcript of Ontwikkeling van een HPLC-MS methode voor de bepaling van ...

Vakgroep Organische Chemie
Onderzoeksgroep Scheidingstechnieken
Ontwikkeling van een HPLC-MS methode voor
de bepaling van ceramiden in de huid
Proefschrift voorgelegd tot het behalen van
de graad van Master in de Chemie door
Barbara DHOOP
Academiejaar 2009-2010
Promotor: prof. dr. P. Sandra
Copromotor: dr. F. Lynen
Begeleider: dr. B. Bicalho


Dankwoord
Ik wil graag alle mensen bedanken die rechtstreeks of onrechtstreeks hebben
meegeholpen aan het tot stand brengen van deze thesis.
Op de eerste plaats wil ik graag prof. dr. Sandra bedanken om mij de kans te geven mijn
thesis uit te voeren in zijn onderzoeksgroep en wil ik hem daarbij ook danken voor het
grondig nalezen van mij thesis. In het bijzonder wil ik dr. Bicalho bedanken voor alle steun
en hulp die zij verleend heeft gedurende dit jaar.
Ook de mensen van het RIC in Kortrijk wil ik bedanken, voor het aanbieden van de
instrumentatie die een gedetailleerde analyse mogelijk maakte, waarvan de resultaten in
het addendum aanwezig zijn.
Daarnaast wil ik in dit dankwoord zowel Seppe als Peejay vermelden die tijdens dit
thesisjaar steeds voor een vrolijke noot zorgden en steeds paraat stonden als
‘proefkonijn’. Ook alle andere personen van het PARC wil ik graag danken voor de
aangename sfeer op het vierde verdiep in S4bis.
Alle vrienden: Annelies, Liselotte, Lin, Hanne, Jos, Maarten, Sander,... en familieleden wil
graag bedanken voor de nodige steun en ontspanning.
Tot slot wil ik mijn ouders speciaal bedanken omdat ze mij de kans hebben gegeven te
studeren wat ik graag wou.
En als allerlaatste wil ik mijn vriend Jan-Baptist bedanken voor zijn hulp,
onvoorwaardelijke steun en vertrouwen.

Inhoudsopgave
1 Inleiding ................................................................................................................. 1
2 Geschiedenis van de chromatografie ..................................................................... 2
2.1 Beginselen van chromatografie....................................................................... 2
2.2 Verdelingschromatografie ............................................................................... 2
2.3 Papierchromatografie ..................................................................................... 3
2.4 Dunlaagchromatografie .................................................................................. 4
2.5 Vloeistofchromatografie (HPLC) ..................................................................... 4
3 Fundamentele aspecten van de vloeistofchromatografie ...................................... 6
3.1 HPLC-modes .................................................................................................. 6
3.2 Theoretische aspecten van een HPLC-systeem ............................................. 7
3.3 Instrumentatie van een HPLC-systeem ......................................................... 17
3.3.1 Mobiele fase .......................................................................................... 18
3.3.2 Injector................................................................................................... 19
3.3.3 Kolom .................................................................................................... 19
3.3.4 Detector ................................................................................................. 20
3.3.4.1 Corona-ontladingsdetector ................................................................. 20
3.3.4.2 Massaspectrometrie ........................................................................... 20
4 Fundamentele aspecten van de massaspectrometrie .......................................... 21
4.1 LC-MS .......................................................................................................... 21
4.1.1 Electrospray ionisatie ............................................................................. 21
4.1.2 Massaspectrometer: quadrupool............................................................ 23
4.1.3 Detector : elektronenmultiplicator ........................................................... 24
4.2 MS-modes .................................................................................................... 24
4.3 Tandem massaspectrometrie (MS/MS) ......................................................... 25
4.4 MS/MS modes .............................................................................................. 25
5 Ceramiden ........................................................................................................... 27
5.1 Structuur en voorkomen in de huid ............................................................... 27
5.2 Overzicht van onderzoek van ceramiden in de huid ...................................... 28
6 Experimenteel gedeelte ....................................................................................... 32
6.1 Algemene aspecten ...................................................................................... 32
6.1.1 Chemicaliën ........................................................................................... 32
6.1.2 Instrumentatie ........................................................................................ 32

6.2 Ontwikkeling en optimalisatie van een NPLC-methode voor analyse van
ceramiden ............................................................................................................... 32
6.3 Karakterisatie van ceramiden door MS-analyse ............................................ 36
6.4 Monstervoorbereiding ................................................................................... 38
6.4.1 Tape stripping-techniek en tape-extractie .............................................. 38
6.4.2 Vaste fase extractie ............................................................................... 40
6.5 NPLC-analyse van ceramiden in de huid ...................................................... 48
6.6 Ontwikkeling van een RPLC-methode voor analyse van ceramiden ............. 48
6.7 RPLC-analyse van ceramiden in de huid ...................................................... 51
7 Conclusie ............................................................................................................. 55
8 Referenties .......................................................................................................... 56

Inleiding
1
1 Inleiding
De hoornlaag van de menselijke huid (stratum corneum) zorgt voor bescherming van het
lichaam tegen waterverlies en schadelijke stoffen uit de omgeving dankzij een unieke
structuur, bestaande uit keratinocyten en intercelullaire lipide lamellen. De huidlipiden,
voornamelijk bestaande uit vrije vetzuren, cholesterol en ceramiden, vormen samen als
het ware het ‘cement’ die de ‘bakstenen’ (de keratinocyten) samenhouden (Elias 1983).
De ceramiden spelen een cruciale rol bij allerlei functies van de huid; een gewijzigde
ceramide-samenstelling is reeds gelinkt aan bepaalde huidziektes zoals psoriasis en
atopische dermatitis (eczeem) (Di Nardo, Wertz et al. 1998; Holleran, Takagi et al. 2006).
In tegenstelling tot de ceramiden die te vinden zijn binnenin cellen, bezitten ceramiden in
de hoornlaag een grote variabiliteit in structuur wat onderzoek en analyse van deze
ceramiden een uitdaging maakt.
In dit werk wordt een methode ontwikkeld om ceramiden in de huid te analyseren, er
wordt tevens gezocht naar een vaste fase extractie (SPE) om ceramiden aanwezig in de
huid selectief te extraheren uit de complexe hoornlaagmatrix. Hierbij is het echter niet
realiseerbaar alle interferenties te verwijderen, maar wordt gezocht naar een methode die
de ceramiden van cruciale interferenties afzondert. De componenten worden gescheiden
met behulp van hoge performantie vloeistofchromatografie (HPLC) in de omkeerfase
mode en in de normaalfase mode. Detectie van de componenten gebeurt door tandem-
massaspectrometrie (MS/MS).
Daar de fractionatie van de hoornlaag ceramiden van de cholesterylesters en triglyceriden
succesvol was, werden extracten onderzocht in samenwerking met Metablys, een afdeling
van het Research Institute for Chromatography, Kortrijk, België met behulp van een recent
ontwikkeld lipodomic platform bestaande uit HPLC bij hoge resolutie en een Q-TOFMS
apparaat dat tevens hoge resolutie spectra oplevert (K.Sandra et al. 2010).

Geschiedenis van de chromatografie
2
2 Geschiedenis van de chromatografie
2.1 Beginselen van chromatografie
Chromatografie vindt reeds in het begin van de 20e eeuw zijn oorsprong. Het was Mikhail
Semyonovich Tswett (1872-1919) die een nieuwe categorie van adsorptieanalyses
ontwikkelde en voor het eerst het begrip chromatografie gebruikte.
In 1903 wist Tswett plantpigmenten te scheiden met behulp van een calcium carbonaat–
kolom. In zijn werk ‘ Adsorptionanalyse und Chromatographische Methode. Anwendungen
auf die Chemie des Chlorophylls’ beschreef hij hoe eerst pigmenten op de kolom werden
gebracht, solvent over de kolom gegoten werd en er zo verschillende zones ontstaan in
de kolom. Pigmenten die minder sterk aangetrokken worden zullen zich makkelijker en
dus verder in de kolom kunnen verplaatsen dan een sterk aangetrokken pigment. De
kolom vertoonde hierdoor verschillende gescheiden en gekleurde zones: een gele zone
werd toegekend aan de carotinoïden uit het extract en twee groene zones werden aan
chlorofylen a en b toegeschreven. Uit deze observaties leidde hij de naam chromatografie
af uit de twee griekse woorden ‘chroma’ en ‘graphien’, wat letterlijk vertaald kan worden
als ‘in kleur schrijven’.
Willstätter, tevens actief in chlorofylonderzoek, concludeerde uit eigen onderzoek dat de
twee verschillende chlorofyl-zones bij Tswett, toe te kennen waren aan decompositie van
het chlorofyl en de chromatografische methode bijgevolg geen goed procedé kon zijn.
Hoewel Tswett het bij het recht eind had, werd zijn theorie hierdoor voor een lange tijd uit
het oog verloren. Het is pas rond 1930 dat chromatografie herondekt werd, verschillende
organische chemisten gebruikten de methode toen voornamelijk bij onderzoek en
opzuiveren van natuurproducten zoals bijvoorbeeld carotenen (Ettre 2000; Ardrey 2003;
Engelhardt 2004).
2.2 Verdelingschromatografie
De volgende mijlpaal in de geschiedenis van chromatografie volgde ongeveer 10 jaar
later. De ontwikkeling van een nieuwe techniek door A. J. P. Martin en R. M. L. Synge
kreeg de naam verdelingschromatografie (ook vloeistof-vloeistof chromatografie
genoemd). Hierbij werd getracht aminozuren te scheiden door twee vloeistoffen in
tegengestelde richting te laten stromen. Deze methode bracht echter weinig resultaten op
en werd aangepast: één van de twee vloeibare fasen werd stationair gemaakt door

Geschiedenis van de chromatografie
3
silicagel als ondersteuning te gebruiken en de andere, mobiele fase werd door deze
stilstaande fase heen gelopen. Hierna kon de keuze gemaakt worden ofwel de
verschillende gescheiden zones te visualiseren door een geschikte indicator toe te
voegen, ofwel een ‘flow through chromatogram’ (‘Durchflusschromatogramm’) te
genereren waarbij de verschillende zones geëlueerd en apart gecollecteerd kunnen
worden.
Martin en Synge benadrukten dat de verdeling van een component over de twee fasen,
de cruciale parameter is en dat deze techniek niet enkel toepasbaar is op aminozuren.
Verder is ook naast een vloeibare mobiele fase ook een gas te gebruiken als mobiele
fase. Deze vaststelling zal later ook de aanleiding blijken tot de ontwikkeling van
gaschromatografie.
Eén van de belangrijkste vaststellingen die bij verdelingschromatografie gemaakt werd, is
dat het concept van theoretische schotels bij destillatieprocessen eveneens toepasbaar
kon gemaakt worden voor chromatografische processen. Door opdelen van de kolom in
denkbeeldige segmenten die de naam theoretische schotels kreeg, werd gepostuleerd dat
net zoals bij een destillatieproces het mogelijk was in ieder segment een evenwicht te
vormen tussen stationaire en mobiele fase. Deze hypothese gaf voornamelijk een idee
over het scheidend vermogen van een kolom: hoe meer schotels een kolom bezit, hoe
beter de scheiding. In het theoretische aspect van HPLC wordt dit model verder
uitgewerkt.
Vloeistof-vloeistof chromatografie werd evenwel weinig in gebruik genomen door gebrek
aan robuustheid. De stationaire fase werd namelijk gaandeweg door de mobiele fase
weggespoeld waardoor resultaten niet reproduceerbaar waren (Ettre 2000; Engelhardt
2004).
2.3 Papierchromatografie
Een techniek die aantrekkelijker en voornamelijk in de praktijk eenvoudiger bleek, werd
eveneens door Martin ontwikkeld, namelijk papierchromatografie.
Een monster werd gespot onderaan het filterpapier waarna het papier in een vat geplaatst
werd. Het vat werd voorzien van een solvent, bijvoorbeeld water, dat door capillaire
krachten door het filterpapier werd opgenomen. Met behulp van een kleurreactie konden
de gescheiden componenten uit het monster gevisualiseerd worden op het filterpapier. Dit
werd de eerste chromatografische methode op micro-analytischniveau.
Hierbij werd de ‘retardation factor’ of RF-waarde gedefinieerd als de verhouding tussen de
afgelegde afstand op het papier door een component gedeeld door de afstand afgelegd

Geschiedenis van de chromatografie
4
door het solvent. Daarnaast gaf Martin ook aan dat het mogelijk was het scheidend
vermogen te vergroten door een 2D-analyse uit te voeren, waarbij twee verschillende
fase-systemen na elkaar worden gebruikt en het tweede eluent het filterpapier doorloopt
90° ten opzichte van de richting van het eerste eluent.
De opkomst van een andere planaire chromatografische methode, namelijk
dunlaagchromatografie of ‘Thin Layer Chromatography ’ (TLC) zorgde ervoor dat ook op
zijn beurt populariteit en gebruik van papierchromatografie afnam (Ettre 2000; Engelhardt
2004).
2.4 Dunlaagchromatografie
Het principe van TLC is analoog aan dit van papierchromatografie: het monster wordt
onderaan de plaat gespot en daarna in een vat met solvent geplaatst.
In tegenstelling tot bij papierchromatografie, kon bij dunlaagchromatografie de stationaire
fase aangepast worden. Daarnaast werd ook de snelheid van een analyse verhoogd
doordat de stationaire fase nu een poreuze laag is, voornamelijk silicagel, in plaats van
een celluloselaag bij papierchromatografie. Dankzij de eenvoud en snelheid van TLC
wordt ze tot op de dag van vandaag nog steeds in gebruik genomen om bijvoorbeeld een
chemische reactie op te volgen of als kwalitatieve of semi-kwantitatieve analyse (Ettre
2000; Engelhardt 2004).
2.5 Vloeistofchromatografie (HPLC)
Eind jaren ’50 had vloeistofchromatografie reeds een aanzienlijke achterstand ten
opzichte van gaschromatografie. Vloeistofchromatografie verliep nog steeds manueel
terwijl bij gaschromatografie reeds vrij gesofisticeerde toestellen werden gebruikt.
Door de kennis verworven voor gaschromatografie toe te passen op
vloeistofchromatografie werd duidelijk dat vooral een trager diffusie-proces een nadeel is
indien een vloeistof als mobiele fase gekozen wordt. Dit probleem kon deels omzeild
worden door niet voor een open capillair te kiezen, maar de kolom te pakken met kleine
deeltjes. Om de snelheid waarmee de mobiele fase de kolom doorloopt te optimaliseren
dienden hoge drukken gebruikt te worden, vandaar werd de term ‘High Pressure Liquid
Chromatography’ geïntroduceerd. Deze naam werd niet veel later gewijzigd in ‘High
Performance Liquid Chromatography’. Kolommen die korter gemaakt werden en uitgerust
met uniforme en kleinere deeltjes zorgen ervoor dat de prestatie van
vloeistofchromatografie aanzienelijk verhoogd werd.

Geschiedenis van de chromatografie
5
Een geautomatiseerde aminozuur-analyzer, een toestel dat via ionuitwisselings
chromatografie aminozuren kon analyseren en gelpermeatie chromatografie, die
scheiding uitvoert van synthetische polymeren op basis van moleculair gewicht, zijn de
voorlopers van een HPLC-systeem. Het eluaat loopt bij beide methodes onder hoge druk
door een (herbruikbare) kolom die gepakt is met kleine deeltjes. Beiden precursor-
systemen waren echter beperkt aangezien ze slechts één soort specimen kunnen
scheiden en analyseren.
Een HPLC-systeem geschikt om universele monsters te analyseren werd pas enkele
jaren later, rond 1965, ontwikkeld door twee onafhankelijke onderzoeksgroepen in Europa
en de Verenigde Staten (Ettre 2000; Engelhardt 2004; Dolan, Kirkland et al. 2010).

Fundamentele aspecten van de vloeistofchromatografie
6
3 Fundamentele aspecten van de vloeistofchromatografie
3.1 HPLC-modes
De analysemethode HPLC bestaat uit een aantal modes die gekarakteriseerd worden
naargelang de eigenschappen van mobiele en stationaire fase: omkeerfase
vloestofchromatografie (Reverse Phase Liquid Chromatography: RPLC), normaalfase
vloeistofchromatografie (Normal Phase Liquid Chromatography: NPLC), hydrofiele
interactie chromatografie (Hydrophilic Interaction Chromatography: HILIC),
ionpaarchromatografie (Ion-pair Chromatography: IPC), ionuitwisselings-chromatografie
(Ion-exchange Chromatography: IEC), size-exclusion chromatografie of
gelchromatografie (Size-exclusion Chromatography: SEC) en chirale chromatografie.
Bij omkeerfase wordt gebruik gemaakt van een apolaire kolom, bijvoorbeeld een C18-
stationaire fase en een mobiele fase die doorgaans een polair mengsel is van water en
organisch solventen, zoals acetonitril of methanol. De scheiding bij RPLC gebeurt op
basis van hydrofobiciteit van de componenten. RPLC is de meest gebruikte methode,
voornamelijk gebruikt bij analyse van wateroplosbare monsters. De voorkeur om de
RPLC-methode te gebruiken, is te verklaren via de vele voordelen tenopzichte van de
overige methodes. Zowel robuustheid en veelzijdigheid van de techniek als meer
reproduceerbare en efficiëntere analyses dragen bij tot de populariteit van deze methode.
Bovendien zijn de gebruikte solventen minder ontvlambaar en toxisch in vergelijking met
solventen gebruikt bij bijvoorbeeld normaalfase chromatografie.
Bij normaalfase chromatografie wordt gebruik gemaakt van een polaire kolom, zoals niet-
gebonden silicagel of een diol stationaire fase en een apolaire mobiele fase bestaande uit
organische solventen zoals hexaan en isopropanol. De scheiding bij een NPLC-analyse
gebeurt op basis van polariteit van de componenten: een polaire component zal een
langere retentie ervaren dan een minder polaire component. Ook bijdrage van sterische
hinder zal invloed hebben op de retentie, hierdoor is het mogelijk met een NPLC-analyse
structurele isomeren te scheiden.
HILIC is een methode afgeleid van NPLC. De mobiele fase bestaat uit een mengsel van
water en organisch solvent en de kolom is meer polair in vergelijking tot kolommen
gebruikt bij NPLC.
Ionpaarchromatografie is een aangepaste vorm van RPLC. Door toevoegen van een
reagens die ionparing veroorzaakt, kunnen ionaire componenten (zoals zure of basische
componenten) in het monster met dit reagens interageren. Bij deze mode is het mogelijk

Fundamentele aspecten van de vloeistofchromatografie
7
de retentie van ionaire componenten te wijzigen, zonder de pH van de mobiele fase te
veranderen. Gebruik van extreme pH-waarden kan hierdoor vermeden worden.
Ionuitwisselingschromatografie maakt gebruik van een kolom voorzien van geioniseerde
of ioniseerbare functionele groepen die ionen in het monster met een tegenovergestelde
lading binden. De mobiele fase bestaat meestal uit een bufferoplossing.
Size-exclusion chromatografie is een scheidingsmethode gebaseerd op moleculaire
massa’s van de componenten, voornamelijk gebruikt bij synthetische polymeren of grote
biomoleculen. De stationaire fase bestaat uit een inerte kolom gepakt met deeltjes met
specifieke poriën. De mobiele fase kan zowel een waterige als organische oplossing zijn.
Chirale chromatografie zorgt voor een scheiding van enantiomere componenten. De
stationaire fase zal hierbij chirale knooppunten bevatten zodat interacties en dus
retentietijden van enantiomeren verschillen (Dolan, Kirkland et al. 2010).
3.2 Theoretische aspecten van een HPLC-systeem
Een HPLC-kolom is gepakt met poreuze deeltjes met een grootteorde van enkele µm,
gemaakt uit silicagel of een polymeer en bij voorkeur sferisch. Het oppervlak van de
deeltjes wordt door ‘endcapping’ voorzien van een gewenste functionele groep die zo de
stationaire fase vormt. Vermits de deeltjes klein en poreus zijn, beschikt elk deeltje over
een groot oppervlak in verhouding tot zijn volume. Analytmolecules, die door de mobiele
fase door de poriën worden gestuurd, kunnen interactie ondergaan met de aanwezige
functionele groep.
De voorkeur van een component om in de stationaire of mobiele fase te verblijven wordt
bepaald door interacties, zijnde specifieke adsorptie veroorzaakt door dipool-dipool of
dipool-geïnduceerde dipool interacties en niet-specifieke adsorptie op de stationaire fase,
veroorzaakt door van der Waals-krachten.
Figuur 1 geeft een voorbeeld weer van elutie van een monster die drie verschillende
analyten/componenten bevat.
Molecules afkomstig van analyt X worden voorgesteld door symbool ●, van analyt Y door
□, van analyt Z door ▲en molecules afkomstig van solvent waarin het monster is
opgelost, worden voorgesteld door +. Iedere component die in het systeem wordt
geïntroduceerd zal zowel een tijd in de mobiele fase als in de stationaire fase verblijven,
enkel de solventmolecules (+) ondergaan weinig tot geen interactie met de stationaire
fase en elueren dus meteen uit de kolom. Tijdens de elutie zullen alle andere
componenten onder invloed van een continu dynamisch proces zich een aantal maal
verplaatsten van mobiele fase naar stationaire fase en vice versa. De totale tijd is de som

Fundamentele aspecten van de vloeistofchromatografie
8
van een aantal verblijftijden in iedere fase en zal voor elke component verschillend zijn,
aangezien elke component andere interacties ondergaat. Omdat enkel de mobiele fase
zich verplaatst zullen componenten slechts in beweging zijn indien ze in mobiele fase
vertoeven. Door dit proces kan scheiding van componenten plaatsvinden.
Naarmate de molecules in de kolom diffunderen zullen ze zich meer en meer spreiden ten
opzichte van elkaar en dus ook een groter volume innemen dan bij aanvang, zoals met
een pijl is aangeduid voor component X in figuur 1. Dit bezette volume door analyt X wordt
ook aangeduid als een band met een zekere breedte. Bij verlaten van de kolom zal deze
band door het detectiesysteem worden geregistreerd en een signaal weergegeven
worden verspreid over een welbepaalde tijdsduur in het chromatogram.
Figuur 1. Illustratie van een scheidingsproces (boven) en het bijhorende chromatogram van het scheidingsproces (onder) (Dolan, Kirkland et al. 2010).
De hoogte of oppervlak van een piek is recht evenredig met de analytconcentratie, met
andere woorden hoe meer molecules van analyt X aanwezig in een monster, hoe groter
het signaal in het chromatogram.
Solventmolecules waarin het monster opgelost werd, zullen zoals reeds vermeld niet
weerhouden worden door de kolom. Elutie van dit solvent wordt dus enkel bepaald door

Fundamentele aspecten van de vloeistofchromatografie
9
de snelheid waarmee de mobiele fase door de kolom loopt en door de lengte en interne
diameter van de kolom. De eerste component die de kolom verlaat, is steeds het monster-
solvent, de tijd die nodig is om de kolom te doorlopen en gedetecteerd te worden, wordt
de dode tijd t0 genoemd.
Algemeen gezien wordt de tijd verlopen na injectie van een component tot en met de
elutie ervan de retentietijd tR genoemd. Dit is dus de som van de tijd doorgebracht in de
mobiele fase (t0) en de tijd doorgebracht in de stationaire fase (tR’).
(1)
Interacties van een component met de stationaire en mobiele fasen, snelheid van mobiele
fase, lengte van de kolom en temperatuur spelen elk een rol bij scheiding van de
componenten.
De snelheid u van de mobiele fase in de kolom wordt berekend door de lengte van de
kolom L te delen door de dode tijd t0.
(2)
Een andere manier om elke component te karakteriseren dan de retentietijd is via de
retentiefactor. Dit is de verhouding van de hoeveelheid component in de stationaire fase
tegenover de hoeveelheid component in de mobiele fase.
(3)
Aangezien
(4)
waarbij c de concentratie van de component is en V het ingenomen volume in de fase is,
volgt hieruit dat:
(5)

Fundamentele aspecten van de vloeistofchromatografie
10
De distributie tussen twee fasen wordt bepaald door het continu dynamisch evenwicht
tussen de fasen. Dit evenwicht is onder invloed van onder andere de moleculaire structuur
van de componenten, van de chemische eigenschappen van mobiele en stationaire fase,
van de temperatuur en van de druk waarmee de mobiele fase door de kolom wordt
gepompt. Dit evenwicht tussen stationaire en mobiele fase kan beschreven worden aan
de hand van de verdelingscoëfficiënt K
(6)
Daarenboven wordt de fase-ratio β gedefinieerd als de verhouding van het volume
stationaire fase over het volume mobiele fase in de kolom aanwezig.
(7)
Deze twee parameters vertegenwoordigen dus de retentiefactor:
(8)
De retentiefactor kan ook als verhouding van de verblijftijden van de component in de
stationaire en mobiele fase geschreven worden.
(9)
De verblijftijd in de mobiele fase stemt overeen met de dode tijd. Het verschil tussen de
retentietijd tR en de dode tijd t0 stelt de verblijftijd in de stationaire fase voor. Zowel de
retentietijd als de retentiefactor kunnen gebruikt worden om de retentie van een
component te beschrijven. Het voordeel bij gebruik van de retentiefactor is dat deze
waarde dimensieloos is en dus onafhankelijk is van de kolomlengte en van de snelheid
van de mobiele fase.
De verhouding van retentiefactoren van twee componenten, eveneens te schrijven als de
verhouding van retentietijden van de componenten aangezien de dode tijd een constante
is, drukt de selectiviteitefficiëntie uit en wordt gedefinieerd als scheidings- of
selectiviteitsfactor α .

Fundamentele aspecten van de vloeistofchromatografie
11
(10)
k2 is steeds groter dan k1, anders geformuleerd zal component 1 eerder elueren dan
component 2 waardoor deze waarde altijd groter of gelijk is aan 1. Indien α = 1 geeft dit
aan dat deze twee componenten niet gescheiden kunnen worden met de gegeven
methode. Een groter verschil in retentietijd en dus grotere selectiviteit zal steeds een
betere scheiding geven. De selectiviteitsfactor is afhankelijk van de eigenschappen van
de componenten, pH, temperatuur, kolomdimensies en -eigenschappen en het type
stationare en mobiele fase.
Iedere component zal na scheiding door een piek weergegeven worden (figuur 1, onder).
Een perfecte chromatografische scheiding zou oneindig smalle pieken genereren zodat
geen overlapping van pieken mogelijk zou zijn. Dit zal echter nooit het geval zijn,
aangezien processen zoals onder andere moleculaire diffusie de piek steeds een zekere
breedte geven.
Een chromatografische piek kan het best beschreven worden als een Gaussiaanse curve,
ook normaal verdeling genoemd, die in algemene vorm geschreven wordt als
(11)
De verwachtingswaarde µ en standaardafwijking σ karakteriseren deze functie. Toegepast
op een chromatografische piek wordt de retentietijd tR gelijk aan de verwachtigswaarde µ
en kan de standaardafwijking σ vervangen worden door breedte w, die ofwel gemeten kan
worden aan de basis van de piek wb ofwel op de helft van de maximale piekhoogte wh
(figuur 2).
Een piek zal echter niet steeds een ideale Gaussiaanse vorm hebben. Dit niet-ideaal
gedrag wordt ook soms staartvorming (tailing) genoemd. Dit fenomeen kan uitgedrukt
worden aan de hand van de symmetrie- of staartfactor (tailingfactor).

Fundamentele aspecten van de vloeistofchromatografie
12
Figuur 2. Een ideale Gaussiaanse piek.
Een cruciaal aspect bij de verbreding van een piek is dat de ruimtelijke variantie
rechtevenredig is met de migratie-afstand z.
(12)
De evenredigheidsfactor H wordt de schotelhoogte (hoogte-equivalent van een
theoretische schotel, Height Equivalent of a Theoretical Plate: HETP) genoemd en heeft
de dimensies van een lengte. Deze kan ook geschreven worden als:
(13)
waarin Dax : axiale dispersiecöeffiënt
u : snelheid van de mobiele fase
Analoog met de distillatietheorie kan de HETP vergeleken worden met de schotels in een
distillatiekolom waarbij het aantal theoretische schotels N en de schotelhoogte H
gerelateerd zijn aan de kolomlengte via:
(14)
met L de lengte van de kolom. Dit betekent dat de scheiding efficiënter gebeurt indien een
langere kolom gebruikt wordt vanwege het groter aantal schotels.

Fundamentele aspecten van de vloeistofchromatografie
13
Het schotelgetal N staat in functie van tR en σ (en dus ook w) en kan experimenteel uit het
chromatogram afgeleid worden:
(15)
Een criterium dat wordt gehanteerd om een scheiding in het chromatogram van twee
naast elkaar liggende pieken te bepalen is de resolutie Rs. Dit is het verschil in
retentietijden gedeeld door de gemiddelde breedte van de pieken, gemeten aan de basis
van de piek.
(16)
Een hogere resolutie door een groter verschil in retentietijd en/of smalle pieken
manifesteert zich in een betere scheiding van de twee componenten. Indien meerdere
componenten aanwezig zijn in een mengsel of monster zullen de twee minst gescheiden
componenten in het chromatogram het zogenaamde kritische paar vormen waarvan de
resolutie bepaalt of het scheidend vermogen van de kolom voldoende is. Een resolutie Rs
= 2 zal twee pieken voor 99.99% gescheiden van elkaar geven.
Selectiviteit, retentie, performantie en resolutie zijn samen te brengen in één vergelijking
die de basisvergelijking van de chromatografie wordt genoemd. Rs wordt uitgedrukt in
functie van α, k en N waarbij wordt aangenomen dat de breedte van de twee
aangrenzende pieken gelijk is.
(17)
waarbij Rs : de resolutie
N : het schotelgetal
α : de scheidingsfactor
k : de retentiefactor
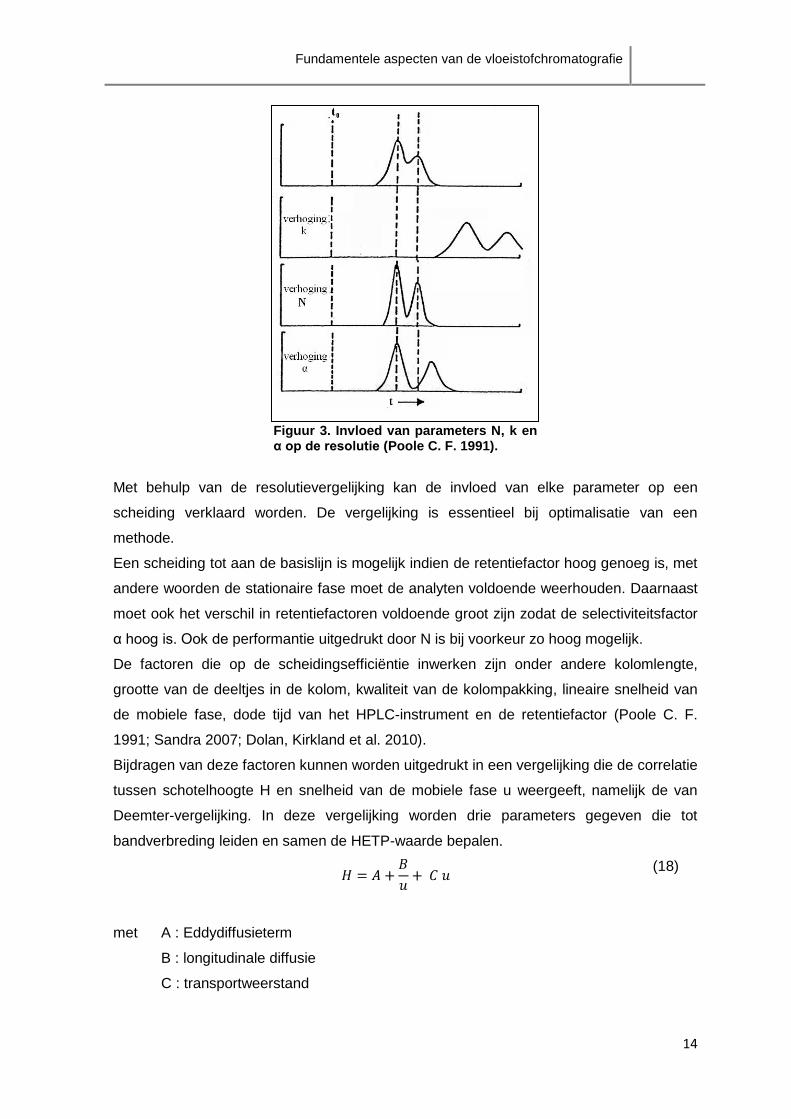
De invloed van de verschillende parameters op Rs wordt geïllustreerd in figuur 3.
Fundamentele aspecten van de vloeistofchromatografie
14
Figuur 3. Invloed van parameters N, k en α op de resolutie (Poole C. F. 1991).
Met behulp van de resolutievergelijking kan de invloed van elke parameter op een
scheiding verklaard worden. De vergelijking is essentieel bij optimalisatie van een
methode.
Een scheiding tot aan de basislijn is mogelijk indien de retentiefactor hoog genoeg is, met
andere woorden de stationaire fase moet de analyten voldoende weerhouden. Daarnaast
moet ook het verschil in retentiefactoren voldoende groot zijn zodat de selectiviteitsfactor
α hoog is. Ook de performantie uitgedrukt door N is bij voorkeur zo hoog mogelijk.
De factoren die op de scheidingsefficiëntie inwerken zijn onder andere kolomlengte,
grootte van de deeltjes in de kolom, kwaliteit van de kolompakking, lineaire snelheid van
de mobiele fase, dode tijd van het HPLC-instrument en de retentiefactor (Poole C. F.
1991; Sandra 2007; Dolan, Kirkland et al. 2010).
Bijdragen van deze factoren kunnen worden uitgedrukt in een vergelijking die de correlatie
tussen schotelhoogte H en snelheid van de mobiele fase u weergeeft, namelijk de van
Deemter-vergelijking. In deze vergelijking worden drie parameters gegeven die tot
bandverbreding leiden en samen de HETP-waarde bepalen.
(18)
met A : Eddydiffusieterm
B : longitudinale diffusie
C : transportweerstand

Fundamentele aspecten van de vloeistofchromatografie
15
u : gemiddelde lineaire snelheid van de mobiele fase
De Eddydiffusieterm houdt rekening met het feit dat niet elke analytmolecule dezelfde weg
zal afleggen in een kolom aangezien niet alle deeltjes in de kolom evengroot of
gelijkvormig zullen zijn en de pakking van de kolom niet uniform zal zijn. Omdat elke
analytmolecule dus een verschillende weg zal afleggen, zal er een zekere spreiding zijn
op de afgelegde weg van de molecules. Daaruit volgende zal dit verschillen in retentie
meebrengen. De Eddydiffusieterm is onafhankelijk van de snelheid waarmee de mobiele
fase de kolom doorloopt. Enkel parameters zoals vorm en grootte van de deeltjes en van
de poriën als ook de kwaliteit van kolompakking zijn hier van belang.
Deze term wordt gegeven als:
(19)
waarbij λ : pakkingsefficiëntie (λmax = 1)
dp : diameter van deeltje
De B-term stelt de bijdrage van longitudinale diffusie voor. Naast diffusie in axiale richting
(loodrecht op de kolomas) is ook longitudinale diffusie of diffusie langsheen de kolomas
mogelijk. Dit proces is het resultaat van concentratieverschillen in de mobiele fase. In het
centrum van de band van analytmolecules zal de concentratie maximaal zijn, hierdoor
zullen molecules, onder invloed van de wetten van Fick, diffunderen en dit zowel in de
richting waarin de mobiele fase vloeit als in de tegenovergestelde richting. Deze diffusie
zal leiden tot een verbreding van de band en is omgekeerd evenredig met de snelheid van
het eluens. Wanneer de snelheid van de mobiele fase verhoogd wordt, zullen de
analytmolecules minder tijd in de kolom doorbrengen en daardoor dus minder onderhevig
zijn aan deze diffusie.
De longitudinale diffusie-term wordt gegeven door
(20)
met DM de diffusiecoëfficiënt in de mobiele fase.
De longitudinale diffusie is gerelateerd aan moleculaire diffusie en wordt dus bepaald door
de viscositeit van de mobiele fase, de temperatuur, de diffusiecoëfficiënt en de
moleculaire massa van het analyt (Hens 2009). Enkel bij lage snelheid van de mobiele

Fundamentele aspecten van de vloeistofchromatografie
16
fase wordt de B-term relevant, aangezien de diffusiecoëficiënt van een component in
vloeistof in de meeste gevallen klein zal zijn in vergelijking met de snelheid van de
mobiele fase.
De transportweerstand geeft aan dat tijdens elutie van analyten de massa van een analyt
niet enkel kan worden getransporteerd van mobiele naar stationaire fase maar ook
opnieuw van stationaire naar mobiele fase. Analyt molecules aanwezig in mobiele fase
zullen diffunderen naar de grens tussen mobiel en stationaire fase en zo overgaan naar
de stationaire fase. Om evenwicht te behouden zullen naast overgang van molecules van
mobiele naar de stationaire fase ook molecules vanuit de stationaire fase terug naar de de
mobiele fase overgaan. Na een zekere tijd zullen alle analytmolecules de stationaire fase
verlaten hebben en terug overgegaan zijn in de mobiele fase.
Het gehele proces zorgt ervoor dat voortdurend massa uitgewisseld wordt tussen de twee
fasen gedurende de analyse. De snelheid waarmee het evenwicht tussen de twee fasen
wordt bereikt zal bepalend zijn voor de bijdrage van de transportweerstand. Zoals reeds
vermeld bevinden molecules van eenzelfde component zich niet allemaal op dezelfde
locatie in de kolom, hierdoor zal ook de transportweerstand voor elke molecule verschillen
en dus een piekverbreding als gevolg hebben.
De transportweerstand kan worden opgedeeld in twee bijdragen: de bijdrage van de
mobiele (CM) en de bijdrage van de stationaire fase (CS) .
De CM-term beschrijft de bijdrage tot band- of piekverbreding in de mobiele fase. Deze
verbreding ontstaat door onderscheid in lineaire viscositeit bij de mobiele fase. Mobiele
fase die dicht langs deeltjes in de kolom vloeit, zal trager bewegen dan mobiele fase die
zich verder van kolomdeeltjes bevindt. Door dit verschil in snelheid van mobiele fase,
zullen ook de analyten aanwezig daarin dit onderscheid ondervinden.
De CS-term beschrijft de bijdrage tot band- of piekverbreding in de stationaire fase. Deze
term wordt onder andere bepaald door de interacties tussen analyt en stationaire fase en
de hoeveelheid stationaire fase.
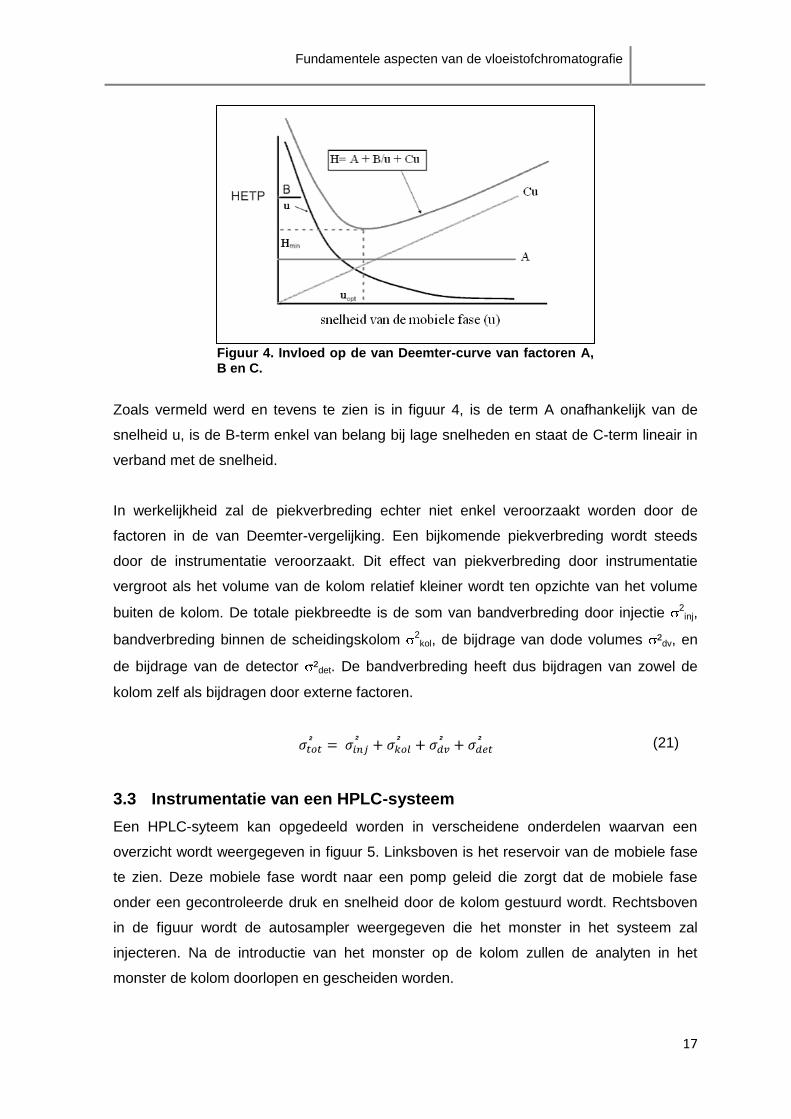
De van Deemter-curve stelt grafisch H in functie van de snelheid van de mobiele fase u
en wordt gebruikt om de optimale snelheid te achterhalen. Deze optimale waarde uopt
komt overeen met de minimale waarde van HETP (figuur 4).
Fundamentele aspecten van de vloeistofchromatografie
17
Figuur 4. Invloed op de van Deemter-curve van factoren A, B en C.
Zoals vermeld werd en tevens te zien is in figuur 4, is de term A onafhankelijk van de
snelheid u, is de B-term enkel van belang bij lage snelheden en staat de C-term lineair in
verband met de snelheid.
In werkelijkheid zal de piekverbreding echter niet enkel veroorzaakt worden door de
factoren in de van Deemter-vergelijking. Een bijkomende piekverbreding wordt steeds
door de instrumentatie veroorzaakt. Dit effect van piekverbreding door instrumentatie
vergroot als het volume van de kolom relatief kleiner wordt ten opzichte van het volume
buiten de kolom. De totale piekbreedte is de som van bandverbreding door injectie 2inj,
bandverbreding binnen de scheidingskolom 2kol, de bijdrage van dode volumes ²dv, en
de bijdrage van de detector ²det. De bandverbreding heeft dus bijdragen van zowel de
kolom zelf als bijdragen door externe factoren.
(21)
3.3 Instrumentatie van een HPLC-systeem
Een HPLC-syteem kan opgedeeld worden in verscheidene onderdelen waarvan een
overzicht wordt weergegeven in figuur 5. Linksboven is het reservoir van de mobiele fase
te zien. Deze mobiele fase wordt naar een pomp geleid die zorgt dat de mobiele fase
onder een gecontroleerde druk en snelheid door de kolom gestuurd wordt. Rechtsboven
in de figuur wordt de autosampler weergegeven die het monster in het systeem zal
injecteren. Na de introductie van het monster op de kolom zullen de analyten in het
monster de kolom doorlopen en gescheiden worden.

Fundamentele aspecten van de vloeistofchromatografie
18
Figuur 5. Schematisch overzicht van instrumentatie bij HPLC.
Een detector zal analyten die uit de kolom elueren registeren onder de vorm van signalen.
De signalen gemeten door de detector corresponderen met de aanwezige
analytconcentratie. Een computer die in verbinding staat met de detector zal de output
van de detector opslaan zodat dataverwerking mogelijk is (Dolan, Kirkland et al. 2010).
3.3.1 Mobiele fase
Solvent gebruikt als mobiele fase wordt in een inert reservoir bewaard, meestal
vervaardigd uit bruin glas. De zuiverheid van het solvent bij een analyse is essentieel, dit
wil zeggen zonder contaminatie van bijproducten of vaste stof deeltjes. Om deeltjes zoals
stof tegen te houden, wordt een filter gekoppeld aan het uiteinde van de verbinding tussen
de solventreservoir en pomp.
Luchtbellen in de mobiele fase kunnen vermeden worden door ontgassen van de
solventen. De ontgassing kan via verschillende technieken gebeuren. Het solvent kan een
tijd in een ultrasoonbad geplaatst worden of een inert gas zoals helium kan door het
solvent borrelen zodat de opgeloste gassen diffunderen naar de heliumbellen en zo uit het
solvent gezuiverd worden. Een derde ontgassingstechniek is vacuümontgassing waarbij
het ontgassen online kan gebeuren en voor de pomp zal geplaatst worden (Dolan,
Kirkland et al. 2010).
Bij een analyse kunnen meerdere solventen (bij meeste HPLC-toestellen tot 4 solventen)
gebruikt worden en indien nodig online gemengd worden in een mengkamer. Elutie kan
op twee manieren gebeuren: isocratische elutie waarbij een constante compositie van de
mobiele fase behouden wordt gedurende de analyse en gradiënt elutie waarbij de
compositie in functie van de elutietijd stapsgewijs of continu wordt gewijzigd.

Fundamentele aspecten van de vloeistofchromatografie
19
3.3.2 Injector
De injector in de autosampler zorgt dat het monster reproduceerbaar en kwantitatief kan
worden toegevoegd aan het systeem. De meest gebruikte injector is de zeswegkraan.
Deze kraan bestaat zoals de naam aantoont uit zes poorten. Naargelang de toestand van
de zeswegkraan: de laadpositie en de injectiepositie, zijn deze poorten op een andere
manier gekoppeld aan elkaar. Zoals schematisch is weergegeven in figuur 6, staat elke
poort in verbinding met een onderdeel van het HPLC-systeem. In de laadpositie zal de
poort in verbinding met de pomp de mobiele fase aanvoeren naar de poort in verbinding
met de kolom. De poort bovenaan, in verbinding met de injectienaald, zal het monster in
de zogenaamde sample loop aanbrengen tot deze loop, met een vast volume, helemaal
gevuld is. De poort die met het einde van de sample loop verbonden is, gaat naar de afval
(waste).
Figuur 6. Laadpositie (links) en injectiepositie (rechts) van een zeswegkraan.
Nadat de sample loop gevuld is (en dus geen lucht meer aanwezig is in de loop) zal van
de laadpositie overgeschakeld worden naar de injectiepositie. Nu zal de poort waar de
mobiele fase uit stroomt gekoppeld worden aan de poort van de sample loop, zodanig dat
het monster nu in de tegenovergestelde richting stroomt in de sample loop. De poort die in
laadpositie de ingang van sample loop aangeeft, zal nu verbonden worden met de poort
die naar de kolom loopt zodat het monster geïntroduceerd wordt op de kolom.
3.3.3 Kolom
Klassiek worden roestvrije stalen kolommen gebruikt met lengte van 5 tot 25 cm en
inwendige diameters van 1 tot 1.6 mm. Deeltjes van 1.8 tot 5 µm met een variëteit van

Fundamentele aspecten van de vloeistofchromatografie
20
geometrie of vorm zijn commercieel beschikbaar. Indien een hogere temperatuur
gewenst is dan kamertemperatuur, kan gebruik worden gemaakt van een kolomoven.
3.3.4 Detector
Direct na de kolom wordt een detector geplaatst. Universele detectoren zoals bijvoorbeeld
een brekingsindexdetector of een corona-ontladingsdetector hebben een lage
detectorselectiviteit aangezien een eigenschap wordt gemeten die aanwezig is bij alle
componenten. Detectoren die componentspecifiek zijn, meten een eigenschap die uniek
is voor bepaalde analyten, zoals bijvoorbeeld een UV-detector. Andere methoden maken
gebruik van onafhankelijke analytische instrumenten als detector. LC-NMR stuurt het
eluens uit de kolom naar een NMR-toestel, LC-MS koppelt een massaspectrometer na de
kolom. In dit werk werd gebruik gemaakt van een massaspectrometer en een corona-
ontladingsdetector.
3.3.4.1 Corona-ontladingsdetector
De corona-ontladingsdetector (corona discharge detector of charged aerosol detector,
CAD) is een universele detector. Het eluaat dat uit de kolom vloeit, wordt verneveld door
een stroom vernevelingsgas, zoals N2, en wordt verdampt in een verwarmde buis. Door
verdamping van de vluchtige mobiele fase blijven analytmolecules over in de gasfase.
Een positief geladen dragersgas, geladen door een corona-ontlading, wordt hierna
gemengd met de analyten, waarbij de lading overgedragen wordt van dragergas naar
analytmolecules. De analytionen worden door een electrometer opgevangen die de
elektrisch lading omzet in een signaal (Dolan, Kirkland et al. 2010).
3.3.4.2 Massaspectrometrie
Massaspectrometrie (MS) is een methode waarbij gasvormige ionen worden gevormd die
kunnen gescheiden worden op basis van verhouding massa tot lading (m/z-ratio). De
uitkomst van een analyse is een spectrum waarin relatieve of absolute abundatie wordt
geplot in functie van de m/z-verhouding. Uit deze spectra kan het moleculaire gewicht en
eventuele informatie over de structuur van een analyt verworven worden.
Een massa spectrometer is op te delen in drie onderdelen: een ionenbron, de massa-
analysator en een detector. De ionenbron genereert ionen van de componenten in het
monster, de massa-analysator scheidt binnenkomende ionen op basis van m/z, waarna
het detectiesysteem elke bundel van ionen omzet in een meetbaar signaal. Gezien het
belang van massaspectrometrie in dit werk wordt dit in detail besproken in het volgende
hoofdstuk met nadruk op de gebruikte opstellingen (Ardrey 2003; Niessen 2006).

Fundamentele aspecten van de massaspectrometrie
21
4 Fundamentele aspecten van de massaspectrometrie
4.1 LC-MS
LC-MS is een techniek waarbij een massaspectrometer gekoppeld wordt na een HPLC-
systeem. Dit is hedendaags een standaardmethode voor onderzoek van organische en
bio-organische monsters.
Vermits een MS-detector ionen meet in gasfase, is er nood aan een interface die naast
ionizatie van de monsteranalyten ook het vloeibare eluens kan verdampen. De twee
meest gebruikte interfaces zijn APCI (Atmospheric Pressure Chemical Ionization) en ESI
(Electrospray Ionization). Beide technieken zorgen ervoor dat fragmentatie van de
analyten zoveel mogelijk wordt vermeden, APCI en ESI worden daarom soms zachte
ionisatietechnieken genoemd.
De massa-analysator scheidt de ionen op basis van de massa-ladingverhouding (m/z) en
registreert tevens de intensiteit van elk ion. Deze scheiding kan, afhankelijk van de
gebruikte massa-analysator, gebeuren op basis van tijd of ruimte. De resolutie en de
accuratesse bepalen de prestatie van een massa-analysator. Mogelijke analysatoren zijn
onder andere de iontrap, de Time of Flight-analyzer (TOF), een quadrupool of een
combinatie van meerdere analysatoren. Indien een koppeling van twee
massaspectometers wordt gebruikt, spreekt men van LC-MS/MS of
tandemmassaspectrometrie.
Een elektronenmultiplicator (electronmultiplier: EM) zet de binnenkomende ionenbundel
om in een meetbaar en versterkt signaal. Er wordt dieper ingegaan op de gebruikte
technieken.
4.1.1 Electrospray ionisatie
Electrospray ionisatie zal achtereenvolgens het eluaat omvormen tot druppels, daarna
deze druppels van een lading voorzien en desolvateren en tenslotte zullen ionen gevormd
worden van de aanwezige analyten.
Het eluaat komende uit de kolom wordt door een capillair gestuurd onder atmosferische
druk waarbij rondom het capillair een verstuivergas stroomt. Het uiteinde van het capillair
is een fijne tip dat onderheven is aan een hoge potentiaal (ca. 3-4 kV). De vloeistof die
door de fijne tip gaat, zal door het elektrisch veld een overmaat lading krijgen en bij
verlaten van de tip zal de vloeistof verneveld worden tot geladen deeltjes, ook aerosol
genoemd.

Fundamentele aspecten van de massaspectrometrie
22
De geladen druppels zullen, onderhevig aan elektrostatische krachten, door een
tegenelektrode worden aangetrokken. Naargelang het teken van de aangebrachte
spanning zullen de resulterende analytionen ofwel positief of negatief geladen zijn.
Eenmaal de druppels gevormd zijn, zal desolvatatie plaatsvinden en solvent uit de
druppels verwijderd worden. Dit wordt bevorderd door een drooggas, bijvoorbeeld N2, in
de tegenovergestelde richten te laten stromen. De druppels worden hierdoor steeds
kleiner zodanig dat gelijke lading in een druppel steeds meer en meer geconcentreerd
wordt. Bij overschrijden van een kritisch punt, de Rayleigh limiet genaamd, zullen de
druppels exploderen. Dit kritisch punt wordt bereikt wanneer in een druppel de
elektrostatische krachten te groot worden ten opzichte van de oppervlaktespanning (figuur
7).
Figuur 7. Desolvatatieproces bij ESI.
Nadat desolvatatie heeft plaatsgevonden, zullen de kleine druppels omgevormd worden
tot ‘naakte’ ionen van de analyten. Het mechanisme van vorming van deze ionen is nog
steeds omstreden en kan door twee verschillende modellen beschreven worden: het
ionevaporatie-model en het ladingresidue-model.
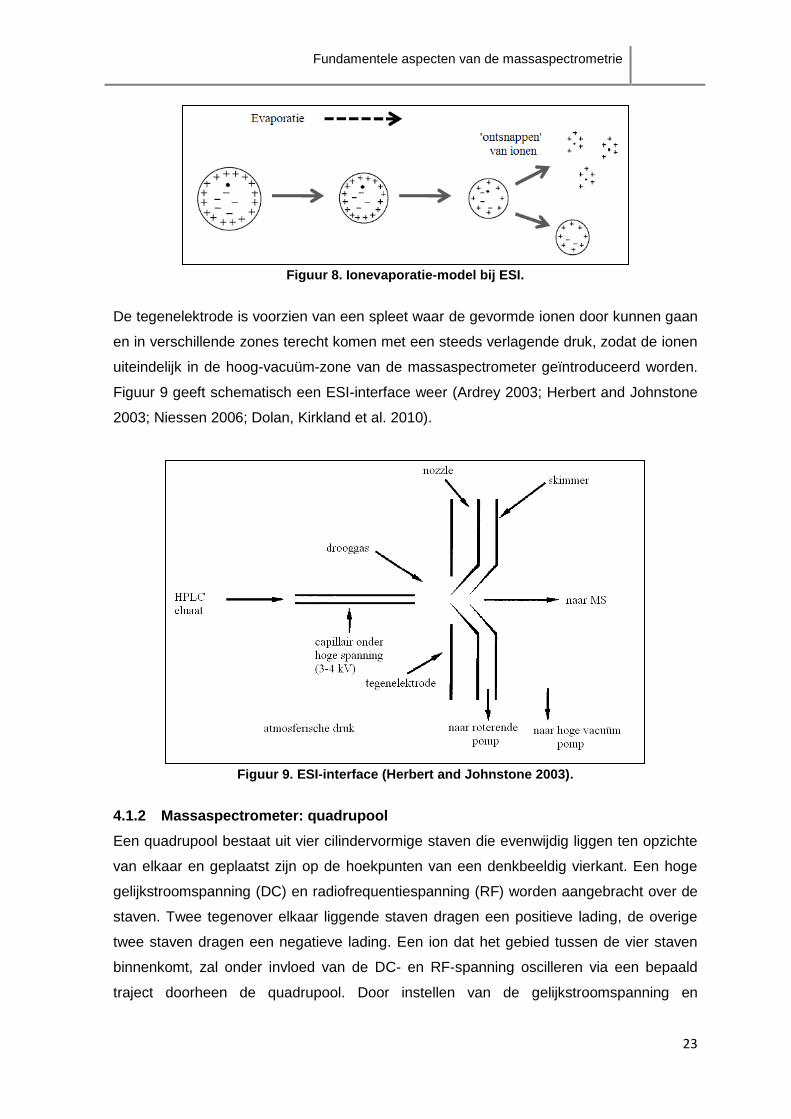
Het ionevaporatie-model stelt dat ionen onmiddelijk uit een druppel vrijgesteld kunnen
worden en in de gasfase overgaan aangezien, door continue verdamping van solvent, het
oppervlak van de druppel voldoende geladen zal zijn. Eenmaal de lading van ionen aan
de oppervlakte van de druppel, en zo dus het elektrisch veld aan de oppervlakte, groter
wordt dan de oppervlaktespanning kunnen naakte ionen uit de druppel ‘ontsnappen’
(figuur 8).
Het ladingresidue-model neemt aan dat de ionen van analytmolecules reeds gevormd zijn
in de vloeibare fase en het proces van desolvatatie doorgaat tot slechts één ion per
druppel overblijft. Tot slot zal ook bij deze druppel desolvatatie plaatsvinden zodat het ion
over kan gaan naar de gasvormige fase.
Fundamentele aspecten van de massaspectrometrie
23
Figuur 8. Ionevaporatie-model bij ESI.
De tegenelektrode is voorzien van een spleet waar de gevormde ionen door kunnen gaan
en in verschillende zones terecht komen met een steeds verlagende druk, zodat de ionen
uiteindelijk in de hoog-vacuüm-zone van de massaspectrometer geïntroduceerd worden.
Figuur 9 geeft schematisch een ESI-interface weer (Ardrey 2003; Herbert and Johnstone
2003; Niessen 2006; Dolan, Kirkland et al. 2010).
Figuur 9. ESI-interface (Herbert and Johnstone 2003).
4.1.2 Massaspectrometer: quadrupool
Een quadrupool bestaat uit vier cilindervormige staven die evenwijdig liggen ten opzichte
van elkaar en geplaatst zijn op de hoekpunten van een denkbeeldig vierkant. Een hoge
gelijkstroomspanning (DC) en radiofrequentiespanning (RF) worden aangebracht over de
staven. Twee tegenover elkaar liggende staven dragen een positieve lading, de overige
twee staven dragen een negatieve lading. Een ion dat het gebied tussen de vier staven
binnenkomt, zal onder invloed van de DC- en RF-spanning oscilleren via een bepaald
traject doorheen de quadrupool. Door instellen van de gelijkstroomspanning en

Fundamentele aspecten van de massaspectrometrie
24
radiofrequentie zullen enkel ionen in een specifiek, klein m/z-interval het einde van de
quadrupool kunnen bereiken. Ionen met een m/z-verhouding die niet in het ingestelde
interval zitten, zullen een onstabiel traject volgen en bijgevolg de detector niet bereiken.
Door aanpassen van DC- en RF-spanning kan het gedetecteerde m/z-venster over een
gewenst massabereik gescanned worden (figuur 10).
Een quadrupool is in staat snel het totale massabereik af te scannen en maakt gebruik
van een lage spanning zodat een relatieve hoge druk, zoals bij LC-MS, kan gebruikt
worden. Daarnaast is ook de lage kostprijs een voordeel van deze massaspectrometer.
Een groot nadeel van de quadrupool is de lage resolutie (Ardrey 2003).
Figuur 10. Een quadrupool.
4.1.3 Detector : elektronenmultiplicator
De EM bestaat uit een aantal dynodes. Dit zijn metalen platen waarbij elke plaat steeds
een hogere potentiaal heeft dan de vorige plaat. Bij invallen van de ionenbundel op de
eerste dynode, komen secundaire elektronen vrij die door de hogere potentiaal van de
tweede plaat versneld naar deze tweede plaat bewegen. Hierdoor komen opnieuw
secundaire elektronen vrij. Dit proces vindt steeds opnieuw plaats bij het bewegen van de
elektronen naar de volgende dynode. Aangezien voor elk elektron dat invalt op een
dynode telkens meerdere elektronen vrijkomen, wordt een vermenigvuldigingsproces
gecreëerd (elektronenlawine) (Herbert and Johnstone 2003).
4.2 MS-modes
De massaspectrometer kan in twee verschillende modes gebruikt worden: Total ion
current (TIC) of Selected ion monitoring (SIM).

Fundamentele aspecten van de massaspectrometrie
25
De scan mode TIC zal het vooraf ingestelde massabereik een aantal maal afscannen,
afhankelijk van de resolutie van de massaspectrometer. Een hogere resolutie zal ervoor
zorgen dat het bereik een aantal keren meer kan afgescanned worden. Verhogen van de
scantijd of verkleinen van het massabereik kan de gevoeligheid verhogen. Dit heeft echter
voor beiden nadelen: scantijd verhogen kan ervoor zorgen dat een te klein aantal
datapunten gecollecteerd wordt, massabereik verkleinen kan ervoor zorgen dat
waardevolle informatie verloren gaat (De Hoffmann E. 1996).
In sommige gevallen is het niet nodig het volledige massabereik te scannen. Wanneer
bijvoorbeeld de structuur van een component gekend is, kunnen ook eigenschappen en
gedrag achterhaald worden. In dit geval zal SIM mode gekozen worden waardoor de
gevoeligheid verhoogd wordt. De massaspectrometer wordt als massafilter gebruikt en zal
enkel ionen binnen het bepaalde m/z-interval naar de detector doorzenden. De detector
zal dus niet een massaspectrum opnemen, maar de intensiteit van de geselecteerde m/z-
waarde registreren (Ardrey 2003; Niessen 2006).
4.3 Tandem massaspectrometrie (MS/MS)
Door een zachte ionisatietechniek zoals ESI te gebruiken, zal er weinig tot geen
fragmentatie van analytionen gebeuren. Soms is echter fragmentatie gewenst om
bijkomende structurele informatie van de analyten te achterhalen. Hiervoor kan tandem
massaspectrometrie als methode gebruikt worden.
Tandem massaspectrometrie (MS/MS) maakt gebruik van een combinatie van massa-
analysatoren (meestal twee): bijvoorbeeld een triple quadrupool (QqQ) of een quadrupool-
TOF combinatie (Q-TOF).
In QqQ worden drie quadrupolen in serie geplaatst. De eerste en derde quadrupool zijn
massa-analysatoren terwijl de tweede quadrupool enkel onder invloed is van een
radiofrequentie en gebruikt wordt als botsingscel. Deze middenste pool kan ook een
hexapool zijn: dit zijn zes parallele staven in plaats van vier. Nadeel van een QqQ is de
lage resolutie. Een Q-TOF kan hoge resolutie opleveren zoals in het addendum
geïllustreerd wordt (De Hoffmann E. 1996; Ardrey 2003).
4.4 MS/MS modes
De MS/MS kan in verschillende modes gebruikt worden:
- Product-ion scan
- Precursor-ion scan
- Constant neutral loss scan
- Selected reaction monitoring

Fundamentele aspecten van de massaspectrometrie
26
Product-ion scan
Bij deze mode, ook daughter ion mode genoemd, zal de eerste quadrupool één of
meerdere ionen selecteren op basis van een gedefinieerde m/z-verhouding. De
doorgelaten ionen zullen in de botsingscel (Collision Induced Dissociation of CID-cel)
gefragmenteerd worden door botsingen met een dragergas, bijvoorbeeld argon. De
tweede massa-analysator zal nu alle m/z-waarden (in het opgegeven massabereik)
scannen zodat de gevormde fragmenten van deze precursor-ionen in een spectrum
worden weergeven.
Precursor-ion scan
In de precursor-ion scan of parent ion mode zal de eerste quadrupool nu het totale
opgegeven massabereik scannen en doorsturen naar de CID-cel. De tweede massa-
analysator wordt ingesteld zodat slechts bepaalde fragmentionen van alle gevormde
fragementen in de botsingcel worden doorgelaten naar de detector.
Constant neutral loss scan
Sommige fragmentaties kunnen een herschikking van elektronen in de ionen teweeg
brengen zodanig dat een (neutrale) massa vrijkomt. Dit verlies van een neutrale massa
kan worden waargenomen voor alle ionen in de constant neutral loss mode. De twee
massa-analysatoren scannen nu m/z-waarden, waarbij een massaverschil wordt ingesteld
tussen beide (een off-set) zodat enkel wanneer dit massaverschil aanwezig is, een
signaal wordt gedetecteerd.
Selected/Multiple-reaction monitoring
Bij multiple reaction monitoring (MRM) zullen beide massa-analysatoren ingesteld worden
zodat enkel specifiek geselecteerde ionen doorgelaten worden. Beide analysatoren zullen
dus als massafilter werken. Deze mode wordt enkel gebruikt indien het analyt en de
eigenschappen, zoals de fragmentatie, van het analyt gekend zijn. Een voordeel van
gebruik van deze mode is de aanzienelijke verhoging van de gevoeligheid.

Ceramiden
27
5 Ceramiden
5.1 Structuur en voorkomen in de huid
Ceramiden vallen onder de klasse sfingolipiden en bestaan uit twee onderdelen, een
vetzuur en een lange amino-alcoholketen (sphingoid base), die covalent verbonden zijn
via een amidebinding. Voor beide onderdelen bestaan echter variaties in aantal OH-
groepen en aantal onverzadigde bindingen. Door deze variaties kunnen de ceramiden
verder opgedeeld worden in verschillende subklassen. De lengte van vetzuurketen en
amino-alcoholketen kan bij elke klasse eveneens variëren. Afhankelijk van de soort cel
waarin de ceramiden zich bevinden, zullen de aanwezigheid en hoeveelheid van de
soorten subklasses en de ketenlengtes binnenin elke subklasse verschillen. Ceramiden in
de meeste menselijke cellen en/of weefsels bevatten slechts 3 soorten klasses, namelijk
Cer[NS], Cer[NDS] en Cer[AS] (zie nomenclatuur 5.2.). Het stratum corneum (hoornlaag)
van de mens bevat daarentegen niet alleen meer klasses maar ook ceramiden met een
grotere variatie in ketenlengtes (Raith, Farwanah et al. 2004; Haynes, Allegood et al.
2009).
De opperste laag van de huid, ongeveer 10-40 µm dik, is de hoornlaag of stratum
corneum. De laag bevat geen levende materie meer en is een onderdeel van de
epidermis die een totale dikte heeft tussen 1.5-0.05 mm, afhankelijk van de locatie op het
lichaam. De lagen van het epidermis zijn van binnen naar buiten achtereenvolgens (figuur
11) het stratum basale, het stratum spinosum, het stratum granulosum, het stratum
lucidum en als buitenste laag het stratum corneum (Brannon 2007).
Figuur 11. Anatomie van de epidermis (Brannon 2007).

Ceramiden
28
Deze stratum corneum-laag bevat ceramiden die een belangrijke fysicochemische rol
spelen bij eigenschappen zoals bescherming tegen waterverlies of afscherming van
(schadelijke) externe omgeving. Daarenboven spelen ze ook een belangrijke rol in
signaaltransductie en celregulatie in de huid. Naast ceramiden bevat de hoornlaag andere
huidlipiden zoals vrije vetzuren en sterolen die samen met ceramiden intercellulaire
ruimtes opvullen tussen de keratinocytcellen en hierbij een lamellaire structuur aannemen
die parallel loopt met het oppervlak van de keratinocyten (Elias 1983; Brooks and Idson
1991).
Het diepteprofiel van de lipidensamenstelling in de epidermis varieert, aangezien de
aanwezigheid van de lipiden in de hoornlaag een gevolg is van de opeenvolgende
processen die doorgaan in dieperliggende epidermislagen. Eerst worden precursorlipiden
gesynthetiseerd in het stratum basale, stratum spinosum en stratum granulosum en
worden overgedragen naar lamellaire granules (keratinosomen) waarin lipiden gevormd
worden; daarna worden de lipiden opnieuw afgescheiden door de keratinosomen. De
lipiden worden omgevormd om uiteindelijk de lamellaire structuur aan te nemen in het
stratum corneum. Bij de overgang naar het stratum corneum ondergaan de lipiden
metabolisch een wijziging waarbij fosfolipiden gedegradeerd worden tot glycerol en vrije
vetzuren en glucosfingolipiden omgezet worden tot ceramides (Coderch, Lopez et al.
2003).
5.2 Overzicht van onderzoek van ceramiden in de huid
Aanvankelijk werden ceramide-klasses opgedeeld op basis van de retentie op een TLC-
plaat waarbij een hoger nummer (hogere retentie) een grotere polariteit betekent van de
klasse. Gaandeweg het onderzoek van ceramiden kon dankzij ondermeer vooruitgang bij
scheidingsmethodes en het daardoor groter inzicht in de structuren van de ceramiden,
een meer specifieke en systematische manier gevonden worden om de verscheidene
ceramiden te classificeren.
Een duidelijk overzicht van de subklasses werd gegeven door Motta (Motta, Monti et al.
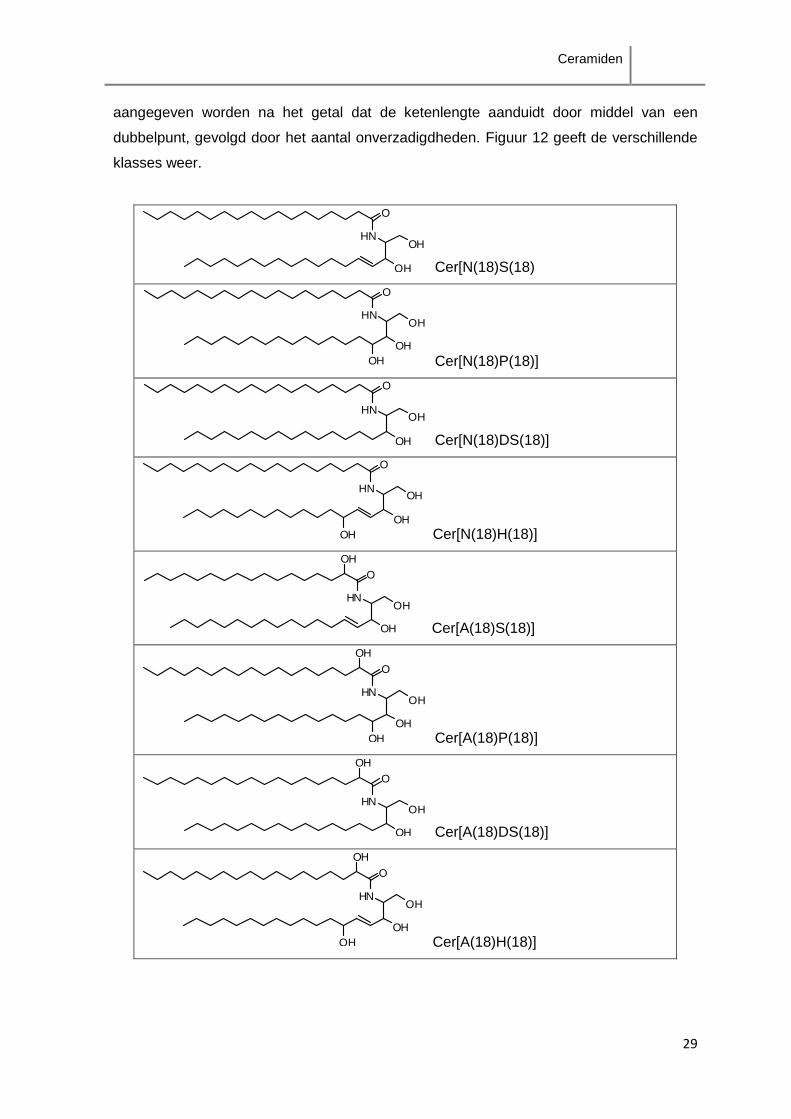
1993) via een systematische naamgeving volgens de gekoppelde delen. Deze
nomenclatuur geeft combinaties van de vetzuurgroep: nonhydroxy-vetzuur (N), alfa-
hydroxy-vetzuur (A), omega-hydroxy-vetzuren (O) veresterd aan een ander vetzuur (E),
met de amino-alcoholketen: sfingosine (S), fytosfingosine (P) dihydrosfingosine (DS) en 6-
hydroxy-sfingosine (H); die via combinatie van de letters het ceramide aangeven, waarbij
de letter gevolgd kan worden door een nummer die de ketenlengte van vetzuur/amino-
alcoholketen aangeeft. Aanwezigheid van onverzadigde bindingen bij N, A, E en O kan
Ceramiden
29
aangegeven worden na het getal dat de ketenlengte aanduidt door middel van een
dubbelpunt, gevolgd door het aantal onverzadigdheden. Figuur 12 geeft de verschillende
klasses weer.
HNOH
OH
O
Cer[N(18)S(18)
HNOH
OH
OH
O
Cer[N(18)P(18)]
HNOH
OH
O
Cer[N(18)DS(18)]
HNOH
OH
O
OH Cer[N(18)H(18)]
HNOH
OH
O
OH
Cer[A(18)S(18)]
HNOH
OH
O
OH
OH Cer[A(18)P(18)]
HNOH
OH
O
OH
Cer[A(18)DS(18)]
HNOH
OH
O
OH
OH Cer[A(18)H(18)]

Ceramiden
30
HNOH
OH
O
OH
OO
Cer[E(18:2)O(18)S(18)]
HNOH
OH
OH
OH
OO
O
Cer[E(18:2)O(18)P(18)]
HNOH
OH
O
OH
OO
Cer[E(18:2)O(18)DS(18)]
HNOH
OH
OH
O
OH
OO
Cer[E(18:2)O(18)H(18)]
Figuur 12. Overzicht van ceramidenklassen.
In de beginjaren van ceramide-onderzoek werden voornamelijk dunlaagchromatografie en
gaschromatografie als scheidingsmethodes gebruikt (Casparrini 1969; Gray and Yardley
1975; Wertz, Miethke et al. 1985). Beide methodes vertonen echter beperkingen: TLC
bood enkel een idee over de polariteit van de ceramiden en slechts weinig tot geen
bijkomende informatie over de diverse structuren, terwijl GC hydrolyse en derivatisatie
vereiste van de componenten en daarbij informatie kon geven over de structuur van zowel
het vetzuur-gedeelte als de amino-alcoholketen maar geen informatie gaf over welke
combinaties van de ketens aan elkaar gekoppeld waren. Daarenboven was scheiding via
een niet-geautomatiseerde en dus weinig reproduceerbare methode zoals TLC moeilijk
om een complex mengsel van ceramiden, zoals in de huid, te analyseren waarin de
structuren nauw verwant zijn. Automatisering van TLC (Automated Multiple Development-
High Performance Thin Layer Chromatography: AMD-HPTLC) kon meer nauwkeurige
resultaten geven.

Ceramiden
31
Ook koppelen van GC na (geautomatiseerde) TLC leidde tot een meer volledige analyse
(Bonte, Saunois et al. 1997; Farwanah, Neubert et al. 2002; Raith, Farwanah et al. 2004;
Farwanah, Raith et al. 2005).
Aanvankelijk werden bij TLC-analyses van ceramiden in de huid zeven klasses
toegekend, corresponderend met zeven spots (Imokawa, Abe et al. 1991). Onderzoek
door zowel Robson en Vietzke wees echter uit dat deze zeven spots overeenkomen met
negen ceramide-klasses vermits twee van de zeven spots niet één maar twee klasses
omvatten. Vijf spots komen overeen met de klasses Cer[NP], Cer[NH], Cer[AP], Cer[AH]
en Cer[EOS]. De overige twee spots omvatten elk twee niet gescheiden klasses, namelijk
Cer[NDS] en Cer[NS] ,en Cer[AS] en Cer[EOH] (Robson, Stewart et al. 1994; Vietzke,
Brandt et al. 2001). Een ander klasse, Cer[EOP], werd door Ponec weergevonden op
spoor-niveau (Ponec, Weerheim et al. 2003). Een recentste toevoeging van de klasse
Cer[ADS] in de hoornlaag werd geïdentificeerd door Masukawa (Masukawa, Narita et al.
2008). Daarnaast werden ook reeds twee klasses die covalent gebonden zijn aan
proteïnen aan het licht gebracht (Cer[OS] en Cer[OH]) (Wertz, Madison et al. 1989).
Bij HPLC-analyse werden in het verleden reeds combinaties van zowel normaalfase als
omkeerfase modes met verschillende detectorsystemen zoals UV-detectie, fluorescentie-
detectie, evaporatie-lichtverstrooiingsdetectie (Evaporative Light Scattering Detector:
ELSD) gebruikt. UV- en fluorescentie-detectie vereisten beiden derivatisatie net zoals bij
GC, terwijl dit bij ELSD niet het geval was.
Ontwikkeling van ESI-MS gekoppeld na een LC-systeem zorgde ervoor dat onmiddelijke
online analyse van ceramiden mogelijk werd. Daarnaast bood HPLC-MS zowel hoge
gevoeligheid als selectiviteit, waardoor deze analysemethode het laatste decennium de
meest gebruikte en meest verkozen techniek is geworden om ceramiden te analyseren
(Vietzke, Strassner et al. 1999; Raith, Farwanah et al. 2004; Masukawa, Narita et al.
2008). Omkeerfase HPLC scheidt ceramiden voornamelijk op basis van ketenlengtes van
vetzuur en amino-alcoholketen. Normaalfase HPLC doet dit op basis van aantal
aanwezige en positie van polaire OH-groepen.

Experimenteel gedeelte
32
6 Experimenteel gedeelte
6.1 Algemene aspecten
6.1.1 Chemicaliën
De solventen isopropanol, chloroform en dicholoromethaan zijn afkomstig van Sigma
Aldrich, water en methanol van Biosolve en hexaan van Fisher Scientific. Alle
bovenvermelde solventen zijn van LC-MS-kwaliteit. Mierenzuur afkomstig van Biosolve,
ammoniumformiaat en azijnzuur afkomstig van Sigma Aldrich werden gebruikt. Van de
ceramiden Cer[N(24)S(18)] en Cer[N(24)P(18)] afkomsting van Larodan en ceramide II
(samengesteld uit Cer[A(18)S(18)], Cer[A(22:0)S(18)], Cer[A(22:2)S(18)],
Cer[A(23:0)S(18)], Cer[A(23:2)S(18)], Cer[A(24:0)S(18)], Cer[A(24:1)S(18)],
Cer[A(25:0)S(18)], Cer[A(25:2)S(18)], Cer[A(26:0)S(18)] en Cer[A(26:1)S(18)]) afkomstig
van Sigma Aldrich, werden stockoplossingen gemaakt in isopropanol-dichloromethaan
(1:1) van respectievelijk 2.5 mg/mL, 2 mg/mL en 5 mg/mL. De concentratie van de
stockoplossingen voor glycerol trioleaat opgelost in dichloromethaan en cholesteryl oleaat
opgelost in hexaan, bedragen respectievelijk 4 mg/mL en 10 mg/mL. Beiden zijn
afkomstig van Sigma Aldrich. Alle standaardoplossingen werden bewaard bij - 4°C.
6.1.2 Instrumentatie
De analyses werden uitgevoerd op een Alliance HPLC-systeem (Waters). Detectie
gebeurde met een Micromass Quattro Micro API LC-MS/MS-systeem (Waters) en een
Corona Charged Aerosol Detector (ESA). Dataverwerking werd uitgevoerd met Masslynx
MS software (Waters) en PeakSimple software (SRI Instruments).
6.2 Ontwikkeling en optimalisatie van een NPLC-methode voor analyse
van ceramiden
Een geschikte NPLC-methode werd gezocht voor de analyse van ceramiden, waarbij
detectie werd uitgevoerd met een Corona Charged Aerosol Detector (ESA). Aangezien
het scheidingsmechanisme bij normaalfase gebaseerd is op polariteit van ceramiden via
aantal en locatie op de koolstofketen van OH-groepen, werd in dit opzicht gepoogd de
ceramidenklasses te scheiden en te isoleren via een fractiecollector.
Enkele methodes met een Zorbax RX-SIL kolom (10 cm L x 3 mm I.D. x 1.8 µm dp;
Agilent Technologies) en verschillende solventen werden getest. Mobiele fase A hexaan-
isopropanol-mierenzuur (95:5:0.1) en mobiele fase B hexaan-isopropanol-50 mM
ammonium formiaat (aq.) (25:65:10) gebruikt door Masukawa (Masukawa, Narita et al.

Experimenteel gedeelte
33
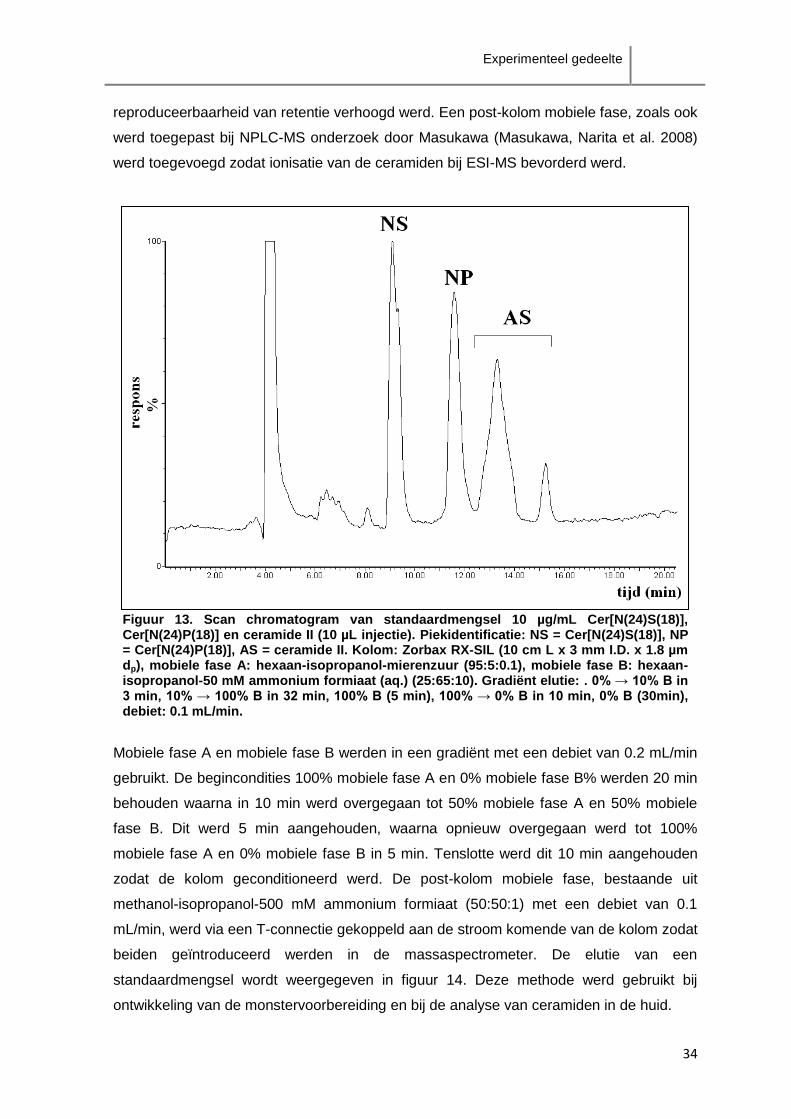
2008) werden in gradiënt elutie bij een debiet van 0.3 mL/min uitgevoerd. In 3 min werd
van de initiële condities 100% mobiele fase A en 0% mobiele fase B lineair overgegaan
naar 90% mobiele fase A en 10% mobiele fase B. Hierna werd overgegaan tot 0%
mobiele fase A en 100% mobiele fase B in 32 min. Deze samenstelling werd 5 min
aangehouden waarna in 10 min opnieuw de begincondities bereikt werden. De
begincondities werden aangehouden gedurende 30 min zodat de kolom voldoende
geconditioneerd werd voor de volgende analyse. Injecties (10 µL) van individuele
standaardoplossingen van 250 µg/mL Cer[N(24)P(18)], Cer[N(24)S(18)] en ceramide II
werden uitgevoerd ter identificatie van de pieken. Daarna werd injectie (10 µL) van een
standaardmengsel van 250 µg/mL Cer[N(24)P(18)], Cer[N(24)S(18)] en ceramide II
uitgevoerd.
Vermits de retentie van de standaarden in het mengsel niet volledig reproduceerbaar was,
waarbij voornamelijk de aanwezigheid van water in mobiele fase B een grote invloed
hierop had, werd een aanpassing ingevoerd. Hierdoor kon de benodigde tijd voor de
kolomconditionering ingekort worden. Mobiele fase B werd gewijzigd tot hexaan-
isopropanol-500 mM ammoniuim formiaat (aq.) (50:50:1). Verschillende gradiënten
werden getest bij een standaardmengsel-injectie (10 µL) van 250 µg/mL Cer[N(24)P(18)],
Cer[N(24)S(18)] en ceramide II. Ook de snelheid van de mobiele fase werd gewijzigd. Een
debiet van 0.1 mL/min gaf problemen voor stabiliteit van de druk, een debiet van 0.2
mL/min bleek wel mogelijk te zijn.
Hierna werd overgegaan tot ontwikkeling van de methode met een Alliance HPLC-
systeem (Waters) gekoppeld aan een Micromass Quattro Micro API LC-MS/MS-systeem
(Waters). De massaspectrometer is in de eerste plaats veel gevoeliger dan een corona-
ontladingsdetector, zodat injectie van 10 µL 10 µg/mL standaardmengsel volstond,
daarenboven wordt structurele informatie verkregen via de massaspectra. De
karakterisatie van de ceramiden wordt beschreven in 6.3. De MS-parameters zoals
capillaire spanning en kegelspanning werden geoptimaliseerd zodat de gewenste ionisatie
plaatsvond. Een capillaire spanning van 3.0 kV en een kegelspanning van 20 V werden
optimaal bevonden bij positieve ionisatie. Bij de ontwikkeling van NPLC-ESI-MS werd ook
hier eerst de methode ontwikkeld door Masukawa getest, ditmaal werd het debiet
ingesteld op 0.1 mL/min, ditmaal zonder problemen met stabiliteit van de druk. Figuur 13
geeft de analyse weer van het standaardmengsel met deze methode.
Vermits de nodige tijd voor conditionering van de kolom het grote nadeel is van deze
methode, werd hierna een andere mobiele fase B gezocht. De finale mobiele fase B heeft
een samenstelling van isopropanol-dichloromethaan-hexaan-mierenzuur (25:25:50:0.05).
De waterige ammonium formiaat-oplossing werd uit deze fase verwijderd zodat de
Experimenteel gedeelte
34
reproduceerbaarheid van retentie verhoogd werd. Een post-kolom mobiele fase, zoals ook
werd toegepast bij NPLC-MS onderzoek door Masukawa (Masukawa, Narita et al. 2008)
werd toegevoegd zodat ionisatie van de ceramiden bij ESI-MS bevorderd werd.
Figuur 13. Scan chromatogram van standaardmengsel 10 µg/mL Cer[N(24)S(18)], Cer[N(24)P(18)] en ceramide II (10 µL injectie). Piekidentificatie: NS = Cer[N(24)S(18)], NP = Cer[N(24)P(18)], AS = ceramide II. Kolom: Zorbax RX-SIL (10 cm L x 3 mm I.D. x 1.8 µm dp), mobiele fase A: hexaan-isopropanol-mierenzuur (95:5:0.1), mobiele fase B: hexaan-isopropanol-50 mM ammonium formiaat (aq.) (25:65:10). Gradiënt elutie: . 0% → 10% B in 3 min, 10% → 100% B in 32 min, 100% B (5 min), 100% → 0% B in 10 min, 0% B (30min), debiet: 0.1 mL/min.
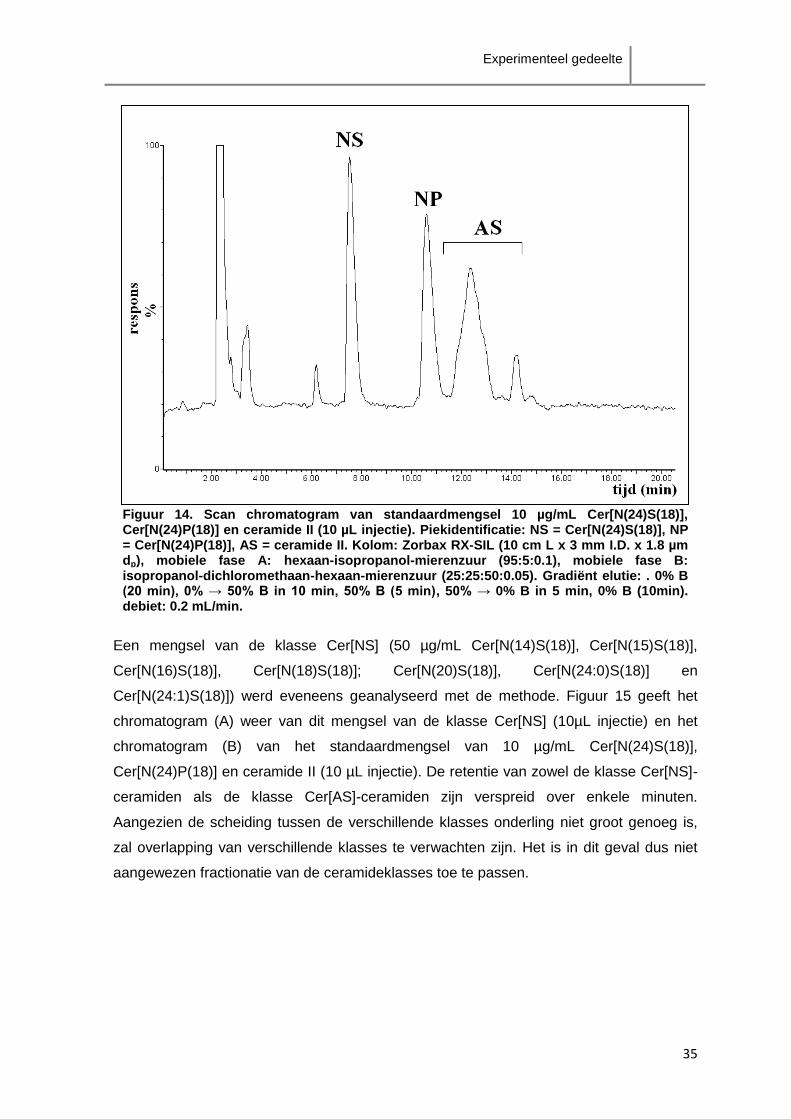
Mobiele fase A en mobiele fase B werden in een gradiënt met een debiet van 0.2 mL/min
gebruikt. De begincondities 100% mobiele fase A en 0% mobiele fase B% werden 20 min
behouden waarna in 10 min werd overgegaan tot 50% mobiele fase A en 50% mobiele
fase B. Dit werd 5 min aangehouden, waarna opnieuw overgegaan werd tot 100%
mobiele fase A en 0% mobiele fase B in 5 min. Tenslotte werd dit 10 min aangehouden
zodat de kolom geconditioneerd werd. De post-kolom mobiele fase, bestaande uit
methanol-isopropanol-500 mM ammonium formiaat (50:50:1) met een debiet van 0.1
mL/min, werd via een T-connectie gekoppeld aan de stroom komende van de kolom zodat
beiden geïntroduceerd werden in de massaspectrometer. De elutie van een
standaardmengsel wordt weergegeven in figuur 14. Deze methode werd gebruikt bij
ontwikkeling van de monstervoorbereiding en bij de analyse van ceramiden in de huid.
Experimenteel gedeelte
35
Figuur 14. Scan chromatogram van standaardmengsel 10 µg/mL Cer[N(24)S(18)], Cer[N(24)P(18)] en ceramide II (10 µL injectie). Piekidentificatie: NS = Cer[N(24)S(18)], NP = Cer[N(24)P(18)], AS = ceramide II. Kolom: Zorbax RX-SIL (10 cm L x 3 mm I.D. x 1.8 µm dp), mobiele fase A: hexaan-isopropanol-mierenzuur (95:5:0.1), mobiele fase B: isopropanol-dichloromethaan-hexaan-mierenzuur (25:25:50:0.05). Gradiënt elutie: . 0% B (20 min), 0% → 50% B in 10 min, 50% B (5 min), 50% → 0% B in 5 min, 0% B (10min). debiet: 0.2 mL/min.
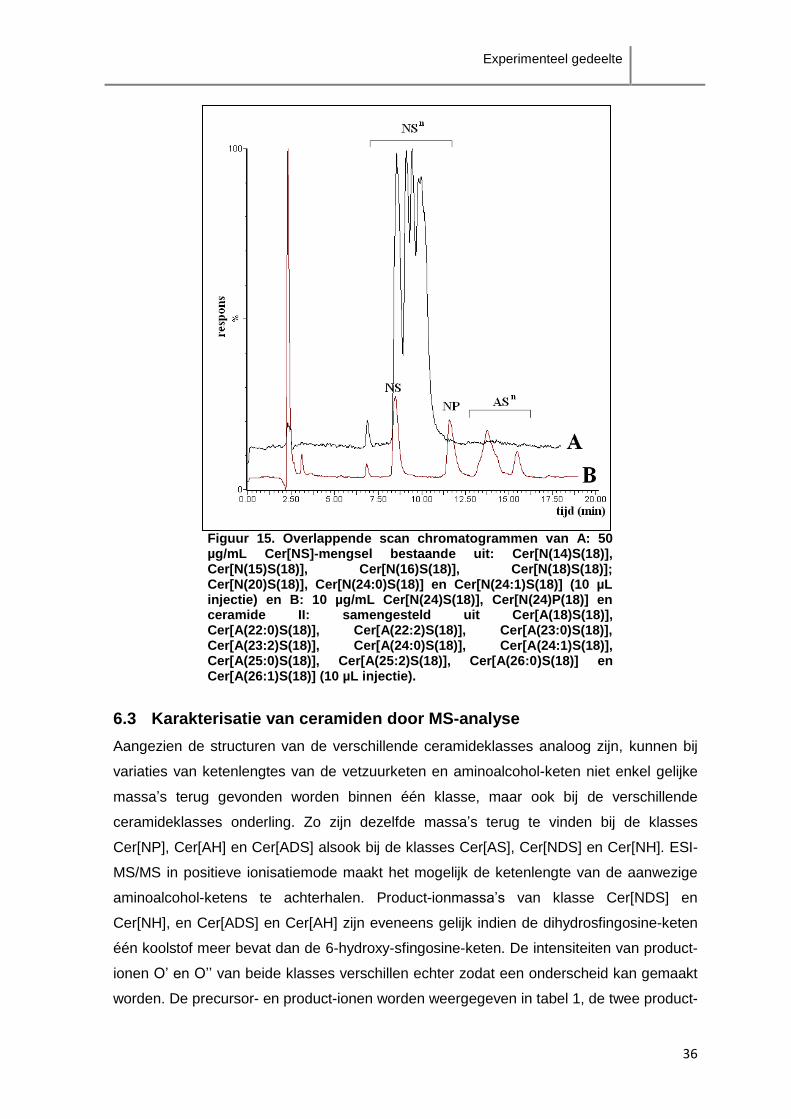
Een mengsel van de klasse Cer[NS] (50 µg/mL Cer[N(14)S(18)], Cer[N(15)S(18)],
Cer[N(16)S(18)], Cer[N(18)S(18)]; Cer[N(20)S(18)], Cer[N(24:0)S(18)] en
Cer[N(24:1)S(18)]) werd eveneens geanalyseerd met de methode. Figuur 15 geeft het
chromatogram (A) weer van dit mengsel van de klasse Cer[NS] (10µL injectie) en het
chromatogram (B) van het standaardmengsel van 10 µg/mL Cer[N(24)S(18)],
Cer[N(24)P(18)] en ceramide II (10 µL injectie). De retentie van zowel de klasse Cer[NS]-
ceramiden als de klasse Cer[AS]-ceramiden zijn verspreid over enkele minuten.
Aangezien de scheiding tussen de verschillende klasses onderling niet groot genoeg is,
zal overlapping van verschillende klasses te verwachten zijn. Het is in dit geval dus niet
aangewezen fractionatie van de ceramideklasses toe te passen.
Experimenteel gedeelte
36
Figuur 15. Overlappende scan chromatogrammen van A: 50 µg/mL Cer[NS]-mengsel bestaande uit: Cer[N(14)S(18)], Cer[N(15)S(18)], Cer[N(16)S(18)], Cer[N(18)S(18)]; Cer[N(20)S(18)], Cer[N(24:0)S(18)] en Cer[N(24:1)S(18)] (10 µL injectie) en B: 10 µg/mL Cer[N(24)S(18)], Cer[N(24)P(18)] en ceramide II: samengesteld uit Cer[A(18)S(18)], Cer[A(22:0)S(18)], Cer[A(22:2)S(18)], Cer[A(23:0)S(18)], Cer[A(23:2)S(18)], Cer[A(24:0)S(18)], Cer[A(24:1)S(18)], Cer[A(25:0)S(18)], Cer[A(25:2)S(18)], Cer[A(26:0)S(18)] en Cer[A(26:1)S(18)] (10 µL injectie).
6.3 Karakterisatie van ceramiden door MS-analyse
Aangezien de structuren van de verschillende ceramideklasses analoog zijn, kunnen bij
variaties van ketenlengtes van de vetzuurketen en aminoalcohol-keten niet enkel gelijke
massa’s terug gevonden worden binnen één klasse, maar ook bij de verschillende
ceramideklasses onderling. Zo zijn dezelfde massa’s terug te vinden bij de klasses
Cer[NP], Cer[AH] en Cer[ADS] alsook bij de klasses Cer[AS], Cer[NDS] en Cer[NH]. ESI-
MS/MS in positieve ionisatiemode maakt het mogelijk de ketenlengte van de aanwezige
aminoalcohol-ketens te achterhalen. Product-ionmassa’s van klasse Cer[NDS] en
Cer[NH], en Cer[ADS] en Cer[AH] zijn eveneens gelijk indien de dihydrosfingosine-keten
één koolstof meer bevat dan de 6-hydroxy-sfingosine-keten. De intensiteiten van product-
ionen O’ en O’’ van beide klasses verschillen echter zodat een onderscheid kan gemaakt
worden. De precursor- en product-ionen worden weergegeven in tabel 1, de twee product-

Experimenteel gedeelte
37
ionen O en O’’’, respectievelijk [M+H-RvetzCO]+ en [M+H-RvetzCO-3H2O]+, worden niet
weergegeven in de tabel.
Tabel 1. Positieve precursor- en productionen van de ceramideklasses.
klasse precursor-ion product-ion product-ion O' product-ion O''
Cer[NS] [M+H]+ > [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ <
[M+H-RvetzCO-2H2O]
+
Cer[NP] [M+H]+ > [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ < [M+H-RvetzCO-2H2O]
+
Cer[NDS] [M+H]+ > [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ <
* [M+H-RvetzCO-2H2O]+
Cer[NH] [M+H]+ < [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ >
* [M+H-RvetzCO-2H2O]
+
Cer[AS] [M+H]+ > [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ < [M+H-RvetzCO-2H2O]
+
Cer[AP] [M+H]+ > [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ < [M+H-RvetzCO-2H2O]
+
Cer[ADS] [M+H]+ > [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ > [M+H-RvetzCO-2H2O]
+
Cer[AH] [M+H]+ < [M+H-H2O]
+ [M+H-RvetzCO-H2O]
+ <
[M+H-RvetzCO-2H2O]
+
*: verschilt van het onderzoek door Masukawa (Masukawa, Narita et al.2009)
Gezien de uitgebreide dataverwerking werden de ω-estergelinkte ceramiden Cer[EOS],
Cer[EOP] en Cer[EOH] en de weinig abundante klasse Cer[AP] niet geanalyseerd in dit
werk (Masukawa, Narita et al. 2009). Figuur 16. toont de spectra van de standaard
Cer[N(24)S(18)] respectievelijk in de scan mode (A) en in de product-ion scan mode (B).
Figuur 16. A: scan mode spectrum van standaard Cer[N(24)S(18)].

Experimenteel gedeelte
38
Figuur 16. B: product-ion scan mode spectrum van standaard Cer[N(24)S(18)].
6.4 Monstervoorbereiding
Het voorbereidend werk van een monster is vaak de belangrijkste maar ook meest
tijdrovende stap van de analyse. Monstervoorbereiding voor analyse van
hoornlaagceramiden kan opgedeeld worden in twee stappen, extractie uit de huid via
tape-stripping en zuiveren van matrixeffecten via vaste fase extractie.
6.4.1 Tape stripping-techniek en tape-extractie
Aanrijken van de ceramiden uit de huid is één van de cruciaalste stappen. Verschillende
in vivo procedures zijn reeds ontwikkeld waaronder oppervlak-extractie met organische
solventen zoals chloroform en methanol. (Imokawa and Hattori 1985; Bonte, Pinguet et al.
1995; Lavrijsen, Bouwstra et al. 1995; Farwanah, Neubert et al. 2002). Nadeel van deze
methode is de mogelijk permanente schade aangericht aan de huid. Bekomen van
huidcellen via het schrapen van de huid is een tweede methode (Lavrijsen, Higounenc et
al. 1994; Gildenast and Lasch 1997; Farwanah, Wohlrab et al. 2005). Een derde methode
bestaat uit collectie van een dunne laag stratum corneum via een glasplaat bedekt met
een cyanoacrylaatpolymeer (Marks and Dawber 1971; Imokawa, Abe et al. 1991; Vietzke,
Strassner et al. 1999). Een vierde en laatste methode is het strippen van de huid via een
tape. Deze methode werd in dit werk gebruikt (Weerheim and Ponec 2001; Masukawa,
Narita et al. 2008).
Hoornlaag-extract werd gecollecteerd door aanbrengen van een tape (Corneofix) van 20 x
20 mm op een specifieke regio van de voorarm. Daarna werd druk uitgeoefend op deze
plaats met behulp van een rol die 10 maal heen en weer werd bewogen. De tape werd
vervolgens in één snelle beweging manueel van de huid verwijderd. Een tweede tape
werd op dezelfde regio aangebracht en werd in de richting tegengesteld aan de richting

Experimenteel gedeelte
39
van de eerste tape-stripping verwijderd, ofwel van pols naar elleboog toe, ofwel van
elleboog naar pols toe (Tassopoulos 2006). De procedure wordt aan de hand van de foto
in figuur 17 (A) geïllustreerd, samen met de tape vóór (B) en na aanbrengen op de huid
(C).
Figuur 17. A: principe van tape aanbrengen op de huid (Tassopoulos 2006), B: tape vóór aanbrengen op de huid en C: na aanbrengen op de huid (B).
In het totaal werden tien tapes verzameld. Extractie van de tapes werd getest via een
aantal extractiesolventen, namelijk methanol, isopropanol, dichloromethaan:methanol
(2:1), dichloromethaan-isopropanol (1:1). Methanol bleek het beste extractiesolvent voor
de tapes. Verzamelde extracten in isopropanol en vooral in dichloromethaan:methanol
(2:1) en in dichloromethaan-isopropanol (1:1) vertoonden na verdampen een kleverige
residu afkomstig van de tape. Heroplossen na verdampen en ook verdere analyse werd
hierdoor bemoeilijkt, onder andere voor de SPE-procedure resulteerde dit in een zeer

Experimenteel gedeelte
40
trage elutie na laden van de monsters. Ook meer dan één tape-extractie uitvoeren leidde
tot loskomen van polymeermateriaal van de tape. Opdat de interferenties van de tape zo
laag mogelijk werden gehouden, werd enkel de eerste tape-extractie uitgevoerd.
De volgende methode werd gebruikt:
- onderdompelen van individuele tapes in 3 mL methanol.
- 10 min sonicatie
- collecteren van extract per 2 tapes
- verdampen bij 90°C
- 2 maal wassen van residu’s met 150 µL dichloromethaan-isopropanol (1:1)
- wasvolumes van alle 10 tapes verzamelen en opnieuw verdampen bij 90°C
- oplossen in 120 µL hexaan-dichloromethaan-isopropanol (100:10:10)
Hierna werd de SPE-procedure, besproken in 6.4.2., toegepast op het extract.
6.4.2 Vaste fase extractie
Vaste fase extractie (solid phase extraction of SPE) is een veel gebruikte
monstervoorbewerkingsmethode voor HPLC. SPE wordt ondermeer toegepast voor
concentreren van een analyt, voor verwijderen van interferenties of verwijderen van
componenten die mogelijks schade aan de kolom aanrichten en voor solventuitwisseling.
Het principe van vaste fase extractie is gelijk aan dat van vloeistofchromatografie.
Componenten vertonen een zekere affiniteit tegenover de stationaire fase (het bed van de
cartridge) en de mobiele fase. Bij SPE wordt enkel isocratische elutie gebruikt. Er kan
gekozen worden om ofwel de interferenties vast te houden op de cartridge en de analyten
die weinig tot geen interactie met de stationaire fase vertonen onmiddellijk te elueren,
ofwel worden eerst interferenties geëlueerd en de analyten op de cartridge gehouden. Na
verwijderen van de interferenties kunnen de analyten geëlueerd worden met een geschikt
solvent. In SPE wordt meestal een kleine (enkele cm’s) uit polymeer vervaardigde kolom
(cartridge) voorzien van een gepakt bed. Elutie gebeurt ofwel onder invloed van de
zwaartekracht ofwel onder vacuüm. Een SPE-methode bestaat in de meeste gevallen uit
vier verschillende stappen: een conditioneringsstap die het sorbent op de cartridge reinigt
en activeert, een laadstap waarbij het monster aangebracht wordt (loading), een wasstap
die interferenties wegspoelt met een geschikt solvent en een elutiestap van de analyten
met een geschikt solvent (figuur 18).
Voordelen ten opzichte van vloeistof-vloeistofextractie zijn onder andere: vlot en
eenvoudig verzamelen van een analytfractie, reductie van het gebruikte organisch

Experimenteel gedeelte
41
solvent, verwijderen van vaste deeltjes, efficiëntere scheiding van interferenties en
volledige extractie van analyten. Daarentegen bevat deze techniek ook enkele nadelige
eigenschappen zoals niet uniform gepakte cartridges, nood aan een (tijdsrovende)
ontwikkeling van de SPE-methode en een soms irreversibele retentie van analyten op de
cartridge (Dolan, Kirkland et al. 2010).
Figuur 18. SPE-procedure (Dolan, Kirkland et al. 2010).
Zoals reeds vermeld, heeft de huid een complexe samenstelling. Indien ceramiden
geanalyseerd wensen te worden, kunnen talrijke andere componenten interferenties
vormen bij analyse. Na de in vivo extractie uit de huid (zie 6.4.1.), zal via vaste fase
extractie gepoogd worden de ceramiden zoveel mogelijk te isoleren van deze mogelijke
interferenties. Er moet eveneens rekening gehouden worden met interferenties afkomstig
van de tape.
Verificatie van de SPE-methode werd hierbij eerst toegepast op een standaardmengsel
van 200 µg/mL Cer[N(24)P(18)], Cer[N(24)S(18)], ceramide II, glycerol trioleaat en
cholesteryl oleaat, waarvan 10 µL opgelost werd in 100 µL hexaan. De verschillende
stappen werden in fracties gecollecteerd en geanalyseerd. Hierna werd ook extract van
tapes die gespiked werden met 5µL van het 200 µg/mL standaarmengsel geanalyseerd.
Elutie gebeurde telkens onder invloed van de zwaartekracht.
De analyse gebeurde met de NPLC-MS methode, besproken in 6.4., in de scan mode en
in de MRM mode. Enkel Cer[A(24)S(18)], analoog aan Cer[N(24)S(18)] en
Cer[N(24)P(18)], werd beschouwd in ceramide II bij de analyse. Bij Cer[N(24)S(18)] werd

Experimenteel gedeelte
42
in MRM mode de massatransitie m/z 650.9 → 263.9 beschouwd, bij Cer[N(24)P(18)] de
transitie 669 → 282 en bij Cer[A(24)S(18)] de transitie 667 → 264. Twee bijkomende
standaarden glycerol trioleaat en cholesteryl oleaat, mogelijke interferenties aanwezig in
de huid, werden toegevoegd aan de oplossing. De transities 903 → 603 en 669 → 369
werden gevolgd respectievelijk voor glycerol trioleaat en cholesteryl oleaat.
Verschillende types cartridges en methodes werden getest. De methode gebruikt door
Masukawa (Masukawa, Narita et al. 2008) werd getest met drie cartridgetypes van 100
mg/1 mL, namelijk een silica-cartridge (Sigma Aldrich), een diol-cartridge (Sigma Aldrich)
en een aminopropyl-cartridge (Alltech). Het protocol houdt in totaal vijf stappen in:
- conditionering: 10 mL chloroform-methanol (99.5:0.5)
- laden van de standaardoplossing
- wassen met 10 mL chloroform-methanol (99.5:0.5)
- elutie met 10 mL chloroform-methanol (99:5)
- een additionele elutie met 10 mL chloroform-methanol (99:5) als controlestap
De 3 laatste stappen werden respectievelijk als fractie 1, 2 en 3 gecollecteerd, verdampt
in een oven (90°C) en terug opgelost in 360 µL hexaan-isopropanol-dichloromethaan
(100:10:10). Uit de analyse bleek dat bij de diol-cartridge zowel Cer[N(24)P(18)] als
Cer[N(24)S(18)] reeds volledig geëlueerd werden bij de wasstap (fractie 1).
Cer[A(24)S(18)] werd teruggevonden zowel in fractie 1 als in fractie 2. Bij de aminopropyl-
cartridge werden Cer[N(24)P(18)] en Cer[A(24)S(18)] enkel in fractie 2 teruggevonden
maar werd Cer[N(24)S(18)] geëlueerd in fractie 1 en 2. De silica-cartridge gaf een
analoog resultaat: Cer[N(24)S(18)] werd eveneens teruggevonden in fractie 1 en 2. Geen
van de drie gebruikte cartridge-types vertoonde het gewenste resultaat, namelijk alle
ceramide-klasses in de elutiestap (fractie 2) elueren.
Andere aanpakken werden getest, waarbij zowel verschillende cartridgetypes als
solventen gebruikt werden. Hierbij bleek voornamelijk, net zoals bij HPLC in normaalfase
mode, voldoende conditionering van de SPE-cartridge van belang te zijn. De beste
resultaten werden bekomen via volgende methode met een 100 mg/1 mL aminopropyl-
cartridge (Alltech):
- conditionering met 20 mL hexaan-0.5% azijnzuur
- laden van standaardoplossing
- wassen met 1.5 mL hexaan
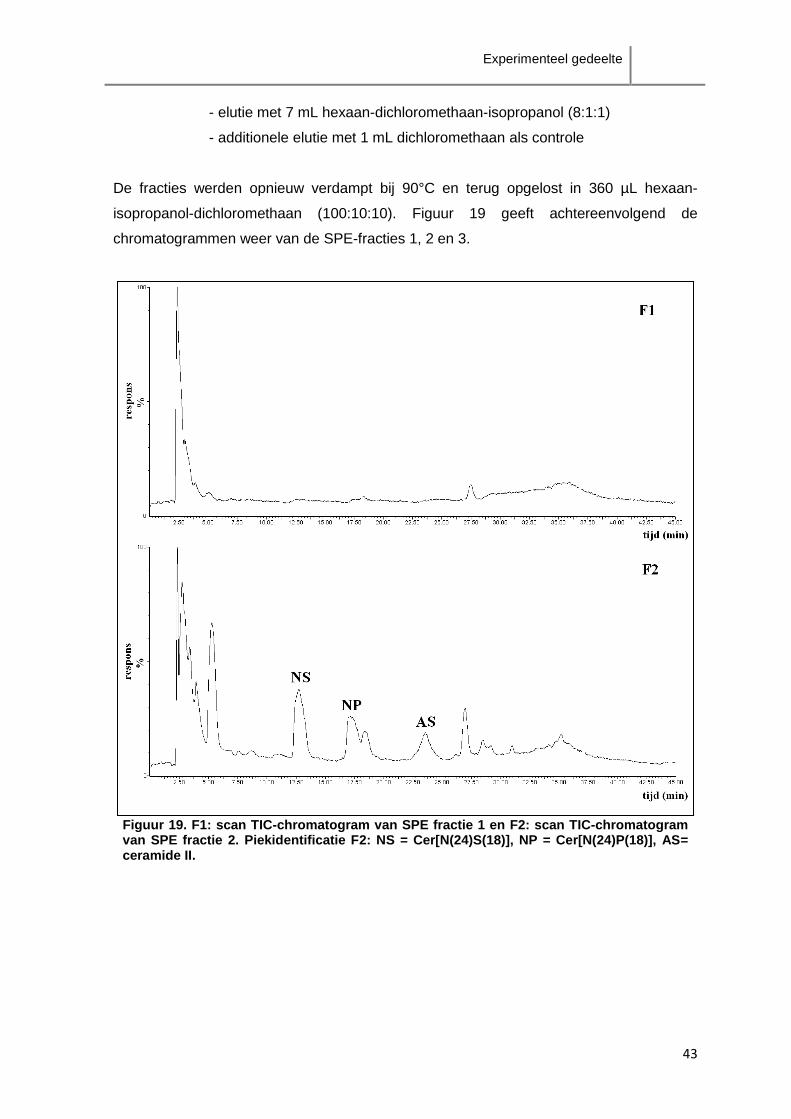
Experimenteel gedeelte
43
- elutie met 7 mL hexaan-dichloromethaan-isopropanol (8:1:1)
- additionele elutie met 1 mL dichloromethaan als controle
De fracties werden opnieuw verdampt bij 90°C en terug opgelost in 360 µL hexaan-
isopropanol-dichloromethaan (100:10:10). Figuur 19 geeft achtereenvolgend de
chromatogrammen weer van de SPE-fracties 1, 2 en 3.
Figuur 19. F1: scan TIC-chromatogram van SPE fractie 1 en F2: scan TIC-chromatogram van SPE fractie 2. Piekidentificatie F2: NS = Cer[N(24)S(18)], NP = Cer[N(24)P(18)], AS= ceramide II.
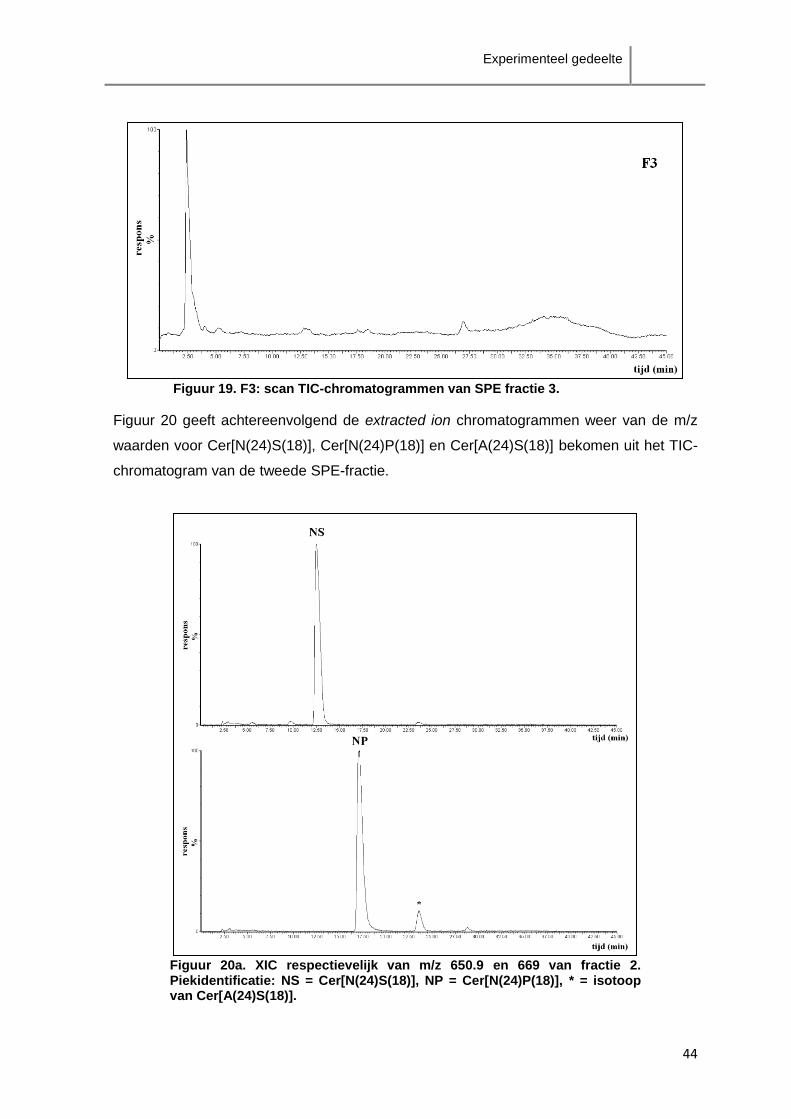
Experimenteel gedeelte
44
Figuur 19. F3: scan TIC-chromatogrammen van SPE fractie 3.
Figuur 20 geeft achtereenvolgend de extracted ion chromatogrammen weer van de m/z
waarden voor Cer[N(24)S(18)], Cer[N(24)P(18)] en Cer[A(24)S(18)] bekomen uit het TIC-
chromatogram van de tweede SPE-fractie.
Figuur 20a. XIC respectievelijk van m/z 650.9 en 669 van fractie 2. Piekidentificatie: NS = Cer[N(24)S(18)], NP = Cer[N(24)P(18)], * = isotoop van Cer[A(24)S(18)].

Experimenteel gedeelte
45
Figuur 20b. XIC van m/z 667 van fractie 2. Piekidentificatie: AS = Cer[A(24)S(18)].
In de scan mode-chromatogrammen van de verschillende fracties is telkens een piek te
zien bij tR = 2.30 min. Analyse van een standaardmengsel van cholesteryl oleaat en
glycerol trioleaat, geeft aan dat beide componenten rond deze retentietijd geëlueerd
worden. Bij nader onderzoek van de massspectra onder de piek (tR= 2,30 min) van elke
fractie, kan worden aangetoond dat alle cholesteryl oleaat en glycerol trioleaat in de
eerste fractie geëlueerd werd. Figuur 21 toont zowel de massaspectra van de
standaardoplossing cholesteryl oleaat en glycerol trioleaat als fractie 1, 2 en 3. Hoewel de
concentratie cholesteryl oleaat en glycerol trioleaat in de standaardoplossing even groot
is, wordt een minder grote piek waargenomen in het massaspectrum voor cholesteryl
oleaat. Dit is toe te kennen aan de lage ioniseerbaarheid van de cholesteryl ester.
Figuur 21a. Massaspectrum van standaardoplossing (10 µg/mL) cholsteryl oleaat en glycerol trioleaat. Piekidentificatie: CE = cholesteryl oleaat, TG = glycerol trioleaat.
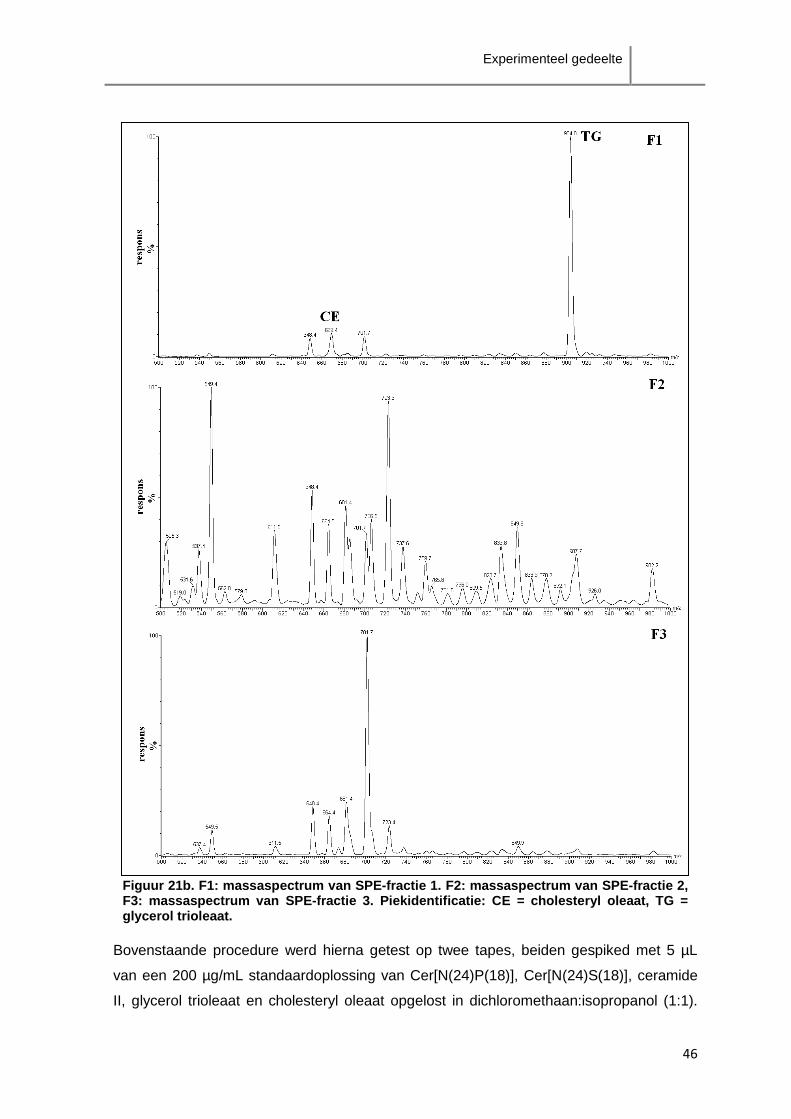
Experimenteel gedeelte
46
Figuur 21b. F1: massaspectrum van SPE-fractie 1. F2: massaspectrum van SPE-fractie 2, F3: massaspectrum van SPE-fractie 3. Piekidentificatie: CE = cholesteryl oleaat, TG = glycerol trioleaat.
Bovenstaande procedure werd hierna getest op twee tapes, beiden gespiked met 5 µL
van een 200 µg/mL standaardoplossing van Cer[N(24)P(18)], Cer[N(24)S(18)], ceramide
II, glycerol trioleaat en cholesteryl oleaat opgelost in dichloromethaan:isopropanol (1:1).

Experimenteel gedeelte
47
Hierbij werd na verdampen, elke fractie terug opgelost in 360 µL hexaan-isopropanol-
dichloromethaan (100:10:10). Dit werd in drievoud uitgevoerd zodat de precisie, meer
bepaald de herhaalbaarheid getest werd. Herhaalbaarheid wordt gedefinieerd als de mate
van spreiding van meetwaarden die onder dezelfde omstandigheden uitgevoerd werden.
Onder dezelfde omstandigheden worden onder andere dezelfde meetmethode en –
apparatuur, dezelfde waarnemer, dezelfde plaats en binnen een kort tijdsbestek verstaan.
De maat voor precisie kan worden uitgedrukt aan de hand van de relatieve
standaardafwijking (relative standard deviation: RSD)
(22)
waarbij de gemiddelde meetwaarde is en de experimentele standaardafwijking is, die
gedefinieerd is als:
(23)
met de ide meetwaarde en de gemiddelde meetwaarde.
Tabel 2 geeft het gemiddelde piekoppervlak en de relatieve standaardafwijking weer en
de verhouding van het piekoppervlak tussen de klasses onderling met de relatieve
standaardafwijking.
Tabel 2. Reproduceerbaarheid getest op 3 tapes (gespiked).
klasse gemiddelde piekoppervlak ± standaardafwijking RSD (%)
Cer[N(24)S(18)] 313300 ± 44650 14.25 Cer[N(24)P(18)] 17770 ± 1687 9.49
Cer[A(24)S(18)] 27500 ± 3386 21.33
verhouding gemiddelde verhouding ± standaardafwijking RSD (%)
0.057 6.33
0.08733 12.61
Onderling vergelijken van de standaarden bij eenzelfde meting, geeft een idee over de
instrumentele fout. Er kan uit de bekomen resultaten opgemaakt worden dat de
instrumentatie een belangrijke rol speelt bij de herhaalbaarheid.

Experimenteel gedeelte
48
6.5 NPLC-analyse van ceramiden in de huid
Zoals eerder vermeld, werden ceramideklasses gespreid over enkele minuten bij de
ontwikkelde methode. In dit opzicht bleek fractionatie van de ceramideklasses niet
adequaat te zijn. Desalniettemin werd de ontwikkelde NPLC-methode toegepast op
extract (50 µL injectie) bekomen na tape-extractie en SPE, in de scan mode. Via extracted
ion chromatogram (XIC) kunnen de m/z-waarden van ceramiden geselecteerd worden uit
het TIC-chromatogram en zo ruis onderdrukt worden. Extract van een blanco tape werd
via dezelfde monstervoorbereiding bekomen en geanalyseerd (50 µL injectie) zodat
mogelijke interferenties van de tape gecontroleerd werden. De data verkregen in scan
mode bij NPLC zijn niet voldoende om identificatie van de ceramiden mogelijk te maken.
Vermits bij de extracted ion chromatogrammen nog steeds een lage signaal-
ruisverhouding werd bekomen, werd een gevoeligere en uitgebreidere analyse in
omkeerfase chromatografie uitgevoerd.
6.6 Ontwikkeling van een RPLC-methode voor analyse van ceramiden
Een chromatografische methode werd ontwikkeld en geverifieerd via standaardceramide-
oplossingen. Ceramide-extract werd in de scan mode geanalyseerd. Om alle klasses te
onderscheiden en de lengte van zowel de amino-alcoholketen als van de vetzuurketen toe
te kennen, werd eveneens een analyse in de product-ion scan mode uitgevoerd.
Op basis van voorgaand onderzoek op ceramiden in bloedplasma, werd een RPLC-
methode ontwikkeld met een Kinetex C18 kolom (5 cm L x 2.1 mm I.D.; 2.6 µm dp;
Phenomenex) en mobiele fase A en B respectievelijk water-0.5 M ammonium formiaat
(100:1) en methanol-mierenzuur (100:0.2). Er werd een gradiënt elutie uitgevoerd met een
debiet van 0.3 mL/min en als begincondities 20% mobiele fase A en 80% mobiele fase B.
Dit werd 1 min aangehouden. Hierna werd een stapsgewijze overgang naar 0% mobiele
fase A en 100% mobiele fase B gemaakt. Deze compositie werd behouden gedurende 10
min. Vervolgens werd opnieuw een stapsgewijze overgang gemaakt naar de beginsituatie
20% mobiele fase A en 80% mobiele fase B. Deze verhouding werd 5 min aangehouden
zodat de kolom geconditioneerd werd voor de daaropvolgende analyse. Figuur 22 geeft
een chromatogram weer van 20 µL injectie van 10 µg/mL Cer[N(24)S(18)] en
Cer[N(24)P(18)] in MRM-mode waarbij respectievelijk massatransitie 650.9 → 263.9 en
669 → 282 werden beschouwd.
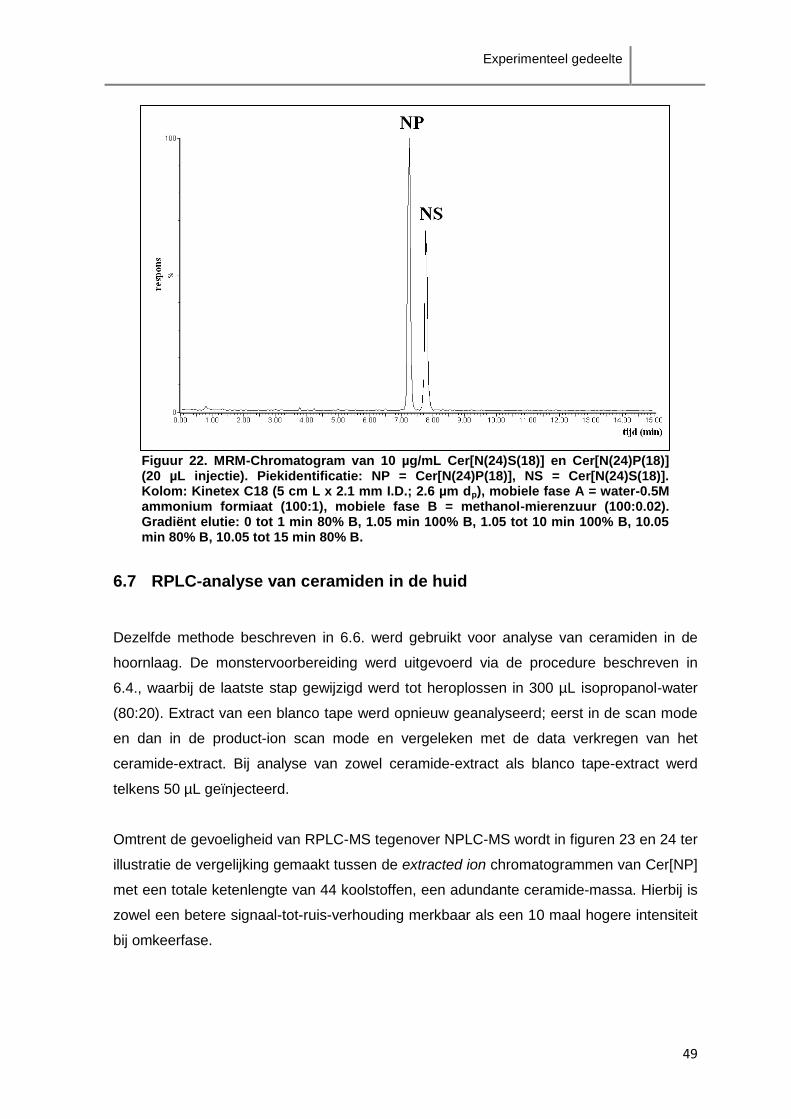
Experimenteel gedeelte
49
Figuur 22. MRM-Chromatogram van 10 µg/mL Cer[N(24)S(18)] en Cer[N(24)P(18)] (20 µL injectie). Piekidentificatie: NP = Cer[N(24)P(18)], NS = Cer[N(24)S(18)]. Kolom: Kinetex C18 (5 cm L x 2.1 mm I.D.; 2.6 µm dp), mobiele fase A = water-0.5M ammonium formiaat (100:1), mobiele fase B = methanol-mierenzuur (100:0.02). Gradiënt elutie: 0 tot 1 min 80% B, 1.05 min 100% B, 1.05 tot 10 min 100% B, 10.05 min 80% B, 10.05 tot 15 min 80% B.
6.7 RPLC-analyse van ceramiden in de huid
Dezelfde methode beschreven in 6.6. werd gebruikt voor analyse van ceramiden in de
hoornlaag. De monstervoorbereiding werd uitgevoerd via de procedure beschreven in
6.4., waarbij de laatste stap gewijzigd werd tot heroplossen in 300 µL isopropanol-water
(80:20). Extract van een blanco tape werd opnieuw geanalyseerd; eerst in de scan mode
en dan in de product-ion scan mode en vergeleken met de data verkregen van het
ceramide-extract. Bij analyse van zowel ceramide-extract als blanco tape-extract werd
telkens 50 µL geïnjecteerd.
Omtrent de gevoeligheid van RPLC-MS tegenover NPLC-MS wordt in figuren 23 en 24 ter
illustratie de vergelijking gemaakt tussen de extracted ion chromatogrammen van Cer[NP]
met een totale ketenlengte van 44 koolstoffen, een adundante ceramide-massa. Hierbij is
zowel een betere signaal-tot-ruis-verhouding merkbaar als een 10 maal hogere intensiteit
bij omkeerfase.
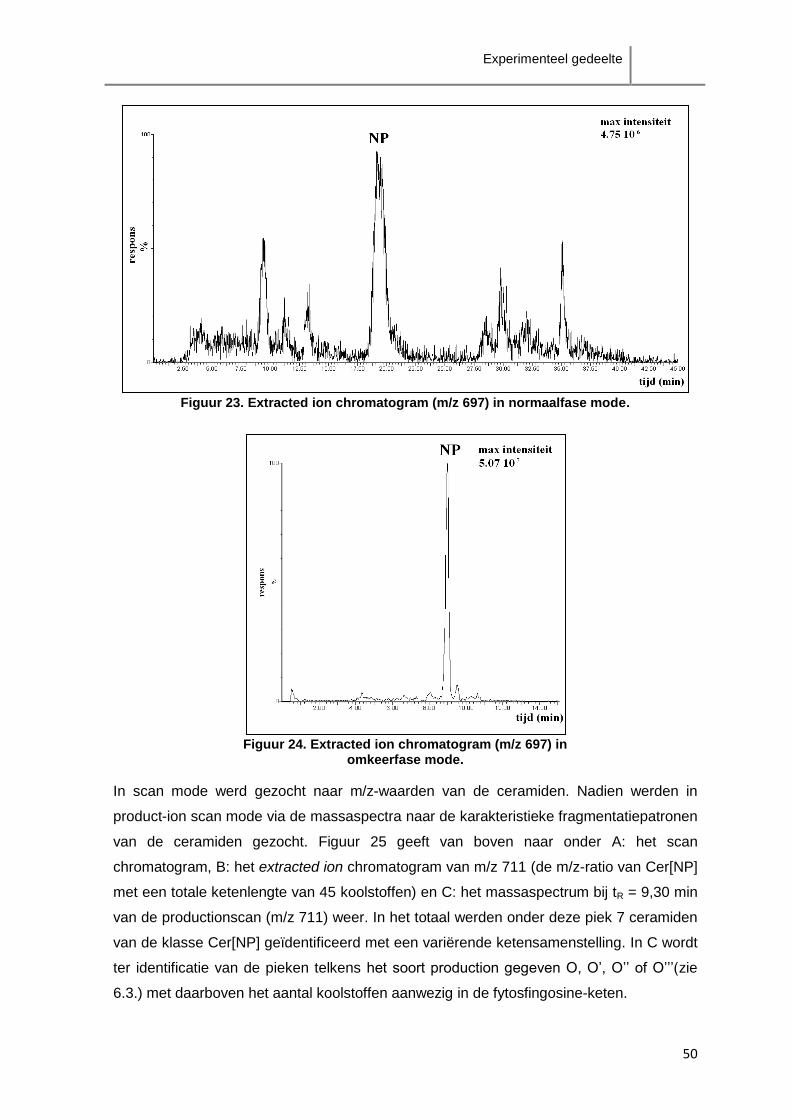
Experimenteel gedeelte
50
Figuur 23. Extracted ion chromatogram (m/z 697) in normaalfase mode.
Figuur 24. Extracted ion chromatogram (m/z 697) in
omkeerfase mode.
In scan mode werd gezocht naar m/z-waarden van de ceramiden. Nadien werden in
product-ion scan mode via de massaspectra naar de karakteristieke fragmentatiepatronen
van de ceramiden gezocht. Figuur 25 geeft van boven naar onder A: het scan
chromatogram, B: het extracted ion chromatogram van m/z 711 (de m/z-ratio van Cer[NP]
met een totale ketenlengte van 45 koolstoffen) en C: het massaspectrum bij tR = 9,30 min
van de productionscan (m/z 711) weer. In het totaal werden onder deze piek 7 ceramiden
van de klasse Cer[NP] geïdentificeerd met een variërende ketensamenstelling. In C wordt
ter identificatie van de pieken telkens het soort production gegeven O, O’, O’’ of O’’’(zie
6.3.) met daarboven het aantal koolstoffen aanwezig in de fytosfingosine-keten.
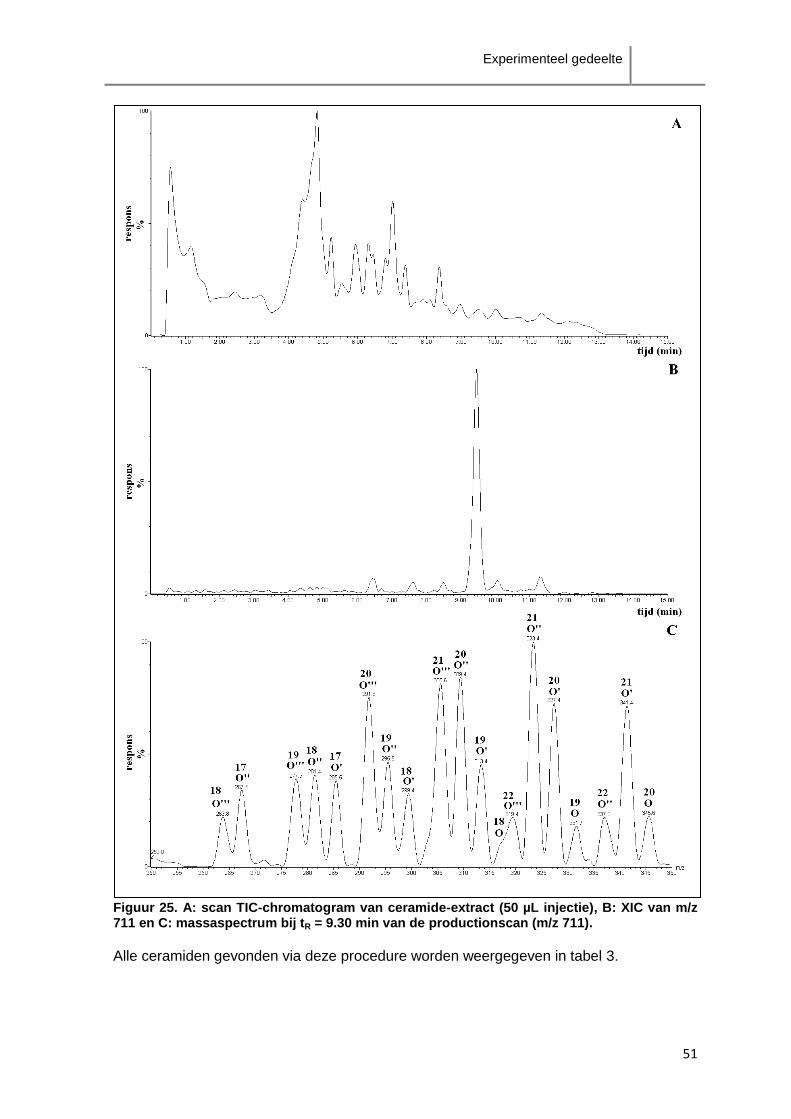
Experimenteel gedeelte
51
Figuur 25. A: scan TIC-chromatogram van ceramide-extract (50 µL injectie), B: XIC van m/z 711 en C: massaspectrum bij tR = 9.30 min van de productionscan (m/z 711).
Alle ceramiden gevonden via deze procedure worden weergegeven in tabel 3.

Experimenteel gedeelte
52
Tabel 3. Ceramiden aanwezig in de hoornlaag.
klasse # C's [M+H]+ ceramiden
Cer[NS] 34 539 [N(16)S(18)]
35 553 [N(18)S(17)]
36 567 [N(16)S(20)] [N(18)S(18)]
38 595 [N(16)S(22)] [N(18)S(20)] [N(20)S(18)] [N(22)S(16)]
39 609 [N(19)S(20)] [N(20)S(19)] [N(21)S(18)] [N(23)S(16)]
40 623 [N(16)S(24)] [N(18)S(22)] [N(20)S(20)] [N(22)S(18)]
41 637 [N(16)S(25)] [N(17)S(24)] [N(18)S(23)] [N(19)S(22)] [N(20)S(21)]
41 637 [N(21)S(20)] [N(22)S(19)] [N(23)S(18)] [N(24)S(17)] [N(25)S(16)]
42 651 [N(18)S(24)] [N(20)S(22)] [N(22)S(20)] [N(24)S(18)]
43 665 [N(21)S(22)] [N(22)S(21)] [N(23)S(20)] [N(24)S(19)] [N(25)S(18)]
44 677 [N(24:1)S(20)] [N(26:1)S(18)]
44 679 [N(22)S(22)] [N(24)S(20)] [N(26)S(18)]
45 693 [N(23)S(22)] [N(24)S(21)] [N(25)S(20)] [N(26)S(19)] [N(27)S(18)]
46 707 [N(24)S(22)] [N(25)S(21)] [N(26)S(20)] [N(27)S(19)] [N(28)S(18)]
47 721 [N(21)S(26)] [N(22)S(25)] [N(23)S(24)] [N(24)S(23)] [N(25)S(22)]
47 721 [N(26)S(21)] [N(27)S(20)] [N(28)S(19)] [N(29)S(18)]
48 735 [N(24)S(24)] [N(26)S(22)] [N(28)S(20)] [N(29)S(19)]
Cer[NP] 39 627 [N(24)P(16)]
40 641 [N(22)P(19)] [N(23)P(18)] [N(24)P(17)] [N(25)P(16)]
41 655 [N(22)P(20)] [N(24)P(18)] [N(25)P(17)] [N(26)P(16)]
42 669 [N(23)P(20)] [N(24)P(19)] [N(25)P(18)] [N(26)P(17)]
43 683 [N(24)P(20)] [N(26)P(18)] [N(27)P(17)] [N(28)P(16)]
44 697 [N(23)P(22)] [N(24)P(21)] [N(25)P(20)] [N(26)P(19)] [N(27)P(18)]
44 697 [N(28)P(17)]
45 711 [N(24)P(22)] [N(26)P(20)] [N(28)P(18)]
46 725 [N(25)P(22)] [N(26)P(21)] [N(27)P(20)] [N(28)P(19)] [N(29)P(18)]
47 739 [N(24)P(24)] [N(26)P(22)] [N(28)P(20)] [N(30)P(18)]
48 753 [N(24)P(25)] [N(25)P(24)] [N(26)P(23)] [N(27)P(22)] [N(28)P(21)]
48 753 [N(29)P(20)] [N(30)P(19)] [N(31)P(18)]
49 767 [N(24)P(26)] [N(26)P(24)] [N(28)P(22)] [N(30)P(20)]
Cer[NDS] 36 569 [N(25)DS(21)]
42 649 [N(23:2)DS(19)]
42 651 [N(23:1)DS(19)]
42 653 [N(26)DS(16)]
43 667 [N(24)DS(19)] [N(27)DS(16)]
44 677 [N(25:2)DS(19)]
44 681 [N(25)DS(19)]
45 695 [N(24)DS(21)]
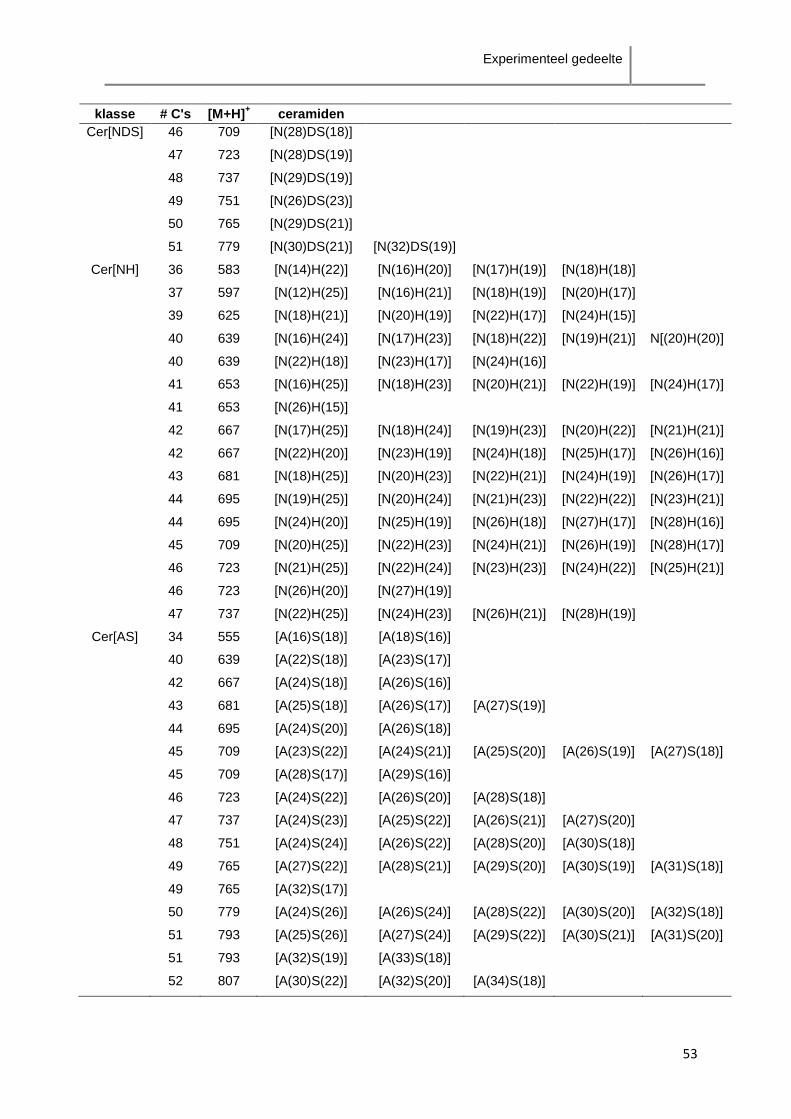
Experimenteel gedeelte
53
klasse # C's [M+H]+ ceramiden
Cer[NDS] 46 709 [N(28)DS(18)]
47 723 [N(28)DS(19)]
48 737 [N(29)DS(19)]
49 751 [N(26)DS(23)]
50 765 [N(29)DS(21)]
51 779 [N(30)DS(21)] [N(32)DS(19)]
Cer[NH] 36 583 [N(14)H(22)] [N(16)H(20)] [N(17)H(19)] [N(18)H(18)]
37 597 [N(12)H(25)] [N(16)H(21)] [N(18)H(19)] [N(20)H(17)]
39 625 [N(18)H(21)] [N(20)H(19)] [N(22)H(17)] [N(24)H(15)]
40 639 [N(16)H(24)] [N(17)H(23)] [N(18)H(22)] [N(19)H(21)] N[(20)H(20)]
40 639 [N(22)H(18)] [N(23)H(17)] [N(24)H(16)]
41 653 [N(16)H(25)] [N(18)H(23)] [N(20)H(21)] [N(22)H(19)] [N(24)H(17)]
41 653 [N(26)H(15)]
42 667 [N(17)H(25)] [N(18)H(24)] [N(19)H(23)] [N(20)H(22)] [N(21)H(21)]
42 667 [N(22)H(20)] [N(23)H(19)] [N(24)H(18)] [N(25)H(17)] [N(26)H(16)]
43 681 [N(18)H(25)] [N(20)H(23)] [N(22)H(21)] [N(24)H(19)] [N(26)H(17)]
44 695 [N(19)H(25)] [N(20)H(24)] [N(21)H(23)] [N(22)H(22)] [N(23)H(21)]
44 695 [N(24)H(20)] [N(25)H(19)] [N(26)H(18)] [N(27)H(17)] [N(28)H(16)]
45 709 [N(20)H(25)] [N(22)H(23)] [N(24)H(21)] [N(26)H(19)] [N(28)H(17)]
46 723 [N(21)H(25)] [N(22)H(24)] [N(23)H(23)] [N(24)H(22)] [N(25)H(21)]
46 723 [N(26)H(20)] [N(27)H(19)]
47 737 [N(22)H(25)] [N(24)H(23)] [N(26)H(21)] [N(28)H(19)]
Cer[AS] 34 555 [A(16)S(18)] [A(18)S(16)]
40 639 [A(22)S(18)] [A(23)S(17)]
42 667 [A(24)S(18)] [A(26)S(16)]
43 681 [A(25)S(18)] [A(26)S(17)] [A(27)S(19)]
44 695 [A(24)S(20)] [A(26)S(18)]
45 709 [A(23)S(22)] [A(24)S(21)] [A(25)S(20)] [A(26)S(19)] [A(27)S(18)]
45 709 [A(28)S(17)] [A(29)S(16)]
46 723 [A(24)S(22)] [A(26)S(20)] [A(28)S(18)]
47 737 [A(24)S(23)] [A(25)S(22)] [A(26)S(21)] [A(27)S(20)]
48 751 [A(24)S(24)] [A(26)S(22)] [A(28)S(20)] [A(30)S(18)]
49 765 [A(27)S(22)] [A(28)S(21)] [A(29)S(20)] [A(30)S(19)] [A(31)S(18)]
49 765 [A(32)S(17)]
50 779 [A(24)S(26)] [A(26)S(24)] [A(28)S(22)] [A(30)S(20)] [A(32)S(18)]
51 793 [A(25)S(26)] [A(27)S(24)] [A(29)S(22)] [A(30)S(21)] [A(31)S(20)]
51 793 [A(32)S(19)] [A(33)S(18)]
52 807 [A(30)S(22)] [A(32)S(20)] [A(34)S(18)]

Experimenteel gedeelte
54
klasse # C's [M+H]+ ceramiden
Cer[AS] 53 821 [A(29)S(24)] [A(30)S(23)] [A(31)S(22)] [A(32)S(21)] [A(33)S(20)]
53 821 [A(34)S(19)] [A(35)S(18)]
Cer[ADS] 39 641 [A(14)H(25)] [A(16)H(23)] [A(18)H(21)] [A(20)H(19)]
49 781 [A(30)H(19)] [A(31)H(18)] [A(32)H(17)] [A(33)H(16)]
Cer[AH] 41 651 [A(21)H(20)] [A(23)H(18)]
42 665 [A(20)H(22)] [A(21)H(21)] [A(22)H(20)] [A(23)H(19)] [A(24)H(18)]
42 665 [A(25)H(17)]
43 677 [A(23:1)H(20)] [A(25:1)H(18)]
43 679 [A(23)H(20)] [A(25)H(18)]
44 693 [A(23)H(21)] [A(24)H20)] [A(25)H(19)] [A(26)H(18)] [A(27)H(17)]
45 707 [A(23)H(22)] [A(25)H(20)] [A(27)H(18)]
47 735 [A(25)H(22)] [A(27)H(20)] [A(29)H(18)]
Bij identificatie van de ceramiden moet echter rekening gehouden worden met
aanwezigheid van onverzadigde bindingen in het vetzuurgedeelte. De massa’s van
verschillende klasses (met verzadigde vetzuurketens) verschillen soms slechts 2 massa-
eenheden. Introductie van een dubbele binding in de vetzuurketen van de ceramide met
de hogere massa geeft dus dezelfde massa als de ceramide met de lagere massa. Bij
klasses waarin de aminoalcohol-keten verschilt, vormt dit bij identificatie geen probleem.
Wanneer echter de vetzuurketen dezelfde is bij beide ceramiden, kan via positieve
ionisatie niet bepaald worden over welke klasse het gaat. Aangezien in de literatuur
voornamelijk wordt aangenomen dat de vetzuurketen verzadigd is, werd ook in dit werk
aangenomen dat in geval van twijfel de verzadigde vetzuurketen aanwezig was. Enkel
analyse in negatieve ionisatie mode zou hierbij uitsluitsel kunnen geven of deze
onderstelling waar is.

Conclusie
55
7 Conclusie
Er werd een normaalfase chromatografische methode ontwikkeld met als opzet
hoornlaagceramiden te analyseren. Daarna werd een monstervoorbereidingprocedure
ontwikkeld om de ceramiden uit de hoornlaag te collecteren via tape-stripping en zoveel
mogelijk te isoleren van interferenties (cholesterylesters en triglyceriden) door gebruik te
maken van vaste fase extractie.
De intentie de ceramideklasses te scheiden bleek niet succesvol, aangezien overlapping
van de verschillende klasses plaatsvond. Het ceramide-extract werd via NPLC-MS
geanalyseerd in de scan mode. Identificatie van de lengte van het vetzuurgedeelte en de
aminoalcohol-keten van de ceramiden vereist daarenboven tandemmassaspectrometrie
vermits variaties in ketenlengte van beide onderdelen resulteren in ceramiden met
dezelfde massa’s.
Wegens de hogere gevoeligheid van RPLC-MS, werd in dit opzicht gekozen de
ceramiden in omkeerfase LC-MS te analyseren. Een methode werd ontwikkeld gebaseerd
op analyse van ceramiden in bloedplasma. De methode werd hierna toegepast op
ceramide-extract, zowel in de scan mode als in de product-ion scan mode. In de scan
mode werd via extracted ion chromatogrammen naar de massa’s van de ceramiden
gezocht. Extract van blanco tape werd tevens in scan en product-ion scan mode
geanalyseerd en mogelijke interferenties werden gecontroleerd. Via de massaspectra van
analyse in product-ion scan mode, werden de fragmentaties van ceramiden onderzocht
en de aanwezige amino-alcoholketens geïdentificeerd. Met kennis van de massa’s van
zowel precursor- als product-ionen, werden hierna de aanwezige ceramiden
geïdentificeerd. Hierbij moet rekening gehouden worden met de aanwezigheid van
eventuele onverzadigde bindingen. Vermits een bijkomende analyse in negatieve ionisatie
mode ter identificatie van de vetzuurketen nodig is, werd in dit werk aangenomen dat bij
geval van twijfel de verzadigde vetzuurketen aanwezig was. Een totaal van 281
ceramiden werd geïdentificeerd met positieve mode ESI tandem quadrupool MS.
In samenwerking met Metablys werden door gebruik te maken van negatieve mode ESI in
combinatie met hoge resolutie Q-TOF MS meer dan 950 ceramiden éénduidig
geïdentificeerd. Dit succes is voornamelijk gebaseerd op een goede
monstervoorbewerkingstechniek, dit is tape-aanrijking en SPE, ontwikkeld in dit werk. De
bekomen resultaten geven nieuwe perspectieven voor onder andere studie van
huidkanker, psoriasis, invloed van cosmetica (antioxidanten) en metabolome studies.

Referenties
56
8 Referenties
Ardrey, R. E. (2003). Liquid chromatography–mass spectrometry: an introduction, John
Wiley & Sons Ltd.
Bonte, F., P. Pinguet, et al. (1995). "Analysis of all stratum corneum lipids by automated
multiple development high-performance thin-layer chromatography." Journal of
Chromatography B-Biomedical Applications 664(2): 311-316.
Bonte, F., A. Saunois, et al. (1997). "Existence of a lipid gradient in the upper stratum
corneum and its possible biological significance." Archives of Dermatological
Research 289(2): 78-82.
Brannon, H. L. (2007). "Epidermis Anatomy." from
http://dermatology.about.com/od/anatomy/ss/epidermis.htm.
Brooks, G. and B. Idson (1991). "Skin lipids." International Journal of Cosmetic Science
13(2): 103-113.
Casparrini, G. H., E. C. ; Horning, M. G. (1969). "Gas phase analytical separation and
structural study of ceramides." Chemistry and Physics of Lipids 3(1): 1-10.
Coderch, L., O. Lopez, et al. (2003). "Ceramides and skin function." American Journal of
Clinical Dermatology 4(2): 107-129.
De Hoffmann E., C. J., Stroobant V. (1996). Mass Spectrometry Principles and
Applications, John Wiley & Sons.
Di Nardo, A., P. Wertz, et al. (1998). "Ceramide and cholesterol composition of the skin of
patients with atopic dermatitis." Acta Dermato-Venereologica 78(1): 27-30.
Dolan, J. W., J. J. Kirkland, et al. (2010). Introduction to Modern Liquid Chromatography,
A John Wiley & Sons, Inc., Publication.
Elias, P. M. (1983). "Epidermal Lipids, Barrier Function, and Desquamation." The Journal
of investigative dermatology 80(6): 44-49.
Engelhardt, H. (2004). "One century of liquid chromatography from Tswett's columns to
modem high speed and high performance separations." Journal of
Chromatography B-Analytical Technologies in the Biomedical and Life Sciences
800(1-2): 3-6.
Ettre, L. S. (2000). "Chromatography: the separation technique of the 20th century."
Chromatographia 51(1-2): 7-17.
Farwanah, H., R. Neubert, et al. (2002). "Improved procedure for the separation of major
stratum corneum lipids by means of automated multiple development thin-layer
chromatography." Journal of Chromatography B-Analytical Technologies in the
Biomedical and Life Sciences 780(2): 443-450.

Referenties
57
Farwanah, H., K. Raith, et al. (2005). "Ceramide profiles of the uninvolved skin in atopic
dermatitis and psoriasis are comparable to those of healthy skin." Archives of
Dermatological Research 296(11): 514-521.
Farwanah, H., J. Wohlrab, et al. (2005). "Profiling of human stratum corneum ceramides
by means of normal phase LC/APCI-MS." Analytical and Bioanalytical Chemistry
383(4): 632-637.
Gildenast, T. and J. Lasch (1997). "Isolation of ceramide fractions from human stratum
corneum lipid extracts by high-performance liquid chromatography." Biochimica Et
Biophysica Acta-Lipids and Lipid Metabolism 1346(1): 69-74.
Gray, G. M. and H. J. Yardley (1975). "Lipid compositions of cells isolated from pig,
human, and rat epidermis." Journal of Lipid Research 16(6): 434-440.
Haynes, C. A., J. C. Allegood, et al. (2009). "Sphingolipidomics: Methods for the
comprehensive analysis of sphingolipids." Journal of Chromatography B-Analytical
Technologies in the Biomedical and Life Sciences 877(26): 2696-2708.
Hens, Z. (2009). Molecular Physical Chemistry. Gent, Universiteit Gent.
Herbert, C. G. and A. A. W. Johnstone (2003). Mass spectrometry basics CRC Press
LLC.
Holleran, W. M., Y. Takagi, et al. (2006). "Epidermal sphingolipids: Metabolism, function,
and roles in skin disorders." Febs Letters 580(23): 5456-5466.
Imokawa, G., A. Abe, et al. (1991). "Decreased Level of Ceramides in Stratum Corneum
of Atopic Dermatitis: An Etiologic Factor in Atopic Dry Skin?" Journal of
Investigative Dermatology 96(4): 523-526.
Imokawa, G. and M. Hattori (1985). "A Possible Function of Structural Lipids in the Water-
Holding Properties of the Stratum Corneum." Journal of Investigative Dermatology
84(4): 282-284.
Lavrijsen, A. P. M., J. A. Bouwstra, et al. (1995). "Reduced Skin Barrier Function Parallels
Abnormal Stratum Corneum Lipid Organization in Patients with Lamellar
Ichthyosis." Journal of Investigative Dermatology 105(4): 619-624.
Lavrijsen, A. P. M., I. M. Higounenc, et al. (1994). "Validation of an in-vivo extraction
method for human stratum-corneum ceramides." Archives of Dermatological
Research 286(8): 495-503.
Marks, R. and R. P. R. Dawber (1971). "Skin Surface Biopsy - improved technique for
examination of horny layer." British Journal of Dermatology 84(2): 117-&.
Masukawa, Y., H. Narita, et al. (2009). "Comprehensive quantification of ceramide species
in human stratum corneum." Journal of Lipid Research 50(8): 1708-1719.

Referenties
58
Masukawa, Y., H. Narita, et al. (2008). "Characterization of overall ceramide species in
human Stratum corneum." Journal of Lipid Research 49(7): 1466-1476.
Motta, S., M. Monti, et al. (1993). "Ceramide composition of the psoratic scale."
Biochimica Et Biophysica Acta 1182(2): 147-151.
Niessen, W. M. A. (2006). Liquid Chromatography–Mass Spectrometry, Taylor and
Francis Group, LLC.
Ponec, M., A. Weerheim, et al. (2003). "New acylceramide in native and reconstructed
epidermis." Journal of Investigative Dermatology 120(4): 581-588.
Poole C. F., P. S. K. (1991). Chromatography today, Elsevier Science.
Raith, K., H. Farwanah, et al. (2004). "Progress in the analysis of Stratum corneum
ceramides." European Journal of Lipid Science and Technology 106(8): 561-571.
Robson, K. J., M. E. Stewart, et al. (1994). "6-hydroxy-4-sphingenine in human epidermal
ceramides." Journal of Lipid Research 35(11): 2060-2068.
Sandra, P. (2007). Analytische Scheidingstechnieken. Gent, Universiteit Gent.
Tassopoulos, T. F. (2006). Evaluation of topical bioavailibility in human stratum corneum
in vivo by tape stripping using a direct spectroscopic method. Basel, University of
Basel.
Vietzke, J. P., O. Brandt, et al. (2001). "Comparative investigation of human stratum
corneum ceramides." Lipids 36(3): 299-304.
Vietzke, J. P., M. Strassner, et al. (1999). "Separation and identification of ceramides in
the human stratum corneum by high-performance liquid chromatography coupled
with electrospray ionization mass spectrometry and electrospray multiple-stage
mass spectrometry profiling." Chromatographia 50(1-2): 15-20.
Weerheim, A. and M. Ponec (2001). "Determination of stratum corneum lipid profile by
tape stripping in combination with high-performance thin-layer chromatography."
Archives of Dermatological Research 293(4): 191-199.
Wertz, P. W., K. C. Madison, et al. (1989). "Covalently Bound Lipids of Human Stratum
Corneum." Journal of Investigative Dermatology 92(1): 109-111.
Wertz, P. W., M. C. Miethke, et al. (1985). "The Composition of the Ceramides from
Human Stratum Corneum and from Comedones." Journal of Investigative
Dermatology 84(5): 410-412.
Woordenlijst
59
Woordenlijst
HPLC High Performance Liquid Chromatography
TLC Thin Layer Chromatography
RPLC Reverse Phase Liquid Chromatography
NPLC Normal Phase Liquid Chromatography
HILIC Hydrophilic Interaction Chromatography
IPC Ion-Pair Chromatography
IEC Ion-Exchange Chromatography
SEC Size Exclusion Chromatography
HETP Height Equivalent of a Theoretical Plate
LC-NMR Liquid Chromatography-Nuclear Magnetic Resonance
LC-MS Liquid Chromatography-Mass Spectrometry
CAD Corona Aerosol Detector
MS Massaspectrometrie
APCI Atmospheric Pressure Chemical Ionization
ESI Electrospray Ionization
TOF Time of Flight
LC-MS/MS Liquid Chromatography-Tandem mass spectrometry
EM Electronmultiplier
DC Direct Current
RF Radio Frequency
TIC Total Ion Current
SIM Selected Ion Monitoring
QqQ triple quadrupole
Q-TOF Quadrupole-Time of Flight
CID Collision Induced Dissociation
MRM Multiple-Reaction Monitoring
GC Gass Chromatography
AMD-HPTLC Automated Multiple Development-High Performance
Thin Layer Chromatography
ELSD Evaporative Light Scattering Detector
NS Nonhydroxy-fatty acid + Sphingosine
NP Nonhydroxy-fatty acid + Phytosphingosine
AS Alpha-hydroxy-fatty acid + Sphingosine

Woordenlijst
60
CE Cholesteryl Ester
TG Triglyceride
RSD Relative Standard Deviation

Symbolenlijst
61
Symbolenlijst
tR retentietijd (s)
t0 dode tijd (s)
tR’ netto retentietijd (s)
u lineaire snelheid van de mobiele fase (m/s)
L kolomlengte (m)
k retentiefactor
mS massa van de component in de stationaire fase (g)
mM massa van de component in de mobiele fase (g)
cS concentratie van de component in de stationaire fase (mol/L)
cM concentratie van de component in de mobiele fase (mol/L)
K verdelingscoëfficiënt
β fase-ratio
VM volume van de component in de mobiele fase (L)
VS volume van de component in de stationaire fase (L)
α selectiviteit of scheidingsfactor
µ verwachtingswaarde
σ standaardafwijking
wb breedte van een piek aan de basis (s)
wh
breedte van een piek op halve hoogte (s)
ruimtelijke variantie
z migratie-afstand (m)
H schotelhoogte (m)
Dax axiale dispersiecöeffiënt (m²/s)
N schotelgetal
Rs resolutie
A Eddydiffusieterm
B longitudinale diffusieterm
C transportweerstand
λ pakkingsefficiëntie
dp diameter van deeltje (m)
DM diffusiecoëfficiënt in de mobiele fase (m²/s)
CM bijdrage tot piekverbreding in de mobiele fase
CS bijdrage tot band- of piekverbreding in de stationaire fase

Symbolenlijst
62
σ2inj bandverbreding door injectie
σ2kol bandverbreding binnen de scheidingskolom
σ²dv bijdrage van dode volumes
σ²det bijdrage van de detector
σ²tot
totale piekbreedte
gemiddelde meetwaarde
experimentele standaardafwijking
xi ide meetwaarde
n aantal metingen

Addendum
63
Addendum
Recent beschreven K. Sandra et al. een lipidomic platform voor totaal analyse in
bloedplasma monsters (J. Chromatogra. A. doi:10.1016/j.chroma.2010.02.039)
De abstract van de publicatie wordt weergegeven:
Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole
time-of-flight mass spectrometry
Koen Sandra, Alberto dos Santos Pereira, Gerd Vanhoenacker,
Frank David and Pat Sandra
Metablys, Kennedypark 26, 8500 Kortrijk, Belgium
Abstract
A lipidomics strategy, combining high resolution reversed-phase liquid chromatography
(RPLC) with high resolution quadrupole time-of-flight mass spectrometry (Q-TOF), is
described. The method has carefully been assessed in both a qualitative and a
quantitative fashion utilizing human blood plasma. The inherent low technical variability
associated with the lipidomics method allows to measure 65% of the features with an
intensity RSD value below 10%. Blood plasma lipid spike-in experiments demonstrate that
relative concentration differences smaller than 25% can readily be revealed by means of a
t-test. Utilizing an advanced identification strategy, it is shown that the detected features
mainly originate from (lyso-)phospholipids, sphingolipids, mono-, di- and triacylglycerols
and cholesterol esters. The high resolution offered by the up-front RPLC step further
allows to discriminate various isomeric species associated with the different lipid classes.
The added value of utilizing a Jetstream electrospray ionization (ESI) source over a
regular ESI source in lipidomics is for the first time demonstrated. In addition, the
application of ultra high performance LC (UHPLC) up to 1200 bar to extend the peak
capacity or increase productivity is discussed.
De methode ontwikkeld voor de monstername en SPE opzuivering van de huid-ceramiden
beschreven in dit werk, werd gecombineerd met de identificatie mogelijkheden van het
beschreven platform. Het resultaat wordt weergegeven in de volgende figuur.
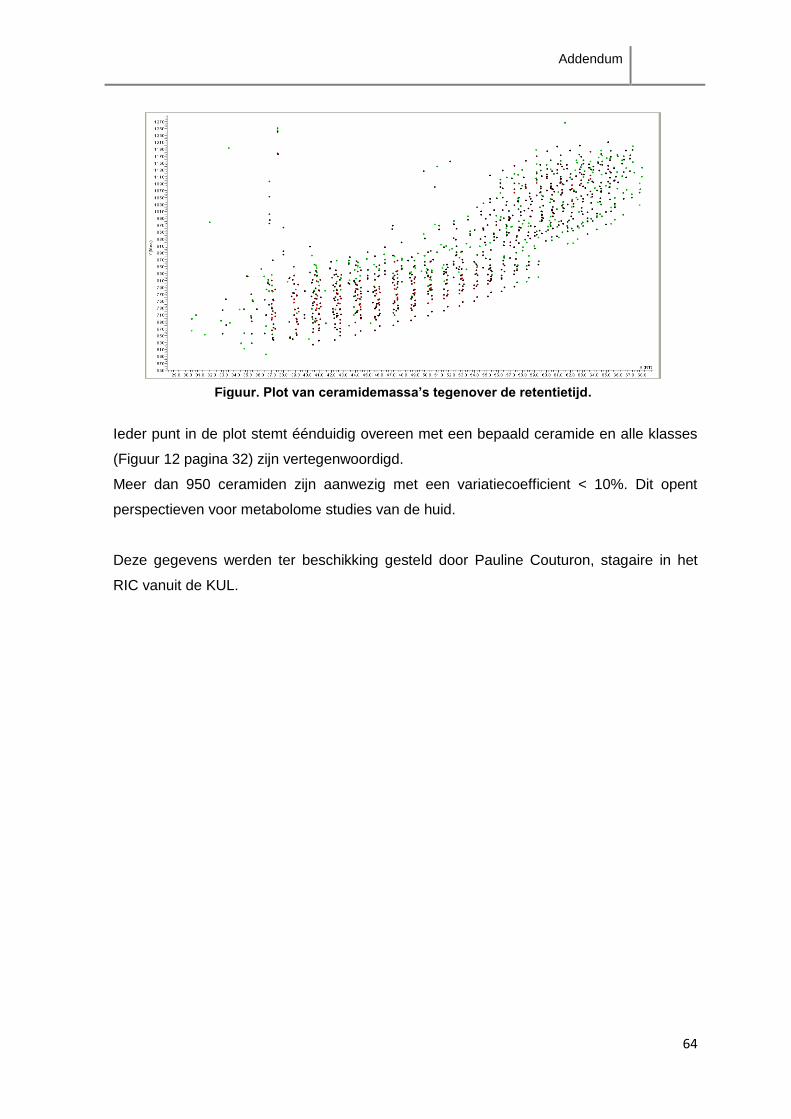
Addendum
64
Ieder punt in de plot stemt éénduidig overeen met een bepaald ceramide en alle klasses
(Figuur 12 pagina 32) zijn vertegenwoordigd.
Meer dan 950 ceramiden zijn aanwezig met een variatiecoefficient < 10%. Dit opent
perspectieven voor metabolome studies van de huid.
Deze gegevens werden ter beschikking gesteld door Pauline Couturon, stagaire in het
RIC vanuit de KUL.
Figuur. Plot van ceramidemassa’s tegenover de retentietijd.

Development of an LC-MS method for the determination of ceramides in the skin
B. Dhoopa*
, B. Bicalhoa, F. Lynen
a and P. Sandra
a&b
a Laboratory Separation Sciences, Department of Organic Chemistry, University of Ghent,
B-9000 Ghent, Belgium. b Metablys, Research Institute of Chromatography, B-8500 Kortrijk, Belgium
* email: [email protected]
Ceramides have been indicated to play a physicochemical and
physiological role in the skin. Compared to intracellular ceramides,
ceramides in the skin are more complex. Not only are there more
classes present in the stratum corneum but the variation in chain
length is also higher. A sample preparation method has been
developed which successfully separated the ceramides from
compounds such as triglycerides and cholesteryl esters, present in the
skin. Since separation in normal phase mode is based on polarity, an
attempt was made to separate/fractionate the different ceramide
classes. However, separation of the different classes was not
sufficient compared to the spreading of 1 class using the developed
method. For a sensitive analysis of the stratum corneum ceramides, a
reverse phase mass spectrometry method was used. Identification of
281 species of ceramides in positive ionization mode was
accomplished.
Introduction
The human horny layer (stratum corneum) provides protection to the human body from a
(harmful) environment and water loss. Due to the unique structure of this skin layer, a
combination of keratinocytes (‘bricks’) and intracellular lipid lamellae (‘mortar’) [1], a
barrier is formed between the body and surroundings. The skin lipids mainly consist of free
fatty acids, cholesterol and ceramides. Ceramides play a crucial physicochemical role in
formation of the barrier, but also physiologically in cell regulation and differentiation and
signal transduction [2]. Several skin diseases, such as psoriasis and atopic dermatitis have
been linked to altered ceramide compositions in the skin [3-4]. Compared to ceramides
present in other human cells or tissues, the ceramide composition in the horny layer is more
complex. Intracellular ceramides consist only of 3 ceramide subclasses, in the skin however
10 classes have already been assigned [5]. Not only are there more classes present in the
stratum corneum, but variation of the chain length can also be large. An overview of the
ceramide structures is given in the following section.
Nomenclature
Ceramides are a subclass of the sphingolipids and are made out of a long chain base
(sphingoid base) and a fatty acid chain linked to each other via an amide bond. In this
report the nomenclature proposed by Motta [6] is used. The different ceramide classes are
termed according to their combination of sphingoid base and fatty acid chain. The
sphingoid base can be a sphingosine (S), phytosphingosine (P), dihydrosphingosine (DS) or
a 6-hydroxy-sphingosine (A). The fatty acid can be a non-hydroxy fatty acid (N), an alpha-
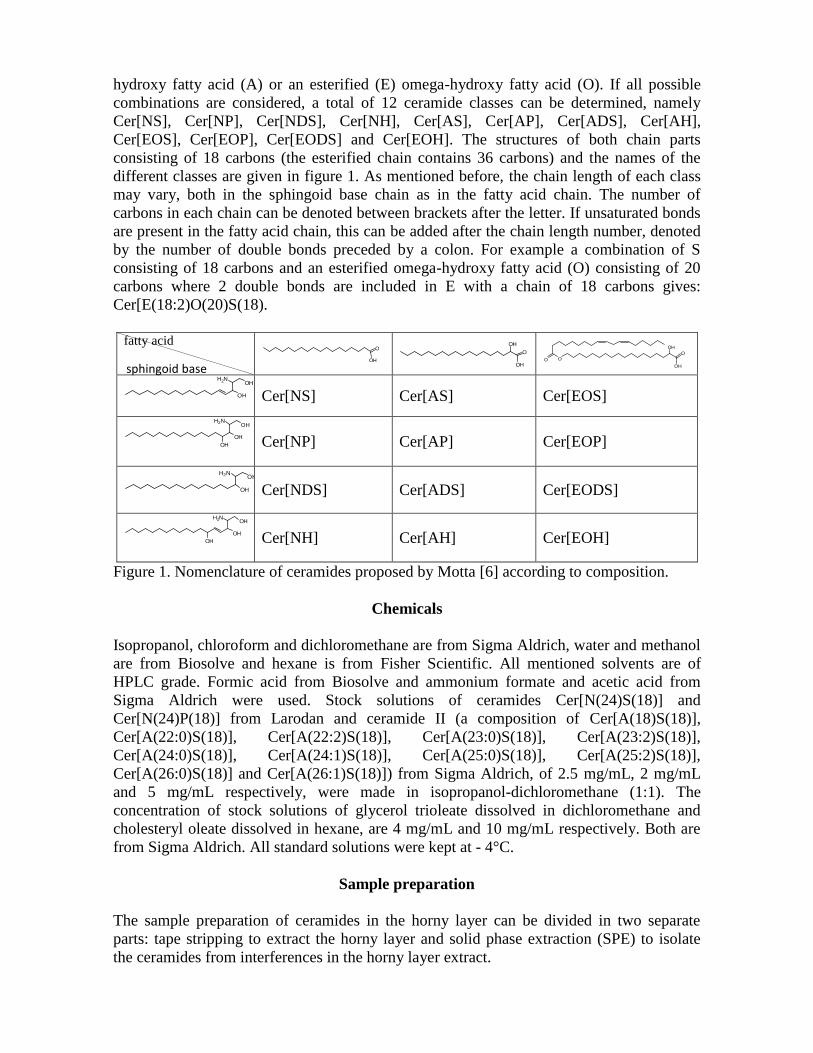
sphingoid base
hydroxy fatty acid (A) or an esterified (E) omega-hydroxy fatty acid (O). If all possible
combinations are considered, a total of 12 ceramide classes can be determined, namely
Cer[NS], Cer[NP], Cer[NDS], Cer[NH], Cer[AS], Cer[AP], Cer[ADS], Cer[AH],
Cer[EOS], Cer[EOP], Cer[EODS] and Cer[EOH]. The structures of both chain parts
consisting of 18 carbons (the esterified chain contains 36 carbons) and the names of the
different classes are given in figure 1. As mentioned before, the chain length of each class
may vary, both in the sphingoid base chain as in the fatty acid chain. The number of
carbons in each chain can be denoted between brackets after the letter. If unsaturated bonds
are present in the fatty acid chain, this can be added after the chain length number, denoted
by the number of double bonds preceded by a colon. For example a combination of S
consisting of 18 carbons and an esterified omega-hydroxy fatty acid (O) consisting of 20
carbons where 2 double bonds are included in E with a chain of 18 carbons gives:
Cer[E(18:2)O(20)S(18).
fatty acid
O
OH O
OH
OH
OH
OO
O
OH H2N OH
OH
Cer[NS] Cer[AS] Cer[EOS]
H2NOH
OH
OH
Cer[NP] Cer[AP] Cer[EOP]
H2NOH
OH
Cer[NDS] Cer[ADS] Cer[EODS]
H2N OH
OH
OH
Cer[NH] Cer[AH] Cer[EOH]
Figure 1. Nomenclature of ceramides proposed by Motta [6] according to composition.
Chemicals
Isopropanol, chloroform and dichloromethane are from Sigma Aldrich, water and methanol
are from Biosolve and hexane is from Fisher Scientific. All mentioned solvents are of
HPLC grade. Formic acid from Biosolve and ammonium formate and acetic acid from
Sigma Aldrich were used. Stock solutions of ceramides Cer[N(24)S(18)] and
Cer[N(24)P(18)] from Larodan and ceramide II (a composition of Cer[A(18)S(18)],
Cer[A(22:0)S(18)], Cer[A(22:2)S(18)], Cer[A(23:0)S(18)], Cer[A(23:2)S(18)],
Cer[A(24:0)S(18)], Cer[A(24:1)S(18)], Cer[A(25:0)S(18)], Cer[A(25:2)S(18)],
Cer[A(26:0)S(18)] and Cer[A(26:1)S(18)]) from Sigma Aldrich, of 2.5 mg/mL, 2 mg/mL
and 5 mg/mL respectively, were made in isopropanol-dichloromethane (1:1). The
concentration of stock solutions of glycerol trioleate dissolved in dichloromethane and
cholesteryl oleate dissolved in hexane, are 4 mg/mL and 10 mg/mL respectively. Both are
from Sigma Aldrich. All standard solutions were kept at - 4°C.
Sample preparation
The sample preparation of ceramides in the horny layer can be divided in two separate
parts: tape stripping to extract the horny layer and solid phase extraction (SPE) to isolate
the ceramides from interferences in the horny layer extract.

Tape-stripping
Several in vivo methods have been developed to extract the horny layer, one of them
being stripping of the skin via a tape [5, 7]. This method has also been used in this report.
The ceramides in the horny layer are collected by applying a tape (20 x 20 mm) on a
specific region of the fore arm. Pressure is applied on this area by moving a roll back and
forth 10 times [8]. Afterwards, the tape was removed rapidly by hand. A second stripping
on the same region was performed, this time stripped in the direction opposite to the
direction of the first stripping (from wrist to elbow or vice versa). In total 10 tapes were
collected using this method. Subsequently, the following procedure was used: immersion of
individual tapes in 3 mL methanol, sonication (10 min), collecting of extract per 2 tapes,
evaporation at 90°C, washing (2x) of residue with 150 µL dichloromethane-isopropanol
(1:1), collecting wash volumes of all 10 tapes, evaporation at 90°C and redissolving in 120
µL hexane-dichloromethane-isopropanol (100:10:10). The SPE procedure discussed in the
next section was applied to this extract.
Solid Phase Extraction
Several procedures have been explored, of which the following, performed on an 100
mg/1 mL aminopropyl-cartridge (Alltech), worked the best; conditioning step: 20 mL of
hexane-0.5% acetic acid, loading of sample, washing step: 1.5 mL of hexane, elution step:
7 mL of hexane-dichloromethane-isopropanol (8:1:1), and an additional elution step: 1 mL
of dichloromethane as a check up. The last three steps were collected as fractions,
evaporated at 90°C and redissolved in 360 µL hexane-dichloromethane-isopropanol
(100:10:10) for normal phase liquid chromatography (NPLC) analysis and in 300 µL
isopropanol-water (80:20) for reverse phase liquid chromatography (RPLC) analysis. The
procedure was first tested on a sample of a standard mix of 200 µg/mL Cer[N(24)S(18)],
Cer[N(24)P(18)], ceramide II, glycerol trioleate and cholesteryl oleate of which 10 µL was
dissolved in 100 µL hexane. Glycerol trioleate (a triglyceride) and cholesteryl oleate (a
cholesteryl ester), two compounds present in the skin, were proven to be eluted in the
washing step. The ceramide classes Cer[NS], Cer[NP] and Cer[AS] were eluted in the
elution step.
LC-MS and LC-MS/MS analysis
An NPLC method was developed using an Alliance HPLC-system (Waters). Detection was
executed with a Micromass Quattro Micro API LC-MS/MS system. Development of a
method was explored using several solvents and a Zorbax RX-SIL column (10 cm L x 3
mm I.D. x 1.8 µm dp; Agilent Technologies). The final method, using mobile phase A
hexane-isopropanol-formic acid (95:5:0.1) and mobile phase B isopropanol-
dichloromethane-hexane-formic acid (25:25:50:0.05), consists of the following steps: initial
conditions 100% A and 0% B were maintained 20 min, afterwards a transition to 50% A
and 50% B was made in 10 min. This composition was maintained 5 min. Afterwards, the
initial conditions were reached again in 5 min. These conditions were maintained 10 min to
condition the column for the next analysis. The flow was set to 0.2 mL/min.
Separation in normal phase mode is based on polarity, for ceramides this is mainly based on
the number and position of OH-groups. In this context, a separation of the different
ceramide classes was attempted. However analysis of the ceramide II standard and a mix of

ceramides of class Cer[NS] both indicated that ceramides of the same class elute over a
time span of several minutes. Furthermore, analysis of a standard mix containing classes
Cer[NS], Cer[NP] and Cer[AS] showed that different classes are not separated enough to
enable fractionation of the classes without class overlapping, as is illustrated in figure 2.
Figure 2. Overlapping chromatograms in normal phase mode of A: a 50 µg/mL mix of
Cer[NS] (10 µL injection) B: a 10 µg/mL mix of Cer[N(24)S(18)], Cer[N(24)P(18)] and
ceramide II (10 µL injection). Peaks for A: NSs = Cer[N(14)S(18)], Cer[N(15)S(18)],
Cer[N(16)S(18)], Cer[N(18)S(18)]; Cer[N(20)S(18)], Cer[N(24:0)S(18)] and
Cer[N(24:1)S(18)]; peaks for B: NS = Cer[N(24)S(18)], NP = Cer[N(24)P(18)] and ASs =
Cer[A(18)S(18)], Cer[A(22:0)S(18)], Cer[A(22:2)S(18)], Cer[A(23:0)S(18)],
Cer[A(23:2)S(18)], Cer[A(24:0)S(18)], Cer[A(24:1)S(18)], Cer[A(25:0)S(18)],
Cer[A(25:2)S(18)], Cer[A(26:0)S(18)] and Cer[A(26:1)S(18)].
For a detailed analysis of the ceramides, reverse phase mode was preferred since a higher
sensitivity is reached when an RPLC coupled to electrospray ionization mass spectrometry
(RPLC-ESI-MS) is used. A method based on previous analysis of ceramides in plasma was
utilized. A Kinetex C18 column (5 cm L x 2.1 mm I.D.; 2.6 µm dp; Phenomenex) and
mobile phases A: water-0.5 M ammonium formate (100:1) and B: methanol-formic acid
(100:0.2) were used. A gradient elution was performed at a flow of 0.3 mL/min starting
from 20% A and 80% B, which was maintained for 1 min. After this a stepwise change was
made to 0% A and 100% B. This composition was kept for 10 min. Subsequently a
stepwise transition was made again to the initial conditions and maintained 10 min to
condition the column for the next analysis.
Results and discussion
Variation in chain length of both the fatty acid and sphingoid base part, give rise to the
same mass. Moreover, the same masses can also be found in some of the different classes.
This makes it necessary in MS analysis to perform tandem mass spectrometry to identify
clearly each ceramide. MS/MS analysis in positive ionization mode will reveal the masses
of the sphingoid bases. Characteristic fragmentations take place: the fatty acid chain
together with 0 to 3 water molecules can be lost. The intensities of these fragmentation ions
will differ for each ceramide class. Since fragmentation ions of sphingoid base H and
sphingoid base DS with one carbon more, give the same masses; these two can only be
distinguished by different intensities of the fragment ions. Another issue differentiating
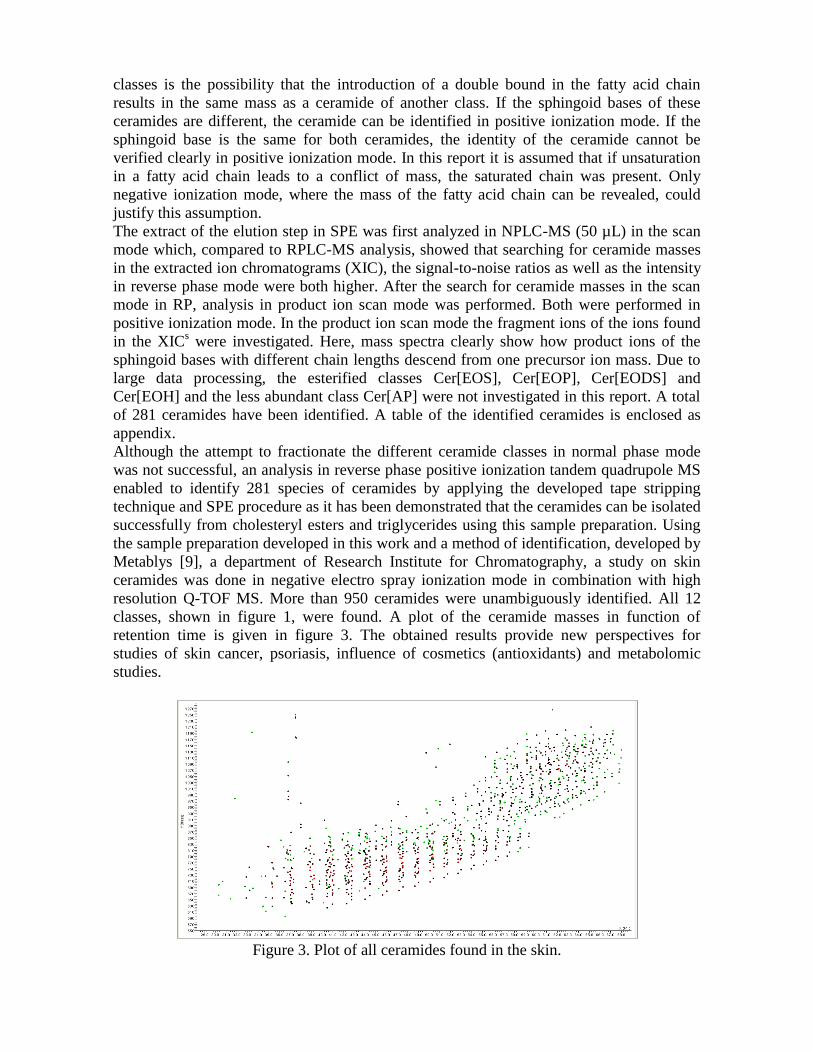
classes is the possibility that the introduction of a double bound in the fatty acid chain
results in the same mass as a ceramide of another class. If the sphingoid bases of these
ceramides are different, the ceramide can be identified in positive ionization mode. If the
sphingoid base is the same for both ceramides, the identity of the ceramide cannot be
verified clearly in positive ionization mode. In this report it is assumed that if unsaturation
in a fatty acid chain leads to a conflict of mass, the saturated chain was present. Only
negative ionization mode, where the mass of the fatty acid chain can be revealed, could
justify this assumption.
The extract of the elution step in SPE was first analyzed in NPLC-MS (50 µL) in the scan
mode which, compared to RPLC-MS analysis, showed that searching for ceramide masses
in the extracted ion chromatograms (XIC), the signal-to-noise ratios as well as the intensity
in reverse phase mode were both higher. After the search for ceramide masses in the scan
mode in RP, analysis in product ion scan mode was performed. Both were performed in
positive ionization mode. In the product ion scan mode the fragment ions of the ions found
in the XICs were investigated. Here, mass spectra clearly show how product ions of the
sphingoid bases with different chain lengths descend from one precursor ion mass. Due to
large data processing, the esterified classes Cer[EOS], Cer[EOP], Cer[EODS] and
Cer[EOH] and the less abundant class Cer[AP] were not investigated in this report. A total
of 281 ceramides have been identified. A table of the identified ceramides is enclosed as
appendix.
Although the attempt to fractionate the different ceramide classes in normal phase mode
was not successful, an analysis in reverse phase positive ionization tandem quadrupole MS
enabled to identify 281 species of ceramides by applying the developed tape stripping
technique and SPE procedure as it has been demonstrated that the ceramides can be isolated
successfully from cholesteryl esters and triglycerides using this sample preparation. Using
the sample preparation developed in this work and a method of identification, developed by
Metablys [9], a department of Research Institute for Chromatography, a study on skin
ceramides was done in negative electro spray ionization mode in combination with high
resolution Q-TOF MS. More than 950 ceramides were unambiguously identified. All 12
classes, shown in figure 1, were found. A plot of the ceramide masses in function of
retention time is given in figure 3. The obtained results provide new perspectives for
studies of skin cancer, psoriasis, influence of cosmetics (antioxidants) and metabolomic
studies.
Figure 3. Plot of all ceramides found in the skin.

References
1. Elias, P.M., Epidermal Lipids, Barrier Function, and Desquamation. The Journal of
investigative dermatology, 1983. 80(6): p. 44-49.
2. Brooks, G. and B. Idson, Skin lipids. International Journal of Cosmetic Science,
1991. 13(2): p. 103-113.
3. Di Nardo, A., et al., Ceramide and cholesterol composition of the skin of patients
with atopic dermatitis. Acta Dermato-Venereologica, 1998. 78(1): p. 27-30.
4. Holleran, W.M., Y. Takagi, and Y. Uchida, Epidermal sphingolipids: Metabolism,
function, and roles in skin disorders. Febs Letters, 2006. 580(23): p. 5456-5466.
5. Masukawa, Y., et al., Characterization of overall ceramide species in human
Stratum corneum. Journal of Lipid Research, 2008. 49(7): p. 1466-1476.
6. Motta, S., et al., Ceramide composition of the psoratic scale. Biochimica Et
Biophysica Acta, 1993. 1182(2): p. 147-151.
7. Weerheim, A. and M. Ponec, Determination of stratum corneum lipid profile by
tape stripping in combination with high-performance thin-layer chromatography.
Archives of Dermatological Research, 2001. 293(4): p. 191-199.
8. Tassopoulos, T.F., Evaluation of topical bioavailibility in human stratum corneum
in vivo by tape stripping using a direct spectroscopic method. 2006, University of
Basel: Basel.
9. Sandra, K.d.S.P., A.; Vanhoenacker, G.; David, F.; Sandra, P., Comprehensive
blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass
spectrometry. Journal of Chromatography A, 2010.
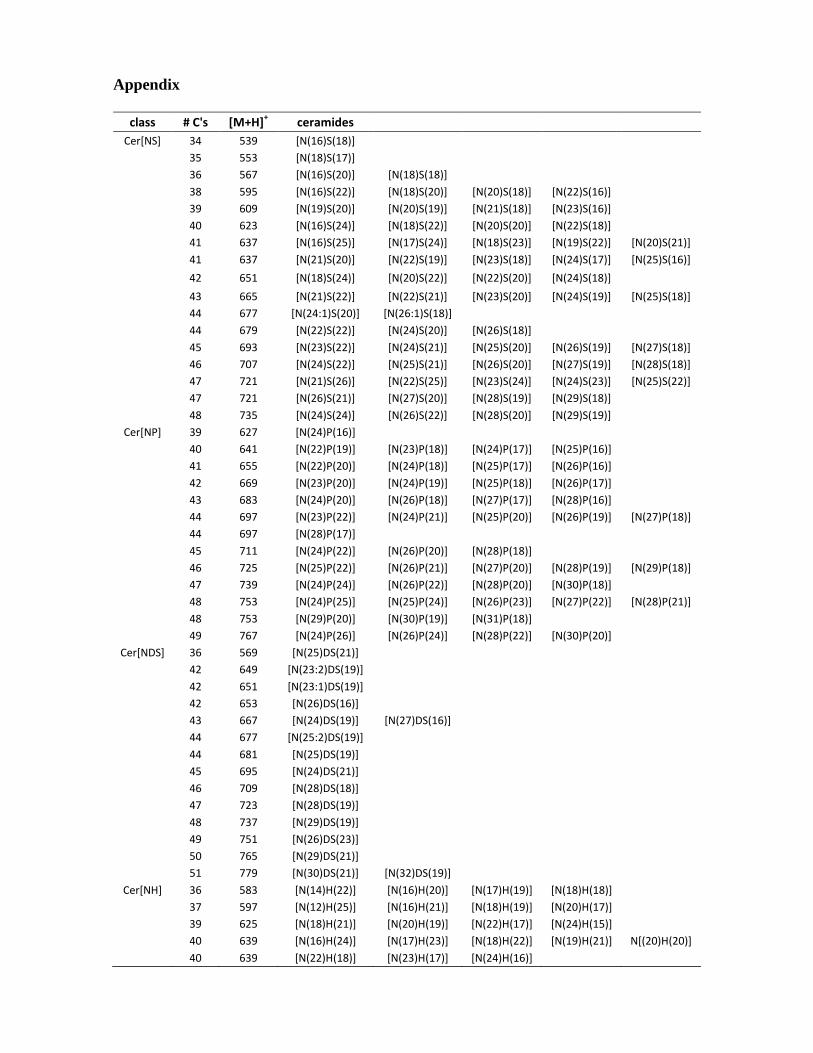
Appendix
class # C's [M+H]+ ceramides
Cer[NS] 34 539 [N(16)S(18)]
35 553 [N(18)S(17)]
36 567 [N(16)S(20)] [N(18)S(18)]
38 595 [N(16)S(22)] [N(18)S(20)] [N(20)S(18)] [N(22)S(16)]
39 609 [N(19)S(20)] [N(20)S(19)] [N(21)S(18)] [N(23)S(16)]
40 623 [N(16)S(24)] [N(18)S(22)] [N(20)S(20)] [N(22)S(18)]
41 637 [N(16)S(25)] [N(17)S(24)] [N(18)S(23)] [N(19)S(22)] [N(20)S(21)]
41 637 [N(21)S(20)] [N(22)S(19)] [N(23)S(18)] [N(24)S(17)] [N(25)S(16)]
42 651 [N(18)S(24)] [N(20)S(22)] [N(22)S(20)] [N(24)S(18)]
43 665 [N(21)S(22)] [N(22)S(21)] [N(23)S(20)] [N(24)S(19)] [N(25)S(18)]
44 677 [N(24:1)S(20)] [N(26:1)S(18)]
44 679 [N(22)S(22)] [N(24)S(20)] [N(26)S(18)]
45 693 [N(23)S(22)] [N(24)S(21)] [N(25)S(20)] [N(26)S(19)] [N(27)S(18)]
46 707 [N(24)S(22)] [N(25)S(21)] [N(26)S(20)] [N(27)S(19)] [N(28)S(18)]
47 721 [N(21)S(26)] [N(22)S(25)] [N(23)S(24)] [N(24)S(23)] [N(25)S(22)]
47 721 [N(26)S(21)] [N(27)S(20)] [N(28)S(19)] [N(29)S(18)]
48 735 [N(24)S(24)] [N(26)S(22)] [N(28)S(20)] [N(29)S(19)]
Cer[NP] 39 627 [N(24)P(16)]
40 641 [N(22)P(19)] [N(23)P(18)] [N(24)P(17)] [N(25)P(16)]
41 655 [N(22)P(20)] [N(24)P(18)] [N(25)P(17)] [N(26)P(16)]
42 669 [N(23)P(20)] [N(24)P(19)] [N(25)P(18)] [N(26)P(17)]
43 683 [N(24)P(20)] [N(26)P(18)] [N(27)P(17)] [N(28)P(16)]
44 697 [N(23)P(22)] [N(24)P(21)] [N(25)P(20)] [N(26)P(19)] [N(27)P(18)]
44 697 [N(28)P(17)]
45 711 [N(24)P(22)] [N(26)P(20)] [N(28)P(18)]
46 725 [N(25)P(22)] [N(26)P(21)] [N(27)P(20)] [N(28)P(19)] [N(29)P(18)]
47 739 [N(24)P(24)] [N(26)P(22)] [N(28)P(20)] [N(30)P(18)]
48 753 [N(24)P(25)] [N(25)P(24)] [N(26)P(23)] [N(27)P(22)] [N(28)P(21)]
48 753 [N(29)P(20)] [N(30)P(19)] [N(31)P(18)]
49 767 [N(24)P(26)] [N(26)P(24)] [N(28)P(22)] [N(30)P(20)]
Cer[NDS] 36 569 [N(25)DS(21)]
42 649 [N(23:2)DS(19)]
42 651 [N(23:1)DS(19)]
42 653 [N(26)DS(16)]
43 667 [N(24)DS(19)] [N(27)DS(16)]
44 677 [N(25:2)DS(19)]
44 681 [N(25)DS(19)]
45 695 [N(24)DS(21)]
46 709 [N(28)DS(18)]
47 723 [N(28)DS(19)]
48 737 [N(29)DS(19)]
49 751 [N(26)DS(23)]
50 765 [N(29)DS(21)]
51 779 [N(30)DS(21)] [N(32)DS(19)]
Cer[NH] 36 583 [N(14)H(22)] [N(16)H(20)] [N(17)H(19)] [N(18)H(18)]
37 597 [N(12)H(25)] [N(16)H(21)] [N(18)H(19)] [N(20)H(17)]
39 625 [N(18)H(21)] [N(20)H(19)] [N(22)H(17)] [N(24)H(15)]
40 639 [N(16)H(24)] [N(17)H(23)] [N(18)H(22)] [N(19)H(21)] N[(20)H(20)]
40 639 [N(22)H(18)] [N(23)H(17)] [N(24)H(16)]
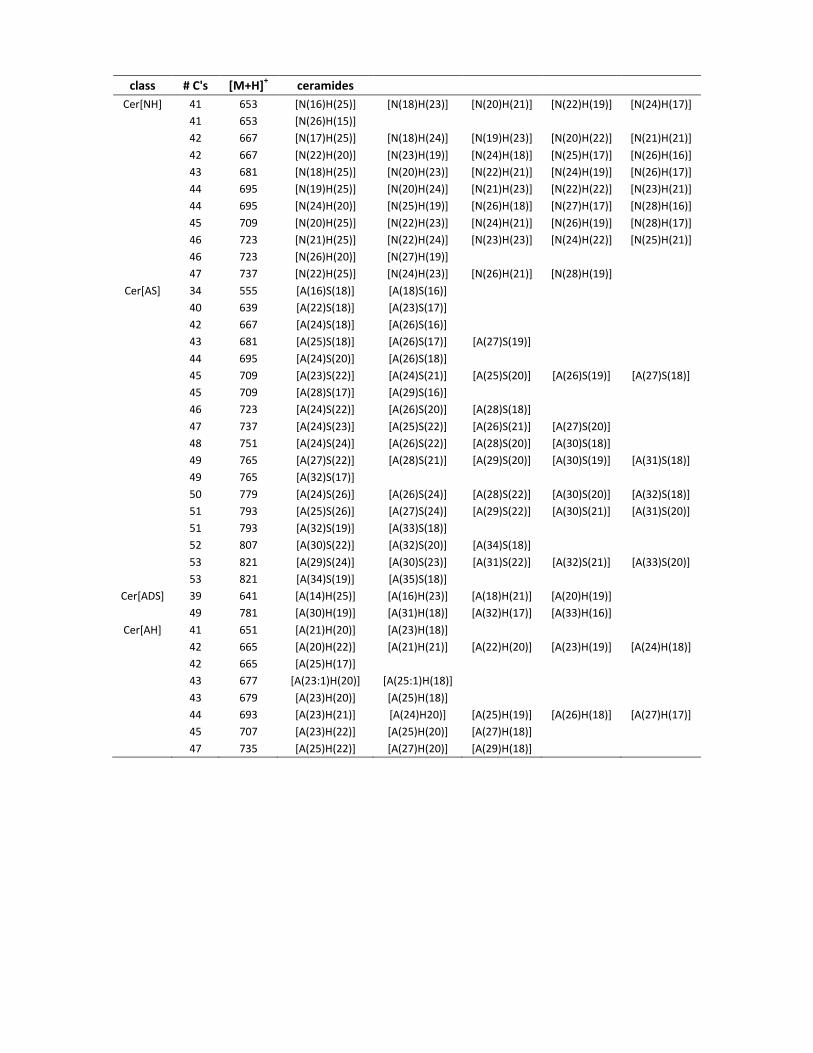
class # C's [M+H]+ ceramides
Cer[NH] 41 653 [N(16)H(25)] [N(18)H(23)] [N(20)H(21)] [N(22)H(19)] [N(24)H(17)]
41 653 [N(26)H(15)]
42 667 [N(17)H(25)] [N(18)H(24)] [N(19)H(23)] [N(20)H(22)] [N(21)H(21)]
42 667 [N(22)H(20)] [N(23)H(19)] [N(24)H(18)] [N(25)H(17)] [N(26)H(16)]
43 681 [N(18)H(25)] [N(20)H(23)] [N(22)H(21)] [N(24)H(19)] [N(26)H(17)]
44 695 [N(19)H(25)] [N(20)H(24)] [N(21)H(23)] [N(22)H(22)] [N(23)H(21)]
44 695 [N(24)H(20)] [N(25)H(19)] [N(26)H(18)] [N(27)H(17)] [N(28)H(16)]
45 709 [N(20)H(25)] [N(22)H(23)] [N(24)H(21)] [N(26)H(19)] [N(28)H(17)]
46 723 [N(21)H(25)] [N(22)H(24)] [N(23)H(23)] [N(24)H(22)] [N(25)H(21)]
46 723 [N(26)H(20)] [N(27)H(19)]
47 737 [N(22)H(25)] [N(24)H(23)] [N(26)H(21)] [N(28)H(19)]
Cer[AS] 34 555 [A(16)S(18)] [A(18)S(16)]
40 639 [A(22)S(18)] [A(23)S(17)]
42 667 [A(24)S(18)] [A(26)S(16)]
43 681 [A(25)S(18)] [A(26)S(17)] [A(27)S(19)]
44 695 [A(24)S(20)] [A(26)S(18)]
45 709 [A(23)S(22)] [A(24)S(21)] [A(25)S(20)] [A(26)S(19)] [A(27)S(18)]
45 709 [A(28)S(17)] [A(29)S(16)]
46 723 [A(24)S(22)] [A(26)S(20)] [A(28)S(18)]
47 737 [A(24)S(23)] [A(25)S(22)] [A(26)S(21)] [A(27)S(20)]
48 751 [A(24)S(24)] [A(26)S(22)] [A(28)S(20)] [A(30)S(18)]
49 765 [A(27)S(22)] [A(28)S(21)] [A(29)S(20)] [A(30)S(19)] [A(31)S(18)]
49 765 [A(32)S(17)]
50 779 [A(24)S(26)] [A(26)S(24)] [A(28)S(22)] [A(30)S(20)] [A(32)S(18)]
51 793 [A(25)S(26)] [A(27)S(24)] [A(29)S(22)] [A(30)S(21)] [A(31)S(20)]
51 793 [A(32)S(19)] [A(33)S(18)]
52 807 [A(30)S(22)] [A(32)S(20)] [A(34)S(18)]
53 821 [A(29)S(24)] [A(30)S(23)] [A(31)S(22)] [A(32)S(21)] [A(33)S(20)]
53 821 [A(34)S(19)] [A(35)S(18)]
Cer[ADS] 39 641 [A(14)H(25)] [A(16)H(23)] [A(18)H(21)] [A(20)H(19)]
49 781 [A(30)H(19)] [A(31)H(18)] [A(32)H(17)] [A(33)H(16)]
Cer[AH] 41 651 [A(21)H(20)] [A(23)H(18)]
42 665 [A(20)H(22)] [A(21)H(21)] [A(22)H(20)] [A(23)H(19)] [A(24)H(18)]
42 665 [A(25)H(17)]
43 677 [A(23:1)H(20)] [A(25:1)H(18)]
43 679 [A(23)H(20)] [A(25)H(18)]
44 693 [A(23)H(21)] [A(24)H20)] [A(25)H(19)] [A(26)H(18)] [A(27)H(17)]
45 707 [A(23)H(22)] [A(25)H(20)] [A(27)H(18)]
47 735 [A(25)H(22)] [A(27)H(20)] [A(29)H(18)]
















![[MS]MySeason 1](https://static.fdocuments.nl/doc/165x107/5599d9cb1a28abff4f8b457f/msmyseason-1.jpg)

