
不斉 Diels-Alder 反応を用いる 天然物の不斉合成研...
50
博士論文 不斉 Diels-Alder 反応を用いる 天然物の不斉合成研究 千葉大学大学院 医学薬学府 創薬生命科学専攻 薬品合成化学研究室 平岡 平岡 平岡 平岡 紫陽 紫陽 紫陽 紫陽 2013 年(平成 25 年)修了
Transcript of 不斉 Diels-Alder 反応を用いる 天然物の不斉合成研...

博士論文
不斉 Diels-Alder 反応を用いる
天然物の不斉合成研究
千葉大学大学院 医学薬学府 創薬生命科学専攻
薬品合成化学研究室
平岡平岡平岡平岡 紫陽紫陽紫陽紫陽
2013 年(平成 25 年)修了
2
略語表 Ac acetyl aq aqueous BINOL 11rsquo-bi-22rsquo-naphthol Bn benzyl Boc t-butyloxycarbonyl Bs benzenesulfonyl BTF benzotrifluoride Bu butyl Bz benzoyl cat catalytic CBS Corey-Bakshi-Shibata dba dibenzylideneacetone DBU 18-diazabicyclo[540]undec-7-ene DDQ 23-dichloro-56-dicyano-p-benzoquinone DMAP 4-dimethylaminopyridine DMF dimethylformamide DMDO dimethyldioxirane DMSO dimethylsulfoxide DMT-MM 4-(46-dimethoxy-135-triazin-2-yl)-4-methylmorpholinium Chloride EDCI 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride ee enantiomeric excess Et ethyl h hour(s) KHMDS potassium bis(trimethylsilyl)amide LAH lithium aluminum hydride LTMP lithium tetramethylpiperidide mCPBA m-chloroperbenzoic acid Me methyl min minute(s) MS molecular sieves Ms mesyl NMO N-methylmorpholine NOE nuclear Overhauser effect Ph phenyl Piv pivaloyl PPTS pyridinium p-toluenesulfonate Pr propyl PTLC preparative thin layer chromatography py pyridine RCM ring closing metathesis rt room temperature TBAI tetrabutylammonium iodide TBDPS t-butyldiphenylsilyl TBS t-butyldimethylsilyl temp temperature TES triethylsilyl Tf trifluoromethanesulfonyl TFA trifluoroacetic acid TFAA trifluoroacetic anhydride THF tetrahydrofuran TIPS triisopropylsilyl TMS trimethylsilyl Tol p-tolyl TPAP tetrapropylammonium perruthenate
3
目次
略語表 2
序論
第一節 Danishefsky 型ジエンの天然物合成への利用 4
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応 5
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応 6
本論
第一章 (ndash)-platyphyllide の触媒的不斉全合成 7
第一節 (ndash)-platyphyllideについて 7
第二節 (+)-platyphyllide の触媒的不斉全合成 7
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定 9
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化 11
第三章 マンザミン B の全合成研究 14
第一節 マンザミン B 及び関連するアルカロイドについて 14
第二節 過去の研究及び合成戦略 14
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討 18
第四節 C 環構築の検討 20
第五節 D 環構築の検討 23
総括 26
実験の部 27
参考文献及び注 47
論文目録及び学会発表 49
審査委員 50
謝辞 50
4
序論
第一節 Danishefsky 型ジエンの天然物合成への利用
天然化合物やその他の生理活性物質がもつ基本的な環構造のひとつに炭素六員環が挙げられるこ
のような化合物においては炭素六員環は多数の置換基をもちまたは縮環しさらには不斉点をもっ
ていることが多いすなわち炭素六員環を不斉合成する手法の開発は光学活性天然物の効率的な合成
において非常に重要である
炭素六員環を構築する一般的な方法としてDiels-Alder 反応が挙げられるDiels-Alder 反応は四
炭素からなるジエンと二炭素からなるジエノフィルに種々の置換基を導入することによって多置換シ
クロヘキセンや縮環した構造を立体位置選択的に得ることができるため天然物合成において広く用
いられてきた置換基の導入されたジエンのうち特に 1974 年に Danishefsky 及び北原によって報告
された 1-methoxy-3-trimethylsilyloxy-13-butadiene (Danishefsky ジエン)1 (1a)をはじめとする高
度に酸化されたジエン(Figure 1)は電子不足オレフ
ィンとの Diels-Alder 反応において高い反応性を示
し酸素官能基化されたシクロヘキセン誘導体を与
えることから天然物合成において非常に有用であ
る (Scheme 1)一例として当研究室の小野は
Danishefsky ジエンを用いる Diels-Alder 反応によって Nakadomarin A の不斉全合成を達成している
(Scheme 2)2
しかしながらDanishefsky 型ジエンはその反応性の高さ故に酸に対して不安定であることからル
イス酸触媒によるジエノフィルの活性化は難しく不斉ルイス酸触媒を用いた触媒的不斉 Diels-Alder
反応の成功例はアルデヒドやイミンに対するヘテロ Diels-Alder 反応の例を除いては非常に少ない
34 これに対し当研究室では 2008 年に Danishefsky ジエンとオレフィンの初の触媒的 Diels-Alder
反応を5a Rawal らは 2000 年に Rawal ジエン(2)とオレフィンの初の触媒的 Diels-Alder 反応を報告し
た6c 次節ではこれら二つの不斉 Diels-Alder 反応について述べる
NMe2
TBSO
OTMS
TMSO
OMe
OMe
TMSO
Danisheskys diene
1a
Rawals diene Brassards diene
Figure 1 Highly oxygenated dienes
2a
OMe
TMSO
REWG
OMe
TMSO
EWG
R
Scheme 1
Danisheskys diene
Scheme 2
1a
BsN
O
O
NBs 2) TFA
BsN
O
OH
O
NBsOMe
OTBS
N
O
N
H
(-)-nakadomarin A
1) 180 oC+
1b
1 Highly functionalized
2 Regioselective
3 Stereoselective (normally endo selective)
5
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応
井口らは 2001 年にランタノイド類
が Danishefsky ジ エ ン を 用 い た
Diels-Alder 反応の有効な触媒となる
ことを報告した(Scheme 3)7 これ
によりキラルランタノイド触媒の開発によって本反応の不斉化を達成できることが示唆された
小林らは Yb(OTf)3BINOL 及び嵩高い三級アミンより調整されるキラル希土類錯体を調製しシク
ロペンタジエンと N-アシルオキサ
ゾリジノンとの不斉 Diels-Alder 反
応において最高 95 ee の不斉収率
で反応を進行させることに成功して
いる(Scheme 4)8
当 研 究 室 の 山 中 は 上 記 の 小 林 ら の 反 応 に な ら い Yb(OTf)3 と 独 自 の 不 斉 配 位 子
BINAMIDE[11rsquo-(22rsquo-bisacylamino)binaphthalene]及び iPr2NEt から光学活性 Yb 錯体を調製し
シクロペンタジエンとクロトニルオキサゾリジノンとの高収率かつ高エナンチオ選択的な不斉
Diels-Alder 反応の開発に成功している(Scheme 5)9
当研究室の須藤は上述の井口らの結果をもとに本錯体を Danishefsky ジエンを用いる Diels-Alder
反応に適用しDanishefsky ジエン 1111 と電子不足オレフィン 3333 との初の触媒的不斉 Diels-Alder 反応の
開発に成功したこれにより有用な合成中間体である光学活性シクロヘキセノン誘導体が高い光学純
度で得られるようになった(Table 1)5a 尚このとき用いた BINAMIDE は 35-difluoro 体 5555 が最
適であり以降5555 を単に BINAMIDE と呼称するさらに当研究室の原田は BINAMIDE を改良し
ウレア側鎖をもつ BINUREA を開発し不斉収率や基質一般性の向上に成功した5b
O
Me
Me H
tBuO2C
OMe
TMSO
O
CO2tBu
Ac
Me
+
Yb(OTf)3(10 mol)
toluene 0 oC87
Scheme 3 Inokuchis work
1a
N
O
O
O
NO O
O
Scheme 4 Kobayashis work
+
chiral Yb complex(20 mol)
with or without additive
MS 4A CH2Cl2 0oC
chiral Yb complex = Yb(OTf)3 + (R)-BINOL + cis-126-trimethylpiperidine (11224)
73 95 ee
3a
N
O
O
O
NO O
O
+
chiral Yb complex(25 mol)
CH2Cl2 0oC
endo (2R 3S)
chiral Yb complex = Yb(OTf)3 + BINAMIDE (R = Ph) + iPr2NEt (11224)
95 gt98 ee (endo)endoexo = 911
NH
NH
O
R
O
R
BINAMIDE
Scheme 5
3a
OMe
SiO
R N O
OOO R
N O
O O
F
F
F
F
N
N
O
O
H
H
+
1) chiral Yb complex
(10 mol)
CH2Cl2
1 eq
2) TFA
chiral Yb complex = Yb(OTf)3 + 5 + DBU (1 12 24) or Yb(OTf)3 + 6 + DBU (1 1 2)
2 eq
5BINAMIDE
3 41
Table 1
R
Me
n-Pr
iBu
iPr
Ph
PhO2C
H
4 ()
quant
93
88
29
26
quant
93
optimized data ofBINAMIDE
optimized data ofBINUREA
linearalkyl chain
branchedalkyl chain
ester
non-substituted
ee
92
97
87
56
60
90
71
4 ()
quant
94
90
84
90
88
21
ee
98
99
97
98
97
86
37 6BINUREA
NH
HN
NH
O
Ph
HN
O
Ph
6
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応
Rawal らは 1997 年に 1-amino-3-siloxybutadiene(2222aaaaRawal ジエン)の合成及びその Diels-Alder 反
応を報告した6a Rawal ジエン(2222aaaa)は Danishefsky ジエンに比べて 3300 倍の反応性を示すほか6b ア
ミノ基を導入できることから含窒素化合物の合成に対し有用であるさらに 2000 年に Rawal らはア
ミノ基に電子吸引基を導入したジエン 2b2b2b2b を用いキラルクロムサレン錯体 7a7a7a7a を触媒とする触媒的不斉
Diels-Alder 反応の開発に成功し報告した(Table 2)6c 本反応ではアミノ基をもつシクロヘキセンま
たは 6-5 員環が縮合した化合物を高立体選択的高エナンチオ選択的に得ることができRawal らはこ
れを用いたアルカロイドの不斉全合成も達成している10
筆者は以下に示す検討を行い以下の結果を得た
(1) 当研究室で開発された Danishefsky ジエンの触媒的不斉 Diels-Alder 反応を鍵工程とし
(ndash)-platyphyllide の触媒的不斉全合成を達成しその絶対配置を改定した
(2) platyphyllide 合成経路において伊藤-三枝酸化を用いた際に少量得られたオレフィンの位置が異
なる副生成物についてこれを選択的に得る条件を求め反応機構を推定した
(3) Rawal らの不斉 Diels-Alder 反応を鍵工程としマンザミン B の全合成研究を行った
本論文は以下の章により構成される
第一章 (ndash)-platyphyllide の触媒的不斉全合成
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
第三章 マンザミン B の全合成研究
以下その研究の経緯について詳細に記述する
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

2
略語表 Ac acetyl aq aqueous BINOL 11rsquo-bi-22rsquo-naphthol Bn benzyl Boc t-butyloxycarbonyl Bs benzenesulfonyl BTF benzotrifluoride Bu butyl Bz benzoyl cat catalytic CBS Corey-Bakshi-Shibata dba dibenzylideneacetone DBU 18-diazabicyclo[540]undec-7-ene DDQ 23-dichloro-56-dicyano-p-benzoquinone DMAP 4-dimethylaminopyridine DMF dimethylformamide DMDO dimethyldioxirane DMSO dimethylsulfoxide DMT-MM 4-(46-dimethoxy-135-triazin-2-yl)-4-methylmorpholinium Chloride EDCI 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride ee enantiomeric excess Et ethyl h hour(s) KHMDS potassium bis(trimethylsilyl)amide LAH lithium aluminum hydride LTMP lithium tetramethylpiperidide mCPBA m-chloroperbenzoic acid Me methyl min minute(s) MS molecular sieves Ms mesyl NMO N-methylmorpholine NOE nuclear Overhauser effect Ph phenyl Piv pivaloyl PPTS pyridinium p-toluenesulfonate Pr propyl PTLC preparative thin layer chromatography py pyridine RCM ring closing metathesis rt room temperature TBAI tetrabutylammonium iodide TBDPS t-butyldiphenylsilyl TBS t-butyldimethylsilyl temp temperature TES triethylsilyl Tf trifluoromethanesulfonyl TFA trifluoroacetic acid TFAA trifluoroacetic anhydride THF tetrahydrofuran TIPS triisopropylsilyl TMS trimethylsilyl Tol p-tolyl TPAP tetrapropylammonium perruthenate
3
目次
略語表 2
序論
第一節 Danishefsky 型ジエンの天然物合成への利用 4
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応 5
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応 6
本論
第一章 (ndash)-platyphyllide の触媒的不斉全合成 7
第一節 (ndash)-platyphyllideについて 7
第二節 (+)-platyphyllide の触媒的不斉全合成 7
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定 9
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化 11
第三章 マンザミン B の全合成研究 14
第一節 マンザミン B 及び関連するアルカロイドについて 14
第二節 過去の研究及び合成戦略 14
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討 18
第四節 C 環構築の検討 20
第五節 D 環構築の検討 23
総括 26
実験の部 27
参考文献及び注 47
論文目録及び学会発表 49
審査委員 50
謝辞 50
4
序論
第一節 Danishefsky 型ジエンの天然物合成への利用
天然化合物やその他の生理活性物質がもつ基本的な環構造のひとつに炭素六員環が挙げられるこ
のような化合物においては炭素六員環は多数の置換基をもちまたは縮環しさらには不斉点をもっ
ていることが多いすなわち炭素六員環を不斉合成する手法の開発は光学活性天然物の効率的な合成
において非常に重要である
炭素六員環を構築する一般的な方法としてDiels-Alder 反応が挙げられるDiels-Alder 反応は四
炭素からなるジエンと二炭素からなるジエノフィルに種々の置換基を導入することによって多置換シ
クロヘキセンや縮環した構造を立体位置選択的に得ることができるため天然物合成において広く用
いられてきた置換基の導入されたジエンのうち特に 1974 年に Danishefsky 及び北原によって報告
された 1-methoxy-3-trimethylsilyloxy-13-butadiene (Danishefsky ジエン)1 (1a)をはじめとする高
度に酸化されたジエン(Figure 1)は電子不足オレフ
ィンとの Diels-Alder 反応において高い反応性を示
し酸素官能基化されたシクロヘキセン誘導体を与
えることから天然物合成において非常に有用であ
る (Scheme 1)一例として当研究室の小野は
Danishefsky ジエンを用いる Diels-Alder 反応によって Nakadomarin A の不斉全合成を達成している
(Scheme 2)2
しかしながらDanishefsky 型ジエンはその反応性の高さ故に酸に対して不安定であることからル
イス酸触媒によるジエノフィルの活性化は難しく不斉ルイス酸触媒を用いた触媒的不斉 Diels-Alder
反応の成功例はアルデヒドやイミンに対するヘテロ Diels-Alder 反応の例を除いては非常に少ない
34 これに対し当研究室では 2008 年に Danishefsky ジエンとオレフィンの初の触媒的 Diels-Alder
反応を5a Rawal らは 2000 年に Rawal ジエン(2)とオレフィンの初の触媒的 Diels-Alder 反応を報告し
た6c 次節ではこれら二つの不斉 Diels-Alder 反応について述べる
NMe2
TBSO
OTMS
TMSO
OMe
OMe
TMSO
Danisheskys diene
1a
Rawals diene Brassards diene
Figure 1 Highly oxygenated dienes
2a
OMe
TMSO
REWG
OMe
TMSO
EWG
R
Scheme 1
Danisheskys diene
Scheme 2
1a
BsN
O
O
NBs 2) TFA
BsN
O
OH
O
NBsOMe
OTBS
N
O
N
H
(-)-nakadomarin A
1) 180 oC+
1b
1 Highly functionalized
2 Regioselective
3 Stereoselective (normally endo selective)
5
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応
井口らは 2001 年にランタノイド類
が Danishefsky ジ エ ン を 用 い た
Diels-Alder 反応の有効な触媒となる
ことを報告した(Scheme 3)7 これ
によりキラルランタノイド触媒の開発によって本反応の不斉化を達成できることが示唆された
小林らは Yb(OTf)3BINOL 及び嵩高い三級アミンより調整されるキラル希土類錯体を調製しシク
ロペンタジエンと N-アシルオキサ
ゾリジノンとの不斉 Diels-Alder 反
応において最高 95 ee の不斉収率
で反応を進行させることに成功して
いる(Scheme 4)8
当 研 究 室 の 山 中 は 上 記 の 小 林 ら の 反 応 に な ら い Yb(OTf)3 と 独 自 の 不 斉 配 位 子
BINAMIDE[11rsquo-(22rsquo-bisacylamino)binaphthalene]及び iPr2NEt から光学活性 Yb 錯体を調製し
シクロペンタジエンとクロトニルオキサゾリジノンとの高収率かつ高エナンチオ選択的な不斉
Diels-Alder 反応の開発に成功している(Scheme 5)9
当研究室の須藤は上述の井口らの結果をもとに本錯体を Danishefsky ジエンを用いる Diels-Alder
反応に適用しDanishefsky ジエン 1111 と電子不足オレフィン 3333 との初の触媒的不斉 Diels-Alder 反応の
開発に成功したこれにより有用な合成中間体である光学活性シクロヘキセノン誘導体が高い光学純
度で得られるようになった(Table 1)5a 尚このとき用いた BINAMIDE は 35-difluoro 体 5555 が最
適であり以降5555 を単に BINAMIDE と呼称するさらに当研究室の原田は BINAMIDE を改良し
ウレア側鎖をもつ BINUREA を開発し不斉収率や基質一般性の向上に成功した5b
O
Me
Me H
tBuO2C
OMe
TMSO
O
CO2tBu
Ac
Me
+
Yb(OTf)3(10 mol)
toluene 0 oC87
Scheme 3 Inokuchis work
1a
N
O
O
O
NO O
O
Scheme 4 Kobayashis work
+
chiral Yb complex(20 mol)
with or without additive
MS 4A CH2Cl2 0oC
chiral Yb complex = Yb(OTf)3 + (R)-BINOL + cis-126-trimethylpiperidine (11224)
73 95 ee
3a
N
O
O
O
NO O
O
+
chiral Yb complex(25 mol)
CH2Cl2 0oC
endo (2R 3S)
chiral Yb complex = Yb(OTf)3 + BINAMIDE (R = Ph) + iPr2NEt (11224)
95 gt98 ee (endo)endoexo = 911
NH
NH
O
R
O
R
BINAMIDE
Scheme 5
3a
OMe
SiO
R N O
OOO R
N O
O O
F
F
F
F
N
N
O
O
H
H
+
1) chiral Yb complex
(10 mol)
CH2Cl2
1 eq
2) TFA
chiral Yb complex = Yb(OTf)3 + 5 + DBU (1 12 24) or Yb(OTf)3 + 6 + DBU (1 1 2)
2 eq
5BINAMIDE
3 41
Table 1
R
Me
n-Pr
iBu
iPr
Ph
PhO2C
H
4 ()
quant
93
88
29
26
quant
93
optimized data ofBINAMIDE
optimized data ofBINUREA
linearalkyl chain
branchedalkyl chain
ester
non-substituted
ee
92
97
87
56
60
90
71
4 ()
quant
94
90
84
90
88
21
ee
98
99
97
98
97
86
37 6BINUREA
NH
HN
NH
O
Ph
HN
O
Ph
6
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応
Rawal らは 1997 年に 1-amino-3-siloxybutadiene(2222aaaaRawal ジエン)の合成及びその Diels-Alder 反
応を報告した6a Rawal ジエン(2222aaaa)は Danishefsky ジエンに比べて 3300 倍の反応性を示すほか6b ア
ミノ基を導入できることから含窒素化合物の合成に対し有用であるさらに 2000 年に Rawal らはア
ミノ基に電子吸引基を導入したジエン 2b2b2b2b を用いキラルクロムサレン錯体 7a7a7a7a を触媒とする触媒的不斉
Diels-Alder 反応の開発に成功し報告した(Table 2)6c 本反応ではアミノ基をもつシクロヘキセンま
たは 6-5 員環が縮合した化合物を高立体選択的高エナンチオ選択的に得ることができRawal らはこ
れを用いたアルカロイドの不斉全合成も達成している10
筆者は以下に示す検討を行い以下の結果を得た
(1) 当研究室で開発された Danishefsky ジエンの触媒的不斉 Diels-Alder 反応を鍵工程とし
(ndash)-platyphyllide の触媒的不斉全合成を達成しその絶対配置を改定した
(2) platyphyllide 合成経路において伊藤-三枝酸化を用いた際に少量得られたオレフィンの位置が異
なる副生成物についてこれを選択的に得る条件を求め反応機構を推定した
(3) Rawal らの不斉 Diels-Alder 反応を鍵工程としマンザミン B の全合成研究を行った
本論文は以下の章により構成される
第一章 (ndash)-platyphyllide の触媒的不斉全合成
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
第三章 マンザミン B の全合成研究
以下その研究の経緯について詳細に記述する
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

3
目次
略語表 2
序論
第一節 Danishefsky 型ジエンの天然物合成への利用 4
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応 5
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応 6
本論
第一章 (ndash)-platyphyllide の触媒的不斉全合成 7
第一節 (ndash)-platyphyllideについて 7
第二節 (+)-platyphyllide の触媒的不斉全合成 7
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定 9
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化 11
第三章 マンザミン B の全合成研究 14
第一節 マンザミン B 及び関連するアルカロイドについて 14
第二節 過去の研究及び合成戦略 14
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討 18
第四節 C 環構築の検討 20
第五節 D 環構築の検討 23
総括 26
実験の部 27
参考文献及び注 47
論文目録及び学会発表 49
審査委員 50
謝辞 50
4
序論
第一節 Danishefsky 型ジエンの天然物合成への利用
天然化合物やその他の生理活性物質がもつ基本的な環構造のひとつに炭素六員環が挙げられるこ
のような化合物においては炭素六員環は多数の置換基をもちまたは縮環しさらには不斉点をもっ
ていることが多いすなわち炭素六員環を不斉合成する手法の開発は光学活性天然物の効率的な合成
において非常に重要である
炭素六員環を構築する一般的な方法としてDiels-Alder 反応が挙げられるDiels-Alder 反応は四
炭素からなるジエンと二炭素からなるジエノフィルに種々の置換基を導入することによって多置換シ
クロヘキセンや縮環した構造を立体位置選択的に得ることができるため天然物合成において広く用
いられてきた置換基の導入されたジエンのうち特に 1974 年に Danishefsky 及び北原によって報告
された 1-methoxy-3-trimethylsilyloxy-13-butadiene (Danishefsky ジエン)1 (1a)をはじめとする高
度に酸化されたジエン(Figure 1)は電子不足オレフ
ィンとの Diels-Alder 反応において高い反応性を示
し酸素官能基化されたシクロヘキセン誘導体を与
えることから天然物合成において非常に有用であ
る (Scheme 1)一例として当研究室の小野は
Danishefsky ジエンを用いる Diels-Alder 反応によって Nakadomarin A の不斉全合成を達成している
(Scheme 2)2
しかしながらDanishefsky 型ジエンはその反応性の高さ故に酸に対して不安定であることからル
イス酸触媒によるジエノフィルの活性化は難しく不斉ルイス酸触媒を用いた触媒的不斉 Diels-Alder
反応の成功例はアルデヒドやイミンに対するヘテロ Diels-Alder 反応の例を除いては非常に少ない
34 これに対し当研究室では 2008 年に Danishefsky ジエンとオレフィンの初の触媒的 Diels-Alder
反応を5a Rawal らは 2000 年に Rawal ジエン(2)とオレフィンの初の触媒的 Diels-Alder 反応を報告し
た6c 次節ではこれら二つの不斉 Diels-Alder 反応について述べる
NMe2
TBSO
OTMS
TMSO
OMe
OMe
TMSO
Danisheskys diene
1a
Rawals diene Brassards diene
Figure 1 Highly oxygenated dienes
2a
OMe
TMSO
REWG
OMe
TMSO
EWG
R
Scheme 1
Danisheskys diene
Scheme 2
1a
BsN
O
O
NBs 2) TFA
BsN
O
OH
O
NBsOMe
OTBS
N
O
N
H
(-)-nakadomarin A
1) 180 oC+
1b
1 Highly functionalized
2 Regioselective
3 Stereoselective (normally endo selective)
5
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応
井口らは 2001 年にランタノイド類
が Danishefsky ジ エ ン を 用 い た
Diels-Alder 反応の有効な触媒となる
ことを報告した(Scheme 3)7 これ
によりキラルランタノイド触媒の開発によって本反応の不斉化を達成できることが示唆された
小林らは Yb(OTf)3BINOL 及び嵩高い三級アミンより調整されるキラル希土類錯体を調製しシク
ロペンタジエンと N-アシルオキサ
ゾリジノンとの不斉 Diels-Alder 反
応において最高 95 ee の不斉収率
で反応を進行させることに成功して
いる(Scheme 4)8
当 研 究 室 の 山 中 は 上 記 の 小 林 ら の 反 応 に な ら い Yb(OTf)3 と 独 自 の 不 斉 配 位 子
BINAMIDE[11rsquo-(22rsquo-bisacylamino)binaphthalene]及び iPr2NEt から光学活性 Yb 錯体を調製し
シクロペンタジエンとクロトニルオキサゾリジノンとの高収率かつ高エナンチオ選択的な不斉
Diels-Alder 反応の開発に成功している(Scheme 5)9
当研究室の須藤は上述の井口らの結果をもとに本錯体を Danishefsky ジエンを用いる Diels-Alder
反応に適用しDanishefsky ジエン 1111 と電子不足オレフィン 3333 との初の触媒的不斉 Diels-Alder 反応の
開発に成功したこれにより有用な合成中間体である光学活性シクロヘキセノン誘導体が高い光学純
度で得られるようになった(Table 1)5a 尚このとき用いた BINAMIDE は 35-difluoro 体 5555 が最
適であり以降5555 を単に BINAMIDE と呼称するさらに当研究室の原田は BINAMIDE を改良し
ウレア側鎖をもつ BINUREA を開発し不斉収率や基質一般性の向上に成功した5b
O
Me
Me H
tBuO2C
OMe
TMSO
O
CO2tBu
Ac
Me
+
Yb(OTf)3(10 mol)
toluene 0 oC87
Scheme 3 Inokuchis work
1a
N
O
O
O
NO O
O
Scheme 4 Kobayashis work
+
chiral Yb complex(20 mol)
with or without additive
MS 4A CH2Cl2 0oC
chiral Yb complex = Yb(OTf)3 + (R)-BINOL + cis-126-trimethylpiperidine (11224)
73 95 ee
3a
N
O
O
O
NO O
O
+
chiral Yb complex(25 mol)
CH2Cl2 0oC
endo (2R 3S)
chiral Yb complex = Yb(OTf)3 + BINAMIDE (R = Ph) + iPr2NEt (11224)
95 gt98 ee (endo)endoexo = 911
NH
NH
O
R
O
R
BINAMIDE
Scheme 5
3a
OMe
SiO
R N O
OOO R
N O
O O
F
F
F
F
N
N
O
O
H
H
+
1) chiral Yb complex
(10 mol)
CH2Cl2
1 eq
2) TFA
chiral Yb complex = Yb(OTf)3 + 5 + DBU (1 12 24) or Yb(OTf)3 + 6 + DBU (1 1 2)
2 eq
5BINAMIDE
3 41
Table 1
R
Me
n-Pr
iBu
iPr
Ph
PhO2C
H
4 ()
quant
93
88
29
26
quant
93
optimized data ofBINAMIDE
optimized data ofBINUREA
linearalkyl chain
branchedalkyl chain
ester
non-substituted
ee
92
97
87
56
60
90
71
4 ()
quant
94
90
84
90
88
21
ee
98
99
97
98
97
86
37 6BINUREA
NH
HN
NH
O
Ph
HN
O
Ph
6
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応
Rawal らは 1997 年に 1-amino-3-siloxybutadiene(2222aaaaRawal ジエン)の合成及びその Diels-Alder 反
応を報告した6a Rawal ジエン(2222aaaa)は Danishefsky ジエンに比べて 3300 倍の反応性を示すほか6b ア
ミノ基を導入できることから含窒素化合物の合成に対し有用であるさらに 2000 年に Rawal らはア
ミノ基に電子吸引基を導入したジエン 2b2b2b2b を用いキラルクロムサレン錯体 7a7a7a7a を触媒とする触媒的不斉
Diels-Alder 反応の開発に成功し報告した(Table 2)6c 本反応ではアミノ基をもつシクロヘキセンま
たは 6-5 員環が縮合した化合物を高立体選択的高エナンチオ選択的に得ることができRawal らはこ
れを用いたアルカロイドの不斉全合成も達成している10
筆者は以下に示す検討を行い以下の結果を得た
(1) 当研究室で開発された Danishefsky ジエンの触媒的不斉 Diels-Alder 反応を鍵工程とし
(ndash)-platyphyllide の触媒的不斉全合成を達成しその絶対配置を改定した
(2) platyphyllide 合成経路において伊藤-三枝酸化を用いた際に少量得られたオレフィンの位置が異
なる副生成物についてこれを選択的に得る条件を求め反応機構を推定した
(3) Rawal らの不斉 Diels-Alder 反応を鍵工程としマンザミン B の全合成研究を行った
本論文は以下の章により構成される
第一章 (ndash)-platyphyllide の触媒的不斉全合成
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
第三章 マンザミン B の全合成研究
以下その研究の経緯について詳細に記述する
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
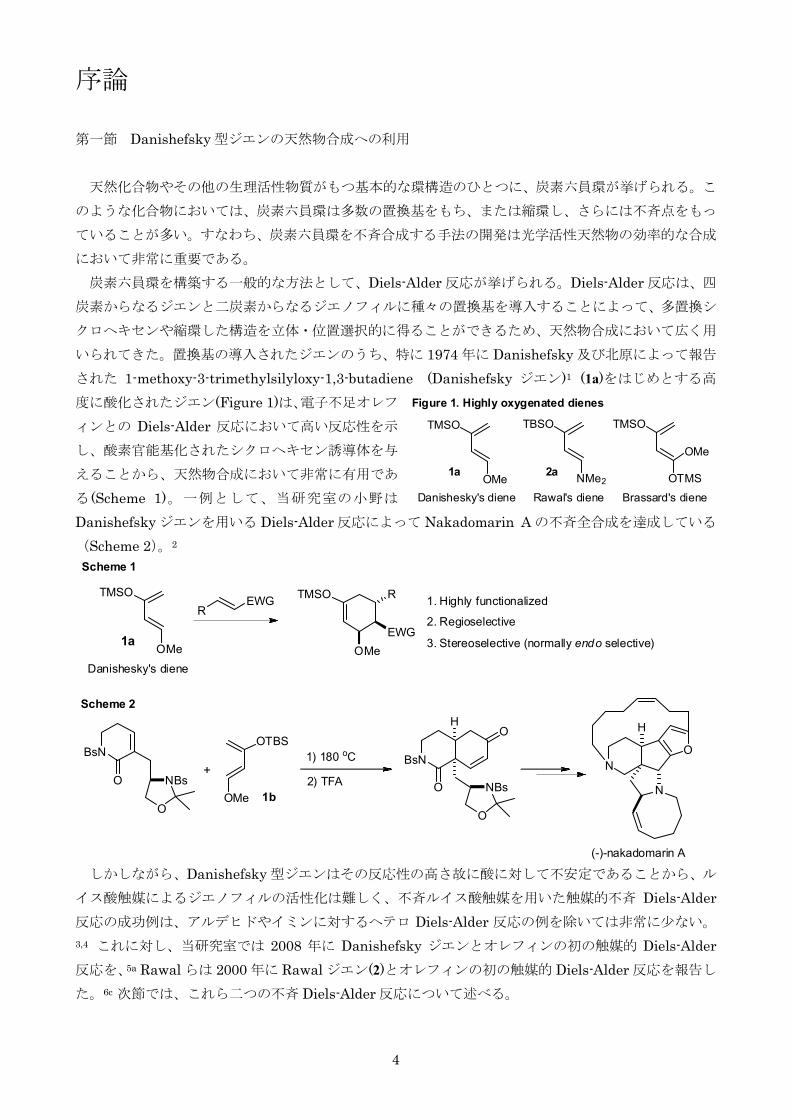
4
序論
第一節 Danishefsky 型ジエンの天然物合成への利用
天然化合物やその他の生理活性物質がもつ基本的な環構造のひとつに炭素六員環が挙げられるこ
のような化合物においては炭素六員環は多数の置換基をもちまたは縮環しさらには不斉点をもっ
ていることが多いすなわち炭素六員環を不斉合成する手法の開発は光学活性天然物の効率的な合成
において非常に重要である
炭素六員環を構築する一般的な方法としてDiels-Alder 反応が挙げられるDiels-Alder 反応は四
炭素からなるジエンと二炭素からなるジエノフィルに種々の置換基を導入することによって多置換シ
クロヘキセンや縮環した構造を立体位置選択的に得ることができるため天然物合成において広く用
いられてきた置換基の導入されたジエンのうち特に 1974 年に Danishefsky 及び北原によって報告
された 1-methoxy-3-trimethylsilyloxy-13-butadiene (Danishefsky ジエン)1 (1a)をはじめとする高
度に酸化されたジエン(Figure 1)は電子不足オレフ
ィンとの Diels-Alder 反応において高い反応性を示
し酸素官能基化されたシクロヘキセン誘導体を与
えることから天然物合成において非常に有用であ
る (Scheme 1)一例として当研究室の小野は
Danishefsky ジエンを用いる Diels-Alder 反応によって Nakadomarin A の不斉全合成を達成している
(Scheme 2)2
しかしながらDanishefsky 型ジエンはその反応性の高さ故に酸に対して不安定であることからル
イス酸触媒によるジエノフィルの活性化は難しく不斉ルイス酸触媒を用いた触媒的不斉 Diels-Alder
反応の成功例はアルデヒドやイミンに対するヘテロ Diels-Alder 反応の例を除いては非常に少ない
34 これに対し当研究室では 2008 年に Danishefsky ジエンとオレフィンの初の触媒的 Diels-Alder
反応を5a Rawal らは 2000 年に Rawal ジエン(2)とオレフィンの初の触媒的 Diels-Alder 反応を報告し
た6c 次節ではこれら二つの不斉 Diels-Alder 反応について述べる
NMe2
TBSO
OTMS
TMSO
OMe
OMe
TMSO
Danisheskys diene
1a
Rawals diene Brassards diene
Figure 1 Highly oxygenated dienes
2a
OMe
TMSO
REWG
OMe
TMSO
EWG
R
Scheme 1
Danisheskys diene
Scheme 2
1a
BsN
O
O
NBs 2) TFA
BsN
O
OH
O
NBsOMe
OTBS
N
O
N
H
(-)-nakadomarin A
1) 180 oC+
1b
1 Highly functionalized
2 Regioselective
3 Stereoselective (normally endo selective)
5
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応
井口らは 2001 年にランタノイド類
が Danishefsky ジ エ ン を 用 い た
Diels-Alder 反応の有効な触媒となる
ことを報告した(Scheme 3)7 これ
によりキラルランタノイド触媒の開発によって本反応の不斉化を達成できることが示唆された
小林らは Yb(OTf)3BINOL 及び嵩高い三級アミンより調整されるキラル希土類錯体を調製しシク
ロペンタジエンと N-アシルオキサ
ゾリジノンとの不斉 Diels-Alder 反
応において最高 95 ee の不斉収率
で反応を進行させることに成功して
いる(Scheme 4)8
当 研 究 室 の 山 中 は 上 記 の 小 林 ら の 反 応 に な ら い Yb(OTf)3 と 独 自 の 不 斉 配 位 子
BINAMIDE[11rsquo-(22rsquo-bisacylamino)binaphthalene]及び iPr2NEt から光学活性 Yb 錯体を調製し
シクロペンタジエンとクロトニルオキサゾリジノンとの高収率かつ高エナンチオ選択的な不斉
Diels-Alder 反応の開発に成功している(Scheme 5)9
当研究室の須藤は上述の井口らの結果をもとに本錯体を Danishefsky ジエンを用いる Diels-Alder
反応に適用しDanishefsky ジエン 1111 と電子不足オレフィン 3333 との初の触媒的不斉 Diels-Alder 反応の
開発に成功したこれにより有用な合成中間体である光学活性シクロヘキセノン誘導体が高い光学純
度で得られるようになった(Table 1)5a 尚このとき用いた BINAMIDE は 35-difluoro 体 5555 が最
適であり以降5555 を単に BINAMIDE と呼称するさらに当研究室の原田は BINAMIDE を改良し
ウレア側鎖をもつ BINUREA を開発し不斉収率や基質一般性の向上に成功した5b
O
Me
Me H
tBuO2C
OMe
TMSO
O
CO2tBu
Ac
Me
+
Yb(OTf)3(10 mol)
toluene 0 oC87
Scheme 3 Inokuchis work
1a
N
O
O
O
NO O
O
Scheme 4 Kobayashis work
+
chiral Yb complex(20 mol)
with or without additive
MS 4A CH2Cl2 0oC
chiral Yb complex = Yb(OTf)3 + (R)-BINOL + cis-126-trimethylpiperidine (11224)
73 95 ee
3a
N
O
O
O
NO O
O
+
chiral Yb complex(25 mol)
CH2Cl2 0oC
endo (2R 3S)
chiral Yb complex = Yb(OTf)3 + BINAMIDE (R = Ph) + iPr2NEt (11224)
95 gt98 ee (endo)endoexo = 911
NH
NH
O
R
O
R
BINAMIDE
Scheme 5
3a
OMe
SiO
R N O
OOO R
N O
O O
F
F
F
F
N
N
O
O
H
H
+
1) chiral Yb complex
(10 mol)
CH2Cl2
1 eq
2) TFA
chiral Yb complex = Yb(OTf)3 + 5 + DBU (1 12 24) or Yb(OTf)3 + 6 + DBU (1 1 2)
2 eq
5BINAMIDE
3 41
Table 1
R
Me
n-Pr
iBu
iPr
Ph
PhO2C
H
4 ()
quant
93
88
29
26
quant
93
optimized data ofBINAMIDE
optimized data ofBINUREA
linearalkyl chain
branchedalkyl chain
ester
non-substituted
ee
92
97
87
56
60
90
71
4 ()
quant
94
90
84
90
88
21
ee
98
99
97
98
97
86
37 6BINUREA
NH
HN
NH
O
Ph
HN
O
Ph
6
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応
Rawal らは 1997 年に 1-amino-3-siloxybutadiene(2222aaaaRawal ジエン)の合成及びその Diels-Alder 反
応を報告した6a Rawal ジエン(2222aaaa)は Danishefsky ジエンに比べて 3300 倍の反応性を示すほか6b ア
ミノ基を導入できることから含窒素化合物の合成に対し有用であるさらに 2000 年に Rawal らはア
ミノ基に電子吸引基を導入したジエン 2b2b2b2b を用いキラルクロムサレン錯体 7a7a7a7a を触媒とする触媒的不斉
Diels-Alder 反応の開発に成功し報告した(Table 2)6c 本反応ではアミノ基をもつシクロヘキセンま
たは 6-5 員環が縮合した化合物を高立体選択的高エナンチオ選択的に得ることができRawal らはこ
れを用いたアルカロイドの不斉全合成も達成している10
筆者は以下に示す検討を行い以下の結果を得た
(1) 当研究室で開発された Danishefsky ジエンの触媒的不斉 Diels-Alder 反応を鍵工程とし
(ndash)-platyphyllide の触媒的不斉全合成を達成しその絶対配置を改定した
(2) platyphyllide 合成経路において伊藤-三枝酸化を用いた際に少量得られたオレフィンの位置が異
なる副生成物についてこれを選択的に得る条件を求め反応機構を推定した
(3) Rawal らの不斉 Diels-Alder 反応を鍵工程としマンザミン B の全合成研究を行った
本論文は以下の章により構成される
第一章 (ndash)-platyphyllide の触媒的不斉全合成
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
第三章 マンザミン B の全合成研究
以下その研究の経緯について詳細に記述する
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
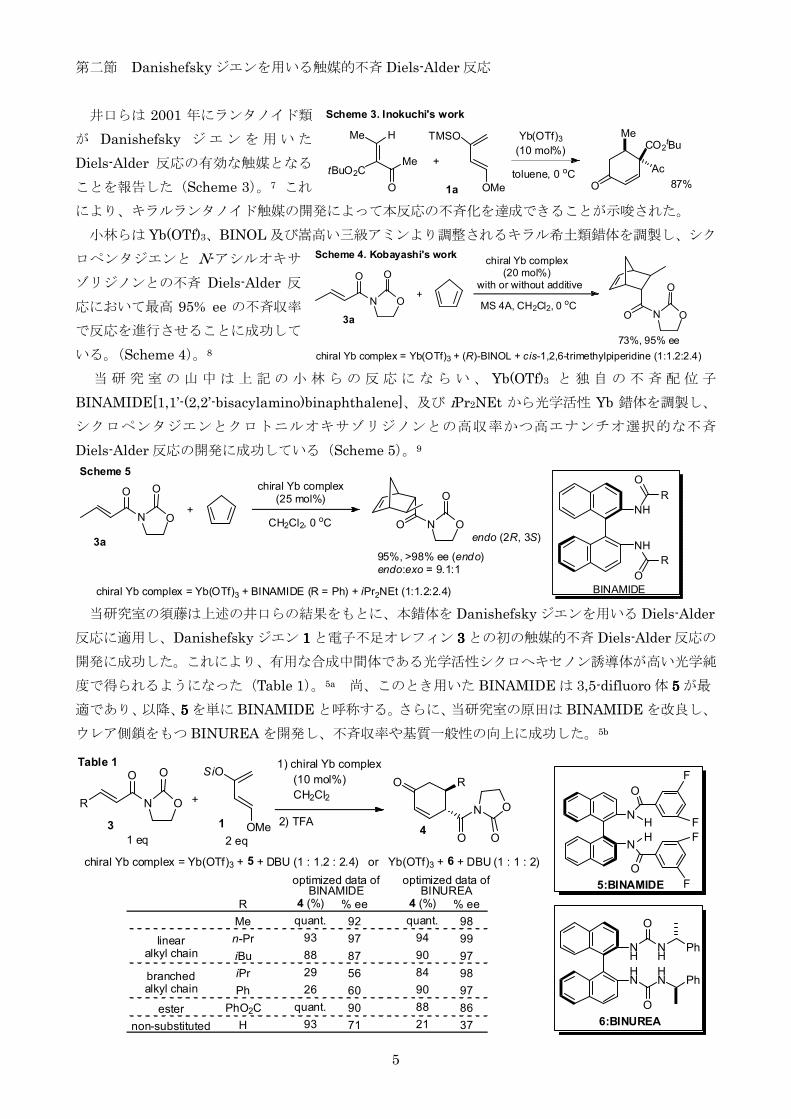
5
第二節 Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応
井口らは 2001 年にランタノイド類
が Danishefsky ジ エ ン を 用 い た
Diels-Alder 反応の有効な触媒となる
ことを報告した(Scheme 3)7 これ
によりキラルランタノイド触媒の開発によって本反応の不斉化を達成できることが示唆された
小林らは Yb(OTf)3BINOL 及び嵩高い三級アミンより調整されるキラル希土類錯体を調製しシク
ロペンタジエンと N-アシルオキサ
ゾリジノンとの不斉 Diels-Alder 反
応において最高 95 ee の不斉収率
で反応を進行させることに成功して
いる(Scheme 4)8
当 研 究 室 の 山 中 は 上 記 の 小 林 ら の 反 応 に な ら い Yb(OTf)3 と 独 自 の 不 斉 配 位 子
BINAMIDE[11rsquo-(22rsquo-bisacylamino)binaphthalene]及び iPr2NEt から光学活性 Yb 錯体を調製し
シクロペンタジエンとクロトニルオキサゾリジノンとの高収率かつ高エナンチオ選択的な不斉
Diels-Alder 反応の開発に成功している(Scheme 5)9
当研究室の須藤は上述の井口らの結果をもとに本錯体を Danishefsky ジエンを用いる Diels-Alder
反応に適用しDanishefsky ジエン 1111 と電子不足オレフィン 3333 との初の触媒的不斉 Diels-Alder 反応の
開発に成功したこれにより有用な合成中間体である光学活性シクロヘキセノン誘導体が高い光学純
度で得られるようになった(Table 1)5a 尚このとき用いた BINAMIDE は 35-difluoro 体 5555 が最
適であり以降5555 を単に BINAMIDE と呼称するさらに当研究室の原田は BINAMIDE を改良し
ウレア側鎖をもつ BINUREA を開発し不斉収率や基質一般性の向上に成功した5b
O
Me
Me H
tBuO2C
OMe
TMSO
O
CO2tBu
Ac
Me
+
Yb(OTf)3(10 mol)
toluene 0 oC87
Scheme 3 Inokuchis work
1a
N
O
O
O
NO O
O
Scheme 4 Kobayashis work
+
chiral Yb complex(20 mol)
with or without additive
MS 4A CH2Cl2 0oC
chiral Yb complex = Yb(OTf)3 + (R)-BINOL + cis-126-trimethylpiperidine (11224)
73 95 ee
3a
N
O
O
O
NO O
O
+
chiral Yb complex(25 mol)
CH2Cl2 0oC
endo (2R 3S)
chiral Yb complex = Yb(OTf)3 + BINAMIDE (R = Ph) + iPr2NEt (11224)
95 gt98 ee (endo)endoexo = 911
NH
NH
O
R
O
R
BINAMIDE
Scheme 5
3a
OMe
SiO
R N O
OOO R
N O
O O
F
F
F
F
N
N
O
O
H
H
+
1) chiral Yb complex
(10 mol)
CH2Cl2
1 eq
2) TFA
chiral Yb complex = Yb(OTf)3 + 5 + DBU (1 12 24) or Yb(OTf)3 + 6 + DBU (1 1 2)
2 eq
5BINAMIDE
3 41
Table 1
R
Me
n-Pr
iBu
iPr
Ph
PhO2C
H
4 ()
quant
93
88
29
26
quant
93
optimized data ofBINAMIDE
optimized data ofBINUREA
linearalkyl chain
branchedalkyl chain
ester
non-substituted
ee
92
97
87
56
60
90
71
4 ()
quant
94
90
84
90
88
21
ee
98
99
97
98
97
86
37 6BINUREA
NH
HN
NH
O
Ph
HN
O
Ph
6
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応
Rawal らは 1997 年に 1-amino-3-siloxybutadiene(2222aaaaRawal ジエン)の合成及びその Diels-Alder 反
応を報告した6a Rawal ジエン(2222aaaa)は Danishefsky ジエンに比べて 3300 倍の反応性を示すほか6b ア
ミノ基を導入できることから含窒素化合物の合成に対し有用であるさらに 2000 年に Rawal らはア
ミノ基に電子吸引基を導入したジエン 2b2b2b2b を用いキラルクロムサレン錯体 7a7a7a7a を触媒とする触媒的不斉
Diels-Alder 反応の開発に成功し報告した(Table 2)6c 本反応ではアミノ基をもつシクロヘキセンま
たは 6-5 員環が縮合した化合物を高立体選択的高エナンチオ選択的に得ることができRawal らはこ
れを用いたアルカロイドの不斉全合成も達成している10
筆者は以下に示す検討を行い以下の結果を得た
(1) 当研究室で開発された Danishefsky ジエンの触媒的不斉 Diels-Alder 反応を鍵工程とし
(ndash)-platyphyllide の触媒的不斉全合成を達成しその絶対配置を改定した
(2) platyphyllide 合成経路において伊藤-三枝酸化を用いた際に少量得られたオレフィンの位置が異
なる副生成物についてこれを選択的に得る条件を求め反応機構を推定した
(3) Rawal らの不斉 Diels-Alder 反応を鍵工程としマンザミン B の全合成研究を行った
本論文は以下の章により構成される
第一章 (ndash)-platyphyllide の触媒的不斉全合成
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
第三章 マンザミン B の全合成研究
以下その研究の経緯について詳細に記述する
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

6
第三節 Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応
Rawal らは 1997 年に 1-amino-3-siloxybutadiene(2222aaaaRawal ジエン)の合成及びその Diels-Alder 反
応を報告した6a Rawal ジエン(2222aaaa)は Danishefsky ジエンに比べて 3300 倍の反応性を示すほか6b ア
ミノ基を導入できることから含窒素化合物の合成に対し有用であるさらに 2000 年に Rawal らはア
ミノ基に電子吸引基を導入したジエン 2b2b2b2b を用いキラルクロムサレン錯体 7a7a7a7a を触媒とする触媒的不斉
Diels-Alder 反応の開発に成功し報告した(Table 2)6c 本反応ではアミノ基をもつシクロヘキセンま
たは 6-5 員環が縮合した化合物を高立体選択的高エナンチオ選択的に得ることができRawal らはこ
れを用いたアルカロイドの不斉全合成も達成している10
筆者は以下に示す検討を行い以下の結果を得た
(1) 当研究室で開発された Danishefsky ジエンの触媒的不斉 Diels-Alder 反応を鍵工程とし
(ndash)-platyphyllide の触媒的不斉全合成を達成しその絶対配置を改定した
(2) platyphyllide 合成経路において伊藤-三枝酸化を用いた際に少量得られたオレフィンの位置が異
なる副生成物についてこれを選択的に得る条件を求め反応機構を推定した
(3) Rawal らの不斉 Diels-Alder 反応を鍵工程としマンザミン B の全合成研究を行った
本論文は以下の章により構成される
第一章 (ndash)-platyphyllide の触媒的不斉全合成
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
第三章 マンザミン B の全合成研究
以下その研究の経緯について詳細に記述する
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
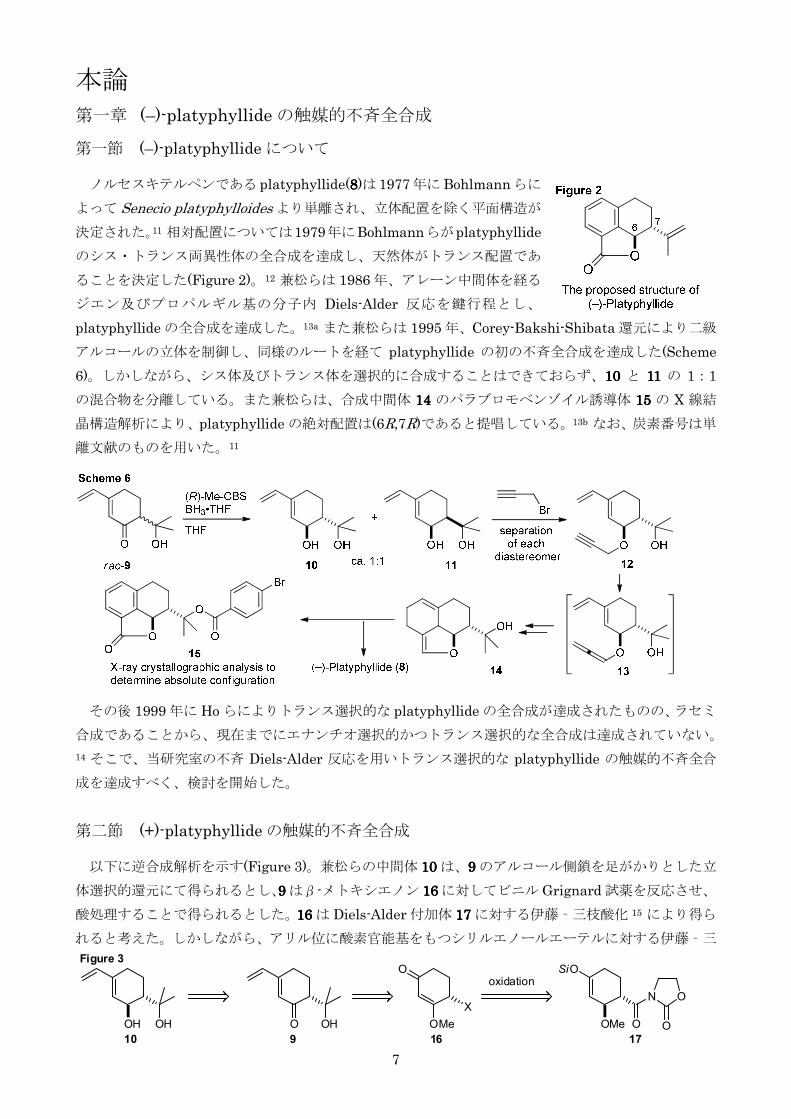
7
本論 第一章 (ndash)-platyphyllide の触媒的不斉全合成
第一節 (ndash)-platyphyllide について ノルセスキテルペンである platyphyllide(8888)は 1977年にBohlmannらに
よって Senecio platyphylloides より単離され立体配置を除く平面構造が
決定された11 相対配置については1979年にBohlmannらがplatyphyllide
のシストランス両異性体の全合成を達成し天然体がトランス配置であ
ることを決定した(Figure 2)12 兼松らは 1986 年アレーン中間体を経る
ジエン及びプロパルギル基の分子内 Diels-Alder 反応を鍵行程とし
platyphyllide の全合成を達成した13a また兼松らは 1995 年Corey-Bakshi-Shibata 還元により二級
アルコールの立体を制御し同様のルートを経て platyphyllide の初の不斉全合成を達成した(Scheme
6)しかしながらシス体及びトランス体を選択的に合成することはできておらず10101010 と 11111111 の 11
の混合物を分離しているまた兼松らは合成中間体 11114444 のパラブロモベンゾイル誘導体 11115555 の X 線結
晶構造解析によりplatyphyllide の絶対配置は(6R7R)であると提唱している13b なお炭素番号は単
離文献のものを用いた11
その後 1999 年に Ho らによりトランス選択的な platyphyllide の全合成が達成されたもののラセミ
合成であることから現在までにエナンチオ選択的かつトランス選択的な全合成は達成されていない
14 そこで当研究室の不斉 Diels-Alder 反応を用いトランス選択的な platyphyllide の触媒的不斉全合
成を達成すべく検討を開始した
第二節 (+)-platyphyllide の触媒的不斉全合成
以下に逆合成解析を示す(Figure 3)兼松らの中間体 10101010 は9999 のアルコール側鎖を足がかりとした立
体選択的還元にて得られるとし9999 はβ-メトキシエノン 16161616 に対してビニル Grignard 試薬を反応させ
酸処理することで得られるとした16161616 は Diels-Alder 付加体 17171717 に対する伊藤‐三枝酸化 15 により得ら
れると考えたしかしながらアリル位に酸素官能基をもつシリルエノールエーテルに対する伊藤‐三
O OH
Figure 3
OMe
O
X
oxidation
OMe
SiO
O
N O
O9 1716
OH OH
10
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
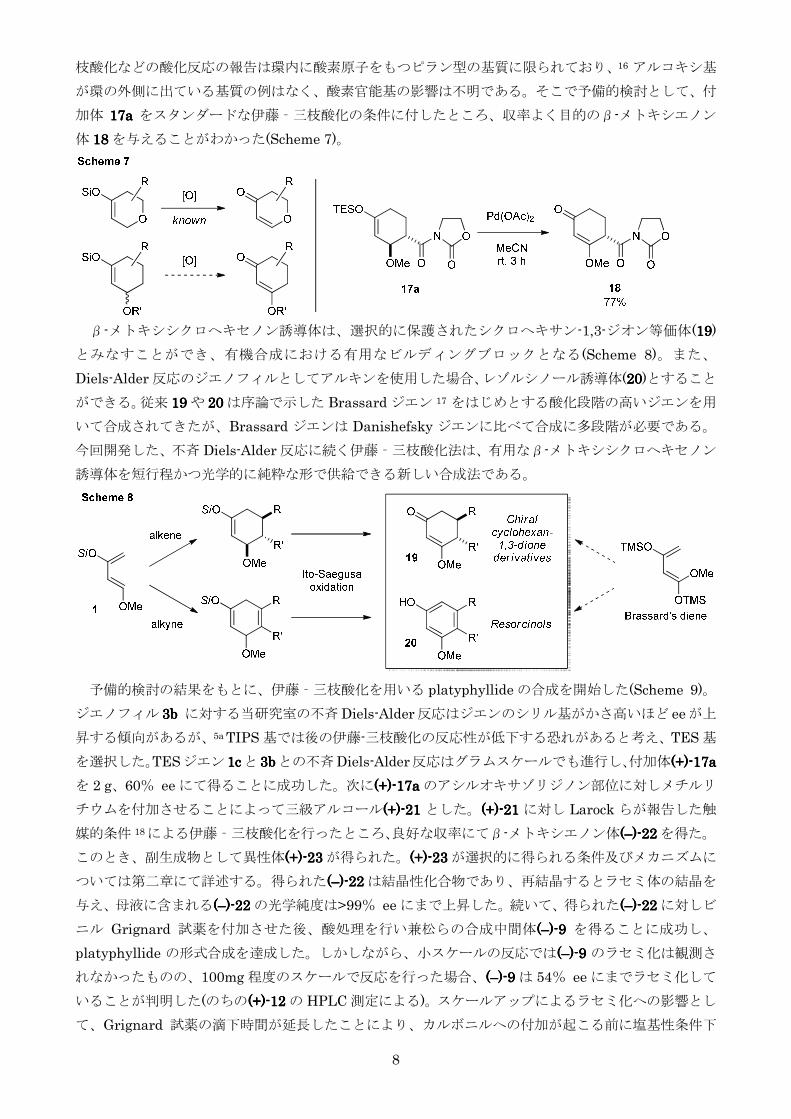
8
枝酸化などの酸化反応の報告は環内に酸素原子をもつピラン型の基質に限られており16 アルコキシ基
が環の外側に出ている基質の例はなく酸素官能基の影響は不明であるそこで予備的検討として付
加体 17171717aaaa をスタンダードな伊藤‐三枝酸化の条件に付したところ収率よく目的のβ-メトキシエノン
体 18181818 を与えることがわかった(Scheme 7)
β-メトキシシクロヘキセノン誘導体は選択的に保護されたシクロヘキサン-13-ジオン等価体(19191919)
とみなすことができ有機合成における有用なビルディングブロックとなる(Scheme 8)また
Diels-Alder 反応のジエノフィルとしてアルキンを使用した場合レゾルシノール誘導体(20202020)とすること
ができる従来 19191919 や 20202020 は序論で示した Brassard ジエン 17 をはじめとする酸化段階の高いジエンを用
いて合成されてきたがBrassard ジエンは Danishefsky ジエンに比べて合成に多段階が必要である
今回開発した不斉 Diels-Alder 反応に続く伊藤‐三枝酸化法は有用なβ-メトキシシクロヘキセノン
誘導体を短行程かつ光学的に純粋な形で供給できる新しい合成法である
予備的検討の結果をもとに伊藤‐三枝酸化を用いる platyphyllide の合成を開始した(Scheme 9)
ジエノフィル 3333bbbb に対する当研究室の不斉 Diels-Alder 反応はジエンのシリル基がかさ高いほど eeが上
昇する傾向があるが5a TIPS 基では後の伊藤-三枝酸化の反応性が低下する恐れがあると考えTES 基
を選択したTESジエン 1111ccccと 3333bbbbとの不斉Diels-Alder反応はグラムスケールでも進行し付加体(+)(+)(+)(+)----17171717aaaa
を 2 g60 ee にて得ることに成功した次に(+)(+)(+)(+)----17171717aaaa のアシルオキサゾリジノン部位に対しメチルリ
チウムを付加させることによって三級アルコール(+)(+)(+)(+)----21212121 とした(+)(+)(+)(+)----21212121 に対し Larock らが報告した触
媒的条件 18による伊藤‐三枝酸化を行ったところ良好な収率にてβ-メトキシエノン体((((ndashndashndashndash))))----22222222 を得た
このとき副生成物として異性体(+)(+)(+)(+)----23232323 が得られた(+)(+)(+)(+)----23232323 が選択的に得られる条件及びメカニズムに
ついては第二章にて詳述する得られた((((ndashndashndashndash))))----22222222 は結晶性化合物であり再結晶するとラセミ体の結晶を
与え母液に含まれる((((ndashndashndashndash))))----22222222 の光学純度はgt99 ee にまで上昇した続いて得られた((((ndashndashndashndash))))----22222222 に対しビ
ニル Grignard 試薬を付加させた後酸処理を行い兼松らの合成中間体((((ndashndashndashndash))))----9999 を得ることに成功し
platyphyllide の形式合成を達成したしかしながら小スケールの反応では((((ndashndashndashndash))))----9999 のラセミ化は観測さ
れなかったものの100mg 程度のスケールで反応を行った場合((((ndashndashndashndash))))----9999 は 54 ee にまでラセミ化して
いることが判明した(のちの(+)(+)(+)(+)----11112222 の HPLC 測定による)スケールアップによるラセミ化への影響とし
てGrignard 試薬の滴下時間が延長したことによりカルボニルへの付加が起こる前に塩基性条件下
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
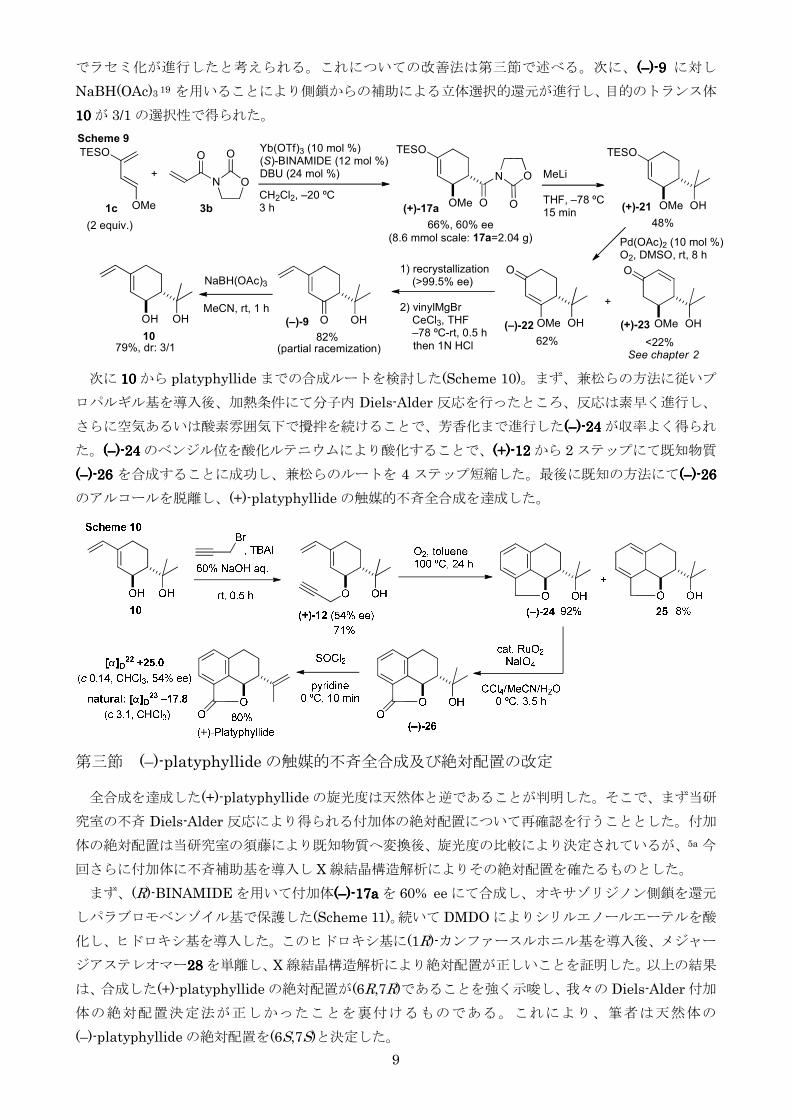
9
でラセミ化が進行したと考えられるこれについての改善法は第三節で述べる次に((((ndashndashndashndash))))----9999 に対し
NaBH(OAc)3 19 を用いることにより側鎖からの補助による立体選択的還元が進行し目的のトランス体
10101010 が 31 の選択性で得られた
TESO
OMe
+
Yb(OTf)3 (10 mol )(S)-BINAMIDE (12 mol )DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) 66 60 ee
(86 mmol scale 17a=204 g)
MeLi
THF ndash78 ordmC15 min
TESO
OHOMe
Scheme 9
1c 3b (+)-17a (+)-21
48
Pd(OAc)2 (10 mol )O2 DMSO rt 8 h
O
OHOMe
1) recrystallization(gt995 ee)
2) vinylMgBrCeCl3 THFndash78 ordmC-rt 05 h
OHO
then 1N HCl82
(partial racemization)
O
OHOMe(+)-23(ndash)-9
lt2262
+
(ndash)-22
NaBH(OAc)3
MeCN rt 1 hOHOH
1079 dr 31
See chapter 2
次に 10101010 から platyphyllide までの合成ルートを検討した(Scheme 10)まず兼松らの方法に従いプ
ロパルギル基を導入後加熱条件にて分子内 Diels-Alder 反応を行ったところ反応は素早く進行し
さらに空気あるいは酸素雰囲気下で攪拌を続けることで芳香化まで進行した((((ndashndashndashndash))))----24242424 が収率よく得られ
た((((ndashndashndashndash))))----24242424 のベンジル位を酸化ルテニウムにより酸化することで(+)(+)(+)(+)----12121212 から 2 ステップにて既知物質
((((ndashndashndashndash))))----26262626 を合成することに成功し兼松らのルートを 4 ステップ短縮した最後に既知の方法にて((((ndashndashndashndash))))----26262626
のアルコールを脱離し(+)-platyphyllide の触媒的不斉全合成を達成した
第三節 (ndash)-platyphyllide の触媒的不斉全合成及び絶対配置の改定
全合成を達成した(+)-platyphyllide の旋光度は天然体と逆であることが判明したそこでまず当研
究室の不斉 Diels-Alder 反応により得られる付加体の絶対配置について再確認を行うこととした付加
体の絶対配置は当研究室の須藤により既知物質へ変換後旋光度の比較により決定されているが5a 今
回さらに付加体に不斉補助基を導入し X 線結晶構造解析によりその絶対配置を確たるものとした
まず(R)-BINAMIDE を用いて付加体((((ndashndashndashndash))))----17a17a17a17a を 60 ee にて合成しオキサゾリジノン側鎖を還元
しパラブロモベンゾイル基で保護した(Scheme 11)続いて DMDO によりシリルエノールエーテルを酸
化しヒドロキシ基を導入したこのヒドロキシ基に(1R)-カンファースルホニル基を導入後メジャー
ジアステレオマー28282828 を単離しX 線結晶構造解析により絶対配置が正しいことを証明した以上の結果
は合成した(+)-platyphyllide の絶対配置が(6R7R)であることを強く示唆し我々の Diels-Alder 付加
体の絶対配置決定法が正しかったことを裏付けるものであるこれにより筆者は天然体の
(ndash)-platyphyllide の絶対配置を(6S7S)と決定した
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
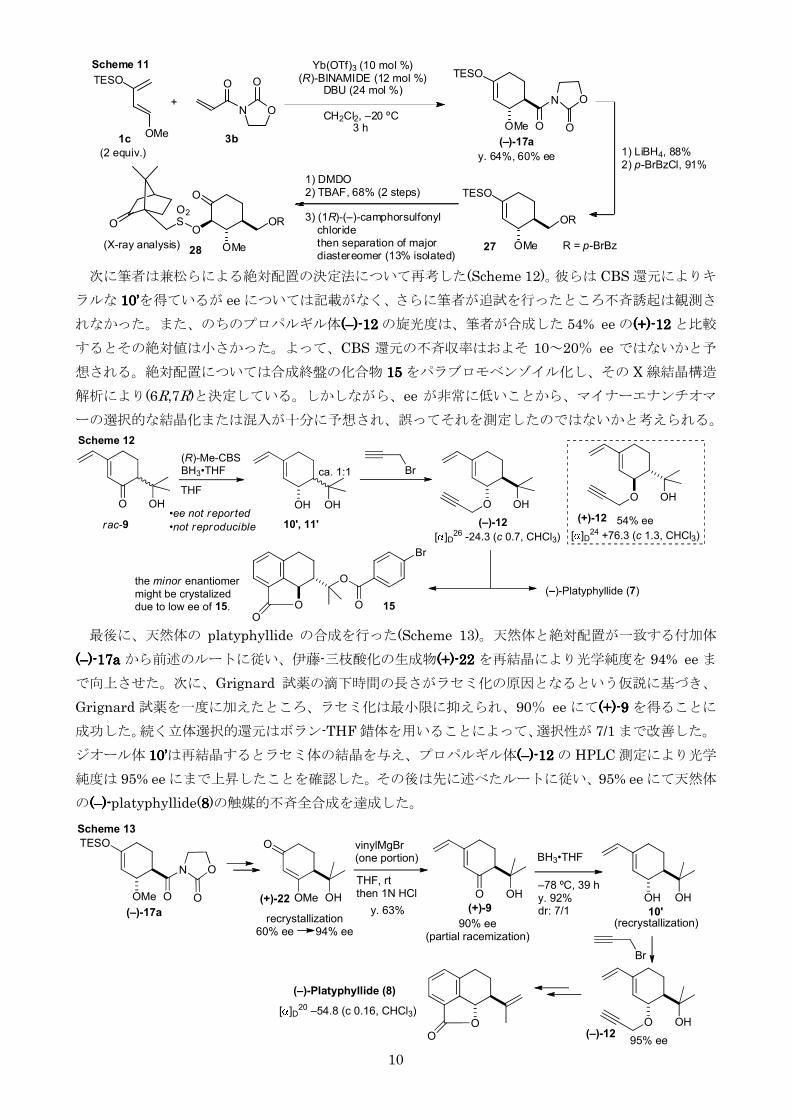
10
次に筆者は兼松らによる絶対配置の決定法について再考した(Scheme 12)彼らは CBS 還元によりキ
ラルな 10101010rsquorsquorsquorsquoを得ているが ee については記載がなくさらに筆者が追試を行ったところ不斉誘起は観測さ
れなかったまたのちのプロパルギル体((((ndashndashndashndash))))----11112222 の旋光度は筆者が合成した 54 ee の(+)(+)(+)(+)----12121212 と比較
するとその絶対値は小さかったよってCBS 還元の不斉収率はおよそ 10~20 ee ではないかと予
想される絶対配置については合成終盤の化合物 11115555 をパラブロモベンゾイル化しその X 線結晶構造
解析により(6R7R)と決定しているしかしながらee が非常に低いことからマイナーエナンチオマ
ーの選択的な結晶化または混入が十分に予想され誤ってそれを測定したのではないかと考えられる
O OH OH OH
(R)-Me-CBSBH3bullTHF
THF
rac-9
ca 11 Br
O OH
(ndash)-Platyphyllide (7)
O
O
OO
Br
the minor enantiomer
might be crystalized
due to low ee of 15
Scheme 12
10 11(+)-12(ndash)-12
15
bullee not repor ted
bullnot reproducible[ ]D
26 -243 (c 07 CHCl3) [ ]D24 +763 (c 13 CHCl3)
O OH
54 ee
最後に天然体の platyphyllide の合成を行った(Scheme 13)天然体と絶対配置が一致する付加体
((((ndashndashndashndash))))----17171717aaaa から前述のルートに従い伊藤-三枝酸化の生成物(+)(+)(+)(+)----22222222 を再結晶により光学純度を 94 ee ま
で向上させた次にGrignard 試薬の滴下時間の長さがラセミ化の原因となるという仮説に基づき
Grignard 試薬を一度に加えたところラセミ化は最小限に抑えられ90 ee にて(+)(+)(+)(+)----9999 を得ることに
成功した続く立体選択的還元はボラン-THF 錯体を用いることによって選択性が 71 まで改善した
ジオール体 10101010rsquorsquorsquorsquoは再結晶するとラセミ体の結晶を与えプロパルギル体((((ndashndashndashndash))))----11112222 の HPLC 測定により光学
純度は 95 ee にまで上昇したことを確認したその後は先に述べたルートに従い95 ee にて天然体
の((((ndashndashndashndash))))----platyphyllide(8888)の触媒的不斉全合成を達成した
O
OHOMe
vinylMgBr(one portion)
THF rtthen 1N HCl
y 63
OHOndash78 ordmC 39 hy 92dr 71
BH3bullTHF
recrystallization60 ee 94 ee
OHOH
90 ee(partial racemization)
Br
OHO
(recrystallization)
95 ee
OO
(ndash)-Platyphyllide (8)
(+)-22(+)-9 10
(ndash)-12
[ ]D20 ndash548 (c 016 CHCl3)
TESO
O
N O
OOMe
(ndash)-17a
Scheme 13
TESO
OMe
+
Yb(OTf)3 (10 mol )(R)-BINAMIDE (12 mol )
DBU (24 mol )
CH2Cl2 ndash20 ordmC3 h
TESO
O
N O
O
N O
O O
OMe
(2 equiv) y 64 60 ee
1c 3b (ndash)-17a
TESO
OMe
OR
OMe
OR
O
O
O2
SO
1) LiBH4 882) p-BrBzCl 91
R = p-BrBz2728(X-ray analysis)
1) DMDO2) TBAF 68 (2 steps)
3) (1R)-(ndash)-camphorsulfonylchloridethen separation of majordiastereomer (13 isolated)
Scheme 11
11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

11
第二章 特殊な基質を用いた伊藤‐三枝酸化におけるオレフィンの異性化
Platyphyllide の合成において TES エノールエーテル 21212121 に伊藤-三枝酸化を用いたところ通常の酸
化体 22222222 の他に本来の反応メカニズムでは説明できない異性化体 23232323 が少量得られた(Scheme 9)そこ
でこのような副生成物が得られる条件及びメカニズムについて精査した
まず21212121 の構造を単純化した一級アルコール 29292929 について量論量の Pd(OAc)2による伊藤-三枝酸化
を行った(Table 3)TES 基を持つ 29a29a29a29a の場合主生成物は通常の酸化体 30a30a30a30a で異性化体 31a31a31a31a はわず
かしかえられなかった(entry 1)シリル基を TIPS 基にしたところ生成比が逆転し異性化体 31a31a31a31a が
主生成物として得られた(entry 2)この比率は基質の立体化学にも影響されシス体 29c29c29c29c を用いるとさ
らに異性化体 31b31b31b31b の割合が上昇した(entry 3)アルコールをメチル基で保護した 29d29d29d29d を用いたところ異
性化体は全く得られず全体としての回収率も大幅に低下した(entry 4)以上より異性化体の生成比
を増加させるにはかさ高い TIPS 基と側鎖の遊離のヒドロキシ基が必要であることがわかった
また基質のメトキシ基は単純なメチル基に置き換えても影響がないことがわかったため32323232aaaa を用
いて反応条件の精査を行った(Table 4 entry 1)基質の濃度を 1M と濃くすると異性化体の生成比は低
下した(entry 2)逆に01Mと薄くした場合比率に変化はなく反応時間の延長のみが観察された(entry
3)反応温度を 60に上昇すると反応は完結せずまた生成比には影響は見られなかった(entry 4)パ
ラジウムを 2 等量用いることで反応時間の短縮は見られたものの比率への影響はなく全体としての収
率が低下した(entry 5)以上より最適条件は伊藤三枝らにより報告されている通りの entry 1 のも
のに決定した尚他のパラジウム種(PdS PdCl2 Pd(CN)2)を用いると酸化は進行せず脱シリル化が進
行したケトン体のみが得られた他の溶媒(DMSODMFTHFCH2Cl2)についても検討したが反応が進
行しないか異性化体の生成比の低下を招くのみであった
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
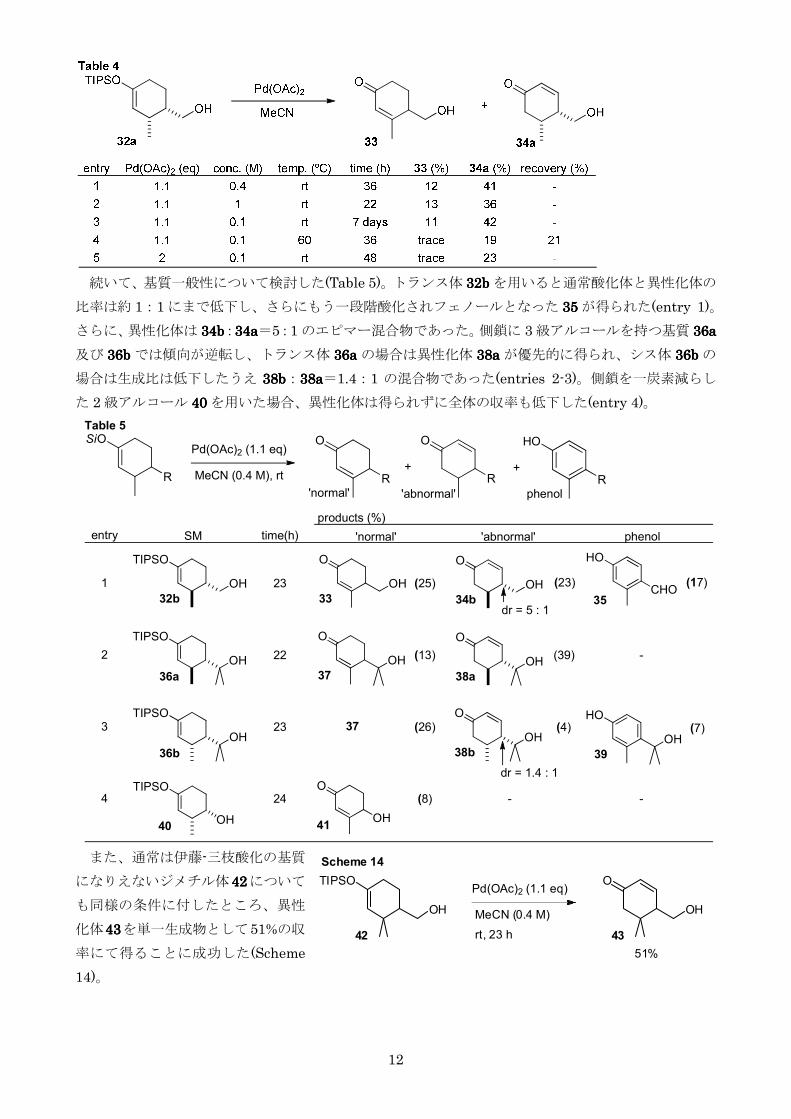
12
続いて基質一般性について検討した(Table 5)トランス体 32b32b32b32b を用いると通常酸化体と異性化体の
比率は約 11 にまで低下しさらにもう一段階酸化されフェノールとなった 35353535 が得られた(entry 1)
さらに異性化体は 34b34b34b34b34a34a34a34a=51 のエピマー混合物であった側鎖に 3 級アルコールを持つ基質 36a36a36a36a
及び 36b36b36b36b では傾向が逆転しトランス体 36a36a36a36a の場合は異性化体 38a38a38a38a が優先的に得られシス体 36b36b36b36b の
場合は生成比は低下したうえ 38b38b38b38b38a38a38a38a=141 の混合物であった(entries 2-3)側鎖を一炭素減らし
た 2 級アルコール 40404040 を用いた場合異性化体は得られずに全体の収率も低下した(entry 4)
R
SiO
SM
MeCN (04 M) rt
Pd(OAc)2 (11 eq)
R
O
R
O
entry
1
2
3
4
time(h)
23
22
23
24
+
TIPSO
OH
TIPSO
OH
TIPSO
OH
OH
TIPSO
CHO
HO
products ()
32b
36a
36b
40
R
HO
phenol
+
-
(25)
(13)
(26)
(8)
-
-
35
(7)
normal phenol
(23)
normal abnormal
abnormal
Table 5
O
OH
33
O
OH
37
37
OH
O
41
HO
OH
39
O
OH
34bdr = 5 1
O
OH
38b
(4)
O
OH
38a
(39)
dr = 14 1
(17)
また通常は伊藤-三枝酸化の基質
になりえないジメチル体 42424242について
も同様の条件に付したところ異性
化体44443333を単一生成物として51の収
率にて得ることに成功した(Scheme
14)
TIPSO
MeCN (04 M)
Pd(OAc)2 (11 eq)
42
OH
O
43
OH
rt 23 h
51
Scheme 14
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
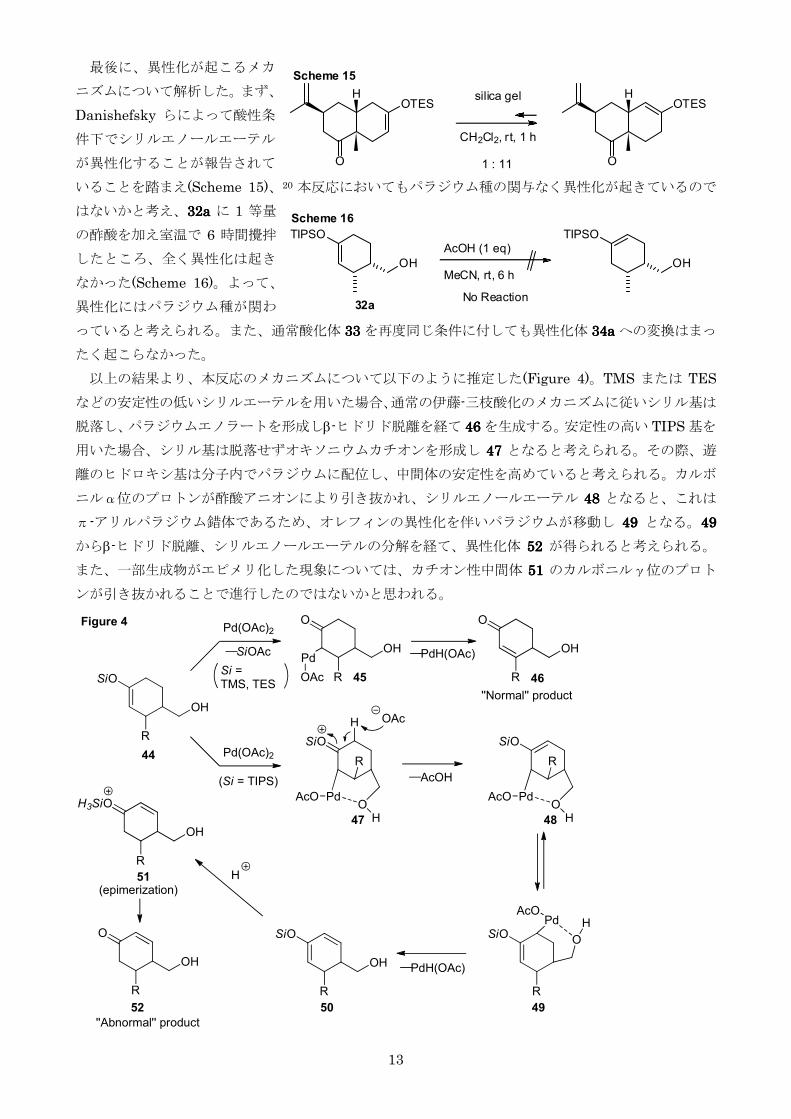
13
最後に異性化が起こるメカ
ニズムについて解析したまず
Danishefsky らによって酸性条
件下でシリルエノールエーテル
が異性化することが報告されて
いることを踏まえ(Scheme 15)20 本反応においてもパラジウム種の関与なく異性化が起きているので
はないかと考え32a32a32a32a に 1 等量
の酢酸を加え室温で 6 時間攪拌
したところ全く異性化は起き
なかった(Scheme 16)よって
異性化にはパラジウム種が関わ
っていると考えられるまた通常酸化体 33333333 を再度同じ条件に付しても異性化体 34a34a34a34a への変換はまっ
たく起こらなかった
以上の結果より本反応のメカニズムについて以下のように推定した(Figure 4)TMS または TES
などの安定性の低いシリルエーテルを用いた場合通常の伊藤-三枝酸化のメカニズムに従いシリル基は
脱落しパラジウムエノラートを形成しβ-ヒドリド脱離を経て 46464646 を生成する安定性の高い TIPS 基を
用いた場合シリル基は脱落せずオキソニウムカチオンを形成し 47474747 となると考えられるその際遊
離のヒドロキシ基は分子内でパラジウムに配位し中間体の安定性を高めていると考えられるカルボ
ニルα位のプロトンが酢酸アニオンにより引き抜かれシリルエノールエーテル 48484848 となるとこれは
π-アリルパラジウム錯体であるためオレフィンの異性化を伴いパラジウムが移動し 49494949 となる49494949
からβ-ヒドリド脱離シリルエノールエーテルの分解を経て異性化体 52525252 が得られると考えられる
また一部生成物がエピメリ化した現象についてはカチオン性中間体 51515151 のカルボニルγ位のプロト
ンが引き抜かれることで進行したのではないかと思われる
SiO
OH
R SiO
O
R
PdAcO
OAcH
SiOO
R
PdAcO
H
SiO
O
R
PdAcO
H
HO
OH
R
Pd(OAc)2
AcOH
SiO
OH
R
H
44
47 48
52 50 49
(Si = TIPS)
Pd(OAc)2
Si =TMS TES
O
Pd
R
OH
OAc
PdH(OAc)
O
OH
R
Normal product
Abnormal product
45 46
SiOAcmdash mdash
mdash
PdH(OAc)mdash
H3SiO
OH
R
51(epimerization)
Figure 4
TIPSO
32a
OH
AcOH (1 eq)
MeCN rt 6 h
No Reaction
TIPSO
OH
Scheme 16
O
OTESH
O
OTESH silica gel
CH2Cl2 rt 1 h
1 11
Scheme 15
14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

14
第三章 マンザミン B の全合成研究
第一節 マンザミン B 及び関連するアルカロイドについて
マンザミンアルカロイドは1986 年に比嘉らによっ
て沖縄産海綿 Haliclona spより単離構造決定されたマ
ンザミン A 21aをはじめとする化合物群であるマンザ
ミン A はその複雑な構造や強力な抗マラリア活性を
もつことから合成化学者の興味を惹き現在までに
我々を含む多くのグループが全合成を報告している
22-26 一方マンザミン B は 1987 年に比嘉らにより同
じ海綿より単離され抗腫瘍活性が報告されているも
のの21b マンザミン A に比べてその単離量は約 140
湿海綿重量の 00016と非常に少ないため現在まで
に比嘉らの 1 例を除き活性評価が行われていないま
た全合成例はないそこで当研究室ではマンザミン B を全合成し十分量を供給することでその活
性解明に貢献すべくこれまで精力的に全合成研究を行ってきた27
マンザミン B の構造的特徴としては以下の二つが挙げられるまずマンザミン A の CD 環をつな
ぐ C-N 結合が開裂し 11 員環を形成していること次にB 環上にエポキシ基をもち B 環を構成する炭
素が全て不斉炭素になっていることである11 員環をもつタイプの他のマンザミンアルカロイドとして
エポキシ基が開環したマンザミン J やその生合成前駆体であるイルシナール B がある(Figure 5)小
林らによりイルシナール B から Pictet-Spengler 反応を用いるマンザミン J の半合成及びマンザミン
B から塩基性条件下でのエポキシ基の開環によるマンザミン J の半合成は報告されているものの28 イ
ルシナール B 及びマンザミン J からのマンザミン B の半合成は報告されていない
第二節 過去の研究及び合成戦略
当研究室では以下の逆合成に従い合成研究を展開してきた(Figure 6)マンザミン B のエポキシ基及
びβカルボリンの立体選択的な構築については合成の最終段階で行うとしイルシナール B へと逆合成
される13 員環である D 環は過去のマンザミン A の全合成と同様に RCM にて構築するとした232426
また 11 員環である C 環も同じく RCM で構築するとした1 位炭素ユニットはケトンに対するアルド
ール反応で導入するとしまた三級アルコール部位はケトンに対する立体選択的なアルキル化で得られ
Manzamine B
N
H
HN
O
NNH H
A B
C
D
11
12
Manzamine A
N
H
N
OH
NNH
A B
D
E C
10
1
27
3613
21
8
4
5
9
22
2526
35
15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

15
るものとしAB 環コア骨格の 54545454 とした54545454 は Rawal によって報告された不斉 Diels-Alder 反応 6c に
よりRawal ジエン 2222 と環状ジエノフィル 55555555 から不斉合成するとした
当研究室の秋葉松村らにより検討された不斉 Diels-Alder 反応の結果を以下に示す(Table 6)27bc ク
ロム錯体のカウンターアニオンについてはクロムのルイス酸性度が高くなるものほどジエノフィルを
活性化し反応性が向
上する傾向にあった
が最もルイス酸性
の高い entry 4 のヘ
キサフルオロアンチ
モン錯体ではその酸
性度の高さからジエ
ンの分解を招き付
加体は得られなかっ
た最も適した錯体
は entry 5 のフッ化
物イオンをカウンタ
ーアニオンに持つ錯
体であった一方ジエンの窒素上の保護基についてはカルバメート型(entries 7-8)よりもアミド型
(entries 5-6)のほうが反応時間が短いことがわかったこれはアミド型のほうが電子吸引性の高さに
より窒素の求核性が抑えられ錯体の中心金属への配位による反応の妨害が抑制されたからと考えられ
る最も適したジエンは entry 5 のクロロアセチル基をもつものであり収率 7595 ee という良
好な結果が得られたまた当研究室の御原はこの最適条件をスケールアップし68 mmol19 g の 55a55a55a55a
を用いて反応を行い粗生成物をカラムクロマトグラフィーにより精製後酢酸エチルヘキサン(14)
の溶媒中で攪拌することにより収率 67光学純度 99 ee 以上の付加体 56a56a56a56a を 27 g 得ることに成
功した(Scheme 17)27c
続いて松村御原らは付加体 56a56a56a56a のアリル基を 0 価パラジウム存在下 TolSO2H を用いて脱保護し
アルデヒドを還元後クロロ基を接触還元により除去しアセチル基とし 58585858 を得た(Scheme 18)次にシ
BsN
OTBSH
NOHC
PG
BsNCHO
OTBS
NPG
+
(RR)-salen-Cr(III)-X(50 mol)
Bs = benzenesulfonyl
entry
1
2
3
4
5
6
7
8
time (h)
87
20
160
-
20
42
72
282
X
Cl
BF4
N3
SbF6
F
F
F
F
yield ()
65
65
12
decomp
75
57
70
58
ee
75
88
89
-
95
-
92
-
PG
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
COCH2Cl
Ac
CO2Me
Boc
O
tBu
tBu O
tBu
tBu
N N
Cr
X
(RR)-salen-Cr(III)-X (7)
4A MS BTF rt
Table 6
2 eq
55a2 56
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
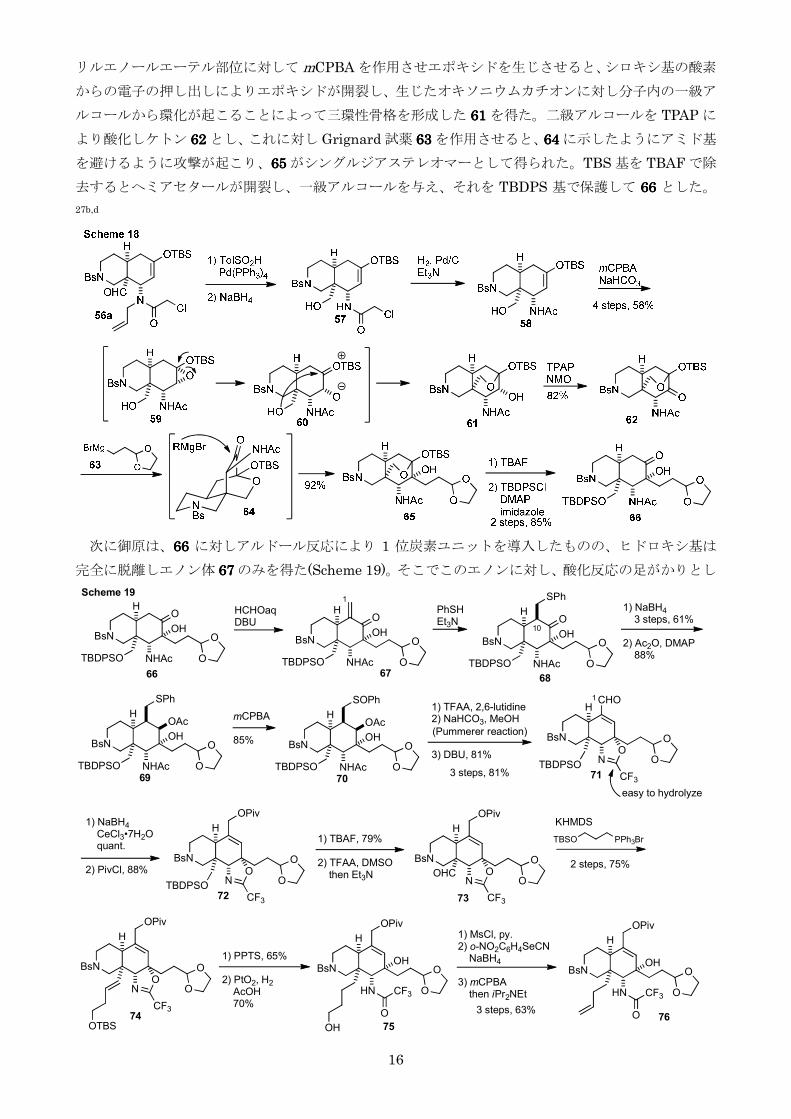
16
リルエノールエーテル部位に対して mCPBA を作用させエポキシドを生じさせるとシロキシ基の酸素
からの電子の押し出しによりエポキシドが開裂し生じたオキソニウムカチオンに対し分子内の一級ア
ルコールから環化が起こることによって三環性骨格を形成した 61616161 を得た二級アルコールを TPAP に
より酸化しケトン 62626262 としこれに対し Grignard 試薬 63636363 を作用させると64646464 に示したようにアミド基
を避けるように攻撃が起こり65656565 がシングルジアステレオマーとして得られたTBS 基を TBAF で除
去するとヘミアセタールが開裂し一級アルコールを与えそれを TBDPS 基で保護して 66666666 とした
27bd
次に御原は66666666 に対しアルドール反応により 1 位炭素ユニットを導入したもののヒドロキシ基は
完全に脱離しエノン体 67676767 のみを得た(Scheme 19)そこでこのエノンに対し酸化反応の足がかりとし
BsN
H
NHAc
OH
O
O
TBDPSO
OHCHOaqDBU
PhSHEt3N
BsN
H
NHAc
OH
O
O
TBDPSO
O
SPh1) NaBH4
3 steps 61
2) Ac2O DMAP88
BsN
H
NHAc
OH
O
O
TBDPSO
SPh
OAc
BsN
H
N O
O
TBDPSO
CHO
mCPBA
85O
CF3
10
1
6768
69 71
Scheme 19
(Pummerer reaction)
BsN
H
NHAc
OH
O
O
TBDPSO
O
66
1
easy to hydrolyze
1) TBAF 79
2) TFAA DMSOthen Et3N
1) NaBH4
CeCl3bull7H2Oquant
2) PivCl 88BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
N O
O
OHC O
CF3
OPiv
TBSO PPh3Br
BsN
H
N O
OO
CF3
OPiv
OTBS
2 steps 75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH1) PPTS 65
2) PtO2 H2
AcOH70
BsN
H
O
O
OPiv
HN
O
CF3
OH
1) MsCl py2) o-NO2C6H4SeCNNaBH4
3) mCPBAthen iPr2NEt
3 steps 63
73
7475
76
KHMDS
1) TFAA 26-lutidine2) NaHCO3 MeOH
3) DBU 81BsN
H
NHAc
OH
O
O
TBDPSO
SOPh
OAc
703 steps 81
17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

17
てチオフェノールを 14-付加にて導入しケトンの還元保護ののち一等量の mCPBA による硫黄原
子の一段階酸化を経て TFAA による Pummerer 転移を用いアルデヒド 71717171 を得ることに成功したまた
このときアセトアミドの窒素と隣接する三級アルコールが TFAA を介して環化しアセチル基が脱落
して 2-トリフルオロメチルオキサゾリンを形成した(Scheme 20)これは通常は加水分解に強塩基性
条件下加熱することを必要
とするアセトアミドを室
温で加水分解可能な官能基
へと穏和な条件下で効率よ
く変換できたことになり
非常に有用な条件である
続いてルーチェ還元によりアルデヒドを還元しアルコールとしピバロイル基で保護し 72727272 とした後
TBDPS 基を脱保護し生じたアルコールを Swern 酸化にてアルデヒド 73737373 とした73737373 に対し当研究室
の寺内が開発した手法に従いWittig 反応にて 4 炭素増炭し 74747474 とした29 次に PPTS による加水分解
条件に付しTBS 基の除去とオキサゾリンの加水分解による開環を同時に進行させた御原は側鎖上の
オレフィンを水素雰囲気下でアダムス触媒のみを用いて還元し 77775555 を得ているが筆者が追試を行った
ところ再現性が得られず原料回収に終わったため酢酸を加えてさらに触媒を活性化することで収率よ
く 75757575 を得ることに成功した最後に末端のアルコールを西沢-Grieco 法を用いてオレフィンとし 76767676 を
得た27d
C 環については松村がモデル基質 77a77a77a77a 及び 77b77b77b77b に対し種々のルテニウム触媒を用い RCM を行い
その構築に成功している(Table 7)27ab 検討の結果基質 77b77b77b77b に対し第一世代 Grubbs 触媒を用いた場
合に最も良い収率Z 選択
性が得られた(entries 4-5)
第二世代Grubbs触媒はE
体が選択的に得られ
(entries 2 and 6)また第
二世代 Hoveyda-Grubbs
触媒では選択性が発現し
なかった(entries 3 and 7)
またこれら二種類の活性
の高い触媒では二量体の
生成も確認された(entries
6-7)
筆者は以上の結果を踏まえマンザミン B の合成計画を次のように大きく二分した
①76767676 から C 環D 環を構築しイルシナール B を全合成しマンザミン B の基本骨格の合成法を確立
②B 環上のエポキシ基及びβ-カルボリンの立体選択的導入法の確立
①でイルシナール B を合成した後に②を達成するためには合成終盤で基質の量が限られる中三級
アルコールからオレフィンに対する分子内環化とそれに続く concave面からの立体選択的プロトン化を
達成しなければならず非常に困難だと考えられる(Figure 7)そこでまず中間体 66666666(Scheme 18)を
鍵中間体に設定しこれを用いて②について検討し①とは別のルートでマンザミン B の合成を目指す
こととした
BsN
HX
HN O
BsN
HX
HN O
entry
1
2
3
4
5
6
7
Z ()
39
23
32
64
64
9
23
conc (mM)
05
05
05
05
10
05
05
time (h)
4
5
5
4
8
7
7
E ()
11
43
28
23
21
22
14
Ru cat
Grubbs I
Grubbs II
Hoveyda-Grubbs II
Grubbs I
Grubbs I
Grubbs II
Hoveyda-Grubbs II
dimer ()
-
-
-
-
-
40
41
X
O
O
O
Ru cat (30 mol )
CH2Cl2 (degassed)reflux
Table 7
77 78
77a
77b
NH
O
OH
N
O
OH
O
CF3
N
O
O
F3COH
N
O
O
CF3
base
NO
CF3NH
O
O
O CF3
TFAA
Scheme 20
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
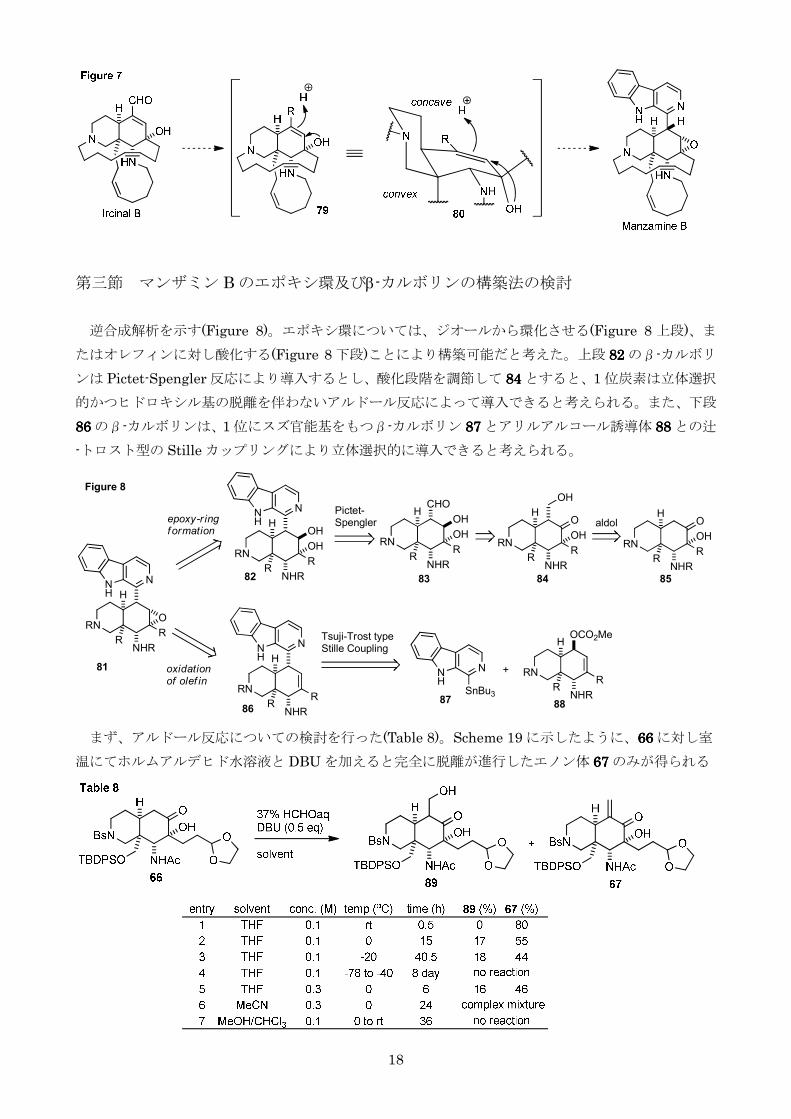
18
第三節 マンザミン B のエポキシ環及びβ-カルボリンの構築法の検討
逆合成解析を示す(Figure 8)エポキシ環についてはジオールから環化させる(Figure 8 上段)ま
たはオレフィンに対し酸化する(Figure 8 下段)ことにより構築可能だと考えた上段 82828282 のβ-カルボリ
ンは Pictet-Spengler 反応により導入するとし酸化段階を調節して 84848484 とすると1 位炭素は立体選択
的かつヒドロキシル基の脱離を伴わないアルドール反応によって導入できると考えられるまた下段
86868686 のβ-カルボリンは1 位にスズ官能基をもつβ-カルボリン 87878787 とアリルアルコール誘導体 88888888 との辻
-トロスト型の Stille カップリングにより立体選択的に導入できると考えられる
RN
H
NHRR
R
O
NNH
epoxy-r ingformation
Pictet-Spengler
oxidationof olef in
aldol
NNH
SnBu3
+
Tsuji-Trost typeStille Coupling
RN
H
NHRR
R
OH
NNH
OHRN
H
NHRR
CHO
R
OH
OH
RN
H
NHRR
R
OH
O
OH
RN
H
NHRR
R
OH
O
RN
H
NHRR
R
NNH
RN
H
NHRR
R
OCO2Me
Figure 8
81
82 83 84 85
8687 88
まずアルドール反応についての検討を行った(Table 8)Scheme 19 に示したように66666666 に対し室
温にてホルムアルデヒド水溶液と DBU を加えると完全に脱離が進行したエノン体 66667777 のみが得られる
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
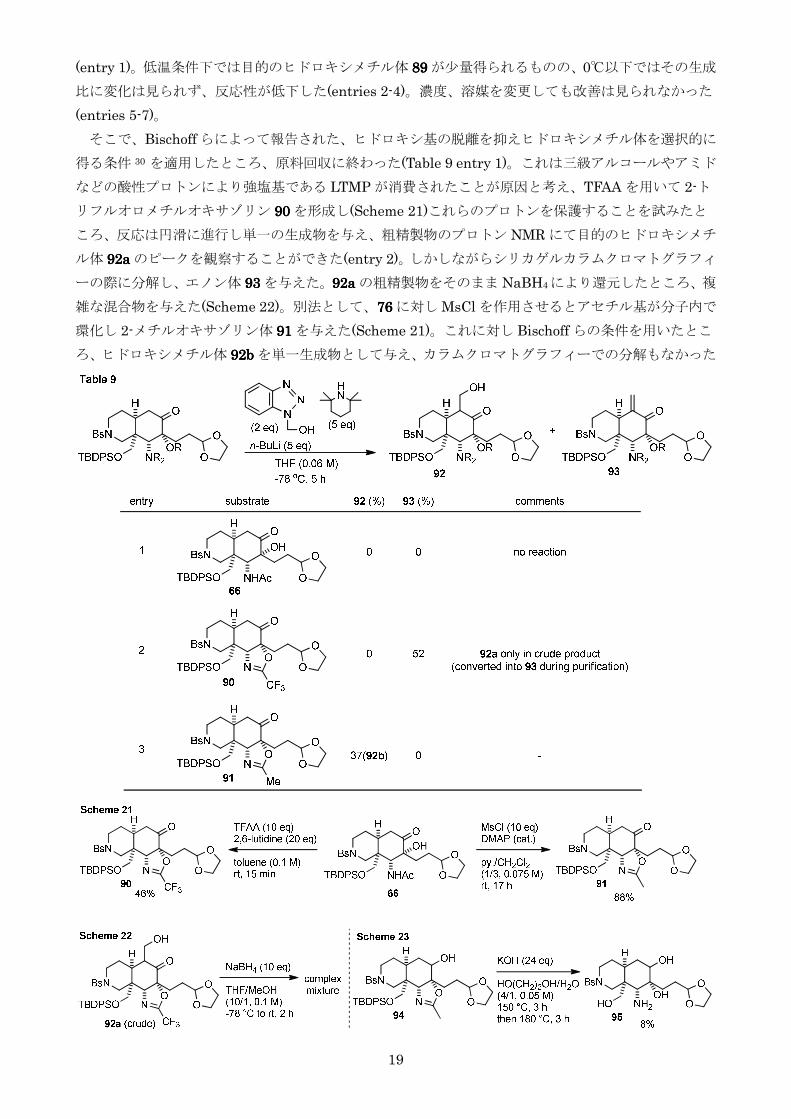
19
(entry 1)低温条件下では目的のヒドロキシメチル体 89898989 が少量得られるものの0以下ではその生成
比に変化は見られず反応性が低下した(entries 2-4)濃度溶媒を変更しても改善は見られなかった
(entries 5-7)
そこでBischoff らによって報告されたヒドロキシ基の脱離を抑えヒドロキシメチル体を選択的に
得る条件 30 を適用したところ原料回収に終わった(Table 9 entry 1)これは三級アルコールやアミド
などの酸性プロトンにより強塩基である LTMP が消費されたことが原因と考えTFAA を用いて 2-ト
リフルオロメチルオキサゾリン 90909090 を形成し(Scheme 21)これらのプロトンを保護することを試みたと
ころ反応は円滑に進行し単一の生成物を与え粗精製物のプロトン NMR にて目的のヒドロキシメチ
ル体 92a92a92a92a のピークを観察することができた(entry 2)しかしながらシリカゲルカラムクロマトグラフィ
ーの際に分解しエノン体 93939393 を与えた92a92a92a92a の粗精製物をそのまま NaBH4により還元したところ複
雑な混合物を与えた(Scheme 22)別法として76767676 に対し MsCl を作用させるとアセチル基が分子内で
環化し 2-メチルオキサゾリン体 91919191 を与えた(Scheme 21)これに対し Bischoff らの条件を用いたとこ
ろヒドロキシメチル体 92b92b92b92b を単一生成物として与えカラムクロマトグラフィーでの分解もなかった
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
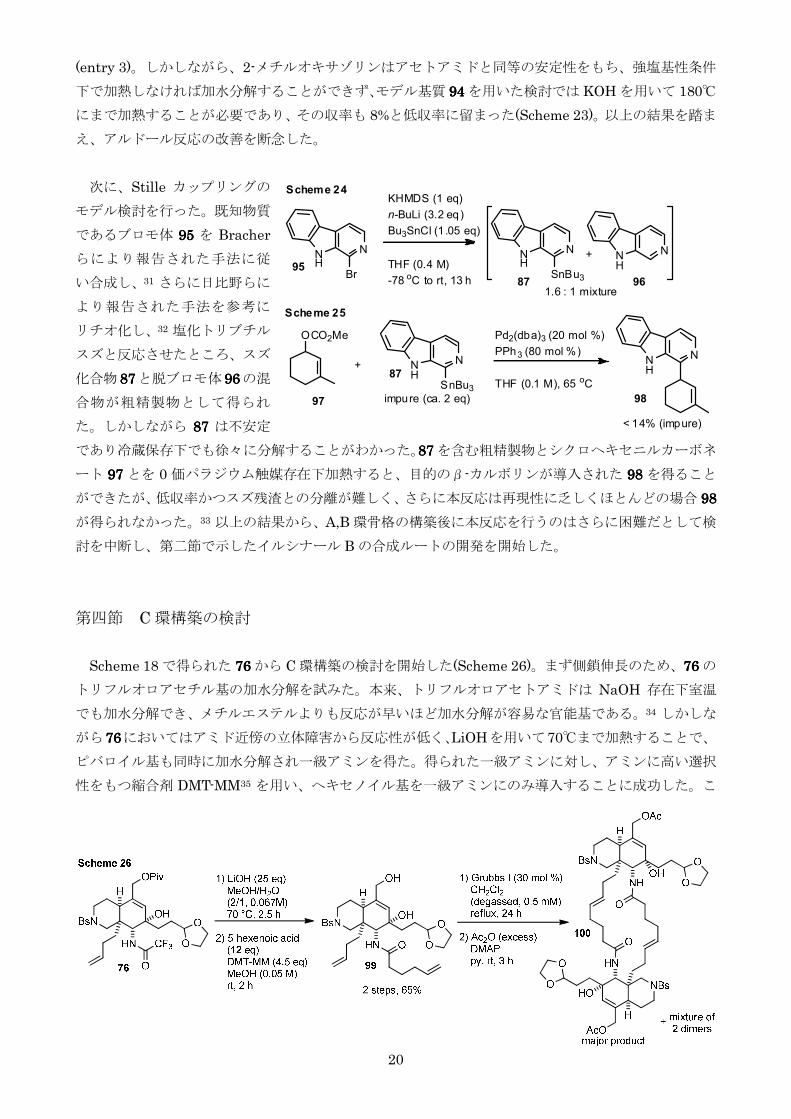
20
(entry 3)しかしながら2-メチルオキサゾリンはアセトアミドと同等の安定性をもち強塩基性条件
下で加熱しなければ加水分解することができずモデル基質 94949494 を用いた検討では KOH を用いて 180
にまで加熱することが必要でありその収率も 8と低収率に留まった(Scheme 23)以上の結果を踏ま
えアルドール反応の改善を断念した
次にStille カップリングの
モデル検討を行った既知物質
であるブロモ体 95959595 を Bracher
らにより報告された手法に従
い合成し31 さらに日比野らに
より報告された手法を参考に
リチオ化し32 塩化トリブチル
スズと反応させたところスズ
化合物 87878787と脱ブロモ体 96969696の混
合物が粗精製物として得られ
たしかしながら 87878787 は不安定
であり冷蔵保存下でも徐々に分解することがわかった87878787 を含む粗精製物とシクロヘキセニルカーボネ
ート 97979797 とを 0 価パラジウム触媒存在下加熱すると目的のβ-カルボリンが導入された 98989898 を得ること
ができたが低収率かつスズ残渣との分離が難しくさらに本反応は再現性に乏しくほとんどの場合 98989898
が得られなかった33 以上の結果からAB 環骨格の構築後に本反応を行うのはさらに困難だとして検
討を中断し第二節で示したイルシナール B の合成ルートの開発を開始した
第四節 C 環構築の検討
Scheme 18 で得られた 76767676 から C 環構築の検討を開始した(Scheme 26)まず側鎖伸長のため76767676 の
トリフルオロアセチル基の加水分解を試みた本来トリフルオロアセトアミドは NaOH 存在下室温
でも加水分解できメチルエステルよりも反応が早いほど加水分解が容易な官能基である34 しかしな
がら76767676においてはアミド近傍の立体障害から反応性が低くLiOHを用いて70まで加熱することで
ピバロイル基も同時に加水分解され一級アミンを得た得られた一級アミンに対しアミンに高い選択
性をもつ縮合剤 DMT-MM35 を用いヘキセノイル基を一級アミンにのみ導入することに成功したこ
N
Br
NH95
N
SnBu3
NH
KHMDS (1 eq)
n-BuLi (32 eq)
Bu3SnCl (1 05 eq)
THF (04 M)
-78 oC to rt 13 h
NNH
+
87 9616 1 mixture
Pd2(dba)3 (20 mol )
PPh3 (80 mol )
THF (01 M) 65 oC
NNH
98
OCO2Me
N
SnBu3
NH
+
impure (ca 2 eq)
lt 14 (impure)
Scheme 24
Scheme 25
97
87
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
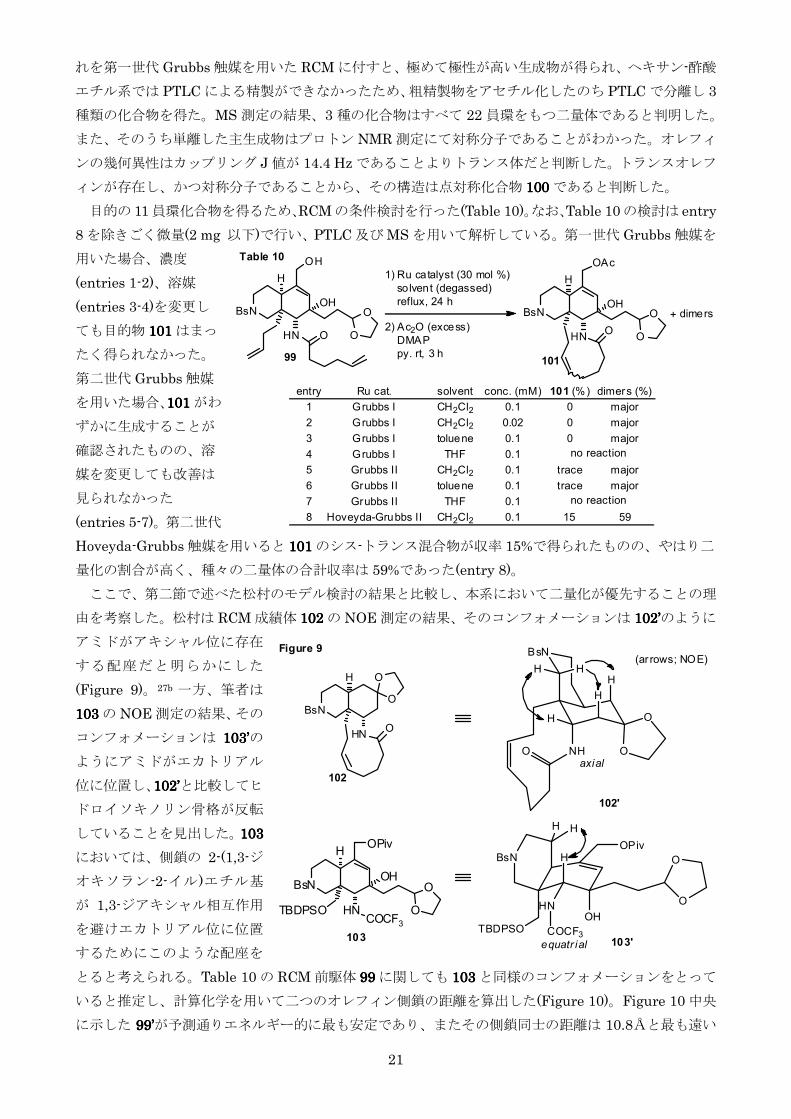
21
れを第一世代 Grubbs 触媒を用いた RCM に付すと極めて極性が高い生成物が得られヘキサン-酢酸
エチル系では PTLC による精製ができなかったため粗精製物をアセチル化したのち PTLC で分離し 3
種類の化合物を得たMS 測定の結果3 種の化合物はすべて 22 員環をもつ二量体であると判明した
またそのうち単離した主生成物はプロトン NMR 測定にて対称分子であることがわかったオレフィ
ンの幾何異性はカップリング J 値が 144 Hz であることよりトランス体だと判断したトランスオレフ
ィンが存在しかつ対称分子であることからその構造は点対称化合物 100100100100 であると判断した
目的の 11 員環化合物を得るためRCMの条件検討を行った(Table 10)なおTable 10の検討は entry
8 を除きごく微量(2 mg 以下)で行いPTLC 及び MS を用いて解析している第一世代 Grubbs 触媒を
用いた場合濃度
(entries 1-2)溶媒
(entries 3-4)を変更し
ても目的物 101101101101 はまっ
たく得られなかった
第二世代 Grubbs 触媒
を用いた場合101101101101 がわ
ずかに生成することが
確認されたものの溶
媒を変更しても改善は
見られなかった
(entries 5-7)第二世代
Hoveyda-Grubbs 触媒を用いると 101101101101 のシス-トランス混合物が収率 15で得られたもののやはり二
量化の割合が高く種々の二量体の合計収率は 59であった(entry 8)
ここで第二節で述べた松村のモデル検討の結果と比較し本系において二量化が優先することの理
由を考察した松村は RCM 成績体 102102102102 の NOE 測定の結果そのコンフォメーションは 102102102102rsquorsquorsquorsquoのように
アミドがアキシャル位に存在
する配座だと明らかにした
(Figure 9)27b 一方筆者は
103103103103 の NOE 測定の結果その
コンフォメーションは 103103103103rsquorsquorsquorsquoの
ようにアミドがエカトリアル
位に位置し102102102102rsquorsquorsquorsquoと比較してヒ
ドロイソキノリン骨格が反転
していることを見出した103103103103
においては側鎖の 2-(13-ジ
オキソラン-2-イル)エチル基
が 13-ジアキシャル相互作用
を避けエカトリアル位に位置
するためにこのような配座を
とると考えられるTable 10 の RCM 前駆体 99999999 に関しても 103103103103 と同様のコンフォメーションをとって
いると推定し計算化学を用いて二つのオレフィン側鎖の距離を算出した(Figure 10)Figure 10 中央
に示した 99999999rsquorsquorsquorsquoが予測通りエネルギー的に最も安定でありまたその側鎖同士の距離は 108Åと最も遠い
entry
1
2
3
4
5
6
7
8
solvent
CH2Cl2
CH2Cl2
toluene
THF
CH2Cl2
toluene
THF
CH2Cl2
Ru cat
Grubbs I
Grubbs I
Grubbs I
Grubbs I
Grubbs II
Grubbs II
Grubbs II
Hoveyda-Grubbs II
1) Ru catalyst (30 mol )
solvent (degassed)
reflux 24 h
2) Ac2O (excess)
DMAP
py rt 3 h
conc (mM)
01
002
01
01
01
01
01
01
Table 10
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
HN
OH
101
O
+ dimers
101 ()
0
0
0
trace
trace
15
dimers ()
major
major
major
major
major
59
no reaction
no reaction
BsN
NH O
O
O
H
H
HH
H
(ar rows NOE)
HN
COCF3
OH
O
OBsNOPiv
H
H
TBDPSO
BsN
H
O
O
OPiv
TBDPSO HNCOCF3
OH
BsN
H
HNO
102
O
O
H
103
102
axial
equatr ial
Figure 9
103
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
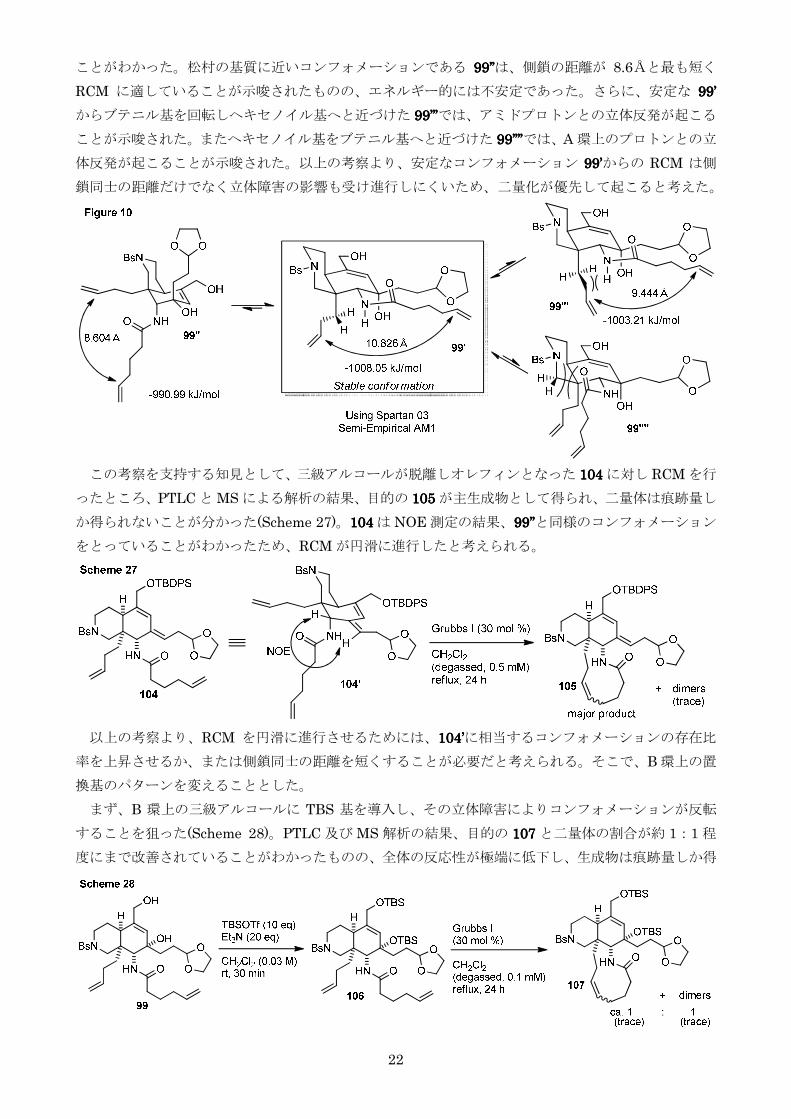
22
ことがわかった松村の基質に近いコンフォメーションである 99999999rsquorsquorsquorsquorsquorsquorsquorsquoは側鎖の距離が 86Åと最も短く
RCM に適していることが示唆されたもののエネルギー的には不安定であったさらに安定な 99999999rsquorsquorsquorsquo
からブテニル基を回転しヘキセノイル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではアミドプロトンとの立体反発が起こる
ことが示唆されたまたヘキセノイル基をブテニル基へと近づけた 99999999rsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquorsquoではA 環上のプロトンとの立
体反発が起こることが示唆された以上の考察より安定なコンフォメーション 99999999rsquorsquorsquorsquoからの RCM は側
鎖同士の距離だけでなく立体障害の影響も受け進行しにくいため二量化が優先して起こると考えた
この考察を支持する知見として三級アルコールが脱離しオレフィンとなった 104104104104 に対し RCM を行
ったところPTLC と MS による解析の結果目的の 105105105105 が主生成物として得られ二量体は痕跡量し
か得られないことが分かった(Scheme 27)104104104104 は NOE 測定の結果99999999rsquorsquorsquorsquorsquorsquorsquorsquoと同様のコンフォメーション
をとっていることがわかったためRCM が円滑に進行したと考えられる
以上の考察よりRCM を円滑に進行させるためには104104104104rsquorsquorsquorsquoに相当するコンフォメーションの存在比
率を上昇させるかまたは側鎖同士の距離を短くすることが必要だと考えられるそこでB 環上の置
換基のパターンを変えることとした
まずB 環上の三級アルコールに TBS 基を導入しその立体障害によりコンフォメーションが反転
することを狙った(Scheme 28)PTLC 及び MS 解析の結果目的の 107107107107 と二量体の割合が約 11 程
度にまで改善されていることがわかったものの全体の反応性が極端に低下し生成物は痕跡量しか得
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
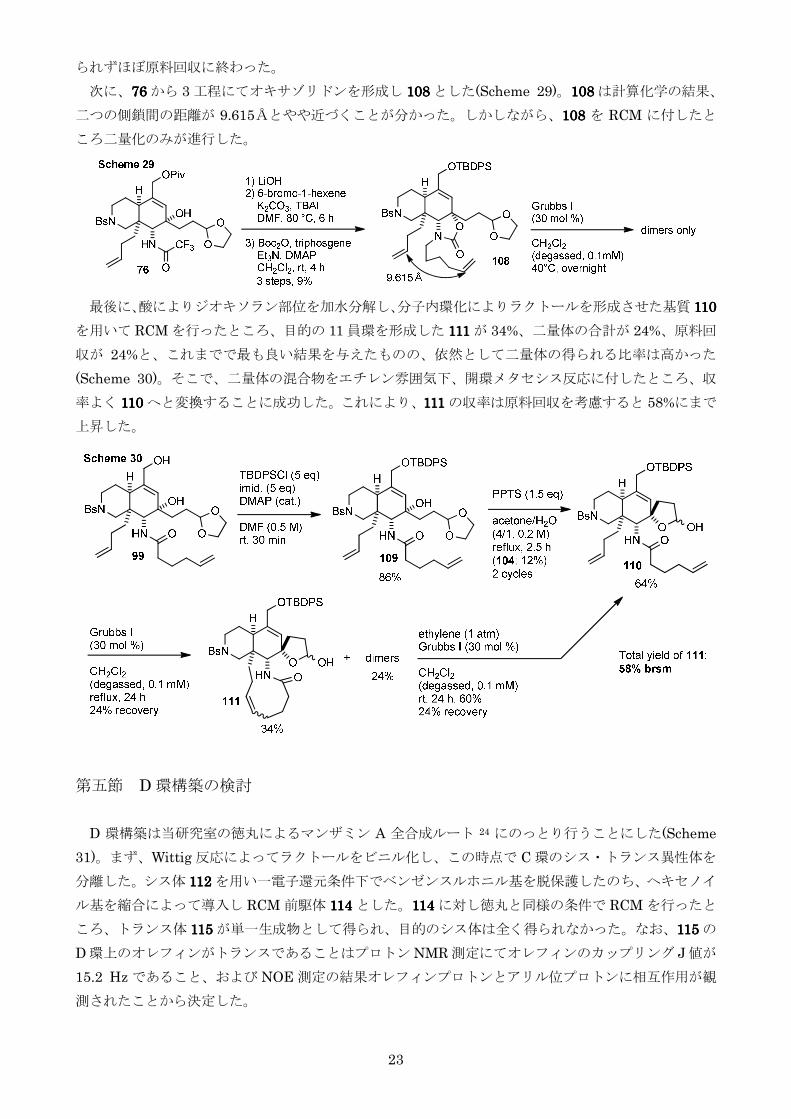
23
られずほぼ原料回収に終わった
次に76767676 から 3 工程にてオキサゾリドンを形成し 108108108108 とした(Scheme 29)108108108108 は計算化学の結果
二つの側鎖間の距離が 9615Åとやや近づくことが分かったしかしながら108108108108 を RCM に付したと
ころ二量化のみが進行した
最後に酸によりジオキソラン部位を加水分解し分子内環化によりラクトールを形成させた基質 110110110110
を用いて RCM を行ったところ目的の 11 員環を形成した 111111111111 が 34二量体の合計が 24原料回
収が 24とこれまでで最も良い結果を与えたものの依然として二量体の得られる比率は高かった
(Scheme 30)そこで二量体の混合物をエチレン雰囲気下開環メタセシス反応に付したところ収
率よく 110110110110 へと変換することに成功したこれにより111111111111 の収率は原料回収を考慮すると 58にまで
上昇した
第五節 D 環構築の検討
D 環構築は当研究室の徳丸によるマンザミン A 全合成ルート 24 にのっとり行うことにした(Scheme
31)まずWittig 反応によってラクトールをビニル化しこの時点で C 環のシストランス異性体を
分離したシス体 112112112112 を用い一電子還元条件下でベンゼンスルホニル基を脱保護したのちヘキセノイ
ル基を縮合によって導入し RCM 前駆体 114114114114 とした114114114114 に対し徳丸と同様の条件で RCM を行ったと
ころトランス体 115115115115 が単一生成物として得られ目的のシス体は全く得られなかったなお115115115115 の
D 環上のオレフィンがトランスであることはプロトン NMR 測定にてオレフィンのカップリング J 値が
152 Hz であることおよび NOE 測定の結果オレフィンプロトンとアリル位プロトンに相互作用が観
測されたことから決定した
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
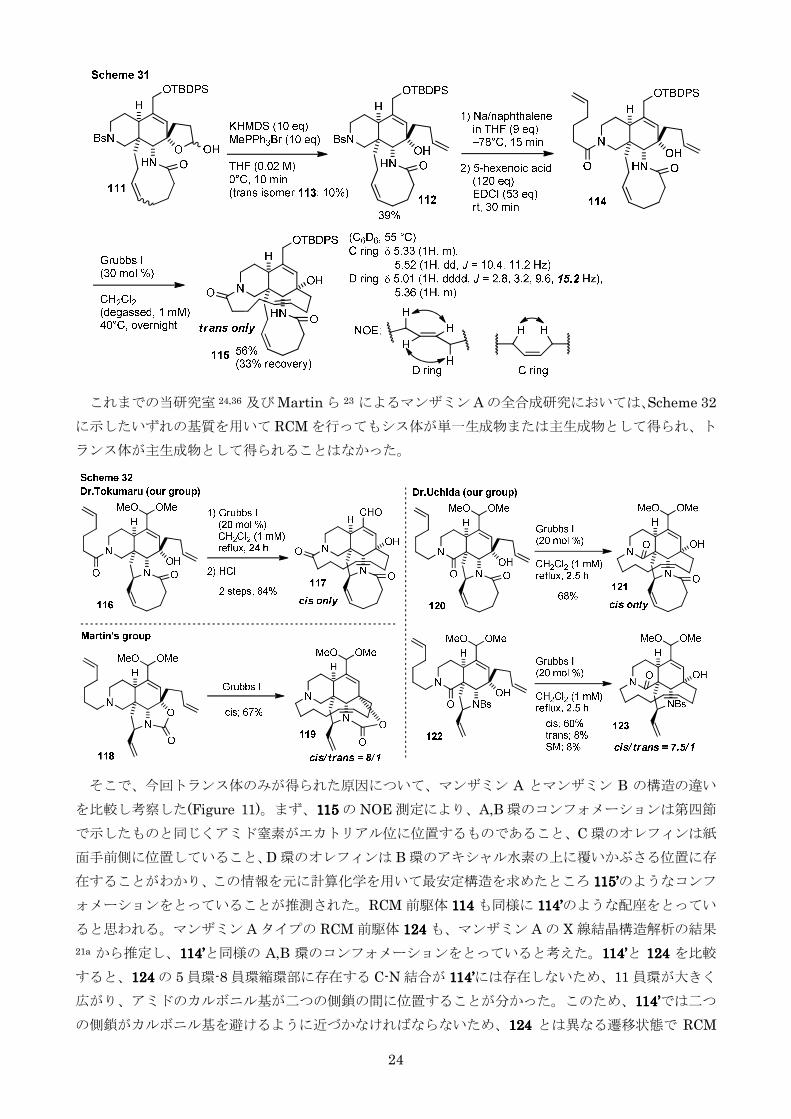
24
これまでの当研究室 2436 及び Martin ら 23 によるマンザミン A の全合成研究においてはScheme 32
に示したいずれの基質を用いて RCM を行ってもシス体が単一生成物または主生成物として得られト
ランス体が主生成物として得られることはなかった
そこで今回トランス体のみが得られた原因についてマンザミン A とマンザミン B の構造の違い
を比較し考察した(Figure 11)まず115115115115 の NOE 測定によりAB 環のコンフォメーションは第四節
で示したものと同じくアミド窒素がエカトリアル位に位置するものであることC 環のオレフィンは紙
面手前側に位置していることD 環のオレフィンは B 環のアキシャル水素の上に覆いかぶさる位置に存
在することがわかりこの情報を元に計算化学を用いて最安定構造を求めたところ 115115115115rsquorsquorsquorsquoのようなコンフ
ォメーションをとっていることが推測されたRCM 前駆体 114114114114 も同様に 114114114114rsquorsquorsquorsquoのような配座をとってい
ると思われるマンザミン A タイプの RCM 前駆体 124124124124 もマンザミン A の X 線結晶構造解析の結果
21a から推定し114114114114rsquorsquorsquorsquoと同様の AB 環のコンフォメーションをとっていると考えた114114114114rsquorsquorsquorsquoと 124124124124 を比較
すると124124124124 の 5 員環-8 員環縮環部に存在する C-N 結合が 114114114114rsquorsquorsquorsquoには存在しないため11 員環が大きく
広がりアミドのカルボニル基が二つの側鎖の間に位置することが分かったこのため114114114114rsquorsquorsquorsquoでは二つ
の側鎖がカルボニル基を避けるように近づかなければならないため124124124124 とは異なる遷移状態で RCM
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
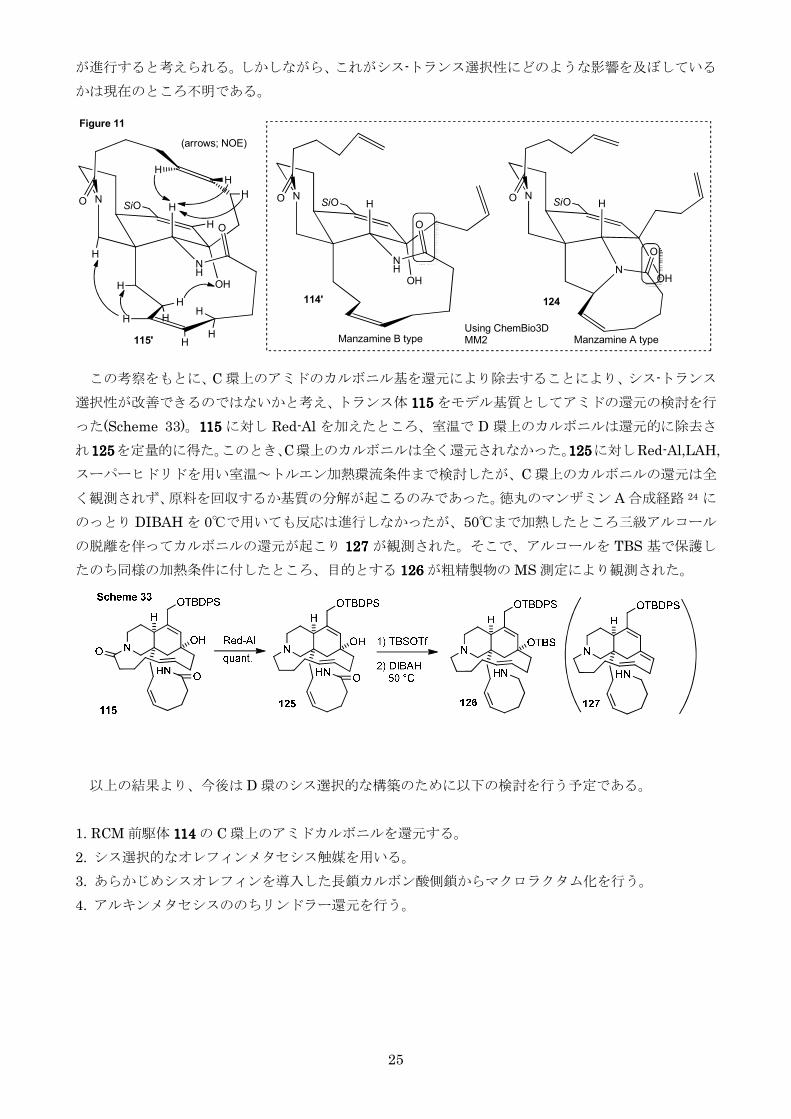
25
が進行すると考えられるしかしながらこれがシス-トランス選択性にどのような影響を及ぼしている
かは現在のところ不明である
N
NH
OH
O
HH
O
SiO
H
H
H
H
H
H
H
H
H
H
(arrows NOE)
N
NOH
O
O
SiO HN
NH
OH
O
O
SiO H
Manzamine B type Manzamine A typeUsing ChemBio3DMM2
H
115
Figure 11
114 124
この考察をもとにC 環上のアミドのカルボニル基を還元により除去することによりシス-トランス
選択性が改善できるのではないかと考えトランス体 115115115115 をモデル基質としてアミドの還元の検討を行
った(Scheme 33)115115115115 に対し Red-Al を加えたところ室温で D 環上のカルボニルは還元的に除去さ
れ125125125125を定量的に得たこのときC環上のカルボニルは全く還元されなかった125125125125に対しRed-AlLAH
スーパーヒドリドを用い室温~トルエン加熱環流条件まで検討したがC 環上のカルボニルの還元は全
く観測されず原料を回収するか基質の分解が起こるのみであった徳丸のマンザミン A 合成経路 24 に
のっとり DIBAH を 0で用いても反応は進行しなかったが50まで加熱したところ三級アルコール
の脱離を伴ってカルボニルの還元が起こり 127127127127 が観測されたそこでアルコールを TBS 基で保護し
たのち同様の加熱条件に付したところ目的とする 111126262626 が粗精製物の MS 測定により観測された
以上の結果より今後は D 環のシス選択的な構築のために以下の検討を行う予定である
1 RCM 前駆体 114114114114 の C 環上のアミドカルボニルを還元する
2 シス選択的なオレフィンメタセシス触媒を用いる
3 あらかじめシスオレフィンを導入した長鎖カルボン酸側鎖からマクロラクタム化を行う
4 アルキンメタセシスののちリンドラー還元を行う
26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

26
総括
筆者は当研究室及び Rawal らが開発した不斉 Diels-Alder 反応を用い天然物の不斉合成について
の研究を行い以下の成果を得た
1 当研究室の Danishefsky ジエンを用いる触媒的不斉 Diels-Alder 反応により天然体の
(-)-platyphyllide の触媒的不斉全合成を達成したまたその絶対配置を報告されていたものとは
逆の(6S7S)と決定した
2 分子内に TIPS エノールエーテルと側鎖にヒドロキシメチル基をもつシクロヘキセン誘導体に対し
て伊藤-三枝酸化を行うとオレフィンの異性化を伴った生成物が優先的に得られることを発見した
3 Rawal らによって開発された Rawal ジエンを用いる触媒的不斉 Diels-Alder 反応を鍵工程としマ
ンザミン B の ABC 環を不斉合成したD 環構築については今後も検討を行う予定である
27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

27
Experimental SectionExperimental SectionExperimental SectionExperimental Section
General Methods General Methods General Methods General Methods All reactions involving air- or moisture-sensitive reagents or intermediates were
performed under an inert atmosphere of argon in glassware Unless otherwise noted solvents and
reagents were reagent grade and used without further purification DBU Et3N and pyridine were
distilled from CaH2 Anhydrous THF CH2Cl2 and toluene were used as received from Kanto
Chemical CO INC Analytical and preparative TLC was carried on E Merck 025 mm silica gel 60
F254 plates Silica gel column chromatography was performed using Fuji Silysia Chemical Ltd silica
gel PSQ 60B Celitereg was used with Celitereg 545 Optical rotations were measured on a JASCO
P-1000 polarimeter at 589 nm Data are reported as follows [α] λtemp concentration (c g100 mL)
and solvent 1H NMR and 13C NMR spectra were taken on 400 MHz 600 MHz 100 MHz and 150
MHz instruments (JEOL LNM-GSX 400α JEOL JMN-ECP 400 JEOL JMN-ECP 600) in the
indicated solvent at rt 1H NMR spectra was recorded with (CH3)4Si (TMS) as internal reference
and 13C NMR spectra was recorded with CDCl3 as internal reference Coupling constants are
reported in hertz (Hz) Spectral splitting patterns are designated as follows s singlet d doublet t
triplet q quartet m multiplet br broad Infrared (IR) spectra were recorded on JASCO FTIR-230
spectrometer MS spectrometry was carried out at the Chemical Analysis Center of Chiba
University (JEOL BU-20 for LR- and HR-EIMS JEOL JMS-AX500 for LR-FABMS JEOL
JMS-AX505 for LR-FABMS JEOL JMS-HX110 for HR-FABMS Thermo Scientific Exactive or
JEOL AccuTOF LC-plus JMS-T100LP for LR- and HR-ESIMS) High performance liquid
chromatography (HPLC) analyses were performed on a Shimadzu LC-2010C (Shimadzu Ind Ltd)
with detection at 254 nm and on a Daicel chiral column (Chiralcel OJ-H Chiralpak AD-H or
Chiralpak IA Daicel Chemical Ind Ltd)
Chapter 1Chapter 1Chapter 1Chapter 1
SSSSection 2ection 2ection 2ection 2
Dienophile 3333bbbb5a and BINAMIDE 55555a were prepared according to the reported procedure
((((EEEE))))----triethyl(4triethyl(4triethyl(4triethyl(4----methoxybutamethoxybutamethoxybutamethoxybuta----13131313----diendiendiendien----2222----yloxy)silaneyloxy)silaneyloxy)silaneyloxy)silane (1(1(1(1cccc)))) To a stirred solution of
(E)-4-methoxybut-3-en-2-one (16 mL 158 mmol) and Et3N (6 mL 430 mmol) in
anhydrous Et2O (30 mL) at ndash20 ordmC was added dropwise TESOTf (4 mL 177 mmol)
The mixture was then warmed to 0 ordmC and stirred for 2 h at the same temperature The mixture was
then diluted with hexane (20 mL) and washed with ice cold NaHCO3 and brine After the volatile
material was removed under reduced pressure bulb-to-bulb distillation of the resulting residue (01
mmHg heating at 90 ordmC) gave 1111cccc (28 mL yield 74) as a colorless liquid 1H NMR (CDCl3 400
MHz) δ 072 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 359 (3H s) 408 (1H s) 409 (1H s) 535
(1H d J = 120 Hz) 689 (1H d J = 120 Hz) 13C NMR (CDCl3 100 MHz) δ 48 66 563 904
1031 1502 1541 IR(neat) 1651 cmndash1 HRMS(FAB) calcd for C11H23O2Si 2151467 found 2151467
3333----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidinenecarbonyl)oxazolidin----2222----
oneoneoneone (((((+)(+)(+)(+)----17171717aaaa)))) A mixture of Yb(OTf)3 (5354 mg 0863 mmol) and
(S)-BINAMIDE (5843 mg 1035 mmol) in a flask with a stirring bar was dried
at 90 ordmC under reduced pressure (lt01 mmHg) for 05 h with stirring After the mixture was allowed
TESO
OMe1c
TESO
O
N O
OOMe(+)-17a
28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

28
to cool to room temperature the flask was charged with argon CH2Cl2 (288 mL) and DBU (316 microL
207 mmol) were successively added and the mixture was stirred for 2 h at room temperature
Dienophile 3333bbbb (122 g 864 mmol) in CH2Cl2 (144 mL) was added at 0 ordmC The mixture was
immediately cooled to ndash20 ordmC and diene 1111cccc (415 mL 172 mmol) was added dropwise and stirred for
3 h at the same temperature Water (10 mL) was then added to quench the reaction and the
insoluble materials were filtered through a pad of Celite The water layer was extracted three times
with CH2Cl2 and the combined organic layers were washed with brine and dried over Na2SO4 After
the volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 51) to give (+)(+)(+)(+)----17171717aaaa (204 g yield 66) as a colorless
oil The enantiomeric excess was determined to be 60 ee by HPLC analysis after conversion to
enone by TFA (Daicel Chiralcel OJ-H flow rate 075 mLmin hexaneiPrOH=6040 retention time
374 min (major) and 390 min (minor)) 1H NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098
(9H t J = 80 Hz) 165-174 (1H m) 200-204 (2H m) 229-237 (1H m) 330 (3H s) 381 (1H
ddd J = 30 88 116 Hz) 398-412 (2H m) 437-444 (2H m) 470 (1H d J = 88 Hz) 500 (1H s)
13C NMR (CDCl3 100 MHz) δ 53 70 249 295 431 440 556 622 766 1032 1533 1534
1749 IR(neat) 1775 1696 1660 cm-1 HRMS(FAB) calcd for C17H30NO5Si 3561893 found 3561879
[α]D25 +458 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----17171717aaaa in Scheme 11 [α]D24 ndash619 (c 024 CHCl3 60 ee)
2222----((1((1((1((1SSSS2222SSSS))))----2222----methoxymethoxymethoxymethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol (((((+)(+)(+)(+)----21212121)))) A
109 M solution of methyllithium in diethyl ether (132 mL 144 mmol) was added
dropwise to a solution of (+)(+)(+)(+)----17171717aaaa (20 g 570 mmol) in THF (58 mL) at ndash78 ordmC The
mixture was stirred for 15 min at the same temperature Saturated aqueous ammonium chloride
(20 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 91) to give (+)(+)(+)(+)----21212121 (838 mg yield 48) as a colorless oil 1H
NMR (CDCl3 400 MHz) δ 069 (6H q J = 80 Hz) 098 (9H t J = 80 Hz) 117 (3H s) 123 (3H s)
125-135 (1H m) 165-171 (1H m) 172-178 (1H m) 196-202 (1H m) 213-225 (1H m) 333
(3H s) 421 (1H d J = 112 Hz) 474 (1H s) 495 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66
237 248 295 303 480 538 729 786 1025 1540 IR(neat) 3464 1666 cm-1 HRMS(FAB)
calcd for C16H32O3SiNa 3232018 found 3232006 [α]D25 +447 (c 10 CHCl3 60 ee)
For the enantiomer ((((ndashndashndashndash))))----21212121 in Scheme 13 [α]D23 ndash828 (c 028 CHCl3 60 ee)
((((RRRR))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----22222222)))) A solution of (+)(+)(+)(+)----21212121
(248 mg 083 mmol) and Pd(OAc)2 (185 mg 0083 mmol) in DMSO (165 mL) was
stirred under O2 for 8 h The solvent was distilled under reduced pressure (01 mmHg)
at 55 ordmC The resulting residue was purified by column chromatography (SiO2 AcOEt) to give ((((ndashndashndashndash))))----22222222
(94 mg yield 62) as a colorless solid and (+)(+)(+)(+)----23232323 (34 mg containing trace inseparable compound
yield lt22) as a colorless oil
((((ndashndashndashndash))))----22222222 (149 mg) was recrystallized from hexaneAcOEt to give racemic 22222222 (53 mg yield 36) as
TESO
OHOMe(+)-21
O
OHOMe(ndash)-22
29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

29
a colorless crystal and chiral ((((ndashndashndashndash))))----22222222 (94 mg yield 63) from mother liquid as a colorless oil The ee
was determined to be gt995 ee by HPLC analysis (Daicel Chiralpak IA hexaneiPrOH = 973 f
10 mLmin tR 487 min ((+)(+)(+)(+)----22222222) and 505 min (((((ndashndashndashndash))))----22222222)) 1H NMR (CDCl3 400 MHz) δ 125 (3H s)
128 (3H s) 179-188 (1H m) 209-217 (1H m) 226-239 (1H m) 250-257 (1H m) 264 (1H dd
J = 58 86 Hz) 308 (1H s) 377 (3H s) 550 (1H s) 13C NMR (CDCl3 100 MHz) δ 241 261 288
354 487 558 732 1046 1783 1992 IR(neat) 3391 1590 cm-1 HRMS(FAB) calcd for C10H17O3
1851178 found 1851170 [α]D23 ndash117 (c 10 CHCl3 gt995 ee)
For the enantiomer (+)(+)(+)(+)----22222222 in Scheme 13 the enantiomeric excess was determined to be 94 ee
by HPLC analysis (Daicel Chiralpak AD-H hexaneiPrOH = 973 f 10 mLmin tR 462 min
(((((ndashndashndashndash))))----22222222)) and 483 min ((+)(+)(+)(+)----22222222)) [α]D20 +167 (c 095 CHCl3 94 ee) mp 925-930 ordmC
(4(4(4(4SSSS5555SSSS))))----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methoxymethoxymethoxymethoxy---- cyclohexcyclohexcyclohexcyclohex----2222----enoneenoneenoneenone (((((+)(+)(+)(+)----23232323)))) 1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 138 (3H s) 244 (1H dd J = 104 160 Hz) 268
(1H dt J = 28 84 Hz) 300 (1H dd J = 40 160 Hz) 341 (3H s) 389 (1H ddd J
= 40 84 104 Hz) 399 (1H s) 609 (1H ddd J = 06 28 104 Hz) 683 (1H dd J = 28 104 Hz)
13C NMR (CDCl3 100 MHz) δ 267 286 422 525 556 725 781 1304 1483 1974 IR(neat)
3420 1670 cm-1 HRMS(FAB) calcd for C10H17O3 1851178 found 1851178 [α]D24 +740 (c 02
CHCl3 60 ee)
((((RRRR))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----2222----enoneenoneenoneenone ((((((((ndashndashndashndash))))----9999)))) CeCl3bull7H2O (66 mg
0018 mmol) in a flask was dried at 150 ordmC under reduced pressure (lt01 mmHg) for
3 h with stirring After cooling to room temperature the flask was charged with dry
argon A solution of ((((ndashndashndashndash))))----22222222 (112 mg 0061 mmol) in THF (26 mL) was added to this flask A 10 M
solution of vinylmagnesium bromide in THF (597 microL 061 mmol) was added dropwise to this
mixture at ndash78 ordmC The mixture was then warmed to room temperature and stirred for 30 min 1N
HCl (2 mL) was slowly added at 0 ordmC and then neutralized with saturated aqueous NaHCO3 (3 mL)
The water layer was extracted three times with AcOEt and the combined organic layers were
washed with brine and dried over Na2SO4 After the volatile material was removed under reduced
pressure the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 31)
to give ((((ndashndashndashndash))))----9999 (90 mg yield 82) as a colorless oil and starting material ((((ndashndashndashndash))))----22222222 (11 mg y 10) 1H
NMR (CDCl3 400 MHz) δ 122 (3H s) 125 (3H s) 173 (1H ddd J = 48 130 264 Hz) 216-221
(1H m) 235-245 (2H m) 270 (1H ddd J = 26 46 178 Hz) 514 (1H s) 552 (1H d J = 108 Hz)
573 (1H d J = 148 Hz) 595 (1H s) 650 (1H dd J = 108 148 Hz) 13C NMR (CDCl3 100 MHz) δ
241 261 288 354 487 558 732 1046 1783 1992 IR(neat) 3438 1637 cm-1 HRMS(FAB)
calcd for C11H17O2 1811229 found 1811232 [α]D22 ndash1636 (c 10 CHCl3)
(+)(+)(+)(+)----9999 in Scheme 21 was obtained by following procedure To a stirred solution of (+)(+)(+)(+)----22222222 (70 mg
039 mmol) in THF (19 mL) at ndash78 ordmC was added a 10 M solution of vinylmagnesium bromide in
THF (38 mL 35 mmol) in one portion The mixture was then warmed to room temperature and
stirred for 15 min 1N HCl (5 mL) was slowly added at 0 ordmC and then neutralized with saturated
aqueous NaHCO3 (7 mL) The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
O
OHOMe(+)-23
OHO(ndash)-9
30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

30
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 31) to give (+)(+)(+)(+)----9999 (50 mg yield 63) as a colorless oil and starting material
(+)(+)(+)(+)----22222222 (94 mg yield 13) [α]D18 +2025 (c 10 CHCl3 90 ee) Ee value for (+)(+)(+)(+)----9999 can be measured as
follows Daicel Chiralpak AD-H hexaneiPrOH = 955 f 10mLmin 254 nm tR 126 min (((((ndashndashndashndash))))----9999)
and 211 min ((+)(+)(+)(+)----9999)
(1(1(1(1SSSS6666SSSS))))----6666----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----vinylcyclovinylcyclovinylcyclovinylcyclo---- hexhexhexhex----2222----enolenolenolenol ((((10101010)))) To a solution of
((((ndashndashndashndash))))----9999 (53 mg 029 mmol) in acetonitrile (27 mL) at rt was added NaBH(OAc)3 (623
mg 29 mmol) After 1 h Saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 31) to give the mixture of 10101010 and 11111111rsquorsquorsquorsquo (42 mg yield 79 1010101011111111rsquorsquorsquorsquo = 31) as a colorless
oil Diastereomeric ratio was determined by 1H NMR spectra of the mixture 1H NMR (CDCl3 400
MHz) δ 123 (3H s) 125-130 (1H m) 133 (3H s) 164 (1H ddd J = 26 94 128 Hz) 174-179
(2H m) 210-218 (1H m) 228 (1H dd J = 56 172 Hz) 307 (1H s) 401 (1H s) 452 (1H d J =
88 Hz) 503 (1H d J = 108 Hz) 515 (1H d J = 176 Hz) 564 (1H s) 638 (1H dd J = 108 176
Hz) 13C NMR (CDCl3 100 MHz) δ 238 240 244 303 514 698 749 1126 1316 1368 1386
IR(neat) 3193 cm-1 HRMS(ESI) calcd for C11H18O2Na 2051199 found 2051199 mp 975-980 ordmC
10101010rsquorsquorsquorsquo in Scheme 21 was obtained by following procedure To a solution of (+)(+)(+)(+)----9999 (119 mg 0067
mmol) in THF (13 mL) at ndash78 ordmC was added BH3bullTHF (526 microL 0533 mmol) After 39 h 1N NaOH
(1 mL) was added to quench the reaction The water layer was extracted three times with AcOEt
and the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
material was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 31) to give the mixture of 10101010rsquorsquorsquorsquo and 11111111 (109 mg yield 92
10101010rsquorsquorsquorsquo11111111 = 71) as a colorless oil Diastereomeric ratio was determined by 1H NMR spectra of the
mixture
The enantiopurity of 10101010rsquorsquorsquorsquo can be raised here by recrystallization Mixture of 10101010rsquorsquorsquorsquo and 11111111 (844 mg)
was recrystallized from hexaneAcOEt to give racemic 10101010 (52 mg yield 6) as a colorless crystal
and mixture of chiral 10101010rsquorsquorsquorsquo and 11111111 (779 mg yield 92) from mother liquid as a colorless oil
2222----((1((1((1((1RRRR2222RRRR))))----2222----(prop(prop(prop(prop----2222----ynyloxy)ynyloxy)ynyloxy)ynyloxy)----4444----vinylcyclohexvinylcyclohexvinylcyclohexvinylcyclohex----3333---- enyl)propanenyl)propanenyl)propanenyl)propan----2222----olololol ((((((((ndashndashndashndash))))----11112222)))) To a
mixture of 10101010rsquorsquorsquorsquo and 11111111 (779 mg 043 mmol 10101010rsquorsquorsquorsquo11111111 = 71) were successively added 60
aqueous NaOH (22 mL) tetrabutylammonium iodide (153 mg 004 mmol) and
propargyl bromide (459 microL 060 mmol) The mixture was stirred for 30 min and then quenched
with saturated aqueous NH4Cl (5 mL) The water layer was extracted three times with AcOEt and
the combined organic layers were washed with brine and dried over Na2SO4 After the volatile
materials were removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 101) to give ((((ndashndashndashndash))))----12121212 (704 mg yield 71) as colorless oil cis
isomer (90 mg yield 9) as colorless oil and starting material 10101010rsquorsquorsquorsquo (67 mg yield 9) The
OHOH10
OHO(ndash)-12
31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

31
enantiomeric excess was determined to be 54 ee by HPLC analysis (Daicel Chiralpak AD-H
hexaneiPrOH = 973 f 10 mLmin 254 nm tR 147 min (((((ndashndashndashndash))))----12121212) and 175 min ((+)(+)(+)(+)----12121212))1H NMR
(CDCl3 400 MHz) δ 124 (3H s) 125 (3H s) 126-136 (1H m) 179 (1H ddd J = 28 96 128 Hz)
188-192 (1H m) 213-220 (1H m) 226-231 (1H m) 249 (1H t J = 24 Hz) 408 (1H s) 421
(1H dd J = 24 120 Hz) 430 (1H dd J = 24 120 Hz) 451 (1H d J = 92 Hz) 507 (1H d J =
108 Hz) 520 (1H d J = 176 Hz) 577 (1H s) 636 (1H dd J = 108 176 Hz) 13C NMR (CDCl3
100 MHz) δ 237 244 248 292 491 543 730 752 769 791 1133 1263 1382 1389
IR(neat) 3487 1732 cm-1 HRMS(ESI) calcd for C14H20O2Na 2431356 found 2431352 [α]D20 ndash1320
(c 094 CHCl3 95 ee)
For the enantiomer (+)(+)(+)(+)----12121212 in Scheme 10 [α]D24 +763 (c 13 CHCl3 54 ee)
2222----((8((8((8((8SSSS8a8a8a8aRRRR))))----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphthonaphthonaphthonaphtho[18[18[18[18----bcbcbcbc]furan]furan]furan]furan----8888----yl)propanyl)propanyl)propanyl)propan----2222----oooollll ((((((((ndashndashndashndash))))----24242424))))
A solution of (+)(+)(+)(+)----12121212 (108 mg 0049 mmol) in toluene (1 mL) was stirred at 100 ordmC for
24 h under O2 After toluene was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexane AcOEt = 101) to give the mixture of
((((ndashndashndashndash))))----24242424 and 25252525 (108 mg quant ((((ndashndashndashndash))))----2424242425252525 = 928) as a colorless oil This mixture can be utilized for
next step without further purification After SiO2 column chromatography ((((ndashndashndashndash))))----24242424 can be isolated as
a pure form 1H NMR (CDCl3 400 MHz) δ 127 (3H s) 132 (3H s) 142-156 (1H m) 174 (1H ddd
J = 28 104 128 Hz) 196-202 (1H m) 275 (1H ddd J = 68 108 176 Hz) 296 (1H dd J = 68
176 Hz) 495 (2H dd J = 80 108 Hz) 508 (1H dd J = 24 124 Hz) 702 (1H d J = 76 Hz) 707
(1H d J = 76 Hz) 720 (1H t J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 238 248 268 281 511
729 730 814 1183 1256 1279 1285 1328 1384 IR(neat) 3404 cm-1 HRMS(FAB) calcd for
C14H18O2Na 2411204 found 2411199 [α]D23 ndash111 (c 055 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----24242424 in Scheme 13 [α]D19 +302 (c 055 CHCl3 95 ee)
(8(8(8(8SSSS8a8a8a8aRRRR))))----8888----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----6788a6788a6788a6788a---- tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]fura]fura]fura]fura
nnnn----2222----oneoneoneone ((((((((ndashndashndashndash))))----26262626)))) A mixture of ((((ndashndashndashndash))))----24242424 and 25252525 (47 mg ((((ndashndashndashndash))))----2424242425252525 = 101 0022 mmol as
((((ndashndashndashndash))))----24242424) was dissolved in CCl4MeCNH2O (22 mL 221 ratio) RuO2bullxH2O (01 mg)
and NaIO4 (167 mg 0080 mmol) were added successively to the mixture at 0 ordmC After 35 h
saturated aqueous sodium thiosulfate (2 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 CH2Cl2 AcOEt = 51) to give ((((ndashndashndashndash))))----26262626
(26 mg yield 58) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 136 (3H s) 142 (3H s)
172-183 (1H m) 216 (1H s) 223 (1H ddd J = 24 72 96 Hz) 279-288 (1H m) 313 (1H dd J
= 36 204 Hz) 528 (1H d J = 124 Hz) 740 (1H d J = 76 Hz) 746 (1H t J = 76 Hz) 769 (1H d
J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 228 260 274 274 496 721 797 1229 1240 1298
1321 1339 1489 1700 IR(neat) 3422 1742 cm-1 HRMS(FAB) calcd for C14H16O3Na 2550997
found 2550987 [α]D24 ndash28 (c 031 CHCl3 54 ee)
For the enantiomer (+)(+)(+)(+)----26262626 in Scheme 13 [α]D20 +62 (c 025 CHCl3 95 ee)
OHO(ndash)-24
OHOO (ndash)-26
32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

32
(8(8(8(8RRRR8a8a8a8aRRRR))))----8888----(prop(prop(prop(prop----1111----enenenen----2222----yl)yl)yl)yl)----6788a6788a6788a6788a----tetrahydrotetrahydrotetrahydrotetrahydro----2222HHHH----naphtho[18naphtho[18naphtho[18naphtho[18----bcbcbcbc]furan]furan]furan]furan----2222----oneoneoneone
(((((+)(+)(+)(+)----8888)))) To a solution of ((((ndashndashndashndash))))----26262626 (27 mg 0012 mmol) in pyridine (483 microL) at 0 ordmC was
added thionyl chloride (6 microL 0082 mmol) After 10 min saturated aqueous
NaHCO3 (1 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material (except pyridine) was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 81) to give (+)(+)(+)(+)----8888
(20 mg yield 80) as a colorless solid 1H NMR (CDCl3 400 MHz) δ 189 (3H s) 194 (1H ddt J =
20 40 100 Hz) 216 (1H ddt J = 16 36 80 Hz) 221-227 (1H m) 281-289 (1H m) 316 (1H
dd J = 80 180 Hz) 498-499 (2H m) 523 (1H d J = 108 Hz) 739 (1H d J = 76 Hz) 745 (1H t
J = 76 Hz) 768 (1H d J = 76 Hz) 13C NMR (CDCl3 100 MHz) δ 206 259 267 463 804 1124
1229 1247 1298 1321 1337 1441 1487 1704 IR(neat) 1760 cm-1 HRMS(ESI) calcd for
C14H14O2Na 2370886 found 2370876 [α]D22 +250 (c 014 CHCl3 54 ee)
For the enantiomer ((((ndashndashndashndash))))----8888 (platyphyllide) in Scheme 13 [α]D20 ndash548 (c 016 CHCl3 95 ee)
SSSSection 3ection 3ection 3ection 3
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex(triethylsilyloxy)cyclohex----3333----enyl)methanolenyl)methanolenyl)methanolenyl)methanol To a solution
of ((((ndashndashndashndash))))----17a17a17a17a (385 mg 11 mmol 60 ee) in THF (10 mL) at 0 ordmC were added MeOH
(65 microL 16 mmol) and LiBH4 in THF (20 M in THF 081 mL 16 mmol) After
45 min at rt water (5 mL) was added at 0 ordmC to quench the reaction The mixture was extracted
three times with AcOEt and combined organic layers were washed with brine and dried over
Na2SO4 After volatile material was removed under reduced pressure resulting residue was
purified by column chromatography (SiO2 hexane AcOEt = 21) to give the title compound (258 mg
yield 88) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 068 (6H q J = 80 Hz) 098 (9H t J =
80 Hz) 135-145 (1H m) 171-176 (1H m) 179-186 (1H m) 199 (1H dt J = 40 152 Hz)
213-221 (1H m) 264 (1H dd J = 32 68 Hz) 333 (3H s) 358-370 (2H m) 391-393 (1H m)
498 (1H s) 13C NMR (CDCl3 100 MHz) δ 50 66 227 288 401 544 663 796 1026 1541
IR(neat) 3413 1660 cm-1 HRMS(ESI) calcd for C14H28O3NaSi 2951700 found 2951689 [α]D17
ndash126 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS))))----2222----MMMMethoxyethoxyethoxyethoxy----4444----(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)(triethylsilyloxy)cyclohexcyclohexcyclohexcyclohex----3333----enyl)methyl enyl)methyl enyl)methyl enyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(27777) ) ) ) To a solution of the above compound (233 mg
085 mmol) in CH2Cl2 (82 mL) were added pyridine (41 mL) and
p-bromobenzoyl chloride (750 mg 34 mmol) at 0 ordmC After 10 min saturated aqueous NaHCO3 (5
mL) was added to quench the reaction The mixture was extracted three times with CH2Cl2 and
combined organic layers were washed with brine and dried over Na2SO4 After volatile material was
removed under reduced pressure resulting residue was purified by column chromatography (SiO2
hexane AcOEt = 101) to give 27272727 (358 mg yield 91) as a colorless oil 1H NMR (CDCl3 400 MHz) δ
069 (6H q J = 80 Hz) 099 (9H t J = 80 Hz) 159-165 (1H m) 190-221 (4H m) 333 (3H s)
383 (1H brs) 429 (1H dd J = 68 112 Hz) 441 (1H dd J = 52 112 Hz) 501 (1H s) 758 (2H d
J = 84 Hz) 790 (2H d J = 84 Hz) 13C NMR (CDCl3 100 MHz) δ 49 66 227 282 375 550
OO
(+)-Platyphyllide
TESO
OMe
OH
TESO
OMe
O
O
Br
27
33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

33
659 761 1024 1279 1291 1310 1316 1542 1658 IR(neat) 1718 1590 cm-1 HRMS(ESI)
calcd for C21H31O4NaSiBr 4771073 found 4771057 [α]D16 ndash133 (c 10 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----HHHHydroxyydroxyydroxyydroxy----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate To a solution of 22227777 (103 mg 022 mmol) in acetone (1
mL) at rt was added DMDO in acetone (9 mL estimated to be 007-009
M 063-081 mmol) After 1 h volatile material was removed under reduced pressure and resulting
residue was used in the next step without purification
To a solution of the crude product in THF (22 mL) at 0 ordmC was added TBAF (10 M in THF 290
microL 290 mmol) After 15 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction
The mixture was extracted three times with CH2Cl2 and combined organic layers were washed with
brine and dried over Na2SO4 After volatile material was removed under reduced pressure
resulting residue was purified by column chromatography (SiO2 hexane AcOEt = 21) to give the
title compound (54 mg yield 68 2 steps) as a colorless oil 1H NMR (CDCl3 400 MHz) δ 154 (1H
dddd J = 40 140 140 140 Hz) 212-219 (1H m) 223-231 (1H m) 247 (1H ddd J = 64 140
140 Hz) 262 (1H ddd J = 24 40 140 Hz) 314 (1H dd J = 92 104 Hz) 362 (3H s) 374 (1H
brs) 424 (1H d J = 92 Hz) 446 (1H dd J = 56 108 Hz) 457 (1H dd J = 32 108 Hz) 760 (2H
d J = 88 Hz) 787 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz) δ 246 374 407 605 648 812
852 1283 1287 1310 1318 1656 2076 IR(neat) 3460 1713 1589 cm-1 HRMS(ESI) calcd for
C15H17O5NaBr 3790157 found 3790147 [α]D16 ndash292 (c 025 CHCl3 60 ee)
((1((1((1((1SSSS2222SSSS3333RRRR))))----3333----(((1(((1(((1(((1RRRR4444SSSS))))----77777777----DDDDimethylimethylimethylimethyl----2222----ooooxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]heptaxobicyclo[221]hepta
nnnn----1111----yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)yl)methylsulfonyloxy)----2222----methoxymethoxymethoxymethoxy----4444----oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl oxocyclohexyl)methyl
4444----bromobenzoatebromobenzoatebromobenzoatebromobenzoate (2(2(2(28888) ) ) ) To a solution of the above compound (53
mg 015 mmol) in CH2Cl2 (15 mL) at 0 ordmC were successively
added Et3N (250 microL) DMAP (75 mg 0061 mmol) and (ndash)-canphorsulfonyl chloride (74 mg 030
mmol) in CH2Cl2 (15 mL) After 10 min saturated aqueous NaHCO3 (1 mL) was added to quench
the reaction The mixture was extracted three times with AcOEt Combined organic layers were
washed with brine and dried over Na2SO4 After volatile material was removed under reduced
pressure diastereomeric ratio was determined to be ca 41 (corresponding to the ee of 17a17a17a17arsquorsquorsquorsquo) by 1H
NMR of the crude product The crude product was purified by column chromatography (SiO2
hexane AcOEt = 351) to give pure 28282828 (115 mg yield 13) as a colorless amorphous and
diastereomixture of 22228888 (47 mg dr 31 yield 55) Recrystallization of 22228888 from etherhexane gave
colorless crystal 1H NMR (CDCl3 400 MHz) δ 092 (3H s) 113 (3H s) 145 (1H ddd J = 40 92
128 Hz) 152-163 (1H m) 175 (1H ddd J = 48 92 140 Hz) 198 (1H d J = 184 Hz) 203-214
(3H m) 228-235 (1H m) 236-252 (3H m) 259 (1H ddd J = 24 40 140 Hz) 340-347 (2H m)
360 (3H s) 393 (1H d J = 152 Hz) 450 (1H dd J = 48 112 Hz) 457 (1H dd J = 32 112 Hz)
516 (1H d J = 96 Hz) 761 (2H d J = 88 Hz) 788 (2H d J = 88 Hz) 13C NMR (CDCl3 100 MHz)
δ 197 198 238 251 268 385 418 425 428 478 492 580 611 643 819 871 1285 1285
1310 1319 1655 2006 2140 IR(neat) 1719 1660 1590 cm-1 HRMS(ESI) calcd for
C25H31O8NaBr 5930821 found 5930775 [α]D20 ndash419 (c 058 CHCl3) mp 1715-1720 ordmC
O
OMe
O
O
Br
HO
OMe
O
O
O
O2
SO
28 O
Br
34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

34
Chapter 2Chapter 2Chapter 2Chapter 2
General General General General Procedure for the ItoProcedure for the ItoProcedure for the ItoProcedure for the Ito----Saegusa OxidationSaegusa OxidationSaegusa OxidationSaegusa Oxidation (29c29c29c29c to 30a30a30a30a and 31b31b31b31b)
To a solution of 29c29c29c29c (94 mg 03 mmol) in MeCN (750 microL) at rt was added Pd(OAc)2 (72 mg 033
mmol) After 18 h the mixture was diluted with CH2Cl2 (3 mL) and filtered through a Celitereg pad
After volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexane AcOEt = 11) to give 30a30a30a30a (45 mg yield 10) as a colorless
solid and 31b31b31b31b (225 mg yield 48) as a colorless solid
4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----3333----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (30303030a)a)a)a) 1H NMR (CDCl3 400 MHz) δ
184 (1H brs) 195-212 (2H m) 234 (1H ddd J = 52 96 148 Hz) 251 (1H ddd
J = 52 58 120 Hz) 267-273 (1H m) 373 (3H s) 384 (2H brs) 543 (1H s) 13C
NMR (CDCl3 100 MHz) δ 238 346 411 558 628 1031 1784 1997 IR(neat) 3384 1631 1583
cm-1 HRMS(ESI) calcd for C16H24O6Na 3351471 found 3351477
ciscisciscis----4444----(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)(Hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enone (enone (enone (enone (31313131b)b)b)b) 1H NMR (CDCl3 400 MHz)
δ 262 (1H dd J = 36 168 Hz) 286 (1H brs) 288 (1H dd J = 64 168 Hz)
288-293 (1H m) 342 (3H s) 388 (1H dd J = 36 112 Hz) 394 (1H dd J = 68
112 Hz) 409 (1H ddd J = 36 36 64 Hz) 613 (1H dd J = 24 104 Hz) 685 (1H dd J = 36
104 Hz) 13C NMR (CDCl3 100 MHz) δ 404 423 566 628 791 1305 1476 1970 IR(neat) 3420
1663 cm-1 HRMS(FAB) calcd for C16H24O6Na 3351471 found 3351483
transtranstranstrans----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methoxycyclohexmethoxycyclohexmethoxycyclohexmethoxycyclohex----2222----enoneenoneenoneenone ((((31a31a31a31a) ) ) ) 1H NMR (CDCl3 400
MHz) δ 222 (1H brs) 241 (1H dd J = 108 160 Hz) 272 (1H dddt J = 28 28
26 84 Hz) 298 (1H dd J = 40 160 Hz) 340 (3H s) 373 (1H ddd J = 40 84
108 Hz) 389 (2H d J = 56 Hz) 610 (1H dd J = 28 60 Hz) 679 (1H dd J = 28 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 421 450 562 633 780 1305 1488 1978 IR(neat) 3421 1669 cm-1
3333----methoxymethoxymethoxymethoxy----4444----(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex(methoxymethyl)cyclohex----2222----enoneenoneenoneenone ((((30b30b30b30b)))) 1H NMR (CDCl3 400 MHz)
δ 200-211 (2H m) 232 (1H ddd J = 56 72 168 Hz) 250 (1H ddd J = 56 88
168 Hz) 269-274 (1H m) 336 (3H s) 357-359 (2H m) 370 (3H s) 540 (1H s)
13C NMR (CDCl3 100 MHz) δ 241 343 390 558 590 721 1032 1777 1994 IR(neat) 1648
cm-1
4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((33333333)))) 1H NMR (CDCl3 400 MHz) δ
148 (1H brs) 202 (3H s) 209-214 (2H m) 234 (1H ddd J = 60 60 172 Hz)
247-257 (1H m) 380-384 (2H m) 594 (1H s) 13C NMR (CDCl3 150 MHz) δ 230
O
OMe30a
OH
O
OMe31b
OH
O
33
OH
O
OMe
OH
31a
O
OMe30b
OMe
35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

35
250 343 422 631 1285 1620 1993 IR(neat) 3393 1652 cm-1 HRMS(ESI) calcd for
C16H24O4Na 3031572 found 3031565
ciscisciscis----4444----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((34a34a34a34a)))) 1H NMR (CDCl3 400 MHz)
δ 101 (3H d J = 68 Hz) 162 (1H brs) 240-253 (3H m) 269-272 (1H m) 381
(2H d J = 64 Hz) 610 (1H dd J = 24 100 Hz) 687 (1H dd J = 32 100 Hz) 13C
NMR (CDCl3 100 MHz) δ 152 307 424 447 624 1301 1495 1996 IR(neat) 3395 1660 cm-1
HRMS(ESI) calcd for C16H24O4Na 3031572 found 3031569
4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((37373737)))) 1H NMR (CDCl3 400 MHz)
δ 131 (3H s) 137 (3H s) 163 (1H s) 201-211 (1H m) 214 (3H s) 214-218 (1H
m) 222-229 (1H m) 229-235 (1H m) 255-265 (1H m) 601 (1H s) 13C NMR
(CDCl3 100 MHz) δ 226 291 358 489 522 617 1292 1498 1997 IR(neat) 3393 1653 cm-1
HRMS(ESI) calcd for C20H32O4Na 3592198 found 3592185
transtranstranstrans----4444----(2(2(2(2----hydroxypropanhydroxypropanhydroxypropanhydroxypropan----2222----yl)yl)yl)yl)----5555----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((38a38a38a38a)))) 1H NMR (CDCl3
400 MHz) δ 111 (3H d J = 68 Hz) 135 (3H s) 136 (3H s) 146 (1H s) 215-221
(2H m) 250-256 (1H m) 283 (1H dd J = 56 168 Hz) 613 (1H d J = 108 Hz)
693 (1H ddd J = 16 52 108 Hz) 13C NMR (CDCl3 100 MHz) δ 227 286 288 289 429 530
739 1303 1484 1992 IR(neat) 3411 1663 cm-1 HRMS(ESI) calcd for C20H32O4Na 3592198
found 3592187
4444----hydroxyhydroxyhydroxyhydroxy----3333----methylcyclohexmethylcyclohexmethylcyclohexmethylcyclohex----2222----enoneenoneenoneenone ((((41414141)))) 1H NMR (CDCl3 400 MHz) δ 171 (1H d J =
64 Hz) 197-213 (1H m) 206 (3H s) 226-241 (2H m) 255-262 (1H m) 435-442
(1H m) 586 (1H s) IR(neat) 3393 1670 cm-1 HRMS(ESI) calcd for C14H20O4Na
2751259 found 2751253
4444----((((hhhhydroxymethyl)ydroxymethyl)ydroxymethyl)ydroxymethyl)----55555555----dimethylcyclohexdimethylcyclohexdimethylcyclohexdimethylcyclohex----2222----enone (enone (enone (enone (44443333)))) 1H NMR (CDCl3 400 MHz) δ
095 (3H s) 114 (3H s) 151 (1H brs) 233 (2H d J = 36 Hz) 240-245 (1H m)
364-370 (1H m) 394-397 (1H m) 611 (1H dd J = 20 100 Hz) 696 (1H dd J =
28 100 Hz) 13C NMR (CDCl3 100 MHz) δ 226 291 358 489 522 616 1292 1499 1997
IR(neat) 3412 1666 cm-1 HRMS(ESI) calcd for C18H28O4Na 3311885 found 3311876
Chapter 3Chapter 3Chapter 3Chapter 3
Section 2Section 2Section 2Section 2
Compounds shown in Scheme 17 18 and compounds 67676767 68686868 69696969 70707070 and 71717171 were prepared by
reported procedure27
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bR)R)R)R)----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluo(trifluo(trifluo(trifluoromethyl)romethyl)romethyl)romethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octoctoctoct
ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54ahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methanolyl)methanolyl)methanolyl)methanol To a solution of 71717171 (193 mg
0247 mmol) and CeCl3bull7H2O (202 mg 0542 mmol) in MeOH (25 mL) at
ndash78 degC was added NaBH4 (19 mg 0494 mmol) After being stirred for 10 min the reaction mixture
was warmed to ndash40 degC and stirred for 16 h The reaction was quenched with water (3 mL) The
water layer was extracted three times with AcOEt and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give
BsN
H
N O
O
TBDPSO
O
CF3
OH
O
43
OH
O
34a
OH
O
OH
37
O
38a
OH
OH
O
41
36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

36
the title compound (195 mg quant) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 091
(9H s) 141 (1H dddd J = 36 120 120 120 Hz) 156 (1H brs) 168-182 (4H m) 187 (1H d J
= 102 Hz) 220 (1H dd J = 108 108 Hz) 238 (1H dd J = 42 132 Hz) 254 (1H d J = 108 Hz)
317 (1H d J = 102 Hz) 340 (1H d J = 102 Hz) 382-386 (4H m) 391 (1H d J = 150 Hz)
394-368 (2H m) 403 (1H d J = 150 Hz) 461 (1H s) 486 (1H t J = 42 Hz) 588 (1H s)
731-743 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 778 (2H d J = 72 Hz) 13C
NMR (CDCl3 150 MHz) δ 193 268 273 350 351 404 465 490 628 644 649 650 684
873 1034 1160 (q J = 273 Hz) 1190 1276 1277 1277 1290 1299 1300 1324 1328 1329
1353 1354 1439 1536 (q J = 40 Hz) IR(neat) 3452 1684 cm-1 [α]D24 +66 (c 100 CHCl3 gt99
ee) HRMS(FAB) calcd for C40H48F3N2O7SSiK 7852904 found 7852884
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((72727272)))) To a solution of the
above compound (195 mg 0247 mmol) and DMAP (15 mg 0124 mmol) in
pyridine (25 mL) at 0 degC was added PivCl (015 mL 124 mmol) The mixture was stirred 18 h at rt
The reaction was quenched with saturated aqueous NaHCO3 (3 mL) The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 72727272 (174 mg yield 81) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 093 (9H s) 121 (9H s) 143 (1H dddd J
= 39 129 129 129 Hz) 162-181 (4H m) 192-195 (1H m) 225 (1H dd J = 110 110 Hz) 239
(1H dd J = 44 127 Hz) 261 (1H d J = 107 Hz) 318 (1H d J = 103 Hz) 340 (1H d J = 103
Hz) 381-399 (6H m) 433 (1H d J = 146 Hz) 452 (1H d J = 144 Hz) 459 (1H s) 486 (1H t J
= 37 Hz) 585 (1H s) 729-744 (10 H m) 751-716 (3H m) 777-779 (2H m) 13C NMR (CDCl3
150 MHz) δ 192 268 270 272 272 350 357 388 405 464 490 627 649 650 683 869
1033 1151 (q J = 273 Hz) 1203 1276 1277 1277 1290 1299 1300 1324 1327 1329 1352
1353 1393 1494 1535 (q J = 40 Hz) 1776 IR(neat) 1685 cm-1 [α]D24 +121 (c 100 CHCl3
gt99 ee) HRMS(FAB) calcd for C45H55F3N2O8SSiNa 8913298 found 8913337
((4a((4a((4a((4aSSSS7777SSSS8888RRRR8a8a8a8aSSSS))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----((((((((((((terttertterttert----butyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)obutyldiphenylsilyl)o
xy)methyl)xy)methyl)xy)methyl)xy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----ttttrifluoroacetamido)rifluoroacetamido)rifluoroacetamido)rifluoroacetamido)----1234123412341234
4a788a4a788a4a788a4a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((103103103103)))) This compound
was obtained as a byproduct when the crude substrate was used in the above
reaction 1H NMR (CDCl3 600 MHz) δ 097 (9H s) 109 (9H s) 141 (1H dd
J = 42 120 Hz) 155 (1H dddd J = 42 120 120 120 Hz) 163-170 (3H m) 179-189 (2H m)
201-205 (2H m) 288 (1H d J = 102 Hz) 306 (1H s) 376 (1H d J = 114 Hz) 386-391 (2H m)
397-400 (2H m) 425 (1H d J = 126 Hz) 432 (1H d J = 102 Hz) 438 (1H d J = 126 Hz) 449
(1H d J = 108 Hz) 475 (1H d J = 96 Hz) 492 (1H t J = 36 Hz) 565 (1H s) 732 (2H t J = 78
Hz) 736 (2H t J = 78 Hz) 742 (1H t J = 78 Hz) 753 (2H t J = 78 Hz) 756 (2H t J = 78 Hz)
763-765 (3H m) 774-779 (3H m) 13C NMR (CDCl3 150 MHz) δ 192 267 270 276 276 341
BsN
H
N O
O
TBDPSO
O
CF372
OPiv
BsN
H
HN O
O
TBDPSO
OH
103
OPiv
CF3
O
37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

37
386 389 401 462 505 508 648 649 650 695 715 1037 1162 (q J = 287 Hz) 1278 1279
1290 1293 1299 1300 1319 1322 1327 1355 1356 1358 1576 (q J = 37 Hz) 1778
((3a((3a((3a((3aSSSS5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----8888----(phen(phen(phen(phen
ylsulfonyl)ylsulfonyl)ylsulfonyl)ylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67899a9b3a5a67899a9b3a5a67899a9b3a5a67899a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoq]isoq]isoq]isoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalayl)methyl pivalayl)methyl pivalayl)methyl pivalatetetete To a solution of 72727272 (304 mg 0350 mmol) in THF
(18 mL) at 0 degC was added TBAF (10 M solution in THF 35 mL 35 mmol)
After 30 min the reaction was quenched with saturated aqueous NH4Cl (5 mL) The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 AcOEt) to give the title compound
(154 mg yield 70) as a colorless amorphous and recovered 72727272 (80 mg 26) This compound was
slowly converted into unknown compound (the alcohol group might attacked the oxazoline) at rt for
several days or at ndash15 degC for 1 month 1H NMR (C6D6 600 MHz 55 degC) δ 107 (9H s) 120-146 (3H
m) 161-183 (5H m) 189 (1H dd J = 44 124 Hz) 238 (1H d J = 122 Hz) 314 (2H s) 344-350
(2H m) 329-335 (2H m) 367 (1H d J = 110 Hz) 415-429 (3H m) 462 (1H t J = 43 Hz) 478
(1H s) 574 (1H s) 693-697 (3H m) 768-771 (2H m) IR(neat) 3522 1728 1685 cm-1 [α]D24
ndash29 (c 100 CHCl3 gt99 ee) HRMS(FAB) calcd for C29H37F3N2O8SNa 6532120 found 6532081
((3a((3a((3a((3aSSSS5a5a5a5aRRRR9a9a9a9aRRRR9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((EEEE))))----4444----((((((((terttertterttert----butyldimebutyldimebutyldimebutyldimetttt
hylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)buthylsilyl)oxy)but----1111----enenenen----1111----yl)yl)yl)yl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----3a5a67893a5a67893a5a67893a5a6789
9a9b9a9b9a9b9a9b----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((74747474)))) To a
solution of TFAA (027 mL 193 mmol) in CH2Cl2 (16 mL) at ndash78 degC was added
dropwise a solution of DMSO (027 mL 386 mmol) in CH2Cl2 (08 mL) over 5
min After 30 min to this mixture was added a solution of the above compound (243 mg 0386
mmol) in CH2Cl2 (16 mL) and the resulting mixture was warmed to ndash60 degC and stirred for 1 h
Et3N (08 mL) was added to the reaction mixture and then warmed to rt The reaction was
quenched with saturated aqueous NaHCO3 (5 mL) The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue (73737373) was used for next
reaction without further purification
To a suspension of bromo(3-((tert-butyldimethylsilyl)oxy)propyl)triphenylphosphorane (995 mg
193 mmol) in THF (20 mL) at 0 degC was added KHMDS (05 M solution in THF 386 mL 193 mmol)
in THF To this mixture a solution of crude aldehyde (73737373) in THF (5 mL) was added After 1 h
saturated aqueous NH4Cl (20 mL) was added to quench the reaction The water layer was extracted
three times with AcOEt and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 41) to give 74747474 (226 mg yield 75 from
the above compound) as a colorless amorphous 1H NMR (CDCl3 600 MHz 55 degC) δ 003 (6H s)
088 (9H s) 119 (9H s) 150 (1H dddd J = 36 120 120 120 Hz) 170-187 (6H m) 195-200
(2H m) 227-235 (2H m) 241-249 (2H m) 352-357 (1H m) 365 (1H td J = 60 96 Hz)
BsN
H
N O
OO
CF3
OPiv
OTBS74
BsN
H
N O
O
HO
O
CF3
OPiv
38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

38
385-391 (2H m) 397-403 (3H m) 450-453 (2H m) 456 (1H d J = 126 Hz) 463 (1H s) 490
(1H t J = 36 Hz) 499 (1H d J = 132 Hz) 543 (1H td J = 66 132 Hz) 601 (1H s) 757 (2H t J
= 78 Hz) 763 (1H t J = 78 Hz) 784 (2H d J = 78 Hz) 13C NMR (CDCl3 150 MHz 55 degC) δ 150
192 268 281 285 289 340 353 398 430 445 468 524 634 659 659 660 698 880
1044 1175 (q J = 272 Hz) 1245 1284 1285 1285 1299 1300 1327 1337 1380 1413 1550
(q J = 40 Hz) 1786 IR(neat) 1725 cm-1 [α]D24 +249 (c 227 CHCl3 gt99 ee) HRMS(ESI) calcd
for C38H56F3N2O8SSi 7853479 found 7853452
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxyhydroxyhydroxyhydroxy----8a8a8a8a----((((((((EEEE))))----4444----hydroxybuhydroxybuhydroxybuhydroxybu
tttt----1111----enenenen----1111----yl)yl)yl)yl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----ococococ
tahydrotahydrotahydrotahydroisoquinolinisoquinolinisoquinolinisoquinolin----5555----yl)methyl pivalatyl)methyl pivalatyl)methyl pivalatyl)methyl pivalateeee To a solution of 74747474 (226 mg 0228
mmol) in acetonewater (91 50 mL) at room temperature was added PPTS
(217 mg 0864 mmol) After 2 days saturated aqueous NaHCO3 (5 mL) was
added to quench the reaction The water layer was extracted three times with AcOEt and the
combined organic layers were washed with brine and dried over Na2SO4 After the volatile material
was removed under reduced pressure the resulting residue was purified by column
chromatography (SiO2 hexaneAcOEt = 12) to give the title compound (129 mg yield 65) as a
colorless amorphous 1H NMR (CDCl3 400 MHz 55 degC) δ 119 (9H s) 153-171 (4H m) 183-200
(3H m) 218-230 (3H m) 239 (1H brs) 261-271 (1H m) 279 (1H s) 363-371 (2H m)
378-394 (5H m) 441-447 (2H m) 457 (1H d J = 132 Hz) 483-487 (2H m) 542-553 (2H m)
575 (1H s) 718 (1H d J = 103 Hz) 750-760 (3H m) 782-784 (2H m) 13C NMR (CDCl3 100
MHz 55 degC) δ 272 277 279 329 332 399 424 438 459 504 508 616 649 649 652 719
772 1040 1164 (q J = 289 Hz) 1281 1290 1307 1322 1328 1337 1365 1580 (q J = 37 Hz)
1779 IR(neat) 1721 cm-1 [α]D23 ndash657 (c 166 CHCl3 gt99 ee) HRMS(ESI) calcd for
C32H43F3N2O9SNa 7112539 found 7112531
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----7777----hydroxhydroxhydroxhydroxyyyy----8a8a8a8a----(4(4(4(4----hydroxybutyl)hydroxybutyl)hydroxybutyl)hydroxybutyl)----
2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoqoctahydroisoqoctahydroisoqoctahydroisoq
uinolinuinolinuinolinuinolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((75757575) ) ) ) A solution of the above compound (28 mg
0041 mmol) and PtO2 (7 mg) and acetic acid (ca 5 microL) in THF (30 mL) was
stirred under hydrogen atmosphere at room temperature After 5 h the
reaction mixture was filtered through a Celite pad and the pad was washed with EtOAc The
combined filtrates were concentrated in vacuo The residue was purified by column chromatography
(SiO2 hexaneAcOEt = 14) to give 75757575 (19 mg yield 70) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 119 (9H s) 122-135 (2H m) 146-177 (9H m) 187-191 (2H m) 197-201
(1H m) 217-223 (2H m) 333 (1H brs) 358-361 (3H m) 374 (1H d J = 122 Hz) 383-397 (4H
m) 441 (1H d J = 134 Hz) 470 (1H d J = 103 Hz) 486 (1H t J = 37 Hz) 569 (1H s) 667 (1H
d J = 103 Hz) 750-760 (3H m) 784 (2H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 200
272 275 277 325 332 332 346 388 393 402 463 495 511 619 650 710 772 1037
1162 (q J = 289 Hz) 1282 1283 1289 1328 1362 1365 1574 (q J = 37 Hz) 1780 IR(neat)
1717 cm-1 [α]D24 ndash244 (c 117 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H45F3N2O9SNa 7132696
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
75
BsN
H
O
O
OPiv
OH
HN
O
CF3
OH
39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

39
found 7132677
((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----
(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----8888----(222(222(222(222----trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)trifluoroacetamido)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquioctahydroisoquioctahydroisoquioctahydroisoqui
nolinnolinnolinnolin----5555----yl)methyl pivalateyl)methyl pivalateyl)methyl pivalateyl)methyl pivalate ((((76767676)))) To a solution of 75757575 (89 mg 0129 mmol) and
pyridine (04 mL) in CH2Cl2 (12 mL) at 0 degC was added MsCl (44 mg 0387
mmol) After 15 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was used for next reaction without further purification
To a solution of o-NO2C6H4SeCN (146 mg 0645 mmol) in DMF (10 mL) at 0 degC was added
NaBH4 (24 mg 0645 mmol) After 15 h at the same temperature a solution of the crude product in
DMF (15 mL) was added and stirred at 0 degC After 18 h water (2 mL) was added to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a solution of crude product in CH2Cl2 (3 mL) at ndash78 degC was added 69 mCPBA (323 mg 129
mmol) portionwise After 15 h at room temperature iPrNEt (1 mL excess) was added After 19 h
saturated aqueous Na2S2O3 (5 mL) was added to quench the reaction The water layer was extracted
three times with CH2Cl2 and the combined organic layers were washed with brine and dried over
Na2SO4 After the volatile material was removed under reduced pressure the resulting residue was
purified by column chromatography (SiO2 hexaneAcOEt = 31) to give 76767676 (74 mg yield 85 from
75757575) as a yellow amorphous 1H NMR (C6D6 600 MHz 55 degC) δ 119 (9H s) 124 (1H dddd J = 47
127 127 127 Hz) 139 (1H d J = 146 Hz) 150-192 (9H m) 199 (1H dd J = 120 120 Hz)
211 (1H d J = 122 Hz) 326-331 (2H m) 342 (1H d J = 127 Hz) 347-352 (2H m) 395 (1H d
J = 122 Hz) 422 (1H d J = 132 Hz) 433 (1H d J = 133 Hz) 465 (1H t J = 43 Hz) 486 (1H d
J = 102 Hz) 490 (1H d J = 172 Hz) 550 (1H s) 558-565 (1H m) 629 (1H s) 671 (1H d J =
102 Hz) 699-705 (3H m) 789 (1H d J = 73 Hz) 13C NMR (CDCl3 100 MHz 55 degC) δ 273 274
278 319 346 389 393 398 464 494 512 536 650 654 709 772 1037 1152 1163 (q J
= 289 Hz) 1283 1288 1289 1328 1361 1365 1380 1572 1574 (q J = 36 Hz) 1780
IR(neat) 1716 cm-1 [α]D24 ndash291 (c 100 CHCl3 gt99 ee) HRMS(ESI) calcd for C32H43F3N2O8SNa
6952590 found 6952578
SSSSection 3ection 3ection 3ection 3
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----2222----(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)(trifluoromethyl)----55a67899a9b55a67899a9b55a67899a9b55a67899a9b----octaoctaoctaocta
hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54hydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((90909090)))) To a solution of 66666666 (20 mg
00272 mmol) in toluene (272 microL) at rt were added 26-lutidine (64 microL 0544
mmol) and TFAA (37 microL 0272 mmol) After 15 min saturated aqueous NaHCO3 (1 mL) was added
to quench the reaction The water layer was extracted three times with AcOEt and the combined
organic layers were washed with brine and dried over Na2SO4 After the volatile material was
BsN
H
O
O
OPiv
HN
O
CF3
OH
76
BsN
H
NO
O
O
TBDPSO
O
CF390
40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

40
removed under reduced pressure the resulting residue was purified by column chromatography
(SiO2 hexaneAcOEt = 41) to give 90909090 (96 mg yield 46) as a colorless amorphous 1H NMR (CDCl3
400 MHz 55 degC) δ 097 (9H s) 159-166 (1H m) 170-180 (3H m) 184-191 (1H m) 198-203
(1H m) 209 (1H dd J = 24 184 Hz) 229-236 (1H m) 250-257 (1H m) 265 (1H dd J = 96
184 Hz) 282 (1H d J = 120 Hz) 330 (1H d J = 108 Hz) 336 (1H d J = 108 Hz) 343 (1H d J
= 120 Hz) 360 (1H d J = 120 Hz) 382-388 (2H m) 391-399 (2H m) 484 (1H s) 487 (1H t J
= 40 Hz) 735-737 (4H m) 740-743 (2H m) 752-756 (6H m) 761 (1H t J = 72 Hz) 777 (2H
d J = 72 Hz) 13C NMR (CDCl3 100 MHz) δ 192 263 267 307 314 386 402 456 488 632
650 650 723 896 1028 1157 (q J = 273 Hz) 1276 1278 1279 1291 1300 1300 1321
1322 1330 1354 1354 1355 1534 (q J = 40 Hz) 2043 HRMS (ESI) calcd for
C39H45F3N2O7SSiNa 7932567 found 7932580
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6755a6755a6755a67899a9b899a9b899a9b899a9b----octahydrooxazoctahydrooxazoctahydrooxazoctahydrooxaz
olo[54olo[54olo[54olo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((91919191)))) To a stirred solution of 66666666 (325 mg
00442 mmol) and DMAP (one piece) in CH2Cl2 (442 microL) was added MsCl (34 microL 0442 mmol) at
0 degC After 1 h at rt saturated aqueous NaHCO3 (1 mL) was added to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic layers were washed
with brine and dried over Na2SO4 After the volatile material was removed under reduced pressure
the resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 91919191
(28 mg yield 88) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 091 (9H s) 150-160
(1H m) 177-183 (2H m) 184 (3H s) 189-195 (3H m) 210 (1H d J = 188 Hz) 225-237 (2H
m) 253 (1H d J = 112 Hz) 266 (1H dd J = 100 188 Hz) 318 (1H d J = 120 Hz) 325 (1H d J
= 120 Hz) 373 (1H d J = 112 Hz) 380-390 (3H m) 396-399 (2H m) 472 (1H s) 488 (1H t J
= 40 Hz) 734-749 (10H m) 756 (2H t J = 72 Hz) 762 (1H t J = 72 Hz) 777 (2H d J = 72 Hz)
13C NMR (CDCl3 100 MHz) δ 136 192 266 266 299 315 315 399 401 464 496 633 650
650 718 869 1031 1275 1277 1278 1289 1299 1324 1325 1327 1353 1354 1356
1631 2075 HRMS(ESI) calcd for C39H48N2O7SSiNa 7392849 found 7392821
(3a(3a(3a(3aRRRR5a5a5a5aSSSS9a9a9a9aSSSS9b9b9b9bRRRR))))----3a3a3a3a----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----9a9a9a9a----((((((((((((terttertterttert----butyldiphenylsilbutyldiphenylsilbutyldiphenylsilbutyldiphenylsil
yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)yl)oxy)methyl)----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----methylmethylmethylmethyl----8888----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----55a6789955a6789955a6789955a67899
a9a9a9a9bbbb----octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54octahydrooxazolo[54----hhhh]isoquinolin]isoquinolin]isoquinolin]isoquinolin----4(3a4(3a4(3a4(3aHHHH))))----oneoneoneone ((((92b92b92b92b)))) To a stirred
solution of n-butyllithium (123 microL 0307 mmol) in THF (366 microL) was added
dropwise 2266-tetramethylpiperidine (52 microL 0307 mmol) at ndash10 degC The solution was allowed to
be stirred at 0 degC for 30 minutes then cooled to ndash78 degC 91919191 (44 mg 00614 mmol) in THF (148 microL)
was added dropwise and the resulting mixture was stirred for an additional 1 hour
1H-benzotriazole-methanol (18 g 0123 mmol) in THF (546 microL) was added dropwise and kept 15
hours at this temperature The reaction was quenched with water (1 mL) The water layer was
extracted three times with AcOEt and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by column chromatography (SiO2 hexaneAcOEt = 14) to give 92b92b92b92b (17 mg
BsN
H
NO
O
O
TBDPSO
O
91
BsN
H
NO
O
O
TBDPSO
O
OH
92b
41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

41
yield 37) as a colorless amorphous 1H NMR (CDCl3 400 MHz) δ 088 (9H s) 144-153 (1H m)
179-183 (1H m) 186 (3H s) 190-195 (2H m) 198-205 (2H m) 215-221 (1H m) 230 (1H dd
J = 48 84 Hz) 240 (1H d J = 116 Hz) 263 (1H brs) 302 (1H d J = 120 Hz) 312 (1H d J =
120 Hz) 338 (1H brs) 373 (1H d J = 116 Hz) 383-397 (7H m) 474 (1H s) 486 (1H t J = 40
Hz) 734-746 (10H m) 754 (2H t J = 72 Hz) 761 (1H t J = 72 Hz) 775 (2H d J = 72 Hz)
HRMS(ESI) calcd for C40H51N2O8SSiNa 7473135 found 7473130
SSSSection 4ection 4ection 4ection 4
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----7777----hydroxyhydroxyhydroxyhydroxy
----5555----(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)(hydroxymethyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolinoctahydroisoquinolin----
8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((99999999)))) A solution of 76767676 (630 mg 0938 mmol) and LiOH
(570 mg 235 mmol) in MeOHwater (21 131 mL) was stirred at 70 degC After
25 h saturated aqueous NH4Cl (20 mL) was added at 0 degC to quench the
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction without further purification
To a stirred solution of this crude amine and 5-hexenoic acid (104 g 112 mmol) in MeOH (15
mL) at rt was added DMT-MM (952 mg 422 mmol) After 2 h water (10 mL) was added to quench
the reaction The water layer was extracted three times with AcOEt and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was purified by column chromatography (SiO2
AcOEt) to give 99999999 (352 mg yield 65 from 76767676) as a colorless amorphous 1H NMR (CDCl3 600 MHz
55 degC) δ 150-167 (2H m) 168-180 (4H m) 183-190 (4H m) 195-203 (3H m) 221-229 (4H m)
260-267 (1H m) 287 (1H brs) 365 (1H d J = 114 Hz) 370-373 (2H m) 378 (1H d J = 108
Hz) 384-387 (2H m) 394-397 (2H m) 440 (1H brs) 486 (1H t J = 42 Hz) 491-495 (2H m)
500 (1H d J = 108 Hz) 511 (1H d J = 168 Hz) 522 (1H d J = 78 Hz) 566-572 (1H m) 579
(1H s) 588-595 (1H m) 759 (2H t J = 73 Hz) 767 (1H d J = 78 Hz) 782 (2H d J = 78 Hz)
13C NMR (CDCl3 150 MHz 55 degC) δ 249 276 281 281 316 333 351 363 386 391 463 492
498 644 647 648 714 768 770 772 1043 1147 1151 1260 1282 1289 1327 1361
1383 1388 1404 1712 1732 IR(neat) 3375 1639 cm-1 [α]D27 ndash401 (c 063 CHCl3 gt99 ee)
HRMS(ESI) calcd for C31H44N2O7SNa 6112767 found 6112759
((4a((4a((4a((4aRRRR7777SSSS7a7a7a7aRRRR13131313EEEE16a16a16a16aRRRR20a20a20a20aRRRR23232323SSSS23a23a23a23aRRRR29292929EEEE32a32a32a32aRRRR))))----723723723723----bis(2bis(2bis(2bis(2----(13(13(13(13----dioxoldioxoldioxoldioxol
anananan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----723723723723----dihydroxydihydroxydihydroxydihydroxy----925925925925----dioxodioxodioxodioxo----218218218218----bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)bis(phenylsulfonyl)----1234123412341234
4a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a242526272831324a77a8910111215161718192020a2323a24252627283132----
octacosahydrooctacosahydrooctacosahydrooctacosahydro----[112]diazacyc[112]diazacyc[112]diazacyc[112]diazacyclodocosino[23lodocosino[23lodocosino[23lodocosino[23----iiii1314131413141314----iiii]diisoquinoline]diisoquinoline]diisoquinoline]diisoquinoline----521521521521
----diyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetatediyl)bis(methylene) diacetate ((((100100100100)))) A solution of 99999999 (86 mg 00145
mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (29 mL) containing
Grubbs 1st generation catalyst (36 mg 000434 mmol) was stirred
overnight at 40 degC After the volatile material was removed under
reduced pressure the resulting residue was used for next reaction
BsN
H
O
O
OH
HN
OH
99
O
BsN
H
O
O
OAc
NHOH
NBs
H
O
O
AcO
HN
HO
O
O
100
42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

42
without further purification
To a stirred solution of the crude product in pyridine (1 mL) were added DMAP (one piece) and
Ac2O (50 microL) After 3 h NaHCO3 (1 mL) was added to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the resulting
residue was purified by preparative TLC (SiO2 hexaneAcOEt = 110) to give 100100100100 (lt1 mg) and
mixture of other dimers 1H NMR (CDCl3 600 MHz 55 degC) δ 153-161 (8H m) 165-173 (6H m)
175-185 (4H m) 189-197 (4H m) 197-211 (8H m) 204 (6H s) 218-221 (4H m) 226 (2H d J
= 120 Hz) 238-240 (2H m) 344 (2H s) 348 (2H d J = 138 Hz) 371 (2H d J = 120 Hz)
383-385 (4H m) 394-396 (4H m) 444 (2H d J = 126 Hz) 452 (2H d J = 126 Hz) 476 (2H d
J = 108 Hz) 487 (2H t J = 42 Hz) 529 (2H ddd J = 60 78 144 Hz) 537 (2H ddd J = 60 78
144 Hz) 565 (2H s) 567 (2H d J = 108 Hz) 754 (4H t J = 72 Hz) 759 (2H t J = 72 Hz) 793
(4H d J = 72 Hz) LRMS (ESI) mz 1227 [M+Na]+
NNNN----((4a((4a((4a((4aRRRR7777SSSS8888RRRR8a8a8a8aRRRR))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethyl)yl)ethyl)yl)ethyl)yl)ethyl)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----bbbb
utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)utyldiphenylsilyl)oxy)methyl)----7777----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----oooo
ctahydroisoquinolinctahydroisoquinolinctahydroisoquinolinctahydroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((109109109109)))) To a stirred solution of 99999999 (352
mg 0598 mmol) and imidazole (163 mg 239 mmol) and DMAP (15 mg 0123
mmol) in DMF (12 mL) was added TBDPSCl (617 microL 239 mmol) at 0 degC
After 20 min saturated aqueous NH4Cl (5 mL) was added to quench the reaction The water layer
was extracted three times with AcOEt and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was purified by column chromatography (SiO2 hexaneAcOEt = 11) to give 109109109109
(426 mg yield 86) as a colorless amorphous 1H NMR (CDCl3 600 MHz) δ 103 (9H s) 138-141
(2H m) 150-155 (1H m) 159-170 (3H m) 181-197 (7H m) 209-220 (4H m) 228 (1H ddd J
= 66 84 150 Hz) 237 (1H ddd J = 66 84 150 Hz) 274 (1H s) 336 (1H d J = 114 Hz) 371
(1H d J = 132 Hz) 380-385 (2H m) 391-395 (2H m) 400-409 (2H m) 466 (1H d J = 102 Hz)
484 (1H t J = 42 Hz) 491-498 (4H m) 504 (1H d J = 114 Hz) 566 (1H s) 566-572 (1H m)
580-585 (1H m) 587 (1H d J = 102 Hz) 735 (1H t J = 78 Hz) 739-741 (2H m) 753 (1H t J
= 78 Hz) 757-759 (1H m) 761-763 (4H m) 789 (1H d J = 78 Hz) 13C NMR (CDCl3 150 MHz)
δ 191 249 268 273 280 281 316 333 351 362 382 389 461 488 497 647 648 652
714 1042 1146 1150 1255 1276 1276 1277 1282 1287 1288 1297 1297 1326 1330
1331 1354 1383 1385 1395 1729 IR(neat) 3377 1654 cm-1 [α]D26 ndash130 (c 128 CHCl3 gt99
ee) HRMS(ESI) calcd for C47H63N2O7SSi 8274125 found 8274132
NNNN----((2((2((2((2SSSS4a4a4a4aRRRR8888RRRR8a8a8a8aRRRR))))----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methylbutyldiphenylsilyl)oxy)methyl
))))----5555----hydroxyhydroxyhydroxyhydroxy----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----23444a588a23444a588a23444a588a23444a588a----octahydrooctahydrooctahydrooctahydro----1111HHHH3333HHHH----spiro[spiro[spiro[spiro[furafurafurafura
nnnn----27272727----isoquinolin]isoquinolin]isoquinolin]isoquinolin]----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((110110110110 mixture of 5 mixture of 5 mixture of 5 mixture of 5RRRR and 5and 5and 5and 5SSSS))))
To a stirred solution of 109109109109 (451 mg 0545 mmol) in acetonewater (41 26 mL)
was added PPTS (205 mg 0817 mmol) The mixture was heated under reflux
for 25 h Saturated aqueous NaHCO3 (1 mL) was added to this mixture at 0 degC to quench the
BsN
H
OTBDPS
HN O
O OH
110
BsN
H
O
O
OTBDPS
HN
OH
109
O
43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

43
reaction The water layer was extracted three times with AcOEt and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 32) to separate starting material 109109109109 from products 110110110110 and 104104104104 This reaction
was repeated 2 cycles and the combined mixture of products was purified by column
chromatography (SiO2 CH2Cl2AcOEt = 81) to give 110110110110 (273 mg yield 64) as a colorless
amorphous and 104104104104 (52 mg yield 12) as a colorless oil The diastereomers could not be separated
1H NMR analysis (CDCl3 400 MHz 55 degC) of diastereomixture of 110110110110 indicated disappearance of
the 13-dioxolan group and formation of a lactol because no aldehyde peak was observed IR(neat)
3382 1646 cm-1 HRMS (ESI) calcd for C45H58N2O6SSiNa 8053683 found 8053694
NNNN----((4a((4a((4a((4aRRRR8888SSSS8a8a8a8aRRRREEEE))))----7777----(2(2(2(2----(13(13(13(13----dioxolandioxolandioxolandioxolan----2222----yl)ethylideyl)ethylideyl)ethylideyl)ethylidene)ne)ne)ne)----8a8a8a8a----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----5555----((((((((((((tetetete
rtrtrtrt----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----2222----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----12344a788a12344a788a12344a788a12344a788a----octahydoctahydoctahydoctahyd
roisoquinolinroisoquinolinroisoquinolinroisoquinolin----8888----yl)hexyl)hexyl)hexyl)hex----5555----enamideenamideenamideenamide ((((104104104104)))) 1H NMR (CDCl3 400 MHz) δ 106 (9H
s) 162-175 (2H m) 178-187 (2H s) 192-205 (3H m) 209-217 (4H m)
220-231 (3H m) 243-250 (2H m) 379-384 (2H m) 388-393 (2H m)
406-416 (3H m) 485 (1H t J = 48 Hz) 491-506 (5H m) 522 (1H d J = 76 Hz) 543 (1H brs)
565-575 (1H m) 575-585 (1H m) 638 (1H s) 732-745 (6H m) 749-763 (8H m) 782 (2H
brd) 13C NMR (CDCl3 100 MHz 55 degC) δ 141 192 248 266 269 275 312 324 332 362 381
392 602 650 658 1038 1148 1151 1201 1277 1278 1281 1288 1298 1326 1333 1334
1344 1355 1370 1381 1384 1723 IR(neat) 1646 cm-1 HRMS (ESI) calcd for C47H61N2O6SSi
8094020 found 8094024 [α]D26 +405 (c 150 CHCl3 gt99 ee)
(2(2(2(2SSSS9a9a9a9aRRRR13a13a13a13aRRRR16a16a16a16aRRRR))))----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)butyldiphenylsilyl)oxy)methyl)----5555----hydroxyhydroxyhydroxyhydroxy----11111111----(ph(ph(ph(ph
enylsulfonyl)enylsulfonyl)enylsulfonyl)enylsulfonyl)----4454454454455891011121313a16a5891011121313a16a5891011121313a16a5891011121313a16a----dodecahydrododecahydrododecahydrododecahydro----1111HHHH3333HHHH----spiro[[1]azacspiro[[1]azacspiro[[1]azacspiro[[1]azac
ycloundecino[23ycloundecino[23ycloundecino[23ycloundecino[23----iiii]isoquinoline]isoquinoline]isoquinoline]isoquinoline----162162162162----furan]furan]furan]furan]----2(32(32(32(3HHHH))))----oneoneoneone ((((111111111111 mixture of 5mixture of 5mixture of 5mixture of 5RRRR and and and and
5555SSSS 41 41 41 41 mixture of mixture of mixture of mixture of ZZZZEEEE isomers)isomers)isomers)isomers) A solution of 110110110110 (458 mg 00584 mmol) in
degassed (three freeze-thaw cycles) CH2Cl2 (584 mL) containing Grubbs 1st
generation catalyst (168 mg 00175 mmol) was stirred overnight at 40 degC After cooled to rt ca 550
mL of CH2Cl2 was removed in vacuo The resulting solution was roughly purified by column
chromatography (SiO2 AcOEt) to give mixture of 111111111111 110110110110 and dimers This reaction was taken 6
times with the same scale (using 458 mg of 110110110110 for each experiment total 275 mg) and the
combined mixture of 111111111111 110110110110 and dimers was purified by column chromatography (SiO2
hexaneAcOEt = 11 to 13) to give 111111111111 (90 mg yield 34) as a colorless amorphous dimers (685 mg
yield 24) and 110110110110 (728 mg yield 24 ) The 4 isomers of 111111111111 could not be separated
A solution of the dimers (118 mg 00145 mmol) in degassed (three freeze-thaw cycles) CH2Cl2
(148 mL) containing Grubbs 1st generation catalyst (4 mg 000416 mmol) was stirred vigorously
under ethylene atmosphere at rt After 24 h ca 120 mL of CH2Cl2 was removed in vacuo The
resulting solution was roughly purified by column chromatography (SiO2 AcOEt) to give mixture of
110110110110 and dimers This mixture was purified by column chromatography (SiO2 hexaneAcOEt = 11)
to give 110110110110 (71 mg yield 60) and dimers (28 mg yield 24) 1H NMR analysis (CDCl3 400 MHz) of the mixture of 111111111111 indicated formation of an internal
BsN
H
O
O
OTBDPS
HN
104
O
BsN
H
OTBDPS
HN O
O OH
111
44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

44
olefin IR(neat) 1651 cm-1 HRMS (ESI) calcd for C43H54N2O6SSiNa 7773370 found 7773366
MS analysis of the mixture of dimers indicated that all dimers have 22-membered ring LRMS
(ESI) mz 1531 [M+Na]+
(9a(9a(9a(9aRRRR11113a3a3a3aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methybutyldiphenylsilyl)oxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((112112112112)))) The lactol 111111111111 (30 mg 00397
mmol) was dissolved in THF (1 mL) and added dropwise at 0 degC to a stirred
solution of methylene triphenylphosphorane which was prepared by stirring a
mixture of methyl triphenylphosphonium bromide (139 mg 0397 mmol) and KHMDS (05 M
solution in toluene 760 microl 0397 mmol) in THF (1 mL) at rt for 1 h After 10 min saturated aqueous
NH4Cl (1 mL) was added to quench the reaction The water layer was extracted three times with
AcOEt and the combined organic layers were washed with brine and dried over Na2SO4 After the
volatile material was removed under reduced pressure the resulting residue was purified by
column chromatography (SiO2 hexaneAcOEt = 11) to give ZE mixture This mixture was purified
by preparative TLC (SiO2 hexaneAcOEt = 11) to give 112112112112 (118 mg yield 39) as a colorless oil
and 113113113113 (30 mg yield 10) 1H NMR analysis of 112112112112 gave an unclear chart due to its rotamer
HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753550
(9a(9a(9a(9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRREEEE))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)butyldiphenylsilyl)oxy)methyoxy)methyoxy)methyoxy)methy
l)l)l)l)----16161616----hydroxyhydroxyhydroxyhydroxy----11111111----(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)(phenylsulfonyl)----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[1[1[1[1
]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23]azacycloundecino[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((113113113113)))) 1H NMR (CDCl3 600 MHz)
δ 105 (9H s) 134-138 (1H m) 148 (1H d J = 144 Hz) 160-163 (2H m)
191-196 (2H m) 201-209 (4H m) 211-217 (4H m) 230 (1H dd J = 120 120
Hz) 235-243 (2H m) 254 (1H dd J = 78 138 Hz) 260-270 (1H m) 341 (1H d J = 120 Hz)
405 (2H s) 406 (1H d J = 150 Hz) 436 (1H d J = 90 Hz) 494 (1H d J = 96 Hz) 501 (1H d J
= 174 Hz) 556 (1H ddd J = 72 78 150 Hz) 561 (1H s) 569 (1H ddd J = 54 90 150 Hz)
577-581 (1H m) 637 (1H d J = 90 Hz) 734 (4H t J = 72 Hz) 740-742 (2H m) 755-763 (7H
m) 795 (1H d J = 72 Hz) HRMS (ESI) calcd for C44H56N2O5SSiNa 7753577 found 7753592
((((9a9a9a9aRRRR13a13a13a13aRRRR16161616SSSS16a16a16a16aRRRRZZZZ))))----16161616----(but(but(but(but----3333----enenenen----1111----yl)yl)yl)yl)----14141414----((((((((((((terttertterttert----butyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)metbutyldiphenylsilyl)oxy)met
hyl)hyl)hyl)hyl)----11111111----(hex(hex(hex(hex----5555----enoyl)enoyl)enoyl)enoyl)----16161616----hydroxyhydroxyhydroxyhydroxy----345891011121313a1616a345891011121313a1616a345891011121313a1616a345891011121313a1616a----dodecahydrododecahydrododecahydrododecahydro----[[[[
1]azacycloundecin1]azacycloundecin1]azacycloundecin1]azacycloundecino[23o[23o[23o[23----iiii]isoquinolin]isoquinolin]isoquinolin]isoquinolin----2(12(12(12(1HHHH))))----oneoneoneone ((((114114114114)))) A 04 M solution of sodium
naphthalenide in THF was prepared by sonicating a mixture of sodium metal
(32 mg 14 mmol) and naphthalene (180 mg 14 mmol) in THF (35 mL) at rt for
1 min and stirred for 2 h This naphthalenide solution (368 microL 0137 mmol) was added dropwise to
a solution of 112112112112 (118 mg 00152 mmol) in THF (100 microL) at ca ndash70 degC (methanol-dry ice bath)
After 30 min saturated aqueous NH4Cl (1 mL) was added to quench the reaction The water layer
was extracted three times with CH2Cl2 and the combined organic layers were washed with brine
and dried over Na2SO4 After the volatile material was removed under reduced pressure the
resulting residue was used for next reaction without further purification
This amine and 5-hexenoic acid (174 mg 0152 mmol) and EDCI (29 mg 0152 mmol) were
N
H
OTBDPS
HN O
OHO
114
BsN
H
OTBDPS
HN O
OH
112
BsN
H
OTBDPS
HN O
OH
113
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽
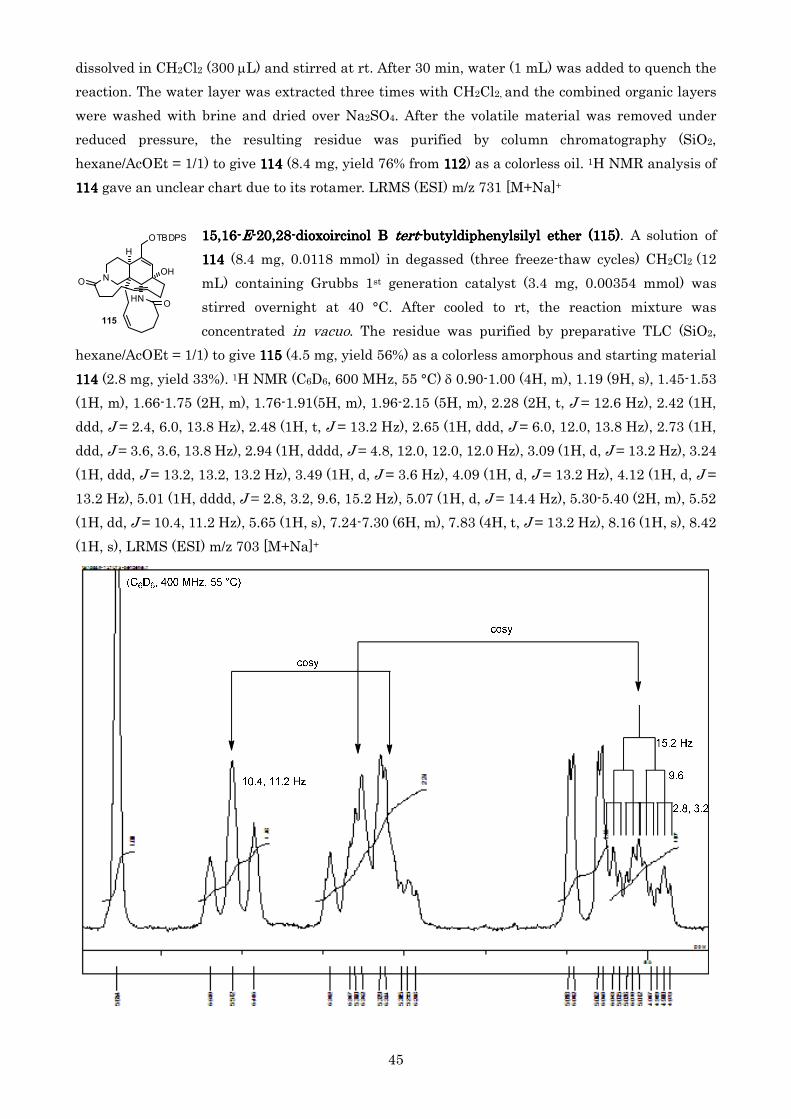
45
dissolved in CH2Cl2 (300 microL) and stirred at rt After 30 min water (1 mL) was added to quench the
reaction The water layer was extracted three times with CH2Cl2 and the combined organic layers
were washed with brine and dried over Na2SO4 After the volatile material was removed under
reduced pressure the resulting residue was purified by column chromatography (SiO2
hexaneAcOEt = 11) to give 114114114114 (84 mg yield 76 from 112112112112) as a colorless oil 1H NMR analysis of
114114114114 gave an unclear chart due to its rotamer LRMS (ESI) mz 731 [M+Na]+
1516151615161516----EEEE----2020202028282828----dioxoircinol B dioxoircinol B dioxoircinol B dioxoircinol B terttertterttert----butyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl etherbutyldiphenylsilyl ether ((((115115115115)))) A solution of
114114114114 (84 mg 00118 mmol) in degassed (three freeze-thaw cycles) CH2Cl2 (12
mL) containing Grubbs 1st generation catalyst (34 mg 000354 mmol) was
stirred overnight at 40 degC After cooled to rt the reaction mixture was
concentrated in vacuo The residue was purified by preparative TLC (SiO2
hexaneAcOEt = 11) to give 115115115115 (45 mg yield 56) as a colorless amorphous and starting material
114114114114 (28 mg yield 33) 1H NMR (C6D6 600 MHz 55 degC) δ 090-100 (4H m) 119 (9H s) 145-153
(1H m) 166-175 (2H m) 176-191(5H m) 196-215 (5H m) 228 (2H t J = 126 Hz) 242 (1H
ddd J = 24 60 138 Hz) 248 (1H t J = 132 Hz) 265 (1H ddd J = 60 120 138 Hz) 273 (1H
ddd J = 36 36 138 Hz) 294 (1H dddd J = 48 120 120 120 Hz) 309 (1H d J = 132 Hz) 324
(1H ddd J = 132 132 132 Hz) 349 (1H d J = 36 Hz) 409 (1H d J = 132 Hz) 412 (1H d J =
132 Hz) 501 (1H dddd J = 28 32 96 152 Hz) 507 (1H d J = 144 Hz) 530-540 (2H m) 552
(1H dd J = 104 112 Hz) 565 (1H s) 724-730 (6H m) 783 (4H t J = 132 Hz) 816 (1H s) 842
(1H s) LRMS (ESI) mz 703 [M+Na]+
N
H
HN
OH
O
O
OTBDPS
115
46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

46
1516151615161516----EEEE----20202020----oxoircinol B oxoircinol B oxoircinol B oxoircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl)))) ether (ether (ether (ether (125125125125)))) To a solution of 115115115115
(3 mg 000441 mmol) in toluene (800 microL) at 0 degC was added Red-Al solution (70
in toluene 20 microL) The mixture was warmed to rt and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The
water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the crude product 125125125125 (39 mg) was obtained LRMS (ESI) mz 667 [M+H]+
1516151615161516----EEEE----ircinol B ircinol B ircinol B ircinol B 1111----((((terttertterttert----butyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilylbutyldiphenylsilyl))))----12121212----((((terttertterttert----butyldimbutyldimbutyldimbutyldimeeeeththththylsilylylsilylylsilylylsilyl)))) ether ether ether ether
((((126126126126)))) To a solution of crude 125125125125 (1 mg ca 000149 mmol) and Et3N (20 microL) in
CH2Cl2 (100 microL) was added dropwise TBSOTf (3 microL 00129 mmol) at 0 ordmC After 1
h saturated aqueous NaHCO3 (1 mL) was added at 0 degC to quench the reaction
The water layer was extracted three times with CH2Cl2 and the combined organic
layers were washed with brine and dried over Na2SO4 After the volatile material was removed
under reduced pressure the resulting residue was used for next reaction without further
purification
To a solution of the crude product in CH2Cl2 (272 microL) at 0 degC was added DIBAH solution (10 M
in toluene 634 microL 00634 mmol) The mixture was heated at 50 degC and stirred for 1 h Then the
mixture was cooled to 0 degC and added water (1 mL) to quench the reaction The water layer was
extracted three times with CH2Cl2 and the combined organic layers were washed with brine and
dried over Na2SO4 After the volatile material was removed under reduced pressure the crude
product 126126126126 (lt1 mg) was obtained LRMS (ESI) mz 767 [M+H]+
N
H
HN
OH
O
OTBDPS
125
N
H
HN
OTBS
OTBDPS
126
47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

47
参考文献及び脚注参考文献及び脚注参考文献及び脚注参考文献及び脚注
1 Danishefsky S Kitahara T J Am Chem Soc 1974197419741974 96 7807
2 (a) Ono K Nakagawa M Nishida A Angew Chem Int Ed 2004200420042004 43 2020 (b) Nakagawa
M Ono K Nagata T Nishida A J Synth Org Chem Jpn 2005200520052005 63 200
3 (a) Joslashrgensen K A Angew Chem Int Ed 2002002002000000 39 3558 For aldehydes and ketones see a
review (b) Pellissier H Tetrahedron 2009200920092009 65 2839 For imines see reviews (c) Buonora P
Olsen J -C Oh T Tetrahedron 2001200120012001 57 6099 (d) Rowland G B Rowland E B Zhang Q
Antilla J C Curr Org Chem 2006200620062006 10 981
4 An example of catalytic asymmetric Diels-Alder reaction using a Danishefsky-type diene see
Ward D E Shen J Org Lett 2007200720072007 9 2843
5 (a) Sudo Y Shirasaki D Harada S Nishida A J Am Chem Soc 2008200820082008 130 12588 (b)
Harada S Toudou N Hiraoka S Nishida A Tetrahedron Lett 2009200920092009 50 5652
6 (a) Kozmin S A Iwama T Huang Y Rawal V H J Org Chem 1997199719971997 62 5252 (b) Kozmin
S A Green M T Rawal V H J Org Chem 1999199919991999 64 8045 (c) Huang Y Iwama T Rawal
V H J Am Chem Soc 2002002002000000 122 7843
7 Inokuchi T Okano M Miyamoto T J Org Chem 2001200120012001 66 8059
8 (a) Kobayashi S Hachiya I Ishitani H Araki M Tetrahedron Lett 1993199319931993 34 4535 (b)
Kobayashi S Araki M Hachiya I J Org Chem 1994199419941994 59 3758 (c) Kobayashi S Ishitani
H J Am Chem Soc 1994199419941994 116 4083 (d) Kobayashi S Ishitani H Araki M Hachiya I
Tetrahedron Lett 1994199419941994 35 6325 (e) Kobayashi S Ishitani H Hachiya I Araki M
Tetrahedron 1994199419941994 50 11623
9 Nishida A Yamanaka M Nakagawa M Tetrahedron Lett 1999199919991999 40 1555
10 Kozmin S A Iwama T Huang Y Rawal V H J Am Chem Soc 2002002002002222 124 4628
11 Bohlmann F Knoll K -H Zdero C Mahanta P K Grenz M Suwita A Ehlers D Le Van
N Abraham W -R Natu A A Phytochemistry 1977197719771977 16 965
12 Bohlmann F Eickeler E Chem Ber 1979197919791979 112 2811
13 (a) Hayakawa K Ohsuki S Kanematsu K Tetrahedron Lett 1986198619861986 27 947 (b) Nagashima
S Ontsuka H Shiro M Kanematsu K Heterocycles 1995199519951995 41 245
14 Ho T -L Ho M -F J Chem Soc Perkin Trans 1 1999199919991999 1823
15 Ito Y Hirao T Saegusa T J Org Chem 1978197819781978 43 1011
16 (a) Danishefsky S J Pearson W H J Org Chem 1983198319831983 48 3865 (b) Danishefsky S J Uang
B J Quallich G J Am Chem Soc 1985198519851985 107 1285 (c) Danishefsky S J Phillips G
Ciufolini M Carbohydrate Res 1987198719871987 171 317 (d) Herrinton P M Klotz K L Hartley W M
J Org Chem 1993199319931993 58 678 (e) Evans P A Nelson J D J Org Chem 1996199619961996 43 7600 (f)
Rovis T Orellana A Chem Commun 2008200820082008 730
17 For preparation see (a) Banville J Brassard P J Chem Soc Perkin Trans 1 1919191976767676 1852
Various conversions of the Diels-Alder adduct derived from the diene see (b) Danishefsky S
Singh R K Gammill R B J Org Chem 1919191978787878 43 379
18 Larock R C Hightower T Kraus G A Hahn P Zheng D Tetrahedron Lett 1995199519951995 36
2423
19 Evans D A Chapman K T Carreira E M J Am Chem Soc 1919191988888888 110 3560
20 Angeles A R Waters S P Danishefsky S J J Am Chem Soc 2008200820082008 130 13765
48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

48
21 (a) Sakai R Higa T Jefford C W Bernardinelli G J Am Chem Soc 1986198619861986 108 6404 (b)
Sakai R Kohmoto S Higa T Jefford C W Bernardinelli G Tetrahedron Lett 1919191987878787 28
5493
22 Winkler J D Axten J M J Am Chem Soc 1998199819981998 120 6425
23 (a) Martin S F Humphrey J M Ali A Hillier M C J Am Chem Soc 1999199919991999 121 866 (b)
Humphrey J M Liao Y Ali A Rein T Wong Y-L Chen H-J Courtney A K Martin S
F J Am Chem Soc 2002200220022002 124 8584
24 (a) Tokumaru K Arai S and Nishida A Presented at the 126th Annual Meeting of the
Pharmaceutical Society of Japan Sendai Japan March 28minus30 2006 028[F]-031 (b) Ohfusa T
Master Thesis 2008200820082008 (Chiba University)
25 Toma T Kita Y Fukuyama T J Am Chem Soc 2010201020102010 132 10233
26 Jakubec P Hawkins A Felzmann W Dixon D J J Am Chem Soc 2012201220122012 134 17482
27 (a) Matsumura T Akiba M Arai S Nakagawa M Nishida A Tetrahedron Lett 2007200720072007 48
1265 (b) Matsumura T Doctor Thesis 2002002002007777 (Chiba University) (c) Mihara Y Matsumura T
Terauchi Y Akiba M Arai S Nishida A Bull Chem Soc Jpn 2222009009009009 82 1520 (d) Mihara
Y Doctor Thesis 2002002002009999 (Chiba University)
28 Kondo K Shigemori H Kikuchi Y Ishibashi M Sasaki T Kobayashi J J Org Chem
1992199219921992 57 2480
29 74747474のオレフィンのカップリングJ値は132 Hzであり典型的なシス(8-12 Hz)トランス(14-17 Hz)
の値のどちらにも当てはまらないNOE 測定に
おいても幾何異性を決定づける情報は得られな
かったため74747474 の幾何異性は決定できていない
一方反応の遷移状態を考慮するとシス体を与え
る遷移状態は AB 環骨格との間に強い立体障害
が生じると推測されるためトランス体が優先的
に得られていると考えられる(Figure 12)
30 Deguest G Bischoff L Fruit C Marsais F Org Lett 2007200720072007 9 1165
31 Bracher F Hildebrand D Tetrahedron 1991991991994444 50 12329
32 Kuwada T Fukui M Hata T Choshi T Nobuhiro J Ono Y Hibino S Chem Pharm
Bull 2003200320032003 51 20
33 Dixon らによる最新の研究 26 では塩
基性アルミナシリカゲルで精製した
87878787 と 128128128128 の Stille カップリングを厳
密に 5 回脱気した DMF 溶媒中で行う
ことによりマンザミン A の全合成を
達成している(Scheme 34)よって本
反応は検討の余地があると考えられる
34 Boger D L Yohannes D J Org Chem 1919191989898989 54 2498
35 Kunishima M Kawachi C Iwasaki F Terao K Tetrahedron Lett 1999199919991999 40
5327
36 Uchida H Doctor Thesis 2002002002002222 (Chiba University)
N
R
NO
Bs
CF3O H
OPiv
Ph3P
TBSO
N
R
NO
Bs
CF3O H
OPiv
Ph3P
OTBS
for cis d isfavored for trans favored
Figure 12
Manzamine A
N
H
N
OH
NNH
N
H
N
OTf
OH
Scheme 34
N
SnBu3
NH
+
Pd(PPh3)4(12 mol )
DMF (degassed)
60 degC 1 h
52128
87
49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

49
論文目録
本学位論文の内容は下記の発表論文による
Catalytic Enantioselective Total Synthesis of (ndash)-Platyphyllide and Its Structural Revision
Hiraoka S Harada S Nishida A J Org Chem 2010 75 3871
Abnormal Ito-Saegusa oxidation of TIPS enol ether assisted by a hydroxy group on a side chain
Hiraoka S Harada S Nishida A Tetrahedron Lett 2011 52 3079
学会発表
平岡紫陽白崎大輔須藤幸徳原田真至西田篤司
「キラル Yb-BINAMIDE錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 25回有機合成化学セミナーP-38阿蘇プラザホテル2008年 9月
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「キラルイッテルビウム錯体を用いる触媒的不斉 Diels-Alder反応と天然物合成への応用」
第 95回有機合成シンポジウム2-14慶應義塾大学薬学部(芝共立キャンパス)マルチメディア講堂
2009年 6月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
IKCOC-11 PB148 Rihga Royal Hotel Kyoto Japan November 9-13 2009
平岡紫陽東道のぞみ白崎大輔須藤幸徳原田真至西田篤司
「不斉 DielsndashAlder反応を用いる platyphyllideの触媒的不斉全合成」
第 8回次世代を担う有機化学シンポジウム2-03日本薬学会長井記念ホール2010年 5月
Shiharu Hiraoka Nozomi Toudou Daisuke Shirasaki Yukinori Sudo Shinji Harada and Atsushi Nishida
lsquoAsymmetric Diels-Alder Reaction of Danishefsky-Type Diene Catalyzed by Chiral Yb(III) Complex and Its
Application to the Total Synthesis of Platyphyllidersquo
0th Cutting- Edge Organic Chemistry Junior Workshop J-23 National Tsing Hua University Taiwan November
6-7 2010 Oral Award 受賞
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
第 53 回天然有機化合物討論会P-44 大阪国際交流センター2011 年 9 月
平岡紫陽御原康洋寺内悠樹松村知亮西田篤司
「海洋産アルカロイドマンザミン B の全合成研究」
日本薬学会第 132年会30E10-am13S北海道大学2012年 3月
Shiharu Hiraoka Yasuhiro Mihara Tomoaki Matsumura Yuki Terauchi and Atsushi Nishida
lsquoSynthetic Study of Manzamine Brsquo
IKCOC-12 PC-096 Rihga Royal Hotel Kyoto Japan November 12-16 2012
50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽

50
審査委員
本学位論文の審査は千葉大学大学院薬学研究院で指名された下記の審査委員により行われた
主査 千葉大学大学院教授(薬学研究院) 薬学博士 濱田康正
副査 千葉大学大学院教授(薬学研究院) 薬学博士 高山廣光
副査 千葉大学大学院教授(薬学研究院) 理学博士 石橋正己
謝辞
本研究の遂行に際しこのような機会をくださりまた終始御指導御鞭撻を賜りました西田 篤司 教
授に心より御礼申上げます
著者の研究活動開始時より有益な御指導御助言御討論を頂きました荒井 秀 准教授に厚く御礼
申し上げます
研究の進め方や考え方など研究者としての基礎の多くを直接御指導賜り有益な御助言御討論を
頂きました原田 真至 助教に厚く御礼申し上げます
有益な御助言御討論を頂きまた博士としての在り方を示してくださいました大房 俊行 博士に感
謝致します
各種スペクトルデータの測定及び御指導を頂きました本学分析センター 関 宏子 博士原 律子
氏荷堂 清香 氏に感謝致します
その他にも在学中に御世話になりました千葉大学医学薬学府 薬品合成化学研究室の諸先輩後輩方
ならびに友人に感謝致します
最後に長きに亘る学生生活を支えてくれた妻 美幸と両親に感謝します
2013 年 平岡 紫陽





![MK2T Series - SMC Corporationca01.smcworld.com/.../BEST-5-3-jp/pdf/3-p1389-1403-mk2t.pdf2] 標準アーム MK 不回転精度、回転精度向上! 不回転精度: ±0.9 ⇒±0.5](https://static.fdocuments.nl/doc/165x107/5fd2340f15d3522a3b7c68ae/mk2t-series-smc-2-ff-mk-ececi.jpg)












