
Genetische heterogeniteit bij Osteogenesis …...Inleiding: Osteogenesis imperfecta (OI) is een...
115
Academiejaar 2015 – 2016 Genetische heterogeniteit bij Osteogenesis Imperfecta – Een continue zoektocht naar oorzakelijke mutaties Staf Rokegem Rebecca Van den Brande Promotor: Dr. Sofie Symoens Co-promotor: Prof. ing. Paul Coucke Masterproef voorgedragen in de 2de Master in het kader van de opleiding MASTER OF MEDICINE IN DE GENEESKUNDE FACULTEIT GENEESKUNDE EN GEZONDHEIDSWETENSCHAPPEN
Transcript of Genetische heterogeniteit bij Osteogenesis …...Inleiding: Osteogenesis imperfecta (OI) is een...

Academiejaar 2015 – 2016
Genetische heterogeniteit bij Osteogenesis Imperfecta
–
Een continue zoektocht naar oorzakelijke mutaties
Staf Rokegem
Rebecca Van den Brande
Promotor: Dr. Sofie Symoens
Co-promotor: Prof. ing. Paul Coucke
Masterproef voorgedragen in de 2de Master in het kader van de opleiding
MASTER OF MEDICINE IN DE GENEESKUNDE
FACULTEIT GENEESKUNDE EN GEZONDHEIDSWETENSCHAPPEN

“De auteur en de promotor geven de toelating dit afstudeerwerk voor consultatie beschikbaar
te stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder de
beperkingen van het auteursrecht, in het bijzonder met betrekking tot de verplichting
uitdrukkelijk de bron te vermelden bij het aanhalen van resultaten uit dit afstudeerwerk.”
Datum: 11 april 2016
Staf Rokegem
Rebecca Van den Brande Dr. Sofie Symoens

Dankwoord
De voorbije twee jaar hebben we veel tijd en inspanning toegewijd aan deze masterproef.
Als onderdeel van het curriculum van onze Master of Medicine, doelt een thesis op het
stimuleren van studenten in wetenschappelijk onderzoek en het kennis maken met zowel de
mogelijkheden als de beperkingen die het met zich meebrengt.
Dit leerproces zou nooit mogelijk zijn geweest zonder de bijdrage in tijd, kennis en ervaring
van onze promotor Dr. Sofie Symoens en de waakzame aanwezigheid van copromotor
Prof. Dr. Paul Coucke. Wij hadden hen graag bedankt voor hun tijd, constructieve opmerkingen,
hun enthousiasme en geruststellende woorden dewelke ons ondersteund hebben in het bereiken
van onze doelen. Dankzij hen is deze masterproef wat zij vandaag is.
Ook hadden wij graag Lynn Demuynck bedankt. Haar inzet om ons technieken aan te leren die
we niet of enkel in theorie kenden was onontbeerlijk voor de taak die wij moesten volbrengen.
De kloof tussen theorie en praktijk is er een die haast onmogelijk te overbruggen valt zonder
adequate begeleiding.
Ten slotte hadden wij graag elkaar bedankt. De laattijdige beslissing om samen te werken was
niet vanzelfsprekend. Hoewel weinig tijd voorhanden was om elkaars werkwijzen te leren
kennen, raakten deze toch vlot verzoend. Het kritisch benaderen van elkaars ideeën was een
drijfveer voor verandering, wat zeker heeft bijgedragen tot de kwaliteit van dit werk. Ook de
mentale steun was naar het einde toe een welkom geschenk.
Rebecca Van den Brande en Staf Rokegem

Afkortingenlijst
µl: microliter
ACAN: Aggrecan
ABI: Applied Biosystems
ADAMTS: A disintegrin and metalloproteinase with thrombospondin motifs
ADAMTS20: ADAM Metallopeptidase With Thrombospondin Type 1 Motif, 20
ADD1: Adducin 1
AKT2: V-Akt Murine Thymoma Viral Oncogene Homolog 2
ALPL: Alkaline Phosphatase, Liver/Bone/Kidney
AP: Antarctic Phosphatase
BMP: Bone Morphogenetic Protein
Cas9: CRISPR associated protein 9
CDC27: Cell Division Cycle 27
CLSD: Cranio-lenticulo-sutural dysplasia
CMGG: Centrum voor Medische Genetica Gent
COG3: Component Of Oligomeric Golgi Complex 3
COL1A1: Collagen, Type I, Alpha 1
COL1A2: Collagen, Type I, Alpha 2
COP: Coat Protein
CRISPR: Clustered regularly-interspaced short palindromic repeats
CRS: Chain Recognition Sequence
CRTAC1: Cartilage Acidic Protein 1
CRTAP: Cartilage – associated protein
CTBP2: C-Terminal Binding Protein 2
CUX1: Cut-Like Homeobox 1
CyPB : Cyclophilin B
Cyp : Cyclophiline
ddNTP : Dideoxynucleotide
DLX: Distal-Less Homeobox
DMSO: Dimethylsulfoxide
DNA : Deoxyribonucleic acid (desoxyribonucleïnezuur)

ECM: Extracellulaire matrix
EDTA: Ethyleendiaminetetra-azijnzuur
ELK4: ELK4, ETS-Domain Protein (SRF Accessory Protein 1)
ERCC2: Excision Repair Cross-Complementation Group 2
Exo: Exonuclease
EYA1: EYA Transcriptional Coactivator And Phosphatase 1
Fe2+: Ijzerion
FKBP10: FK506 Binding Protein 10
fs: Frameshift mutatie
FUOM: Fucose Mutarotase
FZD: Frizzled receptor
GIGYF2: GRB10 Interacting GYF Protein 2
GP: GenomiPhi Product
GRIN3B: Glutamate Receptor, Ionotropic, N-Methyl-D-Aspartate 3B
GTP: Guanosine-5'-triphosphate
HSP47: Heat Shock Protein 47
HOX: Homeobox
IFITM5: Interferon Induced Transmembrane Protein 5
ILGF: Insuline-like growth factor
indel_ea: Inserties/deleties
kDa: Kilodalton
KLHL33: Kelch-Like Family Member 33
LRP: Low-density lipoprotein receptor-related protein 5
LEPRE1: Leucine Proline-Enriched Proteoglycan 1
LH: Lysine hydroxylase
MBTPS1: Membrane-Bound Transcription Factor Peptidase, Site 1
miss: Missense mutatie
MLBR: Major Ligand-Binding Region
mRNA: messenger Ribonucleic Acid (ribonucleïnezuur)
MSC: Mesenchymale stamcel
NCBI: National Center for Biotechnology Information
ng: nanogram

NGS: Next Generation Sequencing
NFKBID: Nuclear Factor Of Kappa Light Polypeptide Gene Enhancer In B-Cells Inhibitor,
Delta
NMD: Nonsense mediated decay
NTP: nucleoside triphosphate
O2: Dizuurstof
OH: hydroxyl
OI: Osteogenesis imperfecta
OPA1: Optic Atrophy 1
OSX: Osterix
P3H1: Prolyl 3-hydroxylase-1
P4H: Proly 4-hydroxylase
P4HA1: Prolyl 4-Hydroxylase, Alpha Polypeptide I
PCR: Polymerase Chain Reaction
PEDF: Pigment Epithelium Derived Factor
PhyloP-waarde: Phylogenetische P-waarde
PLEKHM2: Pleckstrin Homology Domain Containing, Family M (With RUN Domain)
Member 2
PPI: Peptidylprolyl cis-trans isomerasen
PPIB: Peptidylprolyl Isomerase B (Cyclophilin B)
PRDM2: PR Domain Containing 2, With ZNF Domain
PTC: Premature termination codon
RECK: Reversion-Inducing-Cysteine-Rich Protein With Kazal Motifs
(r)ER: (Ruw) endoplasmatisch reticulum
RUNX: Runt-Related Transcription Factor 2
RYK: Receptor-Like Tyrosine Kinase
SEC23B: Sec23 Homolog B, COPII Coat Complex Component
SERPINF1: Serpin Peptidase Inhibitor, Clade F, Member 1
SERPINH1: Serpin Peptidase Inhibitor, Clade H, Member 1
SIPA1L1: Signal-Induced Proliferation-Associated 1 Like 1
SMYD1: SET And MYND Domain Containing 1
SNP: Single nucleotide polymorfism
SP7: Sp7 Transcription Factor

splice: Splice site mutaties
stop: Nonsense mutatie
TAPT1: Transmembrane anterior posterior transformation 1
TAS2R45: Taste Receptor, Type 2, Member 45
TBX22: T-Box 22
TCF/LEF: T-cell factor/lymphoid enhancer factor
TGF: Transforming growth factor
TLD: Tolloid
TLL: Tolloid-like
TMEM38B: Transmembrane Protein 38B
TMPRSS11E: Transmembrane Protease, Serine 11E
TRAPPC12: Trafficking Protein Particle Complex 12
TRIC: Trimeric intracellular cation channel
TTN: Titin
UDP: Uridinedifosfaat
UPR: Unfolded Protein Reponse
VPS33B: Vacuolar Protein Sorting 33 Homolog B
VSVG-GFP: Vesicular Stomatitis Virus Glycoprotein with a Green Fluorescent Protein
WES: Whole Exome Sequencing
WGS: Whole Genome Sequencing
WNT1: Wingless-Type MMTV Integration Site Family, Member 1
ZNF595: Zinc Finger Protein 595


Inhoudstafel
1 Abstract 1
2 Inleiding 3
2.1 De extracellulaire matrix 3
2.2 Collageen 3
De Collageen Superfamilie 3
Classificatie en Nomenclatuur 4
Basisstructuur van type I collageen 5
Biosynthese van type I collageen 7
2.3 Osteogenesis imperfecta 14
Inleiding 14
Classificatie 14
Epidemiologie 17
Klinische diagnose 17
Differentiaal diagnose 18
Etiologie 18
Behandeling van OI 25
3 Methodologie 29
3.1 Overzicht patiënten 29
3.2 GenomiPhi DNA amplificatie 29
Uitvoering 30
3.3 Polymerase Chain Reaction (PCR) 30
Principe 30
Primers 31
Werkwijze 32
3.4 Labchip GX 33

Principe 33
Werkwijze 33
3.5 MiSeq Personal Sequencer 34
Principe 34
Werkwijze 34
3.6 Sanger sequencing 34
Principe 34
Werkwijze 36
Analyse 39
3.7 Exoom analyse 39
Variabelen 39
4 Resultaten 42
4.1 Screening patiënten 42
Resultatentabel 42
Bespreking resultaten 48
4.2 Exoomanalyse 50
Exoom 1: P11 51
Exoom 2: P12 61
Exoom 3: P13 65
5 Discussie 67
5.1 Screening gekende OI genen 67
5.2 Whole Exome Sequencing (WES) 68
Exoom 1 (P11) 68
Exoom 2 (P12) 69
Exoom 3 (P13) 70
Sterktes en zwaktes van Whole Exome Sequencing 71
Incidental Findings 71

5.3 Conclusie 73
6 Referenties 74
7 Bijlagen I
7.1 Gebruikte materialen I
Polymerase Chain Reaction (Bijlage 1) I
Sanger sequencing (Bijlage 2) II
7.2 Resultaten MiSeq III
OI panel 1 (Bijlage 3) III
OI panel 2 (Bijlage 4) IV
7.3 Sanger Sequencing Report VII
7.4 Ford – Variantclassificatie VIII
P1 – COL1A2 (Bijlage 5) VIII
P1 en P2 – CRTAP (Bijlage 6) IX
P1 – PLOD2 (Bijlage 7) X
P3, P6, P7 en P9 – TAPT1 – P1 (Bijlage 8) XI
P1, P3, P4, P7 en P9 – TAPT1 – P2 (Bijlage 9) XII
P4 – COL1A1 (Bijlage 10) XIII
P6 – COL1A1 (Bijlage 11) XIV
P7 – COL1A1 (Bijlage 12) XV
P5 – CREB3L1 (Bijlage 13) XVI
P6 – CREB3L1 (Bijlage 14) XVII
P7 – BMP1 (Bijlage 15) XVIII
P8 – PLS3 (Bijlage 16) XIX
P10 – BMP1 (Bijlage 17) XX
7.5 Exoomanalyse (Bijlage 18) XXI


1
1 Abstract
Inleiding: Osteogenesis imperfecta (OI) is een heterogene genetische aandoening waarbij
fragiliteit van het bot op de voorgrond staat. Secundair ziet men ook symptomen zoals scoliose,
toegenomen laxiteit in de gewrichten, blauwe sclerae en dentinogenesis imperfecta. Aan de
hand van klinische, radiologische en genetische bevindingen wordt de ziekte opgedeeld in
verschillende klassen. In 90% van de gevallen wordt OI autosomaal dominant overgeërfd en
wordt het veroorzaakt door mutaties in de genen COL1A1, COL1A2 en in zeldzame gevallen
IFITM5. COL1A1 en COL1A2 coderen voor het eiwit type I collageen, wat het
hoofdbestanddeel vormt van de extracellulaire botmatrix. IFITM5 speelt een rol in de initiële
fase van de mineralisatie.
In de overige 10% van de gevallen wordt osteogenesis imperfecta autosomaal recessief
overgeërfd en wordt het veroorzaakt door een grote verscheidenheid aan genen. Het merendeel
van deze genen speelt een rol in de collageenbiosynthesis. Ondanks de reeds uitgebreide lijst
aan gekende causale genen, werd voor enkele patiënten met de klinische diagnose van OI nog
geen causale mutatie geïdentificeerd. Het doel van deze thesis was dan ook om nieuwe,
potentieel causale genen op te sporen.
Methodologie: In het kader van deze thesis werden 10 patiënten met een klinische diagnose
van OI gescreend voor mutaties in de reeds gekende causale genen. Deze genen waren
opgenomen in OI panel 1 en 2, respectievelijk autosomaal dominant en autosomaal recessief
(met uitzondering van IFITM5 dat bij OI panel 2 hoort). De screening gebeurde door middel
van PCR, Next Generation Sequencing (MiSeq) en Sanger sequencing. Voor de analyse van
deze screening werd gebruik gemaakt van de software-programma’s Seqpilot en Alamut
Visual. Indien geen causale mutaties werden geïdentificeerd, werd overgegaan naar Whole
Exome Sequencing (WES) en analyse. In deze thesis werden 3 exomen geanalyseerd. Deze
waren niet afkomstig van de 10 door ons gescreende patiënten, maar van patiënten waarvoor
de OI screening en exoomsequencing reeds was uitgevoerd in het Centrum voor Medische
Genetica voor de aanvang van deze thesis.
Resultaten: Na de OI screening van de 10 patiënten werden bij deze patiënten door middel van
MiSeq 10 varianten gevonden in OI panel 1 en 30 varianten in OI panel 2. Van deze 40 varianten
waren er 28 vals positief of polymorfismen.

2
De overige 12 varianten werden bevestigd met Sanger sequencing. Aan 5 van deze varianten
werd een klasse 4 (likely pathogenic) toegekend, namelijk in P1, P4, P6, P7 en P8. In P1 ging
het om een mutatie in COL1A2, namelijk c.2314G>A (p.(Gly772Ser)), in P4, P6 en P7 om
mutaties in COL1A1 (respectievelijk c.484delC (p.(Gln162fs), c.370-2A>G (p.(Gly772Ser) en
c.1012G>A (p.(Gly338Ser). In P8 werd een mutatie in PLS3 vastgesteld, c.766C>T
(p.(Arg256*). Klasse 3 (uncertain significance) werd toegekend aan 2 varianten in P7 en P10.
In deze genen werden varianten gevonden in BMP1, respectievelijk c.1757G>A (p.(Arg586His)
en c.433+12A>G (p.(?)). Aan de overige 5 varianten werd een klasse 2 (likely benign)
toegekend. Het ging om varianten in P1, P2, P5 en P6. In P1 werden varianten in CRTAP
(c.558A>G, p.(=)) en PLOD2 (c.132A>G, p.(=)) gevonden. In P2 werd dezelfde variant
teruggevonden in CRTAP als in P1. In P5 en P6 werden varianten gevonden in CREB3L1,
c.288C>T (p.(=)) en c.651C>T (p.(=)) respectievelijk. De gevonden polymorfismen werden
tevens geklassificeerd, met uitzondering van de polymorfismen in CREB3L1 voor P7.
Na exoomanalyse werden RYK, AKT2, TRAPPC12, SEC23B, ACAN en PLEKHM2
geselecteerd als potentiële genen in Exoom 1. Uit Exoom 2 werden RYK, COG3, RAB1A, EYA1,
RECK, CRTAC1 en ADAMTS20 geselecteerd. Uit Exoom 3 werd enkel TAPT1 geselecteerd.
Discussie: Voor 5 van de 10 patiënten werd een causale variant gevonden na screening,
waarvan 4 patiënten een mutatie hadden in COL1A1/COL1A2 (P1, P4, P6, P7) en 1 patiënt een
mutatie in PLS3 (P8). De overige patiënten blijven genetisch onopgehelderd. Deze moeten
klinisch verder geëvalueerd worden. Indien de kliniek bij deze patiënten inderdaad sterk
suggestief is voor OI, kan men overgaan op Whole Exome Sequencing. De kandidaatgenen
voor OI die in deze thesis werden geselecteerd uit de exomen, zullen allereerst bevestigd
worden via Sanger sequencing. Hierna kan men overgaan naar segregatie-onderzoek binnen de
familie en uiteindelijk functionele studies zoals in vitro studies en zebravisstudies. Ondanks het
feit dat WES een krachtig hulpmiddel is, vermelden we ook de nadelen van deze techniek. Niet
alle varianten kunnen gedetecteerd worden met WES en de heterogeniteit van OI compliceert
de detectie van nieuwe causale genen. Ten slotte bespreken we de rapportering van incidental
findings, hetgeen een ethisch vraagstuk vormt.

3
2 Inleiding
2.1 De extracellulaire matrix
Bindweefsel bestaat in tegenstelling tot de andere weefsels vooral uit een extracellulaire matrix
en in mindere maten uit cellen. In de extracellulaire matrix van bindweefsel bevinden zich
meerdere proteïnen uit diverse proteïnefamilies. Deze proteïnen staan in voor de structurele
integriteit en verschillende fysiologische functies. (1)
De samenstelling en structuur van deze matrix verschilt van bindweefsel tot bindweefsel. De
expressie en synthese van structurele proteïnen en glycoproteïnen zijn namelijk
weefselspecifiek en dit resulteert in unieke functionele en biologische kenmerken. De cellen
aanwezig in de extracellulaire matrix zijn verantwoordelijk voor de synthese en het onderhoud
ervan, maar de extracellulaire matrix werkt op zijn beurt ook in op de cellen. Deze cel-matrix
interacties spelen een rol bij celmigratie en –aanhechting, celdifferentiatie en de regulatie van
genexpressie. Bovendien heeft de extracellulaire matrix ook invloed op de morfogenese en het
celmetabolisme. (1,2)
De belangrijkste proteïne in de extracellulaire matrix is het fibreuze proteïne collageen.
Collageen is vooral te vinden in de vervormbare weefsels zoals de huid, pezen en het hart, maar
is ook abundant aanwezig in botten, kraakbeen en tanden. (3)
2.2 Collageen
De Collageen Superfamilie
Tot op heden zijn reeds minstens 28 verschillende types collageen beschreven. Zij werden
opgelijst naar chronologische volgorde van ontdekking. (4) Indien de collagen-like peptiden in
rekening worden genomen, komt dit aantal al snel op 50 verschillende macromoleculen. Samen
worden zij ook wel de collageen superfamilie genoemd. (5)

4
Classificatie en Nomenclatuur
2.2.2.1 Nomenclatuur
Een collageen eiwit kan homotrimerisch1 of heterotrimerisch2 zijn, naargelang de samenstelling
van de triple helix. Een unieke naamgeving geeft de samenstelling van een collageen weer.
Type I collageen, waarvan de trimeer bestaat uit twee α1-ketens (α1(I)) en één α2-keten (α2(I)),
zal neergeschreven worden als [α1(I)]nα2(I). Hierbij slaat het Romeinse cijfer op het
betreffende collageen type, en n op het nummer van de α keten. (5)
2.2.2.2 Classificatie
Alle collagenen zijn trimeren bestaande uit drie α-ketens die een karakteristieke triple
helixstructuur vormen (zie 2.1.3.1). Onderling kunnen de collagenen wel aanzienlijk
verschillen in grootte, functie en distributie over de verschillende weefsels. (6,7)
Collagenen worden onderverdeeld in subfamilies op basis van de suprastructurele organisatie
die zij aannemen. Deze is afhankelijk van de organisatie van de helix domeinen in de
verschillende types. Daarnaast dragen ook andere niet-collageneuze domeinen bij tot de
functionele heterogeniciteit van de collageen familie. Type IV collageen heeft bijvoorbeeld een
soepelere helix waardoor deze meer geschikt wordt voor het verstevigen van basale
membranen. Type I collageen heeft echter een ononderbroken en lang helix domein waardoor
zij veel steviger zijn en zullen instaan voor bijvoorbeeld de botstevigheid. (7)
Er is ook een groep collagenen waarbij er onderbrekingen bestaan in de sequentie van het helix-
domein, Fibril-Associated Collagens with Interrupted Triple helices (FACIT-Collagenen)
genaamd. (4)
1 Homotrimerisch: De collageen-trimeer bestaat uit drie identieke α ketens
2 Heterotrimerisch: De collageen-trimeer bestaat uit een samenstelling van verschillende α ketens

5
Classificatie Collageen types Supramoleculaire structuur Fibril-vormende collagenen I, II, III Gestreepte fibrillen
V, XI Gestreepte fibrillen, behouden N-
terminale regulatorische domeinen
XXIV, XXVII Onbekend
FACIT collagenen IX, XII, XIV Geassocieerd met fibrillen, andere
interracties
FACIT-like collagenen XVI, XIX, XXI, XXII Interfaciale regios, basale membraan
zones
Netwerk-vormende collagenen
Basale membranen IV Kippengaas netwerk met laterale
associatie
Gekraald filament vormend VI Gekraalde filamenten, netwerken
Ankerende fibrillen VII Lateraal geassocieerde anti-parallele
dimeren
Hexagonale netwerken VIII, X Hexagonale roosters
Transmembraan collagenen XIII, XVII, XXIII, XXV Transmembraneus met oplosbare
ectodomeinen Gliomedines, ectodysplasine
Multiplexine collagenen XXVI, XXVIII Basale membranen, gekliefde C-
terminale domeinen beinvloeden
angiogenese
Andere moleculen met
collageen-domeinen
Acetylcholinesterase,
adiponectine, Clq, collectines,
surfactant, anderen
Collageen domeinen in non-
collageen moleculen
Figuur. 1: Classificatie van de tot nu toe gekende collageenmoleculen.
In het kader van OI zijn de fibrilvormende collagenen de belangrijkste subfamilie. De centrale
ononderbroken triple helix regio is bij hen de grootste component van het eiwit.
De manier waarop en verhouding waarmee de verschillende types collageen samen voorkomen
en in interactie gaan met de andere macromoleculen van de ECM, bepaalt de hogere structuur
en dus de functie van het weefsel waarin deze co-polymerisatie zich voordoet. (4)
2.2.2.3 Fibrilvormende collagenen
Deze subfamilie omvat de collageen types I, II, III, V, XI, XXIV en XXVII. Zij maken 90% uit
van het totale collageen. Hun torsiestabiliteit en treksterkte zorgen voor integriteit en stabiliteit
in weefsels zoals bot (type I en V collageen) en gewrichtskraakbeen (type II en XI collageen).
(2) Type I collageen komt het vaakst voor in bindweefsel en was het eerste collageen dat
ontrafeld werd. (8,9) De fibril-geassocieerde collagenen associëren met grote collageenfibrillen
en spelen waarschijnlijk een rol bij de regulatie van de diameter van de fibrillen.
Basisstructuur van type I collageen
De fundamentele structurele eenheid van collageen bestaat uit een lange (300 nm), dunne
(diameter van 1.5 nm) superhelix bestaande uit 2 pro-α1(I) en 1 pro-α-2(I) keten. Deze trimere

6
moleculen associëren tot polymeren van een hogere orde, fibrillen, die op hun beurt aggregeren
tot grote bundels, namelijk collageenvezels. (8,9)
Figuur 2. De moleculaire structuur van fibril vormende collagenen met de verschillende subdomeinen. De klievingsplaatsen
voor de N- en C-procollagenasen zijn aangeduid. Het gaat hier om een type I collageen.
2.2.3.1 Karakteristiek collageendomein
Ondanks de hoge structurele diversiteit binnen de collageenfamilie bezitten ze allemaal
eenzelfde supramoleculaire structuur: een rechtsdraaiende supercoil van 3 polypeptideketens
(‘triple helix’). (2,9) Elke van de drie ketens bevat een linksdraaiende helix met 18 aminozuren
per omwenteling. Dit karakteristieke collageen domein bevat verschillende herhalingen van het
Gly-X-Y tripeptide, het Gly-X-Y domein, waarin X meestal wordt ingevuld door een proline
en Y door 4-hydroxy-proline. Theoretisch kunnen deze plaatsen echter door eender welk
aminozuur worden bezet. (6) Op elke derde positie vindt men een glycine terug. De 3 ketens
draaien zich uiteindelijk rond een centrale axis tot de triple helix. Dit gebeurt zodanig dat alle
glycineresidu’s zich naar het centrum van de triple helix richten. Aangezien glycine het kleinste
aminozuur is, zorgt dit voor een nauw contact langs de centrale axis. De structuur van de triple
helix is sterk geconserveerd. Een normale triple helix is resistent tegen proteasen zoals pepsine,
trypsine en chymotrypsine en kan alleen afgebroken worden door specifieke collagenasen.
Onderbrekingen in de triple helix veroorzaken intramoleculaire flexibiliteit en laten specifieke
proteolytische splijting toe. Afhankelijk van het collageentype worden specifieke proline en
lysineresidu’s post-translationeel gemodificeerd door hydroxylatie. (2)
Deze modificaties zullen de ketens toelaten onderling waterstofbruggen te vormen die de helix
zullen stabiliseren. (6)

7
2.2.3.2 Flankerende niet-collageen domeinen
Hoewel het helix-domein het gebied is dat de collageenmoleculen hun unieke rol verleent,
hebben ook de flankerende non-helix domeinen een belangrijke functie. Het C-propeptide staat
in voor het initiëren van de helix vorming en de selectie van de nodige α-ketens (zie 2.1.4.3).
Het N-propeptide heeft een invloed op de uiteindelijke diameter van de primaire fibrillen
(zie 2.1.4.6). In tegenstelling tot de triple helix waarvan de structuur sterk geconserveerd is,
vindt men bij de niet-collagene domeinen meer structurele en functionele variabiliteit. (7)
Biosynthese van type I collageen
De biosynthese van fibrillaire collagenen werd reeds uitgebreid bestudeerd. Het proces is
complex en omvat meerdere intracellulaire en extracellulaire stappen, die allemaal bijdragen
tot de structurele en biomechanische eigenschappen van collageen. (10) De biosynthese van
collageen gaat van transcriptie van de betrokken genen tot de aggregatie van collageen
heterotrimeren in fibrillen. Elk type collageen wordt gecodeerd door een specifiek gen en deze
genen worden gevonden op een groot aantal verschillende chromosomen. Bij de synthese van
collageen type I coderen de genen COL1A1 en COL1A2, gelegen op chromosomen 17 en 7,
voor de propeptideketens pro-α-1(I) en pro-α-2(I) respectievelijk. (11)
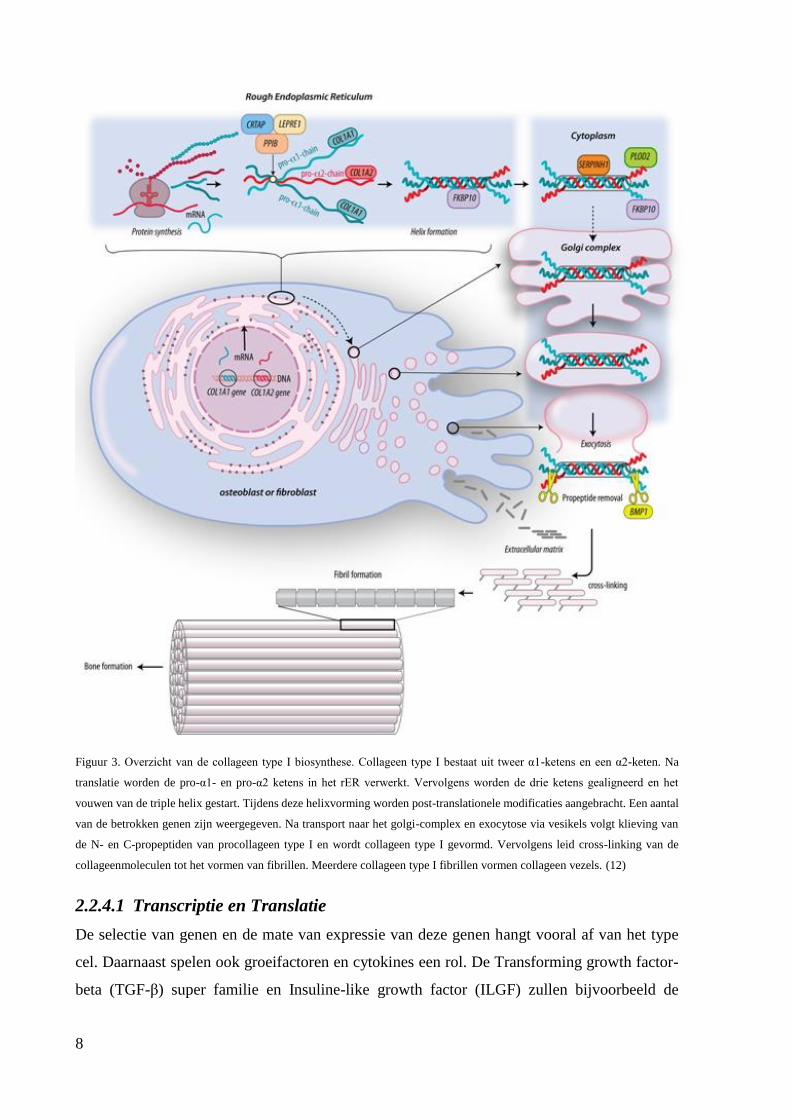
8
Figuur 3. Overzicht van de collageen type I biosynthese. Collageen type I bestaat uit tweer α1-ketens en een α2-keten. Na
translatie worden de pro-α1- en pro-α2 ketens in het rER verwerkt. Vervolgens worden de drie ketens gealigneerd en het
vouwen van de triple helix gestart. Tijdens deze helixvorming worden post-translationele modificaties aangebracht. Een aantal
van de betrokken genen zijn weergegeven. Na transport naar het golgi-complex en exocytose via vesikels volgt klieving van
de N- en C-propeptiden van procollageen type I en wordt collageen type I gevormd. Vervolgens leid cross-linking van de
collageenmoleculen tot het vormen van fibrillen. Meerdere collageen type I fibrillen vormen collageen vezels. (12)
2.2.4.1 Transcriptie en Translatie
De selectie van genen en de mate van expressie van deze genen hangt vooral af van het type
cel. Daarnaast spelen ook groeifactoren en cytokines een rol. De Transforming growth factor-
beta (TGF-β) super familie en Insuline-like growth factor (ILGF) zullen bijvoorbeeld de

9
botvorming stimuleren. De meeste collageen genen bezitten een complex exon-intron patroon.
De messenger RNA’s (mRNA) van fibrillair collageen worden gecodeerd door meer dan 50
exonen, gaande van 54 tot 108 baseparen. Door meerdere transcriptie-initiatie plaatsen,
alternatieve splicing van exonen of een combinatie van beide kunnen binnen één cel meerdere
types mRNA gevonden worden. Naast splicing ondergaat het mRNA ook capping aan het 5’-
uiteinde en polydenylatie aan het 3’-uiteinde. (2) Deze pre-mRNA maturatie vindt plaats in de
nucleus en wordt mRNA processing genoemd. (9)
Dit matuur mRNA wordt naar het cytoplasma overgebracht waar het translatieproces zal starten
op de ribosomen van het ruw endoplasmatisch reticulum (rER). (7)
Het mRNA wordt door de ribosomen getranslateerd tot preprocollageenmoleculen. Deze
moleculen komen in het lumen van het rER terecht dankzij de herkenning van een
signaalpeptide. (2,9)
Initieel wordt een precursormolecule gevormd, procollageen genaamd. Procollageen bevat aan
zijn uiteinden C-en N-propeptiden. (2,13) Deze niet-helicale delen van procollageen maken de
molecule oplosbaar waardoor het zich vlot kan bewegen in de cel. (9)
2.2.4.2 Post-translationele modificatie
Na de synthese van de procollageen ketens op het ribosoom worden deze ketens geïmporteerd
in het endoplasmatisch reticulum. Hier zullen ze een reeks post-translationele modificaties
ondergaan. Deze stappen omvatten onder andere de modificatie van proline residu’s tot
hydroxyprolines, de modificatie van lysines tot hydroxylysines, N- en O-linked glycosylatie,
trimerisatie, zwavelbrugvorming, prolyl cis-trans isomerisatie en het vouwen van de triple
helix. Deze stappen gebeuren na het verwijderen van een signaalpeptide door specifieke
signaal-peptidasen. (14)
2.2.4.2.1 Hydroxylatie van proline
Hydroxylatie gebeurt voornamelijk op de prolines van de Y positie in het Gly-X-Y tripeptide
door het enzyme prolyl 4-hydroxylase, maar in mindere mate ook op de prolines op de X-positie
dankzij prolyl 3-hydroxylases. (10)
In de biosynthese heeft prolyl 4-hydroxylase een centrale rol. De 4-hydroxylproline residu’s
zijn immers essentieel voor de opvouwing van de collageen polypeptide ketens tot triple
helicale molecules (15) en voor het vormen van waterstofbruggen die bijdragen tot de

10
thermische stabiliteit van de triple helix. (7) De proly 4-hydroxylasen (P4H’s) zijn gelokaliseerd
in het lumen van het endoplasmatisch reticulum en katalyseren de vorming van 4-
hydroxyproline door de hydroxylatie van prolines in de Gly-X-Y sequenties van collageen en
meer dan 15 andere collagen–like proteïnen. Ongeveer ¼ van de aminozuren in de α-ketens van
fibrillair collageen zijn prolyl residu’s, waarvan ongeveer 50% een 4-hydroxylatie heeft
ondergaan. (7,16) Inhibitie van deze modificatie zal de triple helix formatie verhinderen en
leiden tot intracellulaire degradatie van de niet-opgevouwde ketens of vertraagde secretie van
het niet-functionele proteïne. (16)
Prolyl 4-hydroxylase is een tetrameer met een moleculair gewicht van 240 kDa en bestaat uit 2
α-units en 2 β-units met elk een gewicht van 64 kDa. Het behoort tot een groep van 2-
oxoglutaraat dioxygenases waartoe ook 2 andere enzymes van de collageen synthese behoren,
namelijk prolyl 3-hydroxylase en lysyl hydroxylases. Deze laatste twee enzymes bezitten dan
ook sterk gelijkaardige katalytische eigenschappen. (16)
Hoewel prolyl hydroxylatie op de Y-positie de norm is, komt ook hydroxylatie van prolines op
de X-positie voor. Dit is echter veel zeldzamer aangezien er per keten maar één zo’n residu
aanwezig is bij collageen type I. Toch heeft ook deze modificatie een belangrijke functie. (10)
Voor prolyl 3-hydroxylase-1 (P3H1), het enzyme dat verantwoordelijk is voor de katalysatie
van de conversie van proline naar 3-hydroxyproline in collageen type I, werd aangetoond dat
het een complex vormt met cartilage-associated protein (CRTAP) en cyclophlin B. Het P3H1
complex zou werkzaam zijn zowel als enzymcomplex en als een chaperone eiwit van de
collageen ketens en zou de premature aggregatie van collageen ketens voorkomen. (15)
2.2.4.2.2 Hydroxylatie van lysine
Ook lysine hydroxylatie is een essentiële post-translationele modificatie bij collageen. (10) Het
patroon en de omvang van deze modificatie beïnvloedt de fibrillogenese, de cross–linking en
de mineralisatie van de matrix. De covalente, intermoleculaire cross-linking is de laatste
modificatie in de biosynthese van collageen en is belangrijk voor de stabiliteit van collageen.
(17) Terzelfdertijd vormen deze residuen een ankerpunt voor het aanhechten van
carbohydraten. Drie vormen van lysyl hydroxylase zijn bekend: LH1, LH2 en LH3. Zij
bevinden zich allen in het endoplasmatisch reticulum. Deze enzymen zijn, net zoals prolyl 4-
hydroxylase en prolyl 3-hydroxylase, 2-oxoglutaraat dioxygenasen. Allen hebben zij het Fe2+
ion, 2-oxoglutaraat, O2 en ascorbaat (vitamine C) als cofactoren. (7,18)

11
De omvang van het aantal lysyl hydroxylaties hangt af van het weefsel. Alleen lysines op de Y
positie van het Gly-X-Y-triplet kunnen gehydroxyleerd worden. Voor LH2 werd aangetoond
dat deze specifiek in de telopeptide regio’s lysine hydroxyleert. Lysines en hydroxylysines in
de telopeptiden zijn substraten voor de enzymen die cross-linking initiëren, namelijk de lysine
oxidasen. De aard van de cross-linking wordt bepaald door de toestand van hydroxylatie van
de telopeptide lysines. (10,17)
2.2.4.2.3 Glycosylatie
O-linked glycosylatie van hydroxylysine is specifiek voor collageenproteïnen. Het gaat om de
covalente binding van galactose en glucose door Uridinedifosfaat (UDP)-carriers. Het enzyme
dat hiervoor verantwoordelijk is, was tot voor kort niet bekend. Recent onderzoek heeft
uitgewezen dat het om het multifunctionele enzym LH3 gaat, dat naast een lysyl hydroxylase
ook een galactosyltransferase en glucosyltransferase is.
LH3 is dus in staat om drie opeenvolgende reacties te katalyseren die nodig zijn voor de
vorming van galactosylhydroxylysine en glucosylgalactosyl-hydroxylysine. (10)
Glysosylatie zou een rol spelen bij collageenfibrillogenese, cross-linking, mineralisatie en
collageen-cel interacties. De helicale cross-linking residu’s α 1-87 zijn belangrijke glycosylatie
plaatsen in type I collageen. (16) Inactivatie van de gehele molecule is embryonaal lethaal. O-
linked glycosylatie is dus essentieel voor normale ontwikkeling. (10) Ondanks de kennis over
type I collageen glycosylatie zijn de precieze loci niet gekend. Deze informatie is belangrijk
om de rol van glycosylatie bij collageen biosynthese verder te begrijpen. (19)
2.2.4.2.4 Peptidyl-prolyl cis-trans isomerisatie
De peptidebinding kan voorkomen in twee conformaties: cis of trans. Voor de meeste
aminozuren is de transconformatie energetisch gunstiger. Voor prolyl- en hydroxyprolyl-
bevattende peptidebindingen bestaat er door hun cyclische zijketens een minder groot
energetisch verschil tussen de twee conformaties. Daarom komen veel proline en
hydroxyprolines in de procollageen ketens voor in cisconformatie. (10) De snelle propagatie
van de triple helix vorming wordt echter verstoord door de aanwezigheid van deze cis-
peptidebinding. Deze peptidebindingen moeten geïsomeriseerd worden tot trans verbindingen
om de triple helix vorming verder te zetten. Deze stap wordt gekataliseerd door peptidylprolyl
cis-trans isomerasen (PPI) of immunophilines, waaronder de cyclophilines en FK506 binding
proteins. (20)

12
2.2.4.2.5 Zwavelbruggen
Protein disulphide isomerase (PDI) heeft als belangrijkste functie de katalysatie van de vorming
en herschikking van zwavelbruggen. PDI is betrokken bij de trimerisatie door de vorming van
intra- en intermoleculaire zwavelbruggen in de propeptide ketens. PDI bindt selectief aan niet-
opgevouwen ketens en is dus ook een chaperone eiwit. (10,21,22)
2.2.4.3 Chain Selection
De C-propeptiden spelen een cruciale rol in de correcte triple helix vorming. De stabiliteit van
de helix is gevoelig voor laterale shifts met één of meer Gly-X-Y repeats. Indien het docken
van de drie α-ketens zou berusten op de herkenning tussen de verschillende helix delen zouden
dergelijke shifts zeer courant gebeuren. Het onderling herkennen van helix domeinen is dan
ook weinig specifiek. (10)
Het C-propeptide treedt op als een trimerisatie-domein om deze shifts te voorkomen. De
aminozuursequenties in het C-propeptide domein zijn sterk geconserveerd behalve een sterk
variabele, discontinue sequentie van 15 aminozuren die de Chain Recognition Sequence (CRS)
wordt genoemd. Deze sequentie bevat de informatie die nodig is voor ketenherkenning. (10,23)
De globulaire structuur van het C-propeptide wordt gestabiliseerd door zwavelbruggen. (7,24)
De C-propeptiden van drie α-helixen verankeren zich d.m.v. zwavelbruggen en vormen op die
manier een zogenaamde zwavelknoop. Deze knoop treedt op als een permanente verbinding,
zelfs nadat de C-propeptiden worden gekliefd. De afgesplitste propeptiden worden uiteindelijk
door trypsine afgebroken. (25)
2.2.4.4 Helixvorming
Enzymen zoals PPI (peptidylprolyl cis-trans-isomerase) en collageen-specifieke chaperone-
eiwitten zoals Heat Shock Protein 47 (HSP47) zorgen ervoor dat de vorming en opvouwing van
de procollageen ketens vlot verloopt. (7) De meeste eiwitten zullen vouwen in een N-C richting,
waarbij het vouwen reeds cotranslationeel zal starten. Dit is niet het geval voor collageen, dat
in een C-N richting vouwt a.d.h.v. de eerder beschreven chain selection. (7,24)
2.2.4.5 Secretie, extracellulaire processing en modificatie
Na de vorming van procollageen worden de molecules verpakt in vesikels en getransporteerd
naar het Golgi network. (7,10) Proteïnen kunnen het endoplasmatisch reticulum (ER) enkel
verlaten indien deze op de correcte wijze zijn opgevouwen. Voor het transport van het ER naar
het Golgi apparaat zijn opeenvolgend zowel Coat Protein I (COPI) en Coat protein (COPII)

13
nodig. COPII staat in voor de export van eiwitten vanuit het ER, terwijl COPI daarentegen het
retrograde transport van resident eiwitten van het ER medieert. COPI zal ook transport van het
ER naar het Golgi apparaat bewerkstelligen. Proteïnen verplaatsen zich over het Golgi-apparaat
zonder ooit het lumen van de Golgi-cisternae te verlaten.
Na het verpakken van de eiwitten in secretoire vesikels in het Golgi-apparaat, worden zij
getransporteerd naar het plasmamembraan waar ze door de vesikels extracellulair worden
vrijgegeven. (24)
2.2.4.6 Fibrilogenese
De fibrilvorming die hierna plaatsvindt, wordt beïnvloed door de propeptiden van de
procollageenmoleculen. Een essentiële stap bij de fibrilvorming is dan ook de verwijdering van
de C-propeptiden en N-propeptiden door specifieke proteasen. C-proteinases zijn leden van de
familie van zink metalloproteïnasen (BMP-1, mTLD en TLL-1). N-proteïnasen zijn leden van
de ADAMTS familie (ADAMTS-2, ADAMTS-3 en ADAMTS-14). (7,2,24) Men gelooft dat
delen van het verwijderde C-propeptide en N-propeptide terugkeren in de cel om de
hoeveelheid collageen te reguleren (feedbackmechanisme). (9)
De functie van de N-propeptiden is nog niet volledig gekend en kan verschillen tussen de
verschillende collageen types, maar deze zou eventueel een rol kunnen spelen bij het reguleren
van de diameter van de fibrillen. (7,2)
Na processing van procollageen schikken de fibrilvormende collagenen zich spontaan in
geordende fibrillaire structuren. In deze ‘self-assembly’ zijn hydrofobische en elektrostatische
interacties tussen de collageenmonomeren betrokken. De oriëntatie van de fibrillen hangt af
van het weefseltype. De fibrilvorming wordt verder gestabiliseerd door covalente cross-linking.
Deze intermoleculaire cross-links zijn essentieel voor de fysieke en mechanische
eigenschappen van de collageen fibrillen en voor een stabiele netwerkvorming. (7,24)

14
2.3 Osteogenesis imperfecta
Inleiding
Osteogenesis imperfecta (OI) is een heterogene groep van ziektebeelden gekarakteriseerd door
gevoeligheid voor botbreuken met bewezen of veronderstelde defecten in de type I collageen
biosynthese. Naast fragiliteit van de beenderen ziet men secundair nog andere symptomen.
Scoliose, toegenomen laxiteit in de gewrichten, blauwe sclerae en dentinogenesis imperfecta
zijn hier enkele voorbeelden van. (16) De ernst van deze ziekte varieert en wordt naargelang
klinische, radiologische en genetische bevindingen onderverdeeld in verschillende types.
(2,11,26,27,3,28)
Classificatie
De huidige nomenclatuur en indeling van OI is nog steeds gebaseerd op het werk dat Prof.
David Sillence in 1979 verrichtte. De Sillence classificatie deelt de ziekte in volgens klinische
en radiologische bevindingen in 4 types. (2,11,3) Later breidde men deze classificatie verder
uit tot 8 types op basis van genetische verschillen. (2,3)
Het classificeren van OI volgens genetische verschillen heeft echter een weerslag op de
klinische praktijk. Met de oorspronkelijke classificatie volgens Sillence was het mogelijk op
basis van kliniek en radiologie de patiënten een plaats te geven in de classificatie. De uitbreiding
van de classificatie op basis van genetische verschillen had echter tot gevolg dat een genetische
screening noodzakelijk was om te beslissen tot welke groep de patiënt behoorde. Het klinische
beeld van de verschillende types was echter niet meer van elkaar te onderscheiden. (12) Een
voorstel werd gedaan om de classificatie te hervormen. (2,12,29)
De Nosology Group of the International Society of Skeletal Dysplasias besloot om de Sillence
classificatie als basis te gebruiken om de verdere ontdekkingen in op te lijsten. Vijf types van
OI werden weerhouden en de verschillende genen verantwoordelijk voor de aandoening werden
onderverdeeld bij het type aan de hand van het fenotype. (12,29,30)
Type 1 geeft een mild fenotype met afwezigheid van ernstige botafwijkingen, maar vaak met
vertebrale fracturen en milde scoliose. Het aantal breuken varieert van patiënt tot patiënt.
Andere symptomen bij OI type 1 zijn blauwe sclerae en vroegtijdige doofheid. Een verdere
onderverdeling in type IA en IB gebeurt op basis van het al dan niet voorkomen van
dentinogenesis imperfecta (niet aanwezig bij IA). (2,11,3,28,31)

15
Type 2 is lethaal door respiratoire insufficiëntie ten gevolge van meerdere ribfracturen (intra-
uterien) of door intracraniale bloedingen bij een vaginale geboorte. Prenataal ziet men het
buigen van de lange beenderen met meerdere breuken. (11,3,28,31) Type 2 wordt op basis van
radiologische bevindingen verder onderverdeeld in subklassen type IIA, IIB en IIC. (2,11)
Patiënten met type 3 overleven de neonatale periode, maar hebben een kleine gestalte en
graduele ledemaat- en ruggengraatafwijkingen door meerdere botbreuken. Dit kan ook leiden
tot de dood door respiratoire insufficiëntie, pneumonie, cor pulmonale of traumata. Deze
patiënten hebben blauwe sclerae, dentinogenesis imperfecta en scoliose. Radiologisch ziet men
‘popcorn epifysen’. (11,3,28,31)
Type 4 resulteert in milde tot matige buiging van de lange beenderen, osteoporose en milde
scoliose met eventueel een kleine gestalte tot gevolg. Het aantal breuken vermindert in de
puberteit. Dit type is vergelijkbaar met mild type 1 of type 3.
Het al dan niet hebben van blauwe sclerae en dentinogenesis imperfecta varieert van patiënt tot
patiënt. Het zijn vaak individuen die niet duidelijk in 1 van de vorige 3 categorieën passen.
(11,3,28,31)
Bij type 5 ziet men matige tot ernstige broze botten en calcificatie van het interosseus membraan
van de voorarm. Dit maakt pro- en supinatie van de voorarm moeilijk. Na een breuk of operatie
krijgt de patiënt ook vaak te maken met hyperplastische callusvorming. (2,26,3,31)
Aangezien de Sillence Classificatie nauw samenhangt met de klinische ernst van de ziekte, zal
men in de praktijk voornamelijk gebruik maken van deze classificatie. Een genetische
classificatie anderzijds kan nuttig zijn aangezien een verschillende genetische etiologie een
andere behandeling vereist. (31)
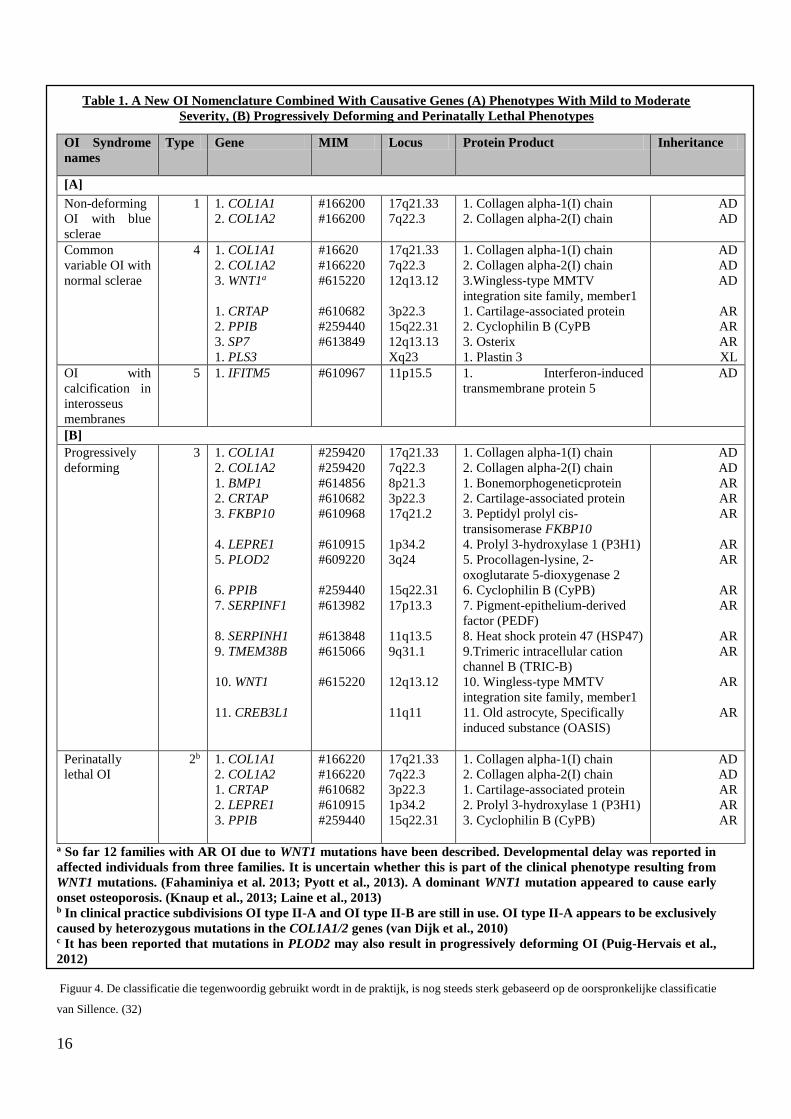
16
Table 1. A New OI Nomenclature Combined With Causative Genes (A) Phenotypes With Mild to Moderate
Severity, (B) Progressively Deforming and Perinatally Lethal Phenotypes
OI Syndrome
names
Type Gene MIM Locus Protein Product Inheritance
[A]
Non-deforming
OI with blue
sclerae
1 1. COL1A1
2. COL1A2
#166200
#166200
17q21.33
7q22.3
1. Collagen alpha-1(I) chain
2. Collagen alpha-2(I) chain
AD
AD
Common
variable OI with
normal sclerae
4 1. COL1A1
2. COL1A2
3. WNT1a
1. CRTAP
2. PPIB
3. SP7
1. PLS3
#16620
#166220
#615220
#610682
#259440
#613849
17q21.33
7q22.3
12q13.12
3p22.3
15q22.31
12q13.13
Xq23
1. Collagen alpha-1(I) chain
2. Collagen alpha-2(I) chain
3.Wingless-type MMTV
integration site family, member1
1. Cartilage-associated protein
2. Cyclophilin B (CyPB
3. Osterix
1. Plastin 3
AD
AD
AD
AR
AR
AR
XL
OI with
calcification in
interosseus
membranes
5 1. IFITM5 #610967 11p15.5 1. Interferon-induced
transmembrane protein 5
AD
[B]
Progressively
deforming
3 1. COL1A1
2. COL1A2
1. BMP1
2. CRTAP
3. FKBP10
4. LEPRE1
5. PLOD2
6. PPIB
7. SERPINF1
8. SERPINH1
9. TMEM38B
10. WNT1
11. CREB3L1
#259420
#259420
#614856
#610682
#610968
#610915
#609220
#259440
#613982
#613848
#615066
#615220
17q21.33
7q22.3
8p21.3
3p22.3
17q21.2
1p34.2
3q24
15q22.31
17p13.3
11q13.5
9q31.1
12q13.12
11q11
1. Collagen alpha-1(I) chain
2. Collagen alpha-2(I) chain
1. Bonemorphogeneticprotein
2. Cartilage-associated protein
3. Peptidyl prolyl cis-
transisomerase FKBP10
4. Prolyl 3-hydroxylase 1 (P3H1)
5. Procollagen-lysine, 2-
oxoglutarate 5-dioxygenase 2
6. Cyclophilin B (CyPB)
7. Pigment-epithelium-derived
factor (PEDF)
8. Heat shock protein 47 (HSP47)
9.Trimeric intracellular cation
channel B (TRIC-B)
10. Wingless-type MMTV
integration site family, member1
11. Old astrocyte, Specifically
induced substance (OASIS)
AD
AD
AR
AR
AR
AR
AR
AR
AR
AR
AR
AR
AR
Perinatally
lethal OI
2b 1. COL1A1
2. COL1A2
1. CRTAP
2. LEPRE1
3. PPIB
#166220
#166220
#610682
#610915
#259440
17q21.33
7q22.3
3p22.3
1p34.2
15q22.31
1. Collagen alpha-1(I) chain
2. Collagen alpha-2(I) chain
1. Cartilage-associated protein
2. Prolyl 3-hydroxylase 1 (P3H1)
3. Cyclophilin B (CyPB)
AD
AD
AR
AR
AR
a So far 12 families with AR OI due to WNT1 mutations have been described. Developmental delay was reported in
affected individuals from three families. It is uncertain whether this is part of the clinical phenotype resulting from
WNT1 mutations. (Fahaminiya et al. 2013; Pyott et al., 2013). A dominant WNT1 mutation appeared to cause early
onset osteoporosis. (Knaup et al., 2013; Laine et al., 2013) b In clinical practice subdivisions OI type II-A and OI type II-B are still in use. OI type II-A appears to be exclusively
caused by heterozygous mutations in the COL1A1/2 genes (van Dijk et al., 2010) c It has been reported that mutations in PLOD2 may also result in progressively deforming OI (Puig-Hervais et al.,
2012)
Figuur 4. De classificatie die tegenwoordig gebruikt wordt in de praktijk, is nog steeds sterk gebaseerd op de oorspronkelijke classificatie
van Sillence. (32)

17
Epidemiologie
De incidentie van OI wordt geschat op 6-7/100 000. (2,11) De prevalentie en incidentie van
types I-IV verschillen onderling. Voor type I zag men in Australië een prevalentie van 3-4 /100
000 en een incidentie van 3.5/100 000. Type II had een incidentie van 1-2/100 000. Wegens
het lethale karakter van type II zijn er geen prevalentiedata beschikbaar. Type III heeft een
prevalentie van 1-2/100 000 en een incidentie van 1.6/100 000. (11)
Van type IV werd lange tijd gedacht dat het om een zeldzaam type ging, maar dit bleek niet het
geval. (11) Types I en IV zijn namelijk verantwoordelijk voor meer dan de helft van de gevallen
van OI. (11,27) De prevalentie van lethaal OI ligt rond de 5/100 000. (2) De prevalentie van
type V is <1/100 000. (33)
Klinische diagnose
De ernst van de kliniek van OI varieert van geen symptomen bij geboorte tot prenatale lethale
botvervormingen. Deze grote variatie maakt het soms moeilijk om een diagnose te stellen. OI
wordt dan wel ingedeeld in verschillende types aan de hand van klinische en radiologische
eigenschappen maar er wordt een continuüm in ernst gezien. (3) De diagnose van OI kan zowel
prenataal als postnataal gebeuren.
2.3.4.1 Prenatale diagnose van OI
OI type II en III kunnen prenataal gediagnosticeerd worden dankzij echografie aangezien men
bij deze types prenatale breuken kan vaststellen. Bij type II ziet men reeds op 14 weken een
verminderde echogeniciteit van de foetale beenderen, gevolgd door meerdere breuken. Type III
is te zien op 18 weken en type IV na 20 weken. Voor type I, IV en V is prenatale echografische
diagnose onbetrouwbaar. (3,12)
2.3.4.2 Postnatale diagnose van OI
Postnatale diagnose kan gesteld worden aan de hand van tekenen en symptomen. Typische
extraskeletale afwijkingen zijn blauwe sclerae, dentinogenesis imperfecta, hyperlaxiteit van de
ligamenten en huid, gehoorverlies en de aanwezigheid van naadbeenderen. Blauwe sclerae zijn
echter ook te zien bij gezonde zuigelingen. (3,12)
Dentinogenesis imperfecta geeft de tanden van deze patiënten een gelig of doorschijnend
blauw-grijs aspect. Vaak zullen deze tanden prematuur uitvallen of zelfs breken.

18
Zij blijken een korte wortel te hebben en een abnormaal dentine kan worden aangetoond. (32)
Gehoorverlies is zeldzaam in de eerste 2 levensdecaden. (11)
De diagnose van OI is eenvoudig bij individuen met een familiale geschiedenis voor deze ziekte
of wanneer typische tekenen aanwezig zijn, maar de diagnose kan moeilijk zijn bij een atypisch
beeld zonder familieleden met dezelfde ziekte. Een bijkomende moeilijkheid in deze situatie is
dat er geen akkoord bestaat over het minimale aantal criteria voor de diagnose van OI. (3)
Differentiaal diagnose
Verschillende skeletaandoeningen kunnen verward worden met OI. Enkele van deze
aandoeningen zijn: Bruck syndroom, idiopathische autosomaal recessieve hyperfosfatemie,
hypofosfatemie, Cole-Carpenter syndroom, idiopathisch juveniele osteoporose,
kindermishandeling, Hajdu-Cheney syndroom of geïsoleerde dentinogenesis imperfecta (11,3)
Etiologie
Hoewel men voornamelijk gebruik maakt van de klinische classificatie (types I-V) van OI, kan
OI ook onderverdeeld worden in klassen volgens genetische verschillen. Voor de diagnose van
OI zal men beroep doen op een DNA analyse van COL1A1/2. Het DNA is hierbij afkomstig
van de huidfibroblasten of uit de witte bloedcellen. Met deze techniek kan men 90% van alle
type I collageen mutaties detecteren. Vervolgens worden ook recessieve genen gelinkt aan OI
onderzocht.(3)
2.3.6.1 Autosomaal dominante OI types (OI panel 1)
2.3.6.1.1 OI Types I-IV (COL1A1/COL1A2)
De dominante OI types I-IV worden veroorzaakt door mutaties in COL1A1 of COL1A2. Er
werden reeds meer dan 1000 varianten in deze genen gevonden. Het type van de mutatie en de
locatie beïnvloeden het fenotype. (3) Deze mutaties kunnen worden onderverdeeld in 2
categorieën: excluderende en includerende mutaties. Excluderende mutaties resulteren in de
exclusie van het product van het gemuteerde allel (nul allel). Slechts de helft van de normale
hoeveelheid type I collageen wordt geproduceerd.
Bij includerende mutaties wordt de abnormale keten mee geïncorporeerd in de heterotrimeer.
Moleculen die ketens bevatten met een mutatie in de triple helix zijn minder stabiel.

19
Type I wordt gekarakteriseerd door een reductie van 50% in type I collageen. Dit gebeurt
meestal door mutaties in één COL1A1 allel die leiden tot mRNA instabiliteit en
haploinsufficiëntie. (11) De mutaties in COL1A1 veroorzaken een prematuur terminatie codon
(PTC) dat nonsense-mediated decay (NMD) van het mRNA van de mutante transcripten
activeert. De meeste transcripten van het gemuteerde COL1A1 worden hierbij afgebroken zodat
slechts de helft van de normale hoeveelheid collageen wordt geproduceerd. (26)
Types II-IV resulteren in de verstrengeling van normaal en gemuteerd type I collageen door
substituties van glycine. Deze mutaties veranderen de structuur van type I collageen. Glycine
substituties in zowel de pro-α-1(I) als de pro-α-2(I)-keten leiden tot een vertraagde opvouwing
van de helix wat post-translationele overmodificatie met zich meebrengt. Het fenotype varieert
tussen mild en lethaal. In een pro-α-1(I)-keten vinden we lethale glycinesubstituties vooral terug
in de major ligand-binding regions (MLBR2 en MLBR3) wat wijst op het belang van interacties
tussen collageen en niet-collagene proteïnen. In de pro-α-2(I)-keten vinden we deze vooral in
proteoglycan binding sites regions. (26)
Een minder voorkomende mutatie die leidt tot OI is die in het domein coderend voor het
carboxyl-terminale propeptide. De meeste van deze mutaties resulteren in een includerende
mutatie en leiden tot een vertraagde opvouwing van de helix met post-translationele
overmodificatie. (11) Er bestaat een zekere homologie in de sequenties van procollageen
(tussen de verschillende types) maar de ketens moeten in een procollageen-specifieke manier
samenkomen. Het vermogen om de verschillende ketens te onderscheiden zit vervat in de
primaire sequentie van het C-propeptide, de chain recognition sequence (CRS) (zie 2.1.4.3).
Het C-propeptide moet correct opgevouwen zijn zodat CRS bloot komt te liggen. Het C-
propeptide bezit ook enkele sterk geconserveerde cysteïneresidu’s. Cys1268 en Cys1285 zijn
de enige cysteïnes die een rol spelen bij zwavelbruggen tussen de verschillende ketens..
Substitutie van deze cysteïneresidu’s resulteert dus in een verstoorde assemblage. Slecht 5%
van de pathologische varianten bij OI bevinden zich in het C-propeptide domein. (34) Mutaties
in het N-terminale propeptide leiden tot Ehler Danlos syndroom. (35)
2.3.6.1.2 OI type V (IFITM5)
Type V gaat gepaard met een mutatie in het gen voor interferon-induced transmembrane protein
5 (IFITM5). IFITM5 behoort tot de interferon-induced transmembrane protein family. Deze
eiwitten worden geïnduceerd door interferon en nemen deel aan een beschermend mechanisme
tegen virale infecties. IFITM5 daarentegen neemt niet deel aan dit mechanisme.

20
IFITM5 codeert voor secretoire en membraaneiwitten in osteoblasten en wordt geactiveerd
tijdens de initiële fase van de mineralisatie. Het OI type V is niet gelinkt aan loss of function
of haploinsufficiëntie van IFITM5. Klinisch ziet men een ernstigere vorm van OI met een kort
gestalte, het buigen van de lange beenderen, buigen van de extremiteiten, vertebrale compressie,
scoliose, blauwe sclerae en dentinogenesis imperfecta. (36)
2.3.6.2 Autosomaal recessief OI
Ongeveer 10% van alle gevallen van OI worden veroorzaakt door recessieve, causatieve
varianten in één van de reeds gekende genen of door varianten in nog niet ontdekte genen voor
OI. Deze genen coderen meestal voor proteïnen die een rol spelen in de biosynthese van type I
collageen.
2.3.6.2.1 OI Type VI (SERPINF1)
Verschillende onderzoeken suggereerden dat OI type VI veroorzaakt wordt door loss of
function mutaties in SERPINF1, het gen dat codeert voor Pigment Epithelium Derived Factor
(PEDF). (37) De mutaties in SERPINF1 bij OI gaven aanleiding tot een PTC (premature
translation-termination codons) met NMD (nonsense-mediated mRNA decay) van de
transcripten als gevolg. (11,26)
In het bot zorgt PEDF voor een upregulatie van osteoprotegerin, hetgeen de osteoclasten
maturatie inhibeert. Mutaties in SERPINF1 resulteren in lagere osteoprotegerin levels en in een
stijging van het aantal osteoclasten, met een verhoogde botresorptie tot gevolg.
OI type VI omvat ongeveer 4 % van alle gevallen. Patiënten met de diagnose van OI type VI
lijken gezond bij de geboorte en hebben geen breuken voor de leeftijd van 6 maanden. Het
histologische kenmerk dat OI type VI onderscheidt van andere vormen van OI is de grote
hoeveelheid van niet-gemineraliseerd osteoid en wazige tetracycline labels, die doen denken
aan osteomalacie. Dit gaat samen met de desorganisatie van de botmatrix waarbij het lamellair
patroon vervangen is door een visschaalpatroon. Patiënten met OI type VI lijken niet te
reageren op bisfosfaten zoals patiënten met een klassiek type I collageen defect.
Zowel de klinische als de histologische bevindingen van OI type VI suggereren dus een ander
uniek mechanisme. (37)

21
2.3.6.2.2 OI types VII, VIII en IX (CRTAP, LEPRE1 en PPIB)
CRTAP codeert voor het cartilage-associated protein, LEPRE1 voor prolyl 3-hydroxylase
(P3H1) en PPIB voor cyclophilin B (CyPB). Samen vormen deze 3 proteïnen een
endoplasmatische reticulum-resident complex (in een 1:1:1 ratio) dat instaat voor de 3-
hydroxylatie van proline 986 van collageen α1(I) en α1(II) en proline 707 in α2(I). (11,26)
Prolyl 3-hydroxylatie is één van de verschillende modificaties van de pro-α-ketens die bijdragen
tot een goede opvouwing, stabiliteit en secretie van het procollageen. CRTAP en LEPRE1
mutaties zijn geassocieerd met posttranslationele overmodificatie van de ketens van type I
collageen. Bij recessieve vormen van OI worden alle moleculen overgemodificeerd, terwijl dit
bij dominante vormen slechts bij de ½ of ¾ van de molecule het geval is. Toch kan het moeilijk
zijn om een onderscheid te maken tussen overmodificatie door structurele mutaties van de type
I collageen gene of door mutaties in het prolyl 3-hydroxylatie complex. (38)
CyPB is een eiwit dat gecodeerd wordt door het PPIB gen en het behoort tot de familie van de
cyclophilines (Cyps). De Cyps zijn een familie van peptidylprolyl cis-trans isomerasen
(PPIases) die instaan voor de cis-trans isomerisatie van peptide bindingen. Vroegere studies
hebben aangetoond dat CyPB direct interageert met procollageen, wat nodig is aangezien
procollageen veel cis-conformers bevat. Enkel trans-peptidebindingen kunnen echter
geïncorporeerd worden in de triple helix van collageen (zie 2.1.4.2.4). CyPB speelt ook een rol
als moleculaire chaperonne in het procollageen export en de secretie samen met HSP47. (39)
Meeste varianten in CRTAP, LEPRE1 en PPIB veroorzaken autosomaal recessieve lethale of
ernstige OI van het type IIB en type III. Als de variant zich in een startcodon bevindt, ziet met
een milder type. (11,26)
2.3.6.2.3 OI types X (SERPINH1)
Mutaties in het gen SERPINH1, dat codeert voor HSP47, geven aanleiding tot het klinische
type III van OI. Mutaties in SERPINH1 beïnvloeden de post-translationele modificatie van type
I procollageen niet. Het lijkt de transit van type I procollageen van het ER naar het Golgi-
apparaat te versnellen, maar de gehele transittijd van intracellulair naar extracellulair te
verlengen. De totale snelheid van type I procollageen productie lijkt echter wel normaal. Men
zag dat de triple helix van het gesecreteerde type I collageen gevoelig bleek voor proteasen op
bepaalde plaatsen. Veranderingen in de triple helix structuur kunnen de fibril assemblage
beïnvloeden en leiden tot een onregelmatige mineralisatie.

22
Bij mutaties van het SERPINH1-gen accumuleert protype I collageen als aggregaten in het ER
en de secretie is vertraagd. Bij muisstudies bleek ook de afsplitsing van de pro-α-1(I) N-
propeptide deficiënt in SERPINH1-/- fibroblasten. De producten van deze genen zullen
waarschijnlijk op een later stadium in de biosynthese met type I procollageen ketens
interageren. (40)
2.3.6.2.4 OI type XI (FKBP10)
Het FKBP10 gen codeert voor een chaperonne eiwit FKBP65 dat betrokken is bij de opvouwing
van protype I collageen. Mutaties in dit gen beïnvloeden de protype I collageen secretie.
Klinisch ziet men een fenotype met matig ernstige vorm van OI met recurrente fracturen van
de lange beenderen, beginnend in de kindertijd, waardoor een rolstoel eventueel kan nodig zijn.
Andere klinische kenmerken zijn progressieve kyfoscoliose, ernstige osteopenie, laxiteit van de
ligamenten, grijze sclerae, normaal gehoor en normale tanden. Het OI fenotype is milder dan
bij mutaties in de genen van het prolyl-3-hydroxylatie complex (CRTAP, LEPRE1 en PPIB)
alsook milder dan OI type 3 veroorzaakt door autosomaal dominante COL1A1/COL1A2
mutaties. Histologische kenmerken waren onder andere een vervormde lamellaire structuur en
een visschaalpatroon samen met verhoogd alkaline fosfatase in het serum. (41)
2.3.6.2.5 OI type XII (SP7)
SP7/OSX codeert voor een osteoblast-specifieke transcriptie factor, die essentieel is voor de
botvorming en osteoblastdifferentiatie. OSX speelt een belangrijke rol in de regulatie van de
differentatie van preosteoblasten naar osteoblasten, downstream van RUNX2, een andere
transcriptiefactor die belangrijk is bij osteoblastdifferentiatie. SP7/OSX komt specifiek in
corticale en trabeculaire osteoblasten tot expressie en in mindere mate ook in de
prehypertrofische chondrocyten in de groeischijf. De klinische aspecten van patiënten met een
mutatie in SP7 zijn recurrente breuken, milde botmisvormingen, vertraagde tanderuptie,
normaal gehoor en witte sclerae. (42)
2.3.6.2.6 OI type XIII (BMP1)
BMP1 codeert voor een gekend protease dat het C-propeptide van type I collageen afsplitst.
Hoewel andere bone morphogenetic proteins (BMP) tot de TGF-beta superfamilie behoren,
codeert dit gen voor een eiwit dat niet nauw samenhangt met andere gekende groeifactoren.

23
Processing van het C-propeptide van protype I collageen wordt uitgevoerd door de familie van
bone morphogenetic protein 1/Tolloid (BMP1/TLD-like) proteïnasen. Hoewel alle leden van
deze familie instaan voor processing van het C-propeptide, is BMP1 hierin het efficiëntst. Een
mutatie in bone morphogenetic protein 1 (BMP1) kan aanleiding geven tot een ernstige vorm
van recessieve OI. Klinisch ziet men een breed voorhoofd, een driehoekig gezicht, prominente
oren zonder gehoorsverlies, recurrente fracturen, buigen van de extremiteiten, hypermobiliteit
van de gewrichten, ernstige afwijkingen van de lange beenderen en een te grote uitrekbaarheid
van de ellebogen, polsen en interphalangeale gewrichten. Er is geen dentinogenesis imperfecta
aanwezig. (26,43)
2.3.6.2.7 OI type XIV (TMEM38B)
TMEM38B codeert voor Trimeric Intracellular Cation Channel B (TRIC-B), een component
van TRIC. TRIC is een monovalent cation-specifiek Ca2+ kanaal dat belangrijk is bij
celdifferentiatie. Het belang van intracellulair Ca2+ bij proliferatie, celdifferentiatie en cellulaire
functie werd reeds meerdere malen bevestigd. Mutaties in TMEM38B kunnen dus leiden tot een
verstoring van de intracellulaire Ca2+ signaling pathways in botcellen. Klinisch ziet men OI
type IV: botfragiliteit, osteoporose, gebogen ledematen, meedere breuken die kunnen leiden tot
pseudo-artrose en wormiaanse botten. Breuken doen zich voor prenataal en in de kindertijd en
verminderen in de puberteit. Ook worden soms blauw-grijze sclerae gezien. (44)
2.3.6.2.8 OI type XV (WNT1)
WNT1 is een gesecreteerd signaaleiwit dat interageert met twee transmembranaire
receptoreiwitten, namelijk Frizzled receptor (FZD) en Low-density lipoprotein receptor-related
protein 5 (LRP5). WNT1 kan dankzij deze interactie de canonical WNT pathway reguleren door
middel van de fosforylatie van β-catenine. Β-catenine kan hierdoor interageren met de
transcriptiefactor T-cell factor/lymphoid enhancer factor (TCF/LEF) in de nucleus. Op die
manier wordt doelgerichte genexpressie gemoduleerd. SP7 en ALPL (dit gen codeert voor een
alkaline fosfatase dat een belangrijke rol speelt bij botmineralisatie) zijn onder andere
downstream targets van WNT-signaling pathway. WNT-signaling is noodzakelijk in osteocyten
om normale bothomeostase te verkrijgen. Bovendien speelt het ook een rol in
osteoblastdifferentiatie en -proliferatie. Homozygote mutaties gaven aanleiding tot een ernstig
fenotype, OI type III. (45) Een heterzygote mutatie in WNT1, hetgeen autosomaal dominant
wordt overgeërfd, gaf echter geen aanleiding tot OI, maar tot early-onset osteoporose. (46)

24
2.3.6.2.9 CREB3L1
Om celschade te voorkomen verwijderen eukaryote cellen ongevouwen eiwitten uit het ER. Dit
systeem wordt de ‘Unfolded Protein Reponse’ (UPR) genoemd. De aanwezigheid van
ongevouwen of verkeerd gevouwen eiwitten wordt opgemerkt door UPR transducers. Zij
transducen dan deze signalen naar de nucleus voor de transcriptie van UPR-target genen, de
attenuatie van globale proteine translatie, en ER-geassocieerde degradatie. (47)
CREB3L1 codeert voor het eiwit OASIS, een transcriptiefactor betrokken bij de UPR. OASIS
is structureel sterk gelijkend op een van de UPR transducers: activating transcription factor 6
(ATF6). OASIS treedt dus op als een weefselspecifieke ER-stress transducer. (47,48)
Indien geen ER-stress aanwezig is, is OASIS ingebed in de ER membranen. Hierbij is het N-
terminale deel cytoplasmatisch gelocaliseerd. Als antwoord op ER-stress wordt OASIS naar
het Golgi getransloceerd waar het gekliefd wordt door resident proteases S1P/MBTPS1 and
S2P/MBTPS2. Het vrijgekomen N-terminaal cytosolische domein gaat dan naar de nucleus,
waar het de transcriptie van COL1A1 zal opreguleren en de secretie van botmatrixproteinen zal
stimuleren. (48,49) Het bindt direct met UPRE (unfolded protein response element) (UPRE-
like sequence) in de osteoblast specifieke COL1A1 promotor regio. Het reguleert geen COL1A1
in andere weefsels zoals bv de huid. (48)
OASIS-/- muizen werden geboren met ernstige osteopenie en spontane fracturen, wat sterk
aanleunt bij menselijke OI. (47,48)
2.3.6.2.10 PLS3
PLS3 (Plastin 3) codeert voor een eiwit dat betrokken is bij de vorming van filamenteuze actine
bundels. Dit eiwit is belangrijk voor de botgezondheid. PLS3 is X-gebonden. Men vond in 5
families een variant in dit gen die bij mannelijke patiënten (die hemizygoot zijn voor deze
variant) aanleiding gaf tot osteogenesis imperfecta type I. Bij oudere vrouwen die heterozygoot
waren voor deze variant zag men dat het risico op breuken tweemaal zo hoog was als bij
vrouwen die geen drager waren van deze variant. (50)
2.3.6.2.11 PLOD2
Hydroxylatie van lysine residuen is essentieel voor de stabiliteit van intermoleculaire cross-
linking in collageen, aangezien zij een bindingsplaats vormen voor carbohydraten. PLOD2

25
codeert voor lysine hydroxylase 2. Reeds 3 PLOD isozymes werden beschreven. Van deze 3
blijkt PLOD2 vooral tot expressie te komen in cellen met een osteoblastische activiteit. (51)
Lysine hydroxylase 2 bevindt zich op de cisternen van het rER, waar het de hydroxylatie zal
verzorgen met ijzerionen en ascorbaat (VitC) als cofactoren. (52)
Behandeling van OI
De huidige behandeling van OI is erop gericht de functionaliteit van de patiënt te maximaliseren
en de frequentie van het optreden van breuken te minimaliseren. Een multidisciplinaire aanpak
met o.a. een orthopedisch chirurg, een kinderarts, een fysiotherapeut en een maatschappelijk
werker is dan ook aangewezen. (53)
2.3.7.1 Farmacologische behandeling
2.3.7.1.1 Bisfosfaten
Orale en intraveneuze bisfosfaten kunnen worden gebruikt voor de meeste OI types (met
uitzondering van SERPINF1) (37), zowel bij kinderen als volwassenen.
Bisfosfaten verstoren de osteoclastvorming, de osteoclastoverleving en de cytoskeletale
dynamiek. (53) Verschillende studies wijzen op verbetering in botdensiteit en een daling van
de chronische botpijn. Het uiteindelijke doel van de behandeling met bisfosfaten is een daling
van het aantal breuken, het voorkomen van afwijkingen van de langere beenderen en
functioneel een verbetering te realiseren. Eventuele bijwerkingen van bisfosfaten zijn een
daling in het calciumserum en een griepreactie bij kinderen. Ook werd aanvankelijk gevreesd
dat het gebruik een mogelijk schadelijk effect op botgroei (longitudinaal) bij kinderen en
adolescenten met zich zou meebrengen, maar dit bleek niet het geval.(11,3,54,55) Andere
bijwerkingen zijn een snelle gewichtstoename, uveïtis en verstoring van botremodellering.
Over de langetermijneffecten van bisfosfaatgebruik is nog maar weinig bekend. (11,3,54)
2.3.7.1.2 Andere farmacologische interventies
Het gebruik van groeihormonen komt ook voor bij de behandeling van OI, zeker bij OI-
patiënten met een kleine gestalte. Toch brengt deze behandeling slechts bescheiden resultaten
met zich mee. Ook zijn de langetermijneffecten niet gekend. Volgens sommige studies zouden
groeihormonen een verhoogde bot turnover met zich meebrengen. (55,56)

26
Ook het parathyroïdhormoon werd overwogen voor de therapie bij OI. Dit resulteerde echter in
osteosacroom bij ratten. (11) Verder moet men bij deze patiënten de inname van Vitamine D
en calcium optimaliseren. (55)
2.3.7.2 Orthopedische behandeling
Intramedullaire staven (vooral bij OI type III en IV) kunnen worden ingebracht in het
beenmergkanaal om breuken te stabiliseren. Bij ernstige scoliose (type III en soms IV) kan men
chirurgie overwegen. Niet-chirurgisch interventies die bij OI kunnen gebruikt worden zijn
bracing en spalken. (11)
2.3.7.3 Fysiotherapie (rehabilitatie)
Een intensief rehabilitatie programma is vaak nodig, zeker bij types III en IV en in de kindertijd,
om de motoriek van de patiënt te verbeteren. (11)
2.3.7.4 Tandheelkundige behandeling
Patiënten met dentinogenesis imperfecta moeten opgevolgd worden door een ervaren tandarts.
Tandheelkundige ingrepen met een hoog risico op het afbreken van tanden moeten vermeden
worden. (55) Het plaatsen van kunstmatige kronen op de tanden van OI-patiënten kan
overwogen worden om infecties en faciale vervormingen te voorkomen. (11)
2.3.7.5 Behandeling voor gehoorverlies
Initieel stelt men bij OI conductief gehoorverlies vast, maar later wordt vaak ook een
neurosensorieel component gediagnosticeerd. OI-patiënten worden daarom best elke 3-5 jaar
audiologisch opgevolgd. In het begin biedt een hoorapparaat enige verbetering, later zal men
een stapedectomie moeten laten uitvoeren. Een cochleair apparaat zou ook eventueel beterschap
kunnen brengen, maar hiervoor bestaat nog onvoldoende bewijs. (11)
2.3.7.6 Toekomstbeeld van OI therapie
Hoewel de huidige behandeling de kwaliteit van leven van de patiënt sterk verbetert, kan OI
niet genezen worden. Het effect van de behandelingstechnieken zoals gentherapie,
beenmergtransplantatie en het gebruik van foetale mesenchymale stamcellen bij OI wordt dan
ook onderzocht (53)

27
2.3.7.6.1 Gentherapie
Gentherapie zou in de toekomst door silencing of vervanging van de causatieve variant een
oplossing kunnen bieden bij OI. (11,28) De meeste varianten bevinden zich in
COL1A1/COL1A2.
Bij de ernstigere types II-IV komen gemuteerde en normale ketens samen, bij het milde type I
is er een verminderde of geen expressie van het gemuteerde COL1A1 allel (haploinsufficiëntie).
Door gebruik van antisense-therapie zou men het allel kunnen silencen en overschakelen op
een milder type van OI. (11,28,55) Het probleem hierbij is het gebrek aan specificiteit voor het
gemuteerde allel. (11,28) Nieuwe technologie zoals het Crispr/Cas9 systeem zou in de toekomst
een volgende stap kunnen zijn in DNA correctie bij OI patiënten. (57) Het Crispr/Cas9 systeem
is afkomstig van bacteriën waar het een rol speelt in de verworven immuniteit tegen virussen
en plasmiden. Het eiwit Cas9 is een endonuclease dat door middel van een guide RNA streng
dat bindt met bepaalde target DNA sequenties site specifieke breuken kan tewerkstelligen in
het dubbelstrengig DNA. Dit mechanisme maakt het mogelijk om het genoom op een efficiënte
en precieze manier te modificiëren. (58)
2.3.7.6.2 Beenmergtransplantatie
Mesenchymale cellen van het beenmerg kunnen differentiëren in verschillende celtypes in
weefsels zoals bot, kraakbeen, spierweefsel en vetweefsel. Een beenmergtransplantatie zou dus
een oplossing kunnen bieden bij aandoeningen waarbij cellen betrokken zijn die voortkomen
uit de mesenchymale cellen. Dit is het geval bij OI. Bij kinderen met OI na een
beenmergtransplantatie zag men een toename in de groei en een verbetering van de botdensiteit.
De langetermijneffecten zijn echter niet gekend. (59)
2.3.7.6.3 Pre- en postnatale transplantatie van foetale mesenchymale stamcellen
Transplantatie van mesenchymale stamcellen kan potentieel de skeletale schade bij OI
verbeteren. Mesenchymale stamcellen (MSC’s) zijn multipotente cellen die kunnen
differentiëren in osteoblasten, chondrocyten en adipocyten en hebben een laag immunogeen
profiel. Daarom worden ze meestal niet afgestoten. In een muismodel zag men dat MSC’s
bijdroegen tot de progenitorpool, hetgeen verbetering van de collageeninhoud en verbetering
van de mineralisatie met zich meebracht. In een klinische studie bij kinderen met OI type III
gaf infusie met allogene MSC’s een verbetering van de mineralisatie en een verhoogde

28
groeisnelheid. Aangezien schade bij ernstige vormen van OI reeds prenataal zich voordoet, valt
er veel te zeggen voor prenatale MSC transplantatie, alvorens er zich bijkomend schade kan
ontwikkelen. Ook ziet men prenataal een hogere succesrate voor de innesteling van de graft,
meer tolerantie voor de donorcellen, een beter systemische distributie dankzij rechts-links
shunting en hogere proportionele celdosissen door de kleine omvang van de foetus. (60)

29
3 Methodologie
Aan de start van ons onderzoek werd ons het genetisch materiaal van tien patiënten aangeboden
waarbij de diagnose van OI klinisch was gesteld. Het startmateriaal is genomische DNA
(gDNA) geëxtraheerd uit EDTA bloed (na extractie uit leukocyten). Via onder andere PCR,
Next Generation Sequencing en Sanger sequencing werd gezocht naar de causale mutatie
verantwoordelijk voor het fenotype bij deze patiënten.
3.1 Overzicht patiënten
Patiënt Concentratie (ng/µl)
P1 398
P2 1282
P3 329
P4 178
P5 550
P6 275
P7 104
P8 117
P9 161
P10 203
3.2 GenomiPhi DNA amplificatie
Aangezien van elke patiënt slechts een beperkte hoeveelheid genetisch materiaal voor handen
was, werd eerst een amplificatie van al het genetisch materiaal gedaan. Dit gebeurde a.d.h.v.
een GenomiPhi™ DNA Amplification Kit. Deze methode maakt gebruik van de unieke
biochemische eigenschappen van Phi29 DNA polymerase om lineair genomisch (g) DNA te
amplificeren. Het geamplificeerde gDNA is representatief voor het originele staal, vertoont zeer
weinig amplificatie bias en bewaart SNP informatie.
Deze kit wordt bewaard op -70°C. De kit bevat volgende componenten:
Sample buffer: 1 x 0.9 ml.
Reactiebuffer: 1 x 0.9 ml.
Enzyme-mix: 1 x 100 μl.

30
Uitvoering
Vul epjes met
- 9µl Sample Buffer
- 1µl Template gDNA
Plaats de epjes vervolgens in de Thermal Cycler :
95°C – 3’
15°C – ∞
Voeg volgende zaken toe aan de epjes :
- 9µl Reactiebuffer
- 1µl Enzyme-mix
Plaats nu de epjes opnieuw in de Thermal Cycler :
30°C – 90’
65°C – 10’
15°C – ∞
De sequeneringsprocedure vraagt om een DNA concentratie van 20ng/µl. Het GenomiPhi
product (GP) wordt door verdunning naar deze concentratie gebracht. De amplificatie
produceert 4-7 µg DNA. De standaardverdunning bedraagt 171,5 µl water en 3,5 µl GP. Indien
bij PCR onvoldoende amplificatie is opgetreden kan men de concentratie van het GP nagaan.
De GenomiPhi procedure kan indien nodig op het oorspronkelijke DNA-materiaal worden
herhaald. De nodige verdunning kan ook manueel berekend worden nadat de concentratie van
het GenomiPhi materiaal is nagegaan.
3.3 Polymerase Chain Reaction (PCR)
Principe
PCR is een techniek om selectief specifieke DNA-fragmenten te amplificeren. Er is slechts een
kleine hoeveelheid DNA nodig om met PCR voldoende kopieën te genereren voor analyse met
conventionele laboratoriumtechnieken. Elke PCR-reactie vereist template DNA, primers,
nucleotiden en DNA polymerase.

31
DNA-polymerase is het enzyme dat de individuele nucleotiden aan elkaar linkt. De primers
bepalen welk DNA product precies wordt geamplificeerd. Het zijn korte DNA fragmenten met
een welbepaalde sequentie die complementair is aan het target DNA. Vanuit deze fragmenten
bouwt het DNA polymerase het PCR product verder uit.
Het mengsel met de 4 bovenstaande componenten wordt in een Thermal Cycler geplaatst
waardoor meerdere cycli van DNA amplificatie plaatsvinden. Dit gebeurt in 3 stappen:
Denaturatie: De machine verhoogt de temperatuur van het mengsel tot smeltpunt
van de twee complementaire DNA strengen van het target DNA zodat deze van
elkaar loskomen.
Hybridisatie: De temperatuur zakt zodat de specifieke primers kunnen binden aan
het target DNA.
Elongatie: De temperatuur stijgt opnieuw waardoor het DNA polymerase in staat is
de primers te verlengen door het toevoegen van nucleotiden.
Bij elke herhaling van deze drie stappen, wordt het DNA verdubbeld. Op die manier verkrijgt
men exponentiële amplificatie van DNA.
Primers
Primers worden ontworpen zodat deze specifiek zijn voor een bepaalde sequentie in het DNA
om te vermijden dat deze aspecifiek binden. De primers voor de screening voor OI waren reeds
aanwezig in het Centrum voor Medische Genetica bij de aanvang van deze thesis.
De primers zijn onderverdeeld in twee verschillende panels. De primers voor de dominante
vorm (COL1A1 en COL1A2, niet IFITM5) van OI zijn gegroepeerd als OI panel I. De primers
voor alle anderen zijn gebundeld in OI panel II. (Zie bijlage 3 en 4). De primers werden
ontworpen en gevalideerd via het Ford protocol. De sequenties van de primers, de ligging van
de primers in het genoom en de nummers van de assays (=primerparen) kunnen worden
teruggevonden op de Ford-website. Voor COL1A1 bevat OI panel I 36 assays, voor COL1A2
43 assays. OI panel II bevat 158 assays.
Voorheen had haast elke primer zijn eigen thermische omstandigheden nodig. Deze
arbeidsintensieve manier van werken is echter historisch. Nu wordt gebruik gemaakt van
zogenaamde ‘universele primers’. De reactie maakt gebruik van drie primers: een
sequentiespecifieke forward primer met een M13 sequentie aan het 5’ uiteinde, de specifieke
reverse primer en ten slotte de universele fluorescent gelabelde M13 primer.

32
Figuur 5. Amplificatieschema voor een enkele PCR reactie dmv universele primers. (A,B) De gearceerde velden duiden de
microsatteliet-specifieke primers aan. (C) Gegolfd veld: de universele M13(-21) sequentie. De ster representeerd het
fluorescente label, in dit geval 6-carboxy-fluorescine (FAM). (D) In de eerste PCR-cycli hybridiseert de forward primer met
de M13 staart op het PCR-product. (E) Deze producten dienen als doelwit voor de FAM-gelabelde universele M13 primer,
welke hybridiseert in latere cycli op lagere temperatuur (53°C). (F) Het eindproduct kan worden geanalyseerd dmv een laser
detectie systeem. Afbeelding afkomstig van (61)
De thermische omstandigheden worden gekozen zodat in de eerste cycli de forward primer met
de M13 sequentie hybridiseert met het opstapelende PCR product. Nadien wordt de temperatuur
verlaagd om het binden van de universele M13 primer te faciliteren. Op dit moment neemt de
universele M13 primer het over van de forward primer en wordt het PCR-product tevens
fluorescent gelabeled. (61) Het Ford-protocol gebruikt in dit onderzoek deze M13 primers.
Werkwijze
De PCR procedure verloopt als volgt. Maak een mengsel van volgende producten:
Componenten reactie Volume (µl)
Kapa2G Robust MM (2x) 5
Forward primer (1µM in TE – buffer) 2,5
Reverse primer (1µM in TE – buffer)
gDNA (20ng/µl) 2,5

33
Hierbij is het gDNA in ons geval het verdunde GenomiPhi product. Vortex het mengsel om een
egale verdeling van de producten te verzekeren. Na centrifugeren wordt de plaat in de Thermal
Cycler geplaatst.
Programma PCR Ford protocol (KAPA ROBUST 60) :
Temperatuur Tijd
95°C 3 minuten
95°C 15 seconden
35 cycli 60°C 10 seconden
72°C 15 seconden
72°C 10 minuten
3.4 Labchip GX
Principe
Om na te gaan of het DNA voldoende geamplificeerd is na PCR, wordt een elektroforese
uitgevoerd. Labchip GX is gebaseerd op traditionele principes van gelelektroforese die
getransfereerd worden naar een microchip. Dit laat toe de DNA- en RNA-fragmenten aan een
hoge snelheid van elkaar te scheiden en de verkregen informatie om te zetten naar een digitaal
formaat. Aan de bekomen banden kan men nagaan of amplificatie voldoende heeft
plaatsgevonden. In geval van onvoldoende intense banden kan men stellen dat de amplificatie
onvoldoende is. Men kan dan trachten de PCR procedure te herhalen.
Werkwijze
Allereerst dient de Labchip ‘geladen’ te worden. Vul voor elke gewenste reactie een
gereserveerde well met
- 17 µl water
- 3 µl van het gewenste PCR-product
Dek opnieuw de plaat af met een seal en centrifugeer. Het resultaat van de Labchip elektroforese
kan bekeken worden via de Labchip GX software.
De PCR-reactie is goed verlopen indien er 1 duidelijke band van de verwachte grootte te zien
is.

34
3.5 MiSeq Personal Sequencer
Principe
De vraag naar een goedkopere en snellere manier om te sequeneren leidde tot het ontwikkelen
van second generation sequencing methoden (Next Generation Sequencing (NGS)). Met NGS
wordt massive parallel sequencing uitgevoerd wat toelaat dat een volledig genoom, of exoom
in dit geval, op minder dan één dag wordt gesequeneerd. Dankzij NGS kan men de sequenties
van verschillende genen van het OI panel 1 en 2 gelijktijdig analyseren en dit voor verschillende
patiënten. In de laatste tien jaar zijn er verschillende NGS platformen ontwikkeld die massive
parallel sequencing aanbieden aan een lage prijs. Eén van de meest gebruikte platformen is
MiSeq Personal Sequencer van Illumina. MiSeq Personal Sequencer maakt gebruikt van de
sequencing-by-synthesis-methode om de amplificatieproducten te sequeneren.
De Miseq Personal Sequencer gebruikt in dit onderzoek is geschikt voor targeted panel
sequencing. Het dient te worden verduidelijkt dat met dit toestel geen exoomscreening kan
gebeuren. De verder genoemde data gebruikt voor de exoomanalyse zijn dan ook niet met dit
toestel verkregen. Hiervoor worden andere sequencingmachines gebruikt of wordt de
sequenering uitbesteed en extern uitgevoerd door bedrijven met een hogere capaciteit.
Werkwijze
Na de controle door de Labchip GX worden de PCR-producten per patiënt en per OI panel
gepoold. d.w.z. van elk PCR-product wordt 2 µl in 1 epje samengevoegd. Dit epje wordt
afgegeven aan de medewerkers die bevoegd zijn voor het bedienen van de MiSeq Personal
Sequencer.
De medewerkers van het Ford-team bereiden het staal voor (library prep) volgens het Nextera
protocol. Na het sequeneren worden de data verwerkt door het team met behulp van de
beschikbare software.
3.6 Sanger sequencing
Principe
De Sanger methode, die ook wel ‘dideoxy sequencing’ of ‘chain termination sequencing’ wordt
genoemd, maakt gebruik van dideoxynucleotiden (ddNTP’s) naast de normale nucleotiden
(NTP’s) die ingebouwd worden in het DNA. De ddNTP’s lijken zeer goed op nucleotiden.
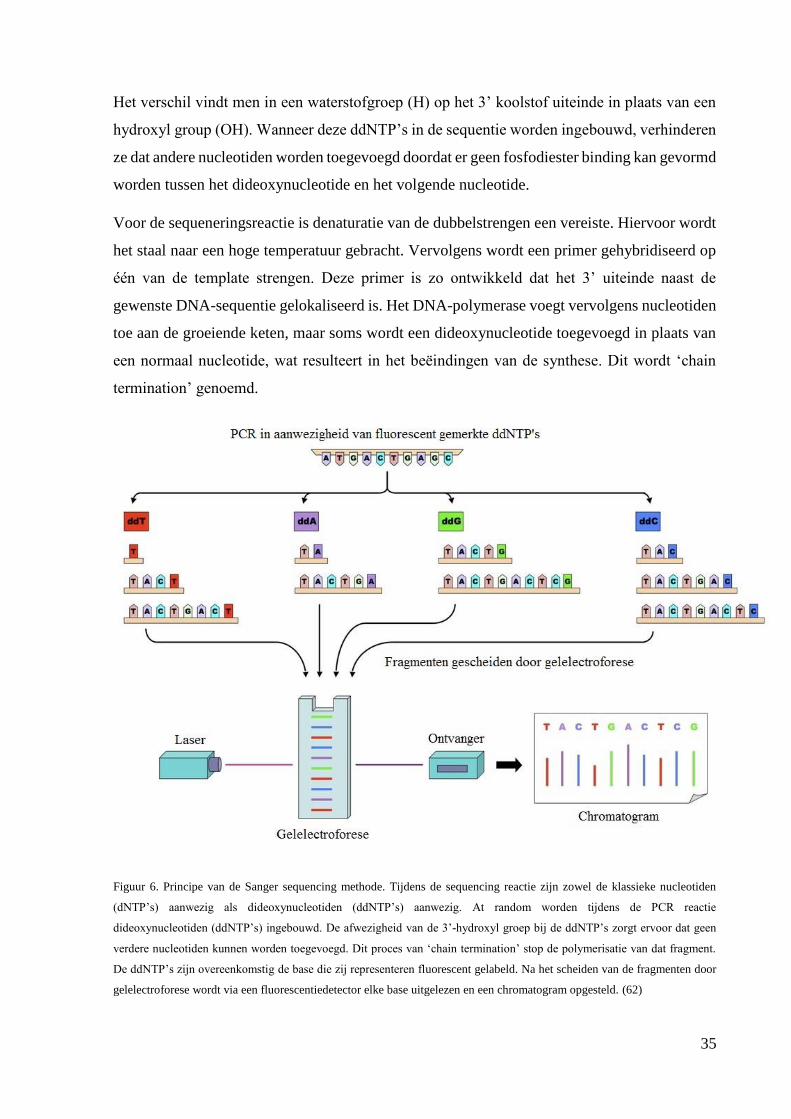
35
Het verschil vindt men in een waterstofgroep (H) op het 3’ koolstof uiteinde in plaats van een
hydroxyl group (OH). Wanneer deze ddNTP’s in de sequentie worden ingebouwd, verhinderen
ze dat andere nucleotiden worden toegevoegd doordat er geen fosfodiester binding kan gevormd
worden tussen het dideoxynucleotide en het volgende nucleotide.
Voor de sequeneringsreactie is denaturatie van de dubbelstrengen een vereiste. Hiervoor wordt
het staal naar een hoge temperatuur gebracht. Vervolgens wordt een primer gehybridiseerd op
één van de template strengen. Deze primer is zo ontwikkeld dat het 3’ uiteinde naast de
gewenste DNA-sequentie gelokaliseerd is. Het DNA-polymerase voegt vervolgens nucleotiden
toe aan de groeiende keten, maar soms wordt een dideoxynucleotide toegevoegd in plaats van
een normaal nucleotide, wat resulteert in het beëindingen van de synthese. Dit wordt ‘chain
termination’ genoemd.
Figuur 6. Principe van de Sanger sequencing methode. Tijdens de sequencing reactie zijn zowel de klassieke nucleotiden
(dNTP’s) aanwezig als dideoxynucleotiden (ddNTP’s) aanwezig. At random worden tijdens de PCR reactie
dideoxynucleotiden (ddNTP’s) ingebouwd. De afwezigheid van de 3’-hydroxyl groep bij de ddNTP’s zorgt ervoor dat geen
verdere nucleotiden kunnen worden toegevoegd. Dit proces van ‘chain termination’ stop de polymerisatie van dat fragment.
De ddNTP’s zijn overeenkomstig de base die zij representeren fluorescent gelabeld. Na het scheiden van de fragmenten door
gelelectroforese wordt via een fluorescentiedetector elke base uitgelezen en een chromatogram opgesteld. (62)

36
Een belangrijk element in deze methode is dat alle reacties starten met hetzelfde nucleotide en
eindigen met een specifieke base. In een oplossing waar dezelfde DNA-streng steeds opnieuw
wordt gesynthetiseerd, zal de nieuwe keten, op alle plaatsen waar het ddNTP kan worden
ingebouwd, beëindigd worden. Er worden dus ketens met verschillende lengtes geproduceerd.
Wanneer de reacties zijn afgerond, wordt het DNA opnieuw gedenatureerd als voorbereiding
op de elektroforese. Na deze run wordt de gel aan een laser blootgesteld. De ddNTP’s zijn
fluorescent gemerkt naargelang de base die zij vertegenwoordigen. Op die manier kan het
eindproduct op een gel worden waargenomen. De uiteindelijk nucleotiden worden afgeleid uit
de frequentie van het signaal vanuit de gemerkte ddNTPs.
Werkwijze
Aan de hand van de resultaten van MiSeq wordt al dan niet overgegaan naar Sanger sequencing.
Van volgende genfragmenten wordt de DNA-sequentie volgens Sanger bepaald:
Een gekende causale mutatie (“CAUSAL MUTATION” in SeqPilot)
Een gekende unclassified variant (“CAUSALITY UNCLEAR” in SeqPilot)
Een variant die nog niet eerder werd gevonden
Een amplicon waarvoor onvoldoende coverage werd behaald
Enkel gekende polymorfismen (voorkomend bij amplicons met voldoende coverage) worden
niet met Sanger DNA-sequenering bevestigd.
Alvorens de Sanger sequencing kan worden gestart, moet voor de geselecteerde genfragmenten
opnieuw PCR en controle met de Labchip GX worden uitgevoerd. Voor genfragmenten met
onvoldoende coverage verloopt dit op dezelfde wijze als hierboven vermeld. Voor causale
mutaties en varianten wordt in plaats van de GenomiPhi verdunning een verdunning van het
originele DNA-staal gebruikt (20ng/µl). De Labchip controle verloopt zoals hierboven
beschreven (zie 3.4.2).
In de eerste stap van Sanger sequencing wordt een mix voorbereid van Exonuclease (Exo),
Antarctic Phosphatase (AP) en water. Deze eerste twee producten moeten op ijs geplaatst
worden. Per staal komen de componenten in volgende hoeveelheden voor:

37
Componenten reactie Volume per staal (µl)
Exonuclease 0,05
Antarctic Phosphatase 0,2
Water 0,75
De mix wordt hierna gevortexed en gecentrifugeerd. Vervolgens wordt 5 µl van de PCR-
producten in een PCR-plaat gepippeteerd. Hieraan wordt 1 µl van bovenstaande mix
toegevoegd. De plaat wordt vervolgens in de Thermal Cycler geplaatst met volgend programma
(Exo(s)AP programma):
Temperatuur Tijd
37°C 15 minuten
80°C 20 minuten
In tweede deel van de Sanger sequencing wordt opnieuw op ijs gewerkt. Twee mixen worden
voorbereid: één met forward primer, één met reverse primer. Per staal worden de componenten
in volgende hoeveelheden toegevoegd:
Componenten reactie Volume per staal (µl)
RR mix 0,5
ABI – Buffer 2
Water 4,5
DMSO 0,5
Primer (forward/reverse) 2
De mixen worden gevortexed en gecentrifugeerd. Vervolgens wordt 9 µl van de forward mix
en 9 µl van de reverse mix in de hiervoor gereserveerde wells van een PCR-plaat gepipetteerd
(elk genfragment dat geselecteerd werd na MiSeq, wordt zowel in forward als in reverse
richting gesequeneerd). Vervolgens wordt 1 µl van het Exo(s)AP-product toegevoegd. De plaat
wordt vervolgens in de Thermal Cycler geplaatst met volgend programma (Sequencing/Abi-
programma):

38
Temperatuur Tijd
95°C 5 minuten
95°C 10 seconden
25 cycli 55°C 5 seconden
60°C 4 minuten
15°C 10 minuten
Het derde deel van de Sanger sequencing heeft als bedoeling het bekomen PCR product te
zuiveren van alle achtergebleven producten en onzuiverheden. Voor deze stap zijn volgende
componenten nodig:
- Vortex de magnetic beads tot een homogene vloeistof is te zien
- Voeg 10 µl magnetic beads toe aan elk epje Sequencing/Abi-product
- Voeg 45 µl ethanol toe en pipetteer op en neer
- Plaats de epjes op een magnetische plaat
- Na 3 minuten: verwijder de ethanol
- Voeg 100 µl ethanol toe
- Plaats opnieuw op de magnetische plaat
- Na 10 seconden: verwijder de ethanol
Laat gedurende 15 minuten de epjes drogen aan de lucht. Op dit punt kunnen de beads, welke
nu het gezuiverde DNA-materiaal bevatten, ingevroren worden in afwachting van de analyse.
Voorafgaan aan de analyse:
- Voeg 10 µl water toe, meng de beads goed door op en neer te pipetteren
- Laat 3 minuten rusten
- Plaats de epjes gedurende 30 seconden op de magnetische plaat
Ten slotte wordt 30 µl van het gezuiverde PCR product in een nieuwe plaat gepipetteerd. De
plaat wordt afgesloten met een seal en zo aan het GSU overgedragen.
Componenten reactie
Magnetic beads
Ethanol 85%
Water

39
Analyse
3.6.3.1 SeqPilot
Analyse van de gesequeneerde DNA-sequenties gebeurt via de SeqPilot software. Deze
software wordt gebruikt om de sequentie te mappen en varianten te decteren. De sequentie van
de patiënt wordt vergeleken met een referentiesequentie uit de Ensembl databank. In het
SeqPilot programma kan men tevens zien of de sequencing niet vroegtijdig is afgebroken. Een
deel van het exon kan bijvoorbeeld niet gesequeneerd zijn of een flankerend intron kan
onvoldoende zijn mee gesequeneerd. Indien dit het geval is, moet het assay overeenkomstig dit
exon opnieuw gesequeneerd worden om varianten te kunnen uitsluiten.
3.6.3.2 Alamut Visual
Alamut Visual is software die de onderzoeker helpt de pathogenische status van de variant,
gevonden werden met MiSeq en bevestigd met Sanger Sequencing en SeqPilot, te bepalen.
Door de graad van conservatie van de sequentie in acht te nemen , door de splice sites te
bepalen, etc. kan de software voorspellen of de variant pathogeen is of niet.
3.7 Exoomanalyse
Bij het klinisch vermoeden van OI wordt een EDTA-bloedstaal afgenomen en verstuurd naar
een genetisch labo. Hier zal men screenen op mutaties in alle gekende genen voor OI. De
huidige screening in het labo van het Centrum voor Medische Genetica Gent (CMGG) bestaat
uit 2 panels zoals hierboven beschreven (zie 3.3.2).
Indien deze screening negatief is, wordt een weefselbiopt aangevraagd en tot Whole Exome
Sequencing (WES) overgegaan. Verscheidene variabelen worden tevens opgesteld die zullen
bijdragen tot het filteren van de data. De lijst van gevonden varianten en hun variabelen
opgesteld door de bio-informaticus wordt manueel doorgenomen. Aan de hand van deze
variabelen en locatie van de varianten wordt een inschatting gemaakt van de kans tot causaliteit.
Variabelen
3.7.1.1 Populatie- en CMGG frequentie
De frequentie van een variant kan reeds belangrijke informatie geven. De kans op causaliteit
bij een variant die in meer dan 2% van de bevolking voorkomt is dan ook klein. Dit is tevens
een gegeven dat wordt gehanteerd bij het classificeren van een variant (Zie Bijlage 5-16).

40
3.7.1.2 Coverage en Readings
De coverage en aantal reads van een variant geven aan hoeveel keer de variant werd herkend
door de sequencingmachine. De foutenmarge van deze apparaten verplicht ons echter tot het
kritisch benaderen van deze waarden. Een lage coverage maakt een vals positief resultaat
waarschijnlijker. Toch zal indien de andere variabelen veelbelovend zijn geprobeerd worden de
variant te bevestigen door opnieuw te sequeneren, ditmaal met Sanger sequencing, met de hoop
op hogere coverage waarden. Op die manier kan men aantonen of de variant daadwerkelijk
aanwezig is en dus een vals positief resultaat uitsluiten.
3.7.1.3 PhyloP Waarde
De PhyloP waarde (Phylogenetische P-waarden) is een maat voor de conservatie van het
aminozuur in de betreffende sequentie. De evolutie van het menselijk genoom heeft door
externe selectiedruk bepaalde genen in meer of mindere mate uitgeselecteerd. Functioneel
essentiële functies zijn dan ook sterk geconserveerd in ons genoom, terwijl andere minder vitale
zaken nog steeds in volle verandering zijn. De PhyloP waarde beschrijft de snelheid waarmee
een sequentie evolueert tegenover een neutraal overervingssysteem. Men spreek van
‘Acceleratie’ als het segment aan meer verandering onderhevig is dan wordt verwacht bij een
ad random overerving. Daartegenover staat een ‘Deceleratie’ welke erop wijst dat het segment
minder verandert dan verwacht wordt.
De PhyloP software maakt gebruik van vier verschillende statistische modellen om haar waarde
te produceren, elk met een vergelijkbare statistische kracht, ondanks hun theoretisch
verschillende benadering. (63)
3.7.1.4 Variant Allele Frequency (Vaf)
Allele frequenctie (af) is de relatieve frequentie van een allel op een specifieke locus in een
populatie, uitgedrukt als een fractie of percentage. Het is de fractie van alle chromosomen in de
populatie die drager zijn van dat allel. Variant allele frequency (Vaf) is dus de fractie allelen
die de variant bevatten tegenover het totaal aantal aanwezige allelen in de populatie. (64)
Hier gaat het echter telkens over één patiënt. Aangezien het menselijk genoom diploïd is, zal
voor elke variant de ‘populatie’ allelen dan ook 2 bedragen. Aangezien gesequeneerd wordt
door dezelfde locus te amplificeren met PCR zal men dus de frequentie van de variant in deze
artificiele populatie berekenen.

41
Uit deze waarde kan men afleiden of de variant homozygoot dan wel heterozygoot aanwezig
is. Waarden dicht bij 50% geven een heterogene variant aan, terwijl waarden die 100% naderen
homozygoot zijn.
Dit is uiteraard essentiële informatie in het zoeken naar causale varianten. In exoom 1 werd
bijvoorbeeld de voorkeur gegeven aan homozygote varianten gezien het overervingspatroon en
de consanguiniteit van de ouders.
3.7.1.5 Reference SNP nummer (rs-nummer)
Het Reference SNP-nummer (rs-nummer) is een nummer dat wordt gebruikt door onderzoekers
en databanken om naar een specifieke SNP te verwijzen. Het nummer staat voor een Reference
SNP cluster ID. (65)
Wanneer onderzoekers een SNP ontdekken, zenden zij een rapport van de SNP samen met de
flankerende sequentie naar de dbSNP database van het NCBI (National Center for
Biotechnology Information). Hier krijgt elk SNP een ss-nummer (submitted SNP). Als een
aantal SNPs op dezelfde locatie van het genoom blijken te liggen, zal het NCBI de
overeenkomende rapporteringen als deel van een groep rapporteren en clusteren in een unieke
reference SNP-cluster. Hieraan wordt dan een uniek rs-nummer toegekend. (65,66)

42
4 Resultaten
4.1 Screening patiënten
Resultatentabel
Legende
- c-notatie: nummering op nucleotide niveau
- p-notatie: nummering op eiwit niveau
- Predictie tools:
o Missense mutaties: SIFT, PolyPhen, Mutation Taster, Align GVGD, Grantham
o Silent, missense en intronische mutaties: splice site predictions (Human Splicing
Finder, Genesplicer, NNSPLICE, MaxEntScan, Splicesite finder like)
o Nonsense mutatie, deleties en inserties: NMD predictie
- NMD: nonsense mediated decay
- Vaf: zie 3.7.1.4
- Kleurencode:
o Groen: benigne variant (klasse 1 of 2)
Klasse 1: Benign
Klasse 2: Likely benign
o Blauw: pathogeniciteit onduidelijk (klasse 3)
Klasse 3: Uncertain significance
o Rood: pathogene variant
Klasse 4: Likely pathogenic
Klasse 5: Pathogenic
o Zwart: Vals positief
Patiënten waarin MiSeq geen varianten aangaf in OI panel 1 of 2 worden in onderstaande tabel
niet vermeld. Hetzelfde geldt voor de genen van OI panel 2 waarin bij geen enkele patiënt een
variant werd gevonden. Voor de volledige lijst van MiSeqresultaten (inclusief assays die te
weinig coverage hadden), zie Bijlage 3 en 4.
43
OI panel 1
Patiënt COL1A1 COL1A2 Exon Vaf
MiSeq Sanger Predictie
tools
Classificatie Commentaar
c-notatie p-notatie c-notatie p-notatie
P1 c.2314G>A p.(Gly772Ser) 38 49,822 CAUSAL MUTATION Bevestigd 5/5 positief Klasse 4:
likely
pathogenic
Glycine in alfa-
helix
P2 c.2867G>T
c.1243C>T
p.(Gly956Val)
p.(Arg415*)
40
19
Unknown variant
CAUSAL MUTATION
Vals
positief
Vals
positief
P4 c.484delC
c.2867G>T
p.(Gln162fs)
p.(Gly956Val)
6
40
44,348 CAUSAL MUTATION
Unknown variant
Bevestigd
Vals
positief
NMD
Voorspeld
Klasse 4:
likely
pathogenic
STOPCODON
P5 c.1283delC
c.1353+8
T>C
p.(Gly428fs)
19
20
Unknown variant
Unknown variant
Vals
positief
Vals
positief
P6 c.370-2A>G 5 61,364 Unknown variant Bevestigd Human Splicing
Finder positief
Klasse 4:
Likely
pathogenic
Verlies van
splice site
P7 c.1012G>A p.(Gly338Ser) 16 46,711 CAUSAL MUTATION Bevestigd 4/5 positief Klasse 4:
Likely
pathogenic
Glycine
verandering
P9 c.2867G>T p.(Gly956Val) 40 Unknown variant Vals
positief
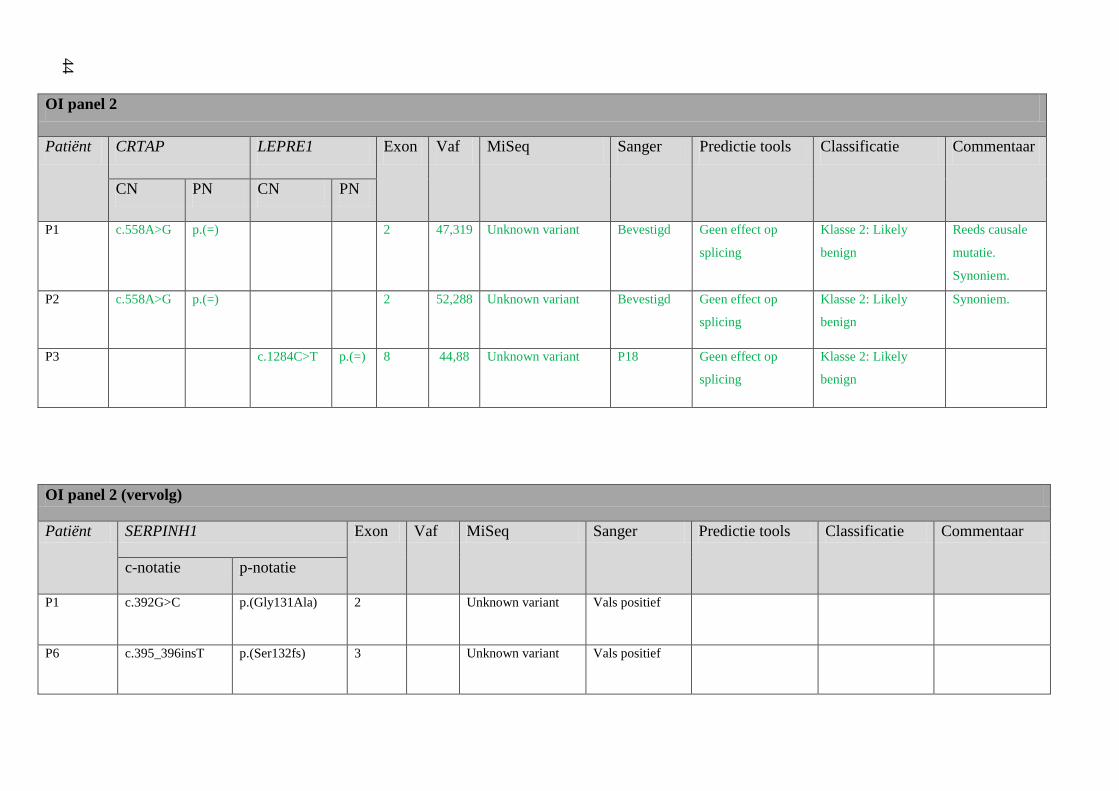
44
OI panel 2
Patiënt CRTAP LEPRE1 Exon Vaf MiSeq Sanger Predictie tools Classificatie Commentaar
CN PN CN PN
P1 c.558A>G p.(=) 2 47,319 Unknown variant Bevestigd Geen effect op
splicing
Klasse 2: Likely
benign
Reeds causale
mutatie.
Synoniem.
P2 c.558A>G p.(=) 2 52,288 Unknown variant Bevestigd Geen effect op
splicing
Klasse 2: Likely
benign
Synoniem.
P3 c.1284C>T p.(=) 8 44,88 Unknown variant P18 Geen effect op
splicing
Klasse 2: Likely
benign
OI panel 2 (vervolg)
Patiënt SERPINH1 Exon Vaf MiSeq Sanger Predictie tools Classificatie Commentaar
c-notatie p-notatie
P1 c.392G>C p.(Gly131Ala) 2 Unknown variant Vals positief
P6 c.395_396insT p.(Ser132fs) 3 Unknown variant Vals positief
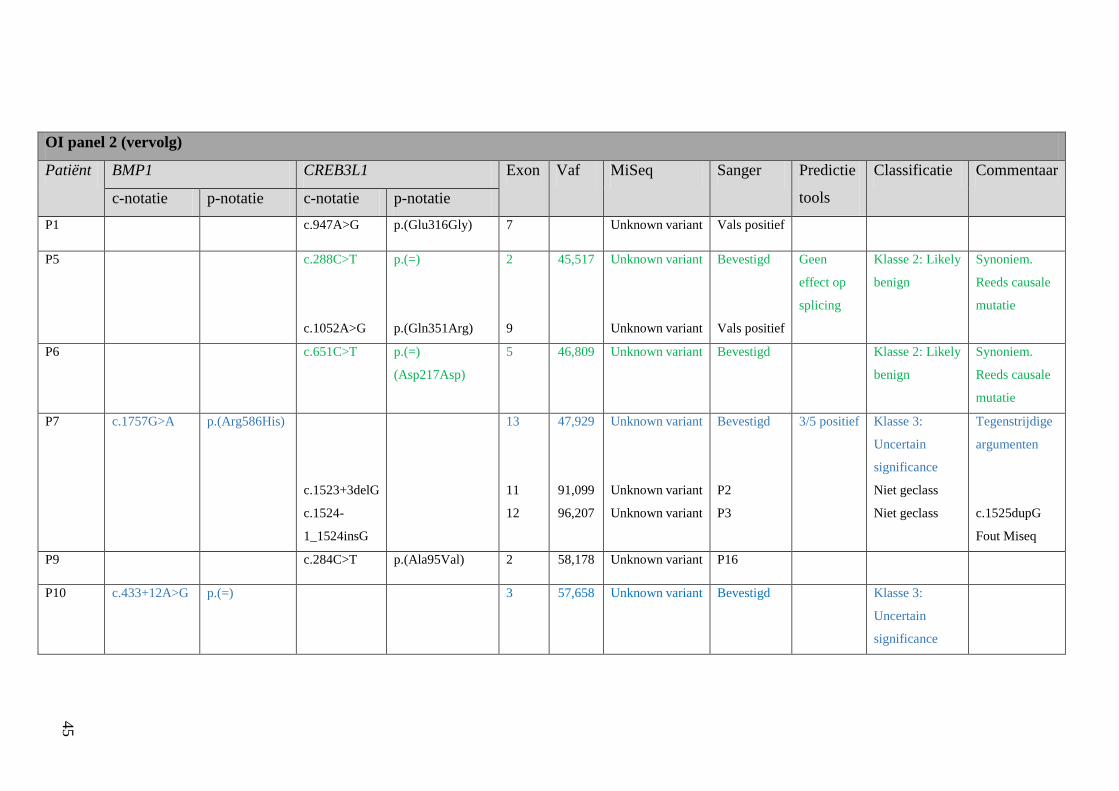
45
OI panel 2 (vervolg)
Patiënt BMP1 CREB3L1 Exon Vaf MiSeq Sanger Predictie
tools
Classificatie Commentaar
c-notatie p-notatie c-notatie p-notatie
P1 c.947A>G p.(Glu316Gly) 7 Unknown variant Vals positief
P5 c.288C>T
c.1052A>G
p.(=)
p.(Gln351Arg)
2
9
45,517 Unknown variant
Unknown variant
Bevestigd
Vals positief
Geen
effect op
splicing
Klasse 2: Likely
benign
Synoniem.
Reeds causale
mutatie
P6 c.651C>T p.(=)
(Asp217Asp)
5 46,809 Unknown variant Bevestigd Klasse 2: Likely
benign
Synoniem.
Reeds causale
mutatie
P7 c.1757G>A
p.(Arg586His)
c.1523+3delG
c.1524-
1_1524insG
13
11
12
47,929
91,099
96,207
Unknown variant
Unknown variant
Unknown variant
Bevestigd
P2
P3
3/5 positief
Klasse 3:
Uncertain
significance
Niet geclass
Niet geclass
Tegenstrijdige
argumenten
c.1525dupG
Fout Miseq
P9 c.284C>T p.(Ala95Val) 2 58,178 Unknown variant P16
P10 c.433+12A>G p.(=) 3 57,658 Unknown variant Bevestigd Klasse 3:
Uncertain
significance
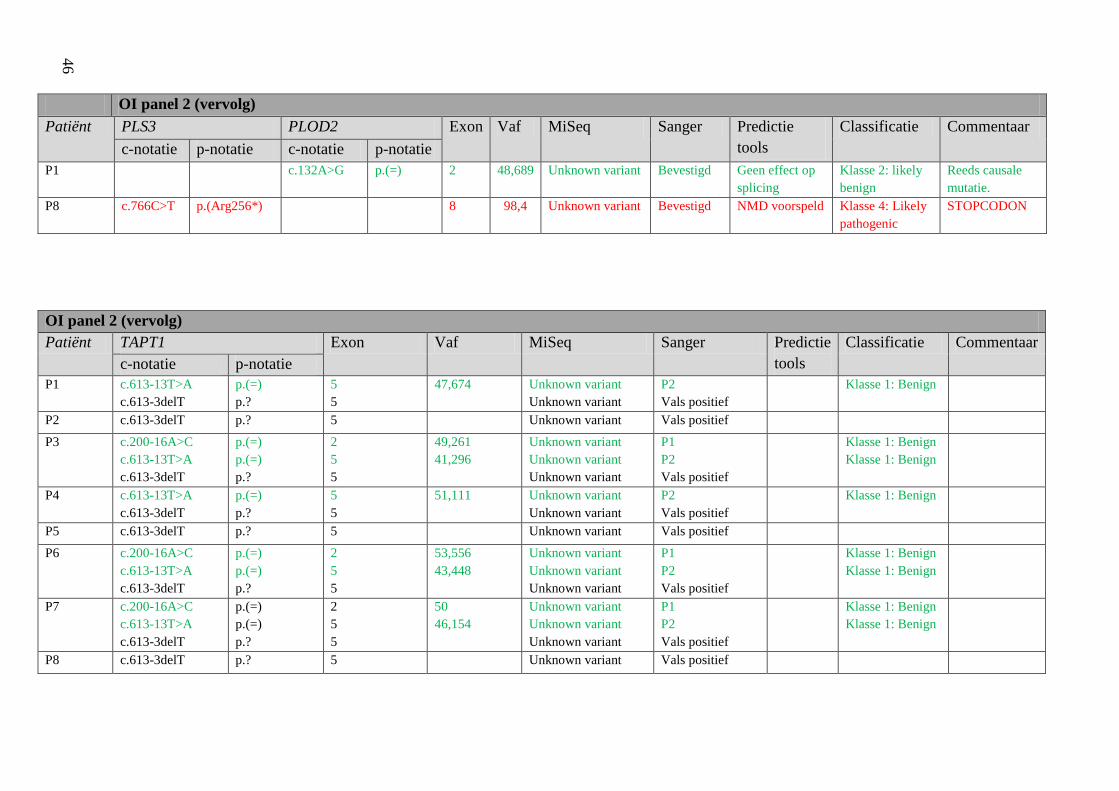
46
OI panel 2 (vervolg)
Patiënt PLS3 PLOD2 Exon Vaf MiSeq Sanger Predictie
tools
Classificatie Commentaar
c-notatie p-notatie c-notatie p-notatie
P1 c.132A>G p.(=) 2 48,689 Unknown variant Bevestigd Geen effect op
splicing
Klasse 2: likely
benign
Reeds causale
mutatie.
P8 c.766C>T p.(Arg256*) 8 98,4 Unknown variant Bevestigd NMD voorspeld Klasse 4: Likely
pathogenic
STOPCODON
OI panel 2 (vervolg)
Patiënt TAPT1 Exon Vaf MiSeq Sanger Predictie
tools
Classificatie Commentaar
c-notatie p-notatie
P1 c.613-13T>A
c.613-3delT
p.(=)
p.?
5
5
47,674 Unknown variant
Unknown variant
P2
Vals positief
Klasse 1: Benign
P2 c.613-3delT p.? 5 Unknown variant Vals positief
P3 c.200-16A>C
c.613-13T>A
c.613-3delT
p.(=)
p.(=)
p.?
2
5
5
49,261
41,296
Unknown variant
Unknown variant
Unknown variant
P1
P2
Vals positief
Klasse 1: Benign
Klasse 1: Benign
P4 c.613-13T>A
c.613-3delT
p.(=)
p.?
5
5
51,111 Unknown variant
Unknown variant
P2
Vals positief
Klasse 1: Benign
P5 c.613-3delT p.? 5 Unknown variant Vals positief
P6 c.200-16A>C
c.613-13T>A
c.613-3delT
p.(=)
p.(=)
p.?
2
5
5
53,556
43,448
Unknown variant
Unknown variant
Unknown variant
P1
P2
Vals positief
Klasse 1: Benign
Klasse 1: Benign
P7 c.200-16A>C
c.613-13T>A
c.613-3delT
p.(=)
p.(=)
p.?
2
5
5
50
46,154
Unknown variant
Unknown variant
Unknown variant
P1
P2
Vals positief
Klasse 1: Benign
Klasse 1: Benign
P8 c.613-3delT p.? 5 Unknown variant Vals positief
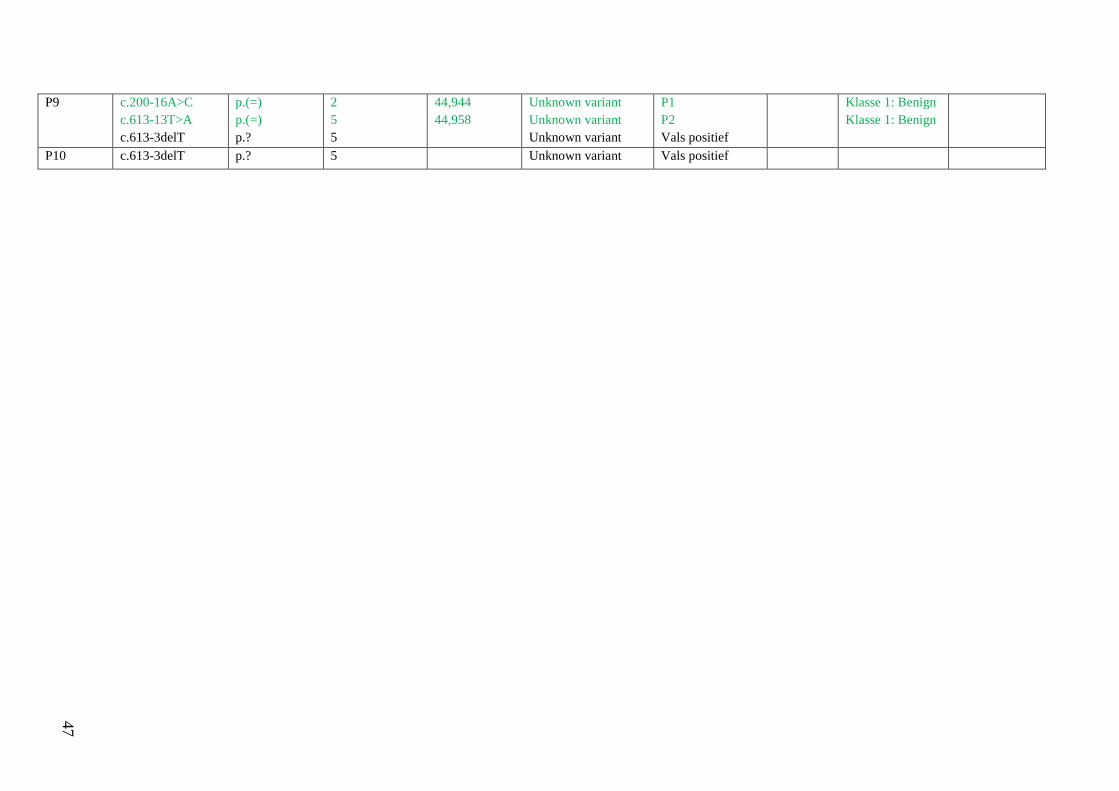
47
P9 c.200-16A>C
c.613-13T>A
c.613-3delT
p.(=)
p.(=)
p.?
2
5
5
44,944
44,958
Unknown variant
Unknown variant
Unknown variant
P1
P2
Vals positief
Klasse 1: Benign
Klasse 1: Benign
P10 c.613-3delT p.? 5 Unknown variant Vals positief

48
Bespreking resultaten
Na screening van 10 patiënten gekend met OI fenotype, werd slechts bij 5 patiënten causale
mutaties (klasse 4 of 5) gevonden in de gekende OI genen, d.w.z. de genen opgenomen in OI
panels 1 en 2.
Voor P1 vonden we een heterozygote mutatie in COL1A2, namelijk c.2314G>A wat aanleiding
gaf tot een eiwitverandering p.(Gly772Ser). Het ging hier om een glycine in de α-helix van type
I collageen, wat een sterk argument is voor pathogeniciteit. De predictie tools voorspelden
bovendien een negatief effect. Deze mutatie werd geclassificeerd als klasse 4. Andere varianten
die bevestigd werden in P1, namelijk in c.558A>G in CRTAP en c.132A>G in PLOD2, waren
heterozygote synonieme mutaties. Aan beide varianten werd klasse 2 toegekend. Aangezien
mutaties in CRTAP en PLOD2 autosomaal recessieve OI veroorzaken, is het onwaarschijnlijk
dat heterozygote mutaties aanleiding geven tot het ziektebeeld (tenzij een 2de mutatie gevonden
wordt in deze genen (compound heterozygoot), maar dat was hier niet het geval). Ook geven
synonieme mutaties zelden aanleiding tot een verandering in het fenotype. Synonieme mutaties
kunnen wel eventueel een effect hebben op de splice site, maar de predictie tools voorspelden
geen splice site verandering. In combinatie met het feit dat voor deze patiënt reeds een causale
mutatie is geïdentificeerd in COL1A2 kunnen we concluderen dat deze varianten het ziektebeeld
niet veroorzaken bij deze patiënt. Ten slotte vonden we ook een polymorfisme in TAPT1, c.613-
13T>A (P2).
Bij P2 werd slecht één variant gevonden, namelijk c.558A>G in CRTAP. Dit is dezelfde
heterozygote synonieme variant als werd vastgesteld in P1. Dezelfde argumenten als
aangehaald bij P1 gelden hier: een heterozygote synonieme variant in CRTAP zonder effect op
de splice site is hoogstwaarschijnlijk niet pathogeen. Ook het feit dat deze variant in
verschillende patiënten werd teruggevonden is suggestief voor niet-causaliteit. Voor deze
patiënt werd dus geen causale mutatie geïdentificeerd binnen de gekende genen voor OI.
Bij P3 werden variant gevonden in de genen van OI panel 1 en 2. Het ging om een polymorfisme
in LEPRE1 (P18) en 2 polymorfismen in TAPT1, c.200-16A>C (P1) en P2 (zie P1).
Bij P4 werd een heterozygote mutatie in COL1A1 vastgesteld, namelijk c.484delC hetgeen
aanleiding gaf tot de eiwitverandering p.(Gln162fs). Deze frameshift gaf geen aanleiding tot
een glycineverandering in de α-helix, maar leidde wel tot nonsense mediated decay van het
mRNA door de introductie van een stopcodon. Dit is een sterk argument voor pathogeniciteit.

49
Aan deze mutatie werd een klasse 4 toegekend. Deze mutatie is waarschijnlijk de oorzaak van
OI bij deze patiënt. Verder werd ook een polymorfisme in TAPT1 vastgesteld, namelijk P2.
Bij P5 werd één variant vastgesteld, namelijk c.228C>T in CREB3L1. Dit is een heterozygote
synonieme variant waaraan klasse 2 werd toegekend. Aangezien mutaties in CREB3L1
aanleiding geven tot autosomaal recessieve OI, is het onwaarschijnlijk dat deze variant
aanleiding geeft tot het ziektebeeld. Bovendien gaat het om een synonieme variant zonder effect
op de splice site. We vermelden dat bij deze patiënt dat voor het gen FKBP10 één assay niet
gesequeneerd kon worden. Een deel van dit gen kon dus niet onderzocht worden en het is
mogelijk dat een mutatie zich in dit gen bevindt. Voor deze patiënt wordt best een nieuw DNA-
staal aangevraagd.
Bij P6 werd een causale mutatie in COL1A1 geïdentificeerd: c.370-2A>G. Dit leidt tot het
verloren gaan van de splice-acceptor site, waardoor een overslaan van exon 5 zeer
waarschijnlijk is. Aan deze variant werd een klasse 4 toegekend. In CREB3L1 werd een
heterozygote, synonieme variant vastgesteld zonder effect op splicing, namelijk c.651C>T
(p.(=)). Aan deze variant werd klasse 2 toegekend. In TAPT1 werden 2 polymorfismen
gevonden, P1 en P2.
Bij P7 werd eveneens een causale mutatie gevonden in COL1A1: c.1012G>A. Op eiwitniveau
gaat hierdoor een glycine verloren in de α-helix regio: p.(Gly338Ser). Aan deze variant werd
klasse 4 toegekend. Verder werd ook heterozygote variant gevonden in BMP1, namelijk
c.1757G>A (p.(Arg586His). De predictie tools voorspelden een negatief effect (3/5). Aan deze
variant werd een klasse 3 toegekend. Aangezien mutaties in BMP1 aanleiding geven tot
autosomaal recessieve OI, is het echter onwaarschijnlijk dat deze variant aanleiding geeft tot
het ziektebeeld. Bovendien werd bij deze patiënt reeds een causale mutatie gevonden in
COL1A1. In CREB3L1 werden tevens 2 varianten beschreven voor deze patiënt: c.1524-
1_1524insG en c.1523+3delG. Voor de eerste variant is echter geweten dat het MiSeq apparaat
deze variant keer op keer verkeerd interpreteert. In werkelijkheid gaat het hier over
c.1525dupG. Deze variant wordt sinds 2013 als polymorfisme gerapporteerd. De variant
c.1523+3delG werd niet geclassificeerd. Ten slotte vermelden we ook 2 polymorfismen in
TAPT1, P1 en P2.
Bij P8 werd een onbekende variant bevestigd in PLS3, welke aanleiding gaf tot een prematuur
stopcodon: c.766C>T , p.(Arg256*). Aangezien het hier gaat om een nonsense-mutatie is de
kans op NMRD groot. De variant werd dan ook geclassificeerd als klasse 4.

50
Voor P9 werden 3 polymorfismen teruggevonden, één in CREB3L1 (P16) en 2 in TAPT1 (P1
en P2).
Bij P10 werd een onbekende variant bevestigd in BMP1: c.433+12A>G. Aangezien aan geen
van de criteria voor classificatie werd voldaan wordt deze dan ook gerapporteerd als een variant
met klasse 3: Uncertain significance. Aangezien de substitutie zich 12 basenparen ver in het
intron bevindt, is de kans op pathogeniteit echter klein.
4.2 Exoomanalyse
De resultaten van de exoomsequencing worden weergegeven in een excelbestand (zie Bijlage
18) dat al snel meer dan 10 000 varianten bevat. Om in dit groot aantal varianten genen te
selecteren die interessant zijn voor OI, werd er gebruik gemaakt van filters. Het excelbestand
bevat ook verschillende tabbladen waarin varianten worden ingedeeld aan de hand van het type
mutatie (nonsense, frameshift, deleties/inserties en splice variants) wat de selectie ook
gemakkelijker maakt.
Analyse van het exoom gebeurt door eerst de nonsense mutaties (stop) te bekijken. Een
nonsense mutatie is een genetische mutatie in de DNA sequentie die resulteert in een stopcodon,
waardoor een korter, onvolledige eiwitproduct wordt geproduceerd. Dit eiwit is meestal niet
functioneel. Vaak is het mRNA na transcriptie van deze DNA sequentie het doelwit van
nonsense mediated decay, waardoor slechts de helft van de normale hoeveelheid van het eiwit
aanwezig is. Dit vormt bij autosomaal recessieve aandoeningen een probleem indien het om
een homozygote mutatie of een compound heterozygote mutatie gaat. (67)
Vervolgens werden de frameshift mutaties (fs) geanalyseerd. Een frameshift mutatie is een
mutatie die veroorzaakt wordt door een deletie of insertie waardoor het aflezen van de sequentie
verschuift. De sequentie wordt verstoord door de deletie of insertie van 1 of meer nucleotiden,
op voorwaarden dat het aantal nucleotiden geen veelvoud van 3 is. Indien het wel gaat om een
veelvoud van 3, veroorzaakt dit geen verschuiving in het leesraam.(68)
Deleties/inserties (indel_ea) worden als derde geanalyseerd. Dit betekent één of meer
nucleotiden verloren gaan (deletie) of worden toegevoegd (insertie). Het kan zelfs gaan tot
volledige gendeletie of genduplicatie. Indien het gaat om een deletie of insertie van 3
nucleotiden of een veelvoud hiervan, blijft het leesraam bewaard en is er dus geen sprake van
een frameshift mutatie.(69)

51
Splice site mutaties (splice) worden hierna onderzocht. Dit zijn mutaties in splice donor of
acceptor sites die interfereren met RNA splicing.(69)
Missense mutaties (miss) worden als laatste onderzocht. Een puntmutatie resulteert hier in een
aminozuursubstitutie. Aangezien hier slechts 1 aminozuur wordt vervangen, leidt dit minder
vaak tot pathogeniciteit als de ander mutaties. (69)(70)
Exoom 1: P11
4.2.1.1 Nonsense mutatie
De volgende filters werden toegepast op de resultaten:
- VAF > 50 (homozygoot)
- rs-nummer : In een eerste selectie zullen we dus enkel kijken naar varianten zonder rs-
nummer.
- Coverage > 30
- PhyloP > 3
Nadat deze filters werden ingeschakeld, werd 1 variant geselecteerd: SMYD1. Dit bleek geen
goede kandidaat voor OI. Wanneer de PhyloP-filter wordt aangepast zodat ook varianten met
onbekende PhyloP worden geïncludeerd, worden 5 extra varianten bekomen in 2 verschillende
genen (CDC27 en ZNF595). Wanneer het excelbestand meerdere varianten aangeeft in
hetzelfde gen, gaat het vaak om dezelfde variant die in verschillende transcripten wordt
teruggevonden. Ook van deze varianten werden geen weerhouden (dit op basis van de
literatuur). Hierbij werd gebruik gemaakt van de databanken OMIM en GeneCards. Na
raadplegen van deze databanken werd bijvoorbeeld duidelijk dat SMYD1 voornamelijk een rol
speelt in de ontwikkeling van het hart waardoor we dit gen konden uitsluiten.
Vervolgens werd de filter voor coverage aangepast zodat ook varianten met coverage < 30
werden weerhouden. Ook heterozygote varianten (vaf < 50) werden nu geïncludeerd. Vijf extra
varianten in 4 verschilledende genen (FUOM, CTBP2, GRIN3B en TAS2R45) werden bekomen.
CTBP2 leek aanvankelijk een goede kandidaat vanwege zijn rol in de WNT-signaling, maar
werd uiteindelijk toch niet weerhouden door het hoog aantal varianten dat werd teruggevonden
in dit gen in de drie exomen. Van de andere varianten werden geen weerhouden op basis van
de literatuur. Uiteindelijk werden ook de rs-nummers onderzocht. Dit leverde 27 extra varianten
op in 17 verschillende genen, waarvan geen enkele weerhouden werd.

52
4.2.1.2 Frameshift mutatie
Na het toepassen van de filters zoals bij nonsense mutaties werden geen varianten geselecteerd.
De filter voor PhyloP werd vervolgens aangepast zodat ook varianten met onbekende PhyloP
werden geïncludeerd. Er werden 9 varianten in 2 verschillende genen (ERCC2 en GIGYF2)
door middel van deze filters geselecteerd. Geen enkel variant werd weerhouden als kandidaat
voor OI na het raadplegen van de databanken.
Na inclusie van de heterozygote varianten werden 17 extra varianten in 12 genen geselecteerd.
Al deze varianten werden verworpen als kandidaat voor OI.
Indien de filter voor coverage werd aangepast zodat ook varianten met coverage < 30 werden
geïncludeerd, werden 23 extra varianten in 13 genen bekomen. Van de overige genen werden
de 2 varianten in het RYK gen weerhouden.
4.2.1.2.1 RYK
Gen Vaf Cov3 FR4 RR5 cNomen pNomen Transcript Frequentie
RYK 100 3 1 2 c.59_60insC p.Ala20fs NP_002949 Populatiefreq: /
CMGG freq: 0
GONL freq: 0
RYK 100 3 1 2 c.59_60insC p.Ala20fs NP_001005861
RYK (Receptor-like tyrosine kinase) is lid van de familie van groeifactor receptor proteïne
tyrosine kinasen. Het is een atypisch lid van de familie. Het verschilt van de andere leden door
een aantal verschillen in de geconserveerde residu’s in de activatiedomeinen en domeinen die
nucleotiden binden (71–73). Het eiwit dat door RYK wordt gecodeerd zou een rol spelen bij het
stimuleren van WNT signaling pathways. (71,72)
Uit onderzoek bleek dat RYK bij zoogdieren de rol vervult van coreceptor (samen met Frizzled)
van WNT liganden.
3 Coverage 4 Forward read 5 Reverse read

53
De WNT liganden zijn extracellulaire eiwitten die andere cellen stimuleren door middel van
specifieke receptoren zoals leden van de Frizzled familie en de coreceptor LRP5/6. Deze 3
elementen (LRP5/6, Frizzled en de WNT ligenden) vormen een receptor-ligand complex. (74)
Het extracellulaire domein van RYK vormt een ternair complex met Frizzled en WNT-1. Het
intracellulaire domein bindt met Dishevelled, hetgeen vereist is voor activatie van de WNT
pathway. (72,74) Stroomafwaarts inhibeert WNT signaling de kinase activiteit van GSK3β
zodat β-catenine accumuleert in het cytoplasma en getransloceerd wordt naar de nucleus.
Nucleaire β-catenine bindt leden van de LEF/TCF familie. Deze transcriptiefactoren activeren
de WNT target genen. (74)
Aangezien er reeds werd aangetoond dat mutaties in WNT-1 aanleiding kunnen geven tot OI, is
het plausibel dat mutaties in het RYK-gen ook tot OI kunnen leiden. RYK knock-out muizen
sterven vlak na de geboorte en vertonen afwijkingen zoals een volledige splijting van het
secundaire gehemelte en karakteristieke craniofaciale abnormaliteiten. (73)
Figuur 7. RYK, een atypisch tyrosine kinase dat een rol speelt in de WNT-signaling. Het is een WNT co-receptor
waarvan het extracellulair deel bindt met Frizzled en WNT-1 en het intracellulair deel met Dishevelled. Deze link
met Dishevelled heeft de inhibitie van GSK-3β tot gevolg, hetgeen leidt tot de opstapeling van β-catenine in het
cytoplasma en de translocatie van hiervan naar de nucleus. Door de daaropvolgende binding van β-catenine op
LEF/TCF activeren deze transcriptiefactoren de WNT target genen met o.a. uitgroei van neurieten tot gevolg (74)

54
4.2.1.3 Deleties/inserties
Indien de filters zoals bij de nonsense mutaties werden toegepast, werden geen varianten
geselecteerd. Na aanpassing van PhyloP zodat ook varianten met onbekende PhyloP
geïncludeerd worden, werden 27 varianten in 22 genen bekomen. Deze varianten bleken geen
goede kandidaten voor OI na het raadplegen van de databanken.
Vervolgens werden ook de heterozygote varianten (vaf < 50) onderzocht. Er werden op deze
manier 61 extra varianten in 38 genen geselecteerd, waarvan we geen weerhouden. Ten slotte
werden ook de rs-nummers bekeken. De 34 varianten in 12 genen die er op deze wijze
bijkwamen, werden verworpen.
4.2.1.4 Splice variants
Bij het toepassen van de filters zoals bij de nonsense mutaties, werd 1 variant bekomen in het
gen TBX22. Dit bleek geen goede kandidaat voor OI. Na aanpassing van PhyloP zodat ook
varianten met onbekende PhyloP geïncludeerd werden, bekwamen we 22 extra varianten in 12
genen, waarvan we geen weerhouden. Het analyseren van de heterozygote varianten (vaf < 50)
leverde 27 extra varianten op in 11 genen.
Van deze varianten werd CUX1 overwogen vanwege zijn rol als inhibitor van de collageen type
1, (75) maar werd uiteindelijk toch verworpen vanwege de diep intronische ligging van de
varianten (met dus minder waarschijnlijk een effect op de splice site). COG3, dat een
component is van het oligomere Golgi complex (76), werd eveneens verworpen vanwege de
diep intronische ligging van de variant. Van de overige varianten werden geen weerhouden.
Vervolgens werden ook varianten met een coverage < 30 geïncludeerd. Dit leverde 12 nieuwe
varianten op in 7 genen. P4HA1 werd weerhouden vanwege argumenten in de literatuur.
Wanneer we in alle varianten op zoek gaan naar andere varianten in P4HA1, bekomen we 5
varianten. De overige varianten met coverage < 30 bleken geen goede kandidaten voor OI.
Wanneer ook de rs-nummers bekeken worden, werden 33 extra varianten in 18 genen
geselecteerd. Geen van deze werden weerhouden.

55
4.2.1.4.1 P4HA1
Gen Vaf Cov FR RR CN PN Transcript Frequentie
P4HA1 100 4 1 4 c.-125-1G>A p.? NP_001136067 Populatiefreq: /
CMGG freq: 0
GONL freq: 0
P4HA1 100 4 1 4 c.-32-2899G>A p.? NP_001017962
P4HA1 100 4 1 4 c.-32-2899G>A p.? NP_001136068
P4HA1 100 4 1 4 c.-32-2899G>A p.? NP_000908
P4HA1 100 4 1 4 / / /
Van deze 5 transcripten werd enkel het eerste weerhouden vanwege zijn locatie ter hoogte van
de splice site acceptor. Posities -2, -1, +1 en +2 zijn namelijk het belangrijkst voor splicing.
Mutaties op deze plaats kunnen resulteren in de inclusie van intronen die normaal door splicing
uit het RNA verdwijnen. (77) P4HA1 codeert voor een component van prolyl 4-hydroxylase.
Dit enzyme speelt, zoals eerder vermeld (zie 2.1.4.2.1), een belangrijke rol in de collageen
synthese door katalysatie van de formatie van 4-hydroxyproline. Het eiwit waarvoor P4HA1
codeert is één van verschillende types van alpha subunits van het enzym. Dit alpha subunit
maakt deel uit van de katalytische site van het enzyme. (78,79)
4.2.1.5 Missense mutaties
Na het toepassen van de filters zoals bij nonsense mutaties werden 46 varianten gevonden. Na
het toepassen van 3 nieuwe filters om het aantal varianten te verminderen, namelijk de filters
voor predictie tools SIFT, PolyPhen en Mapp die op respectievelijk op ‘deleterious’, ‘probably
damaging/possibly damaging’ en ‘bad’ werden gezet, werden 4 varianten in 3 genen (ELK4,
NFKBID en TBX22) geselecteerd. Hiervan werd geen enkele variant weerhouden.
Vervolgens werden ook varianten met een onbekende uitkomst bij de prediction tools
geïncludeerd. Er werden 17 extra varianten in 4 genen (SIPA1L1, VPS33B, TTN, OPA1 en
ADD1) bekomen, waarvan we geen weerhouden. Na het uitschakelen van de predictie tool
filters, werden 25 extra varianten uit 10 genen bekomen. Uit deze varianten werden na
literatuurstudie 2 genen geselecteerd die goede kandidaten leken voor OI: AKT2 en TRAPPC12.
Ook de heterozygote varianten werden onderzocht. Na het inschakelen van de predictie tool
filters en de filters coverage > 30, PhyloP > 3 en rs-nummer = /, werden 15 extra varianten in
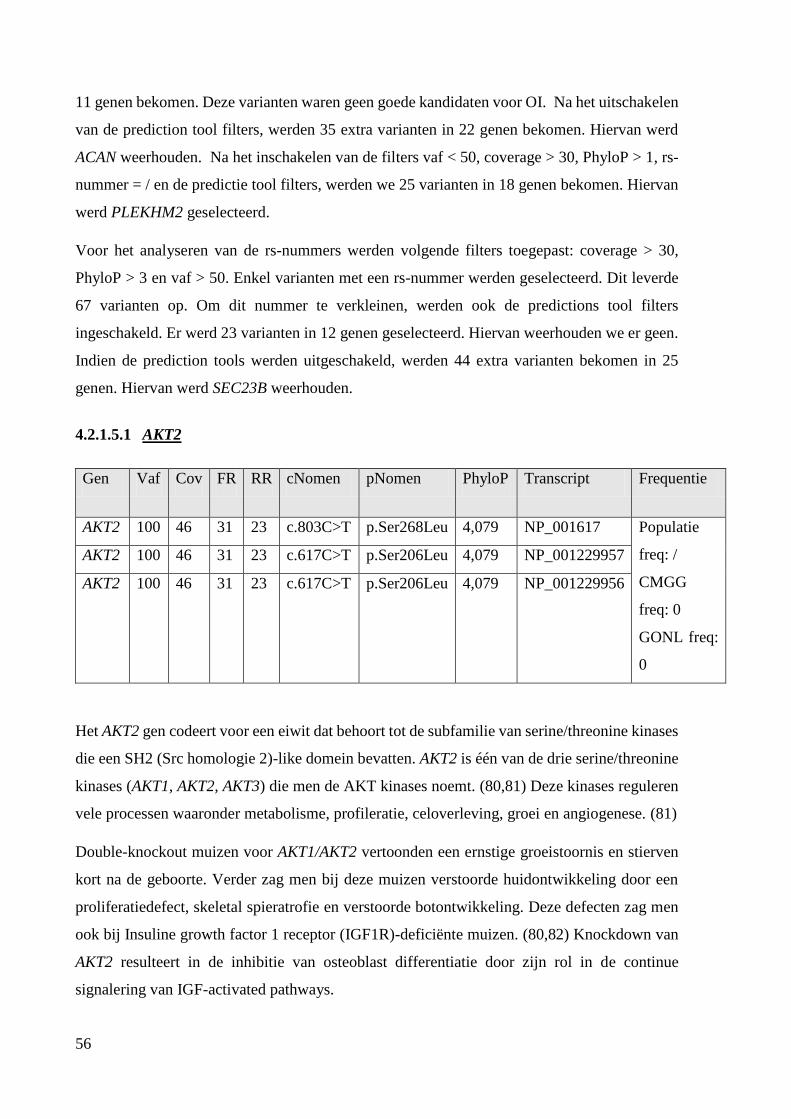
56
11 genen bekomen. Deze varianten waren geen goede kandidaten voor OI. Na het uitschakelen
van de prediction tool filters, werden 35 extra varianten in 22 genen bekomen. Hiervan werd
ACAN weerhouden. Na het inschakelen van de filters vaf < 50, coverage > 30, PhyloP > 1, rs-
nummer = / en de predictie tool filters, werden we 25 varianten in 18 genen bekomen. Hiervan
werd PLEKHM2 geselecteerd.
Voor het analyseren van de rs-nummers werden volgende filters toegepast: coverage > 30,
PhyloP > 3 en vaf > 50. Enkel varianten met een rs-nummer werden geselecteerd. Dit leverde
67 varianten op. Om dit nummer te verkleinen, werden ook de predictions tool filters
ingeschakeld. Er werd 23 varianten in 12 genen geselecteerd. Hiervan weerhouden we er geen.
Indien de prediction tools werden uitgeschakeld, werden 44 extra varianten bekomen in 25
genen. Hiervan werd SEC23B weerhouden.
4.2.1.5.1 AKT2
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
AKT2 100 46 31 23 c.803C>T p.Ser268Leu 4,079 NP_001617 Populatie
freq: /
CMGG
freq: 0
GONL freq:
0
AKT2 100 46 31 23 c.617C>T p.Ser206Leu 4,079 NP_001229957
AKT2 100 46 31 23 c.617C>T p.Ser206Leu 4,079 NP_001229956
Het AKT2 gen codeert voor een eiwit dat behoort tot de subfamilie van serine/threonine kinases
die een SH2 (Src homologie 2)-like domein bevatten. AKT2 is één van de drie serine/threonine
kinases (AKT1, AKT2, AKT3) die men de AKT kinases noemt. (80,81) Deze kinases reguleren
vele processen waaronder metabolisme, profileratie, celoverleving, groei en angiogenese. (81)
Double-knockout muizen voor AKT1/AKT2 vertoonden een ernstige groeistoornis en stierven
kort na de geboorte. Verder zag men bij deze muizen verstoorde huidontwikkeling door een
proliferatiedefect, skeletal spieratrofie en verstoorde botontwikkeling. Deze defecten zag men
ook bij Insuline growth factor 1 receptor (IGF1R)-deficiënte muizen. (80,82) Knockdown van
AKT2 resulteert in de inhibitie van osteoblast differentiatie door zijn rol in de continue
signalering van IGF-activated pathways.
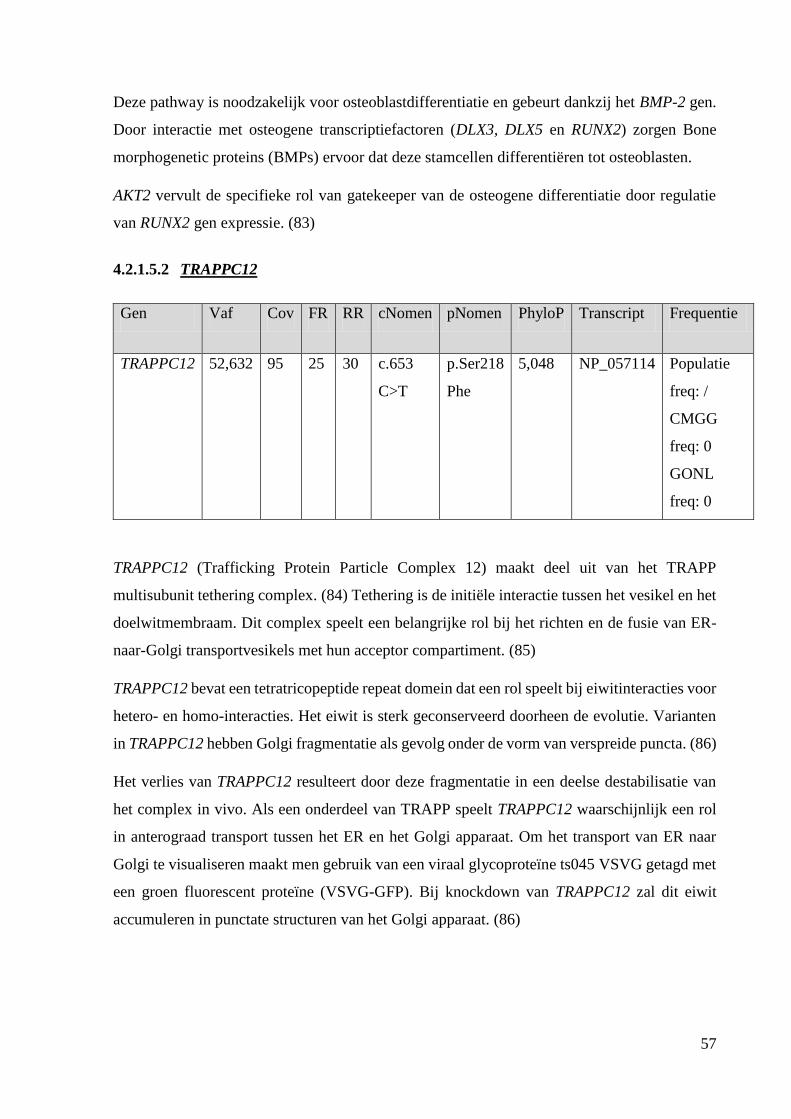
57
Deze pathway is noodzakelijk voor osteoblastdifferentiatie en gebeurt dankzij het BMP-2 gen.
Door interactie met osteogene transcriptiefactoren (DLX3, DLX5 en RUNX2) zorgen Bone
morphogenetic proteins (BMPs) ervoor dat deze stamcellen differentiëren tot osteoblasten.
AKT2 vervult de specifieke rol van gatekeeper van de osteogene differentiatie door regulatie
van RUNX2 gen expressie. (83)
4.2.1.5.2 TRAPPC12
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
TRAPPC12 52,632 95 25 30 c.653
C>T
p.Ser218
Phe
5,048 NP_057114 Populatie
freq: /
CMGG
freq: 0
GONL
freq: 0
TRAPPC12 (Trafficking Protein Particle Complex 12) maakt deel uit van het TRAPP
multisubunit tethering complex. (84) Tethering is de initiële interactie tussen het vesikel en het
doelwitmembraam. Dit complex speelt een belangrijke rol bij het richten en de fusie van ER-
naar-Golgi transportvesikels met hun acceptor compartiment. (85)
TRAPPC12 bevat een tetratricopeptide repeat domein dat een rol speelt bij eiwitinteracties voor
hetero- en homo-interacties. Het eiwit is sterk geconserveerd doorheen de evolutie. Varianten
in TRAPPC12 hebben Golgi fragmentatie als gevolg onder de vorm van verspreide puncta. (86)
Het verlies van TRAPPC12 resulteert door deze fragmentatie in een deelse destabilisatie van
het complex in vivo. Als een onderdeel van TRAPP speelt TRAPPC12 waarschijnlijk een rol
in anterograad transport tussen het ER en het Golgi apparaat. Om het transport van ER naar
Golgi te visualiseren maakt men gebruik van een viraal glycoproteïne ts045 VSVG getagd met
een groen fluorescent proteïne (VSVG-GFP). Bij knockdown van TRAPPC12 zal dit eiwit
accumuleren in punctate structuren van het Golgi apparaat. (86)
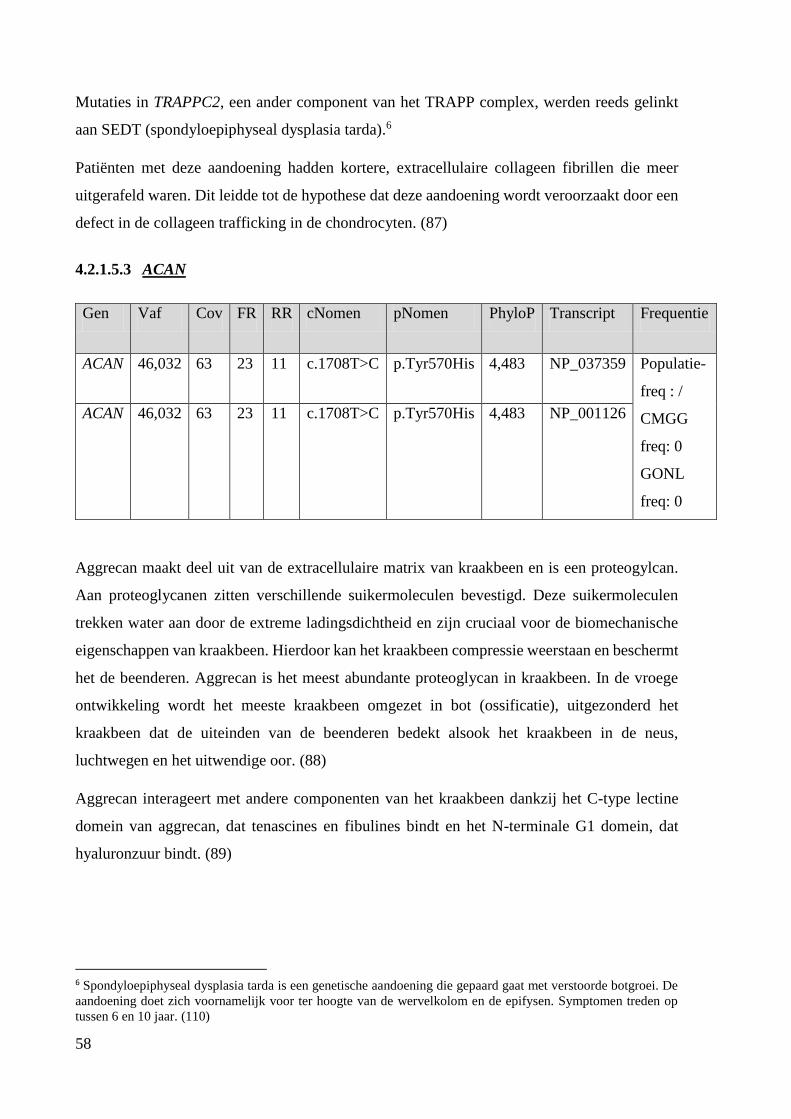
58
Mutaties in TRAPPC2, een ander component van het TRAPP complex, werden reeds gelinkt
aan SEDT (spondyloepiphyseal dysplasia tarda).6
Patiënten met deze aandoening hadden kortere, extracellulaire collageen fibrillen die meer
uitgerafeld waren. Dit leidde tot de hypothese dat deze aandoening wordt veroorzaakt door een
defect in de collageen trafficking in de chondrocyten. (87)
4.2.1.5.3 ACAN
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
ACAN 46,032 63 23 11 c.1708T>C p.Tyr570His 4,483 NP_037359 Populatie-
freq : /
CMGG
freq: 0
GONL
freq: 0
ACAN 46,032 63 23 11 c.1708T>C p.Tyr570His 4,483 NP_001126
Aggrecan maakt deel uit van de extracellulaire matrix van kraakbeen en is een proteogylcan.
Aan proteoglycanen zitten verschillende suikermoleculen bevestigd. Deze suikermoleculen
trekken water aan door de extreme ladingsdichtheid en zijn cruciaal voor de biomechanische
eigenschappen van kraakbeen. Hierdoor kan het kraakbeen compressie weerstaan en beschermt
het de beenderen. Aggrecan is het meest abundante proteoglycan in kraakbeen. In de vroege
ontwikkeling wordt het meeste kraakbeen omgezet in bot (ossificatie), uitgezonderd het
kraakbeen dat de uiteinden van de beenderen bedekt alsook het kraakbeen in de neus,
luchtwegen en het uitwendige oor. (88)
Aggrecan interageert met andere componenten van het kraakbeen dankzij het C-type lectine
domein van aggrecan, dat tenascines en fibulines bindt en het N-terminale G1 domein, dat
hyaluronzuur bindt. (89)
6 Spondyloepiphyseal dysplasia tarda is een genetische aandoening die gepaard gaat met verstoorde botgroei. De
aandoening doet zich voornamelijk voor ter hoogte van de wervelkolom en de epifysen. Symptomen treden op
tussen 6 en 10 jaar. (110)
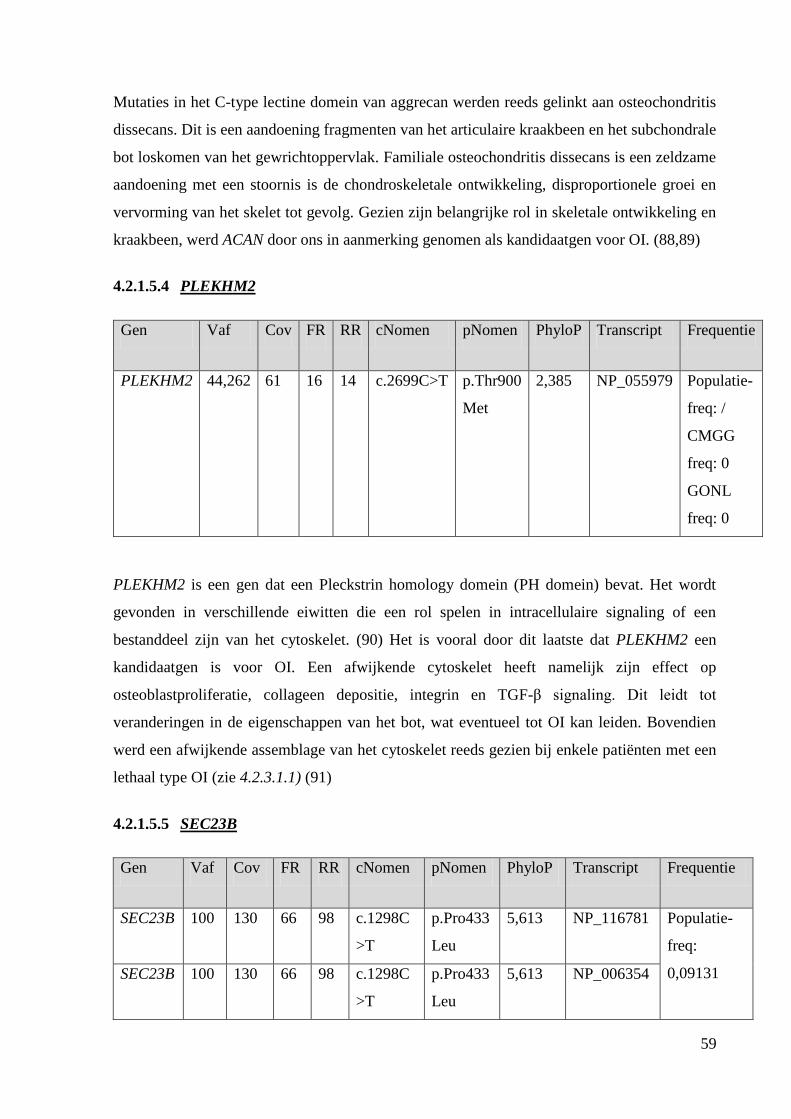
59
Mutaties in het C-type lectine domein van aggrecan werden reeds gelinkt aan osteochondritis
dissecans. Dit is een aandoening fragmenten van het articulaire kraakbeen en het subchondrale
bot loskomen van het gewrichtoppervlak. Familiale osteochondritis dissecans is een zeldzame
aandoening met een stoornis is de chondroskeletale ontwikkeling, disproportionele groei en
vervorming van het skelet tot gevolg. Gezien zijn belangrijke rol in skeletale ontwikkeling en
kraakbeen, werd ACAN door ons in aanmerking genomen als kandidaatgen voor OI. (88,89)
4.2.1.5.4 PLEKHM2
PLEKHM2 is een gen dat een Pleckstrin homology domein (PH domein) bevat. Het wordt
gevonden in verschillende eiwitten die een rol spelen in intracellulaire signaling of een
bestanddeel zijn van het cytoskelet. (90) Het is vooral door dit laatste dat PLEKHM2 een
kandidaatgen is voor OI. Een afwijkende cytoskelet heeft namelijk zijn effect op
osteoblastproliferatie, collageen depositie, integrin en TGF-β signaling. Dit leidt tot
veranderingen in de eigenschappen van het bot, wat eventueel tot OI kan leiden. Bovendien
werd een afwijkende assemblage van het cytoskelet reeds gezien bij enkele patiënten met een
lethaal type OI (zie 4.2.3.1.1) (91)
4.2.1.5.5 SEC23B
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
SEC23B 100 130 66 98 c.1298C
>T
p.Pro433
Leu
5,613 NP_116781 Populatie-
freq:
0,09131 SEC23B 100 130 66 98 c.1298C
>T
p.Pro433
Leu
5,613 NP_006354
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
PLEKHM2 44,262 61 16 14 c.2699C>T p.Thr900
Met
2,385 NP_055979 Populatie-
freq: /
CMGG
freq: 0
GONL
freq: 0
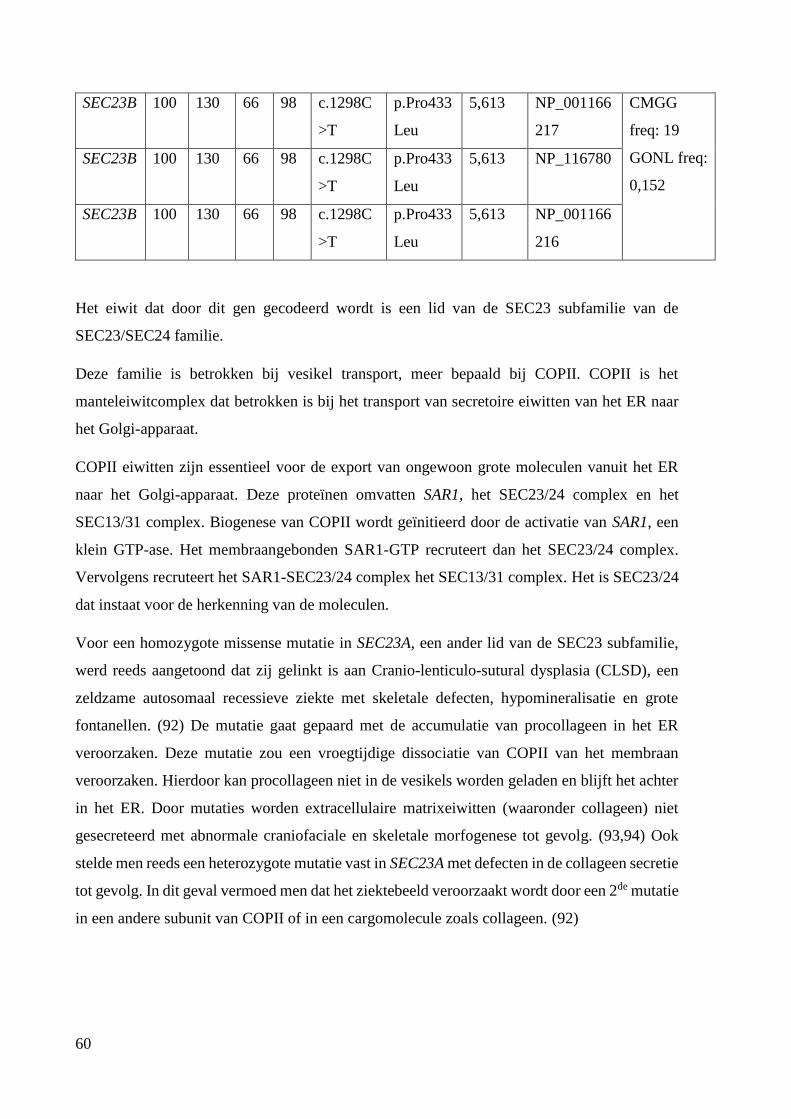
60
SEC23B 100 130 66 98 c.1298C
>T
p.Pro433
Leu
5,613 NP_001166
217
CMGG
freq: 19
GONL freq:
0,152
SEC23B 100 130 66 98 c.1298C
>T
p.Pro433
Leu
5,613 NP_116780
SEC23B 100 130 66 98 c.1298C
>T
p.Pro433
Leu
5,613 NP_001166
216
Het eiwit dat door dit gen gecodeerd wordt is een lid van de SEC23 subfamilie van de
SEC23/SEC24 familie.
Deze familie is betrokken bij vesikel transport, meer bepaald bij COPII. COPII is het
manteleiwitcomplex dat betrokken is bij het transport van secretoire eiwitten van het ER naar
het Golgi-apparaat.
COPII eiwitten zijn essentieel voor de export van ongewoon grote moleculen vanuit het ER
naar het Golgi-apparaat. Deze proteïnen omvatten SAR1, het SEC23/24 complex en het
SEC13/31 complex. Biogenese van COPII wordt geïnitieerd door de activatie van SAR1, een
klein GTP-ase. Het membraangebonden SAR1-GTP recruteert dan het SEC23/24 complex.
Vervolgens recruteert het SAR1-SEC23/24 complex het SEC13/31 complex. Het is SEC23/24
dat instaat voor de herkenning van de moleculen.
Voor een homozygote missense mutatie in SEC23A, een ander lid van de SEC23 subfamilie,
werd reeds aangetoond dat zij gelinkt is aan Cranio-lenticulo-sutural dysplasia (CLSD), een
zeldzame autosomaal recessieve ziekte met skeletale defecten, hypomineralisatie en grote
fontanellen. (92) De mutatie gaat gepaard met de accumulatie van procollageen in het ER
veroorzaken. Deze mutatie zou een vroegtijdige dissociatie van COPII van het membraan
veroorzaken. Hierdoor kan procollageen niet in de vesikels worden geladen en blijft het achter
in het ER. Door mutaties worden extracellulaire matrixeiwitten (waaronder collageen) niet
gesecreteerd met abnormale craniofaciale en skeletale morfogenese tot gevolg. (93,94) Ook
stelde men reeds een heterozygote mutatie vast in SEC23A met defecten in de collageen secretie
tot gevolg. In dit geval vermoed men dat het ziektebeeld veroorzaakt wordt door een 2de mutatie
in een andere subunit van COPII of in een cargomolecule zoals collageen. (92)

61
Exoom 2: P12
Voor Exoom 2 werden bij een eerste analyse geen kandidaatgenen geselecteerd. De genen die
hieronder vermeld zijn, werden gevonden na een meer uitgebreide analyse. De inculsie van
deze genen is gebaseerd op de informatie die werd gevonden in de databanken OMIM en
GeneCards. Verder literatuurstudie omtrent deze kandidaten is vereist voordat men kan
overgaan naar de volgende stap in de analyse.
4.2.2.1 Nonsense mutaties
Na het toepassen van de filters zoals in exoom 1, werden 1 variant geselecteerd, TMPRSS11E.
Dit bleek geen goede kandidaat voor OI. Hierna werd de filter voor PhyloP aangepast zodat
ook varianten met een onbekend PhyloP werden geïncludeerd. Er werden 4 extra varianten
geselecteerd in 1 gen. Deze bleek geen goede kandidaat voor OI.
Na het aanpassen voor vaf zodat zowel homo- als heterozygote varianten werden geïncludeerd,
werd 1 extra variant hieruit gefilterd. Deze bleek geen goede kandidaat voor OI. Vervolgens
werden ook varianten met coverage < 30 geïncludeerd. Er werden 9 extra varianten in 6 genen
geselecteerd. Van deze varianten werd geen enkele weerhouden. Bij de heterozygote varianten
werden opnieuw varianten gevonden in CTBP2, zoals in exoom 1.
4.2.2.2 Frameshift mutatie
Na het toepassen van de filters zoals in exoom 1, werden geen varianten geselecteerd. De
PhyloP filter werd ook aangepast zodat ook varianten met onbekende PhyloP-waarde werden
geïncludeerd. Dit leverde 1 variant op, KLHL33, die werd verworpen.
Wanneer ook varianten met coverage < 30 werden onderzocht, werden 14 nieuwe varianten in
12 genen geselecteerd. De varianten in het gen RYK werden weerhouden. Bij het onderzoeken
van de heterozygote varianten werden 15 extra varianten in 11 genen bekomen. Ook hier duikt
het gen CTBP2 op. De andere varianten bleken geen goede kandidaten.
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
RYK 100 3 1 2 c.9_10insG p.Arg4fs / NP_002949 Populatiefreq: /
CMGG freq: 0
GONL freq: 0
RYK 100 3 1 2 c.9_10insG p.Arg4fs / NP_001005861

62
4.2.2.3 Deleties
Wanneer de filters van exoom 1 werden toegepast, werden geen varianten geselecteerd. Daarom
werd de PhyloP filter aangepast zodat ook varianten met onbekende PhyloP in het exoom
werden geïncludeerd. Er werden 28 varianten in 21 genen bekomen. Van deze varianten werd
COG3 als een potentiële kandidaat geselecteerd.
Ook de varianten met coverage <30 werden onderzocht. Dit leverde 16 extra varianten in 12
genen op, waarvan er geen geselecteerd werden.
4.2.2.3.1 COG3
Gen Vaf Cov FR RR cNomen pNomen Transcript Frequentie
COG3 100 68 20 48 c.2474_2475
delTGinsCA
p.Leu825
Ser
NP_113619 Populatiefreq: /
CMGG freq: 0
GONL freq: 0
COG3 codeert voor een onderdeel van het Golgi complex, wat bestaat uit 8 subunits nodig voor
de normale Golgi morfologie en localisatie. Defecten in COG resulteren in defecten in de
eiwitglycosylatie.(76)
4.2.2.4 Splice site mutaties
Bij het toepassen van de filters zoals in exoom, werd 1 variant geselecteerd, PRDM2. Deze was
geen goede kandidaat voor OI. Vervolgens werd de PhyloP filter toegepast om ook varianten
met een onbekende PhyloP toe te laten. Dit leverde 18 extra varianten in 7 genen op. Uit deze
18 varianten werd COG3 vanwege zijn rol als component in het Golgi complex als mogelijke
kandidaat gezien, maar vanwege zijn locatie diep intronisch bleek dit toch geen goede
kandidaat.
Ook de heterozygote varianten werden onderzocht. Er werden 26 extra varianten in 14 genen
geselecteerd. Geen van deze werden weerhouden.
De PhyloP filter werd vervolgens verlaagd zodat ook waarden met PhyloP>0 werden
geïncludeerd. Er werden zo 5 extra varianten in 4 genen bekomen. Hiervan werd RAB1A
weerhouden.

63
Vervolgens werden ook varianten met coverage < 30 geïncludeerd. Er werden 30 extra
varianten in 12 genen geselecteerd, waarvan geen enkele werd weerhouden.
4.2.2.4.1 RAB1A
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
RAB1A 40,541 37 15 2 c.96A>T / 0,932 NP_056358 Populatiefreq: /
CMGG freq: 0
GONL freq: 0 RAB1A 40,541 37 15 2 c.96A>T / 0,932 NP_004152
RAB1A codeert voor een lid van de Ras superfamilie van GTPasen. Dit GTPase regelt het
vesikeltransport van het ER naar het Golgi-apparaat. (95)
4.2.2.5 Missense mutaties
Wanneer de filters werden toegepast zoals in exoom 1, werden 20 varianten in 13 genen
geselecteerd. Hiervan werden EYA1 en RECK weerhouden. Vervolgens werd de PhyloP filter
aangepast zodat ook varianten met een onbekende PhyloP werden geïncludeerd. Er werden 62
extra varianten in 36 genen bekomen. Hiervan werd CRTAC1 weerhouden.
Vervolgens werd de PhyloP filter aangepast naar PhyloP > 2. Er werden 7 extra varianten in 5
genen geselecteerd. Hiervan werd ADAMTS20 weerhouden.
Ook de heterozygote varianten werden onderzocht. Volgende filters worden toegepast: vaf <
50, coverage > 30, PhyloP > 3 en rs-nummer = /. Er werden 46 varianten geselecteerd. Om dit
aantal terug te dringen werden de filters voor de predictie tools SIFT (deleterious), Polyphen
(probably damaging/possibly damaging) en Mapp (bad) gebruikt. Op deze wijze werden 6
varianten in 6 genen bekomen. Deze waren geen goede kandidaten voor OI.
4.2.2.5.1 EYA1
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
EYA1 55,952 84 25 32 c.901C>
T
p.Arg301
Trp
3,999 NP_742056 Populatie-
freq: /
CMGG
freq: 0
EYA1 55,952 84 25 32 c.898C>
T
p.Arg300
Trp
3,999 NP_001275
503
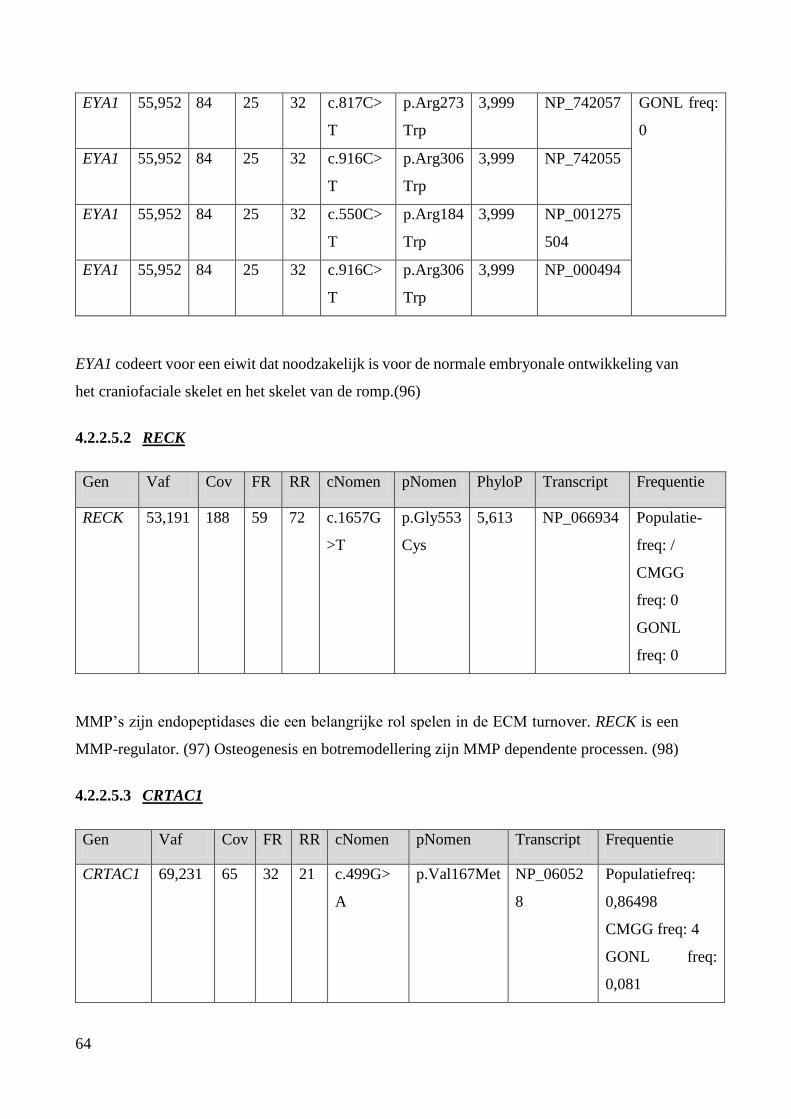
64
EYA1 55,952 84 25 32 c.817C>
T
p.Arg273
Trp
3,999 NP_742057 GONL freq:
0
EYA1 55,952 84 25 32 c.916C>
T
p.Arg306
Trp
3,999 NP_742055
EYA1 55,952 84 25 32 c.550C>
T
p.Arg184
Trp
3,999 NP_001275
504
EYA1 55,952 84 25 32 c.916C>
T
p.Arg306
Trp
3,999 NP_000494
EYA1 codeert voor een eiwit dat noodzakelijk is voor de normale embryonale ontwikkeling van
het craniofaciale skelet en het skelet van de romp.(96)
4.2.2.5.2 RECK
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
RECK 53,191 188 59 72 c.1657G
>T
p.Gly553
Cys
5,613 NP_066934 Populatie-
freq: /
CMGG
freq: 0
GONL
freq: 0
MMP’s zijn endopeptidases die een belangrijke rol spelen in de ECM turnover. RECK is een
MMP-regulator. (97) Osteogenesis en botremodellering zijn MMP dependente processen. (98)
4.2.2.5.3 CRTAC1
Gen Vaf Cov FR RR cNomen pNomen Transcript Frequentie
CRTAC1 69,231 65 32 21 c.499G>
A
p.Val167Met NP_06052
8
Populatiefreq:
0,86498
CMGG freq: 4
GONL freq:
0,081
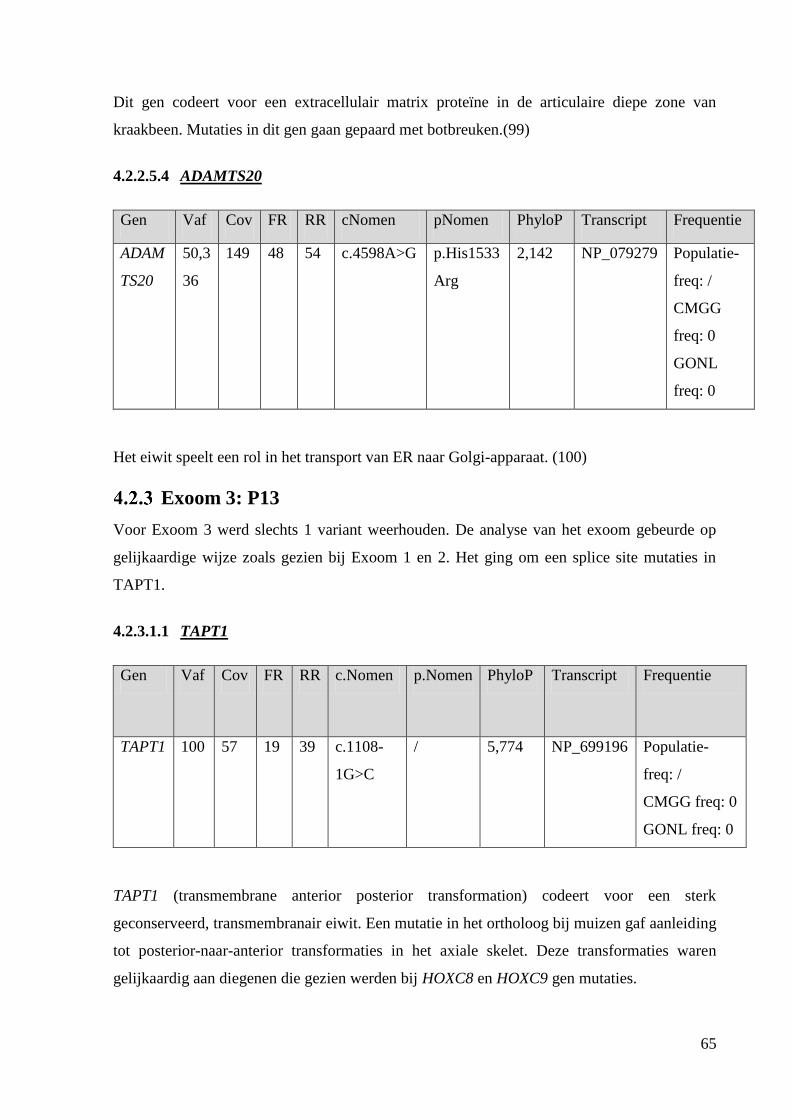
65
Dit gen codeert voor een extracellulair matrix proteïne in de articulaire diepe zone van
kraakbeen. Mutaties in dit gen gaan gepaard met botbreuken.(99)
4.2.2.5.4 ADAMTS20
Gen Vaf Cov FR RR cNomen pNomen PhyloP Transcript Frequentie
ADAM
TS20
50,3
36
149 48 54 c.4598A>G p.His1533
Arg
2,142 NP_079279 Populatie-
freq: /
CMGG
freq: 0
GONL
freq: 0
Het eiwit speelt een rol in het transport van ER naar Golgi-apparaat. (100)
Exoom 3: P13
Voor Exoom 3 werd slechts 1 variant weerhouden. De analyse van het exoom gebeurde op
gelijkaardige wijze zoals gezien bij Exoom 1 en 2. Het ging om een splice site mutaties in
TAPT1.
4.2.3.1.1 TAPT1
Gen Vaf Cov FR RR c.Nomen p.Nomen PhyloP Transcript Frequentie
TAPT1 100 57 19 39 c.1108-
1G>C
/ 5,774 NP_699196 Populatie-
freq: /
CMGG freq: 0
GONL freq: 0
TAPT1 (transmembrane anterior posterior transformation) codeert voor een sterk
geconserveerd, transmembranair eiwit. Een mutatie in het ortholoog bij muizen gaf aanleiding
tot posterior-naar-anterior transformaties in het axiale skelet. Deze transformaties waren
gelijkaardig aan diegenen die gezien werden bij HOXC8 en HOXC9 gen mutaties.

66
Specificatie van de wervelkolom gebeurt vroeg in de ontwikkeling en HOX-genen spelen hier
een belangrijke rol in. Deze genen coderen voor transcriptiefactoren die genen die downstream
gelegen zijn kunnen activeren of onderdrukken. (101) TAPT1 zou stroomafwaarts van
homeobox C8 werken als effector waar het de transductie van extracellulaire informatie, nodig
voor de ontwikkeling van het axiale skelet, bewerkstelligt. (102)
TAPT1 werd onlangs bevestigd als causaal gen voor OI en reeds geïncludeerd in OI panel 2.
Het zou van belang zijn bij het transport van intracellulaire eiwitten en de organisatie van
organellen zoals het Golgi-apparaat. Het speelt waarschijnlijk ook een rol bij de ciliaire
assemblage en signaling. Door deze functies kan het inwerken op de osteogenese door
osteoblastdifferentiatie en chondrocytdifferentiaties te beïnvloeden. In vitro studies toonden
aan dat TAPT1 gelokaliseerd was in het centrosoom en/of in de regio van het ciliair basaal
lichaam en dat defecten in TAPT1 aanleiding gaven tot verandering in het opvouwen en de
secretie, alsook een effect hebben op de morfologie van het Golgi-apparaat en transport van het
plasmamembraan naar het Golgi-apparaat. Met zebravisstudies werd aangetoond dat
knockdown van tapt1b (zebravisortholoog van TAPT1) aanleiding gaf tot ernstige craniofaciale
afwijkingen, microcefalie en veralgemeende vertraagde ossificatie.(103)

67
5 Discussie
Het opzet van deze thesis was om voor Osteogenesis imperfecta, een klinisch en genetisch
heterogene aandoening, nieuwe potentieel causale genen op te sporen. De meest voorkomende
vorm van osteogenesis imperfecta is de dominante vorm met mutaties in COL1A1 en COL1A2,
die ongeveer 90% van de gevallen verklaart. De overige 10% wordt verklaard door mutaties in
recessief overgeërfde genen, met uitzondering van IFITM5, dat dominant overerft.
Ondanks de steeds groeiende lijst van causale genen, worden er elk jaar nog nieuwe causale
varianten geïdentificeerd in de reeds gekende genen. Vooraleer een nieuw gen effectief kan
beschouwd worden als een nieuw OI gen, moeten verschillende stappen doorlopen worden.
Wanneer een patiënt zich aanmeldt met een fenotype van OI, moet allereest een screening van
de gekende varianten gebeuren. Indien deze screening geen mutaties oplevert, kan men
overgaan op Whole Exome Sequencing (WES) of eventueel Whole Genome Sequencing
(WGS). Analyse van het exoom levert vaak kandidaatgenen op voor OI. Het bevestigen van de
causaliteit van deze kandidaatgenen gebeurt later via functionele studies, zoals bijvoorbeeld in
vitro studies en zebravis studies.
5.1 Screening gekende OI genen
De screening bij 10 patiënten gekend met OI fenotype leverde bij 5 patiënten een causale
mutatie op (klasse 4 of 5) in de gekende OI genen (uit OI panel 1 en 2). Van deze 5 causale
mutaties werden 4 gevonden in genen die coderen voor type I collageen (COL1A1/COL1A2).
Eén causale mutatie werd gevonden in PLS3.
In P1 werd een klasse 4 mutatie in COL1A2 vastgesteld, namelijk c.2314G>A wat aanleiding
gaf tot een eiwitverandering p.(Gly772Ser). Dit glycine bevond zich in de α-helix van type I
collageen.
Bij P4 was er sprake van een klasse 4 mutatie in COL1A1, namelijk c.484delC hetgeen
aanleiding gaf tot de eiwitverandering p.(Gln162fs) wat leidde tot nonsense mediated decay
(NMD) van het mRNA door de introductie van een stopcodon.
Bij P6 werd een causale mutatie in COL1A1 geïdentificeerd: c.370-2A>G. Dit leidt tot het
verloren gaan van de splice-acceptor site, waardoor een overslaan van exon 5 zeer
waarschijnlijk is.

68
Bij P7 werd eveneens een klasse 4 mutatie gevonden in COL1A1: c.1012G>A (p.(Gly338Ser)).
Ook hier bevindt het glycine zich in de α-helix.
Bij P8 werd een klasse 4 mutatie bevestigd in PLS3, welke aanleiding gaf tot een prematuur
stopcodon: c.766C>T , p.(Arg256*). Aangezien het hier gaat om een nonsense-mutatie is de
kans op NMD groot.
Het merendeel van de gevonden causale mutaties veroorzaakt een verandering in type I
collageen. Dit wijst op het belang van dit eiwit in de pathogenese. Ook zien we bij 2 van deze
mutaties veranderingen in glycines in de α-helix, wat de importantie van deze glycines
benadrukt voor een goede werking van het eiwit. Toch werd ook één causale variant vastgesteld
in een gen uit OI panel 2. Hoewel minder frequent de oorzaak van OI, mogen deze genen niet
vergeten worden.
P2, P3, P5, P9 en P10 blijven na deze screening genetisch onopgehelderd. De volgende stap
bij deze patënten is het evalueren van de kliniek. Indien de kliniek sterk suggestief is voor OI,
kan overgegaan worden naar Whole Exome Sequencing (WES) om op zoek te gaan naar nieuw
genen voor OI.
5.2 Whole Exome Sequencing (WES)
In het kader van deze thesis werden 3 exomen geanalyseerd. Dit waren niet de exomen van de
gescreende patiënten, maar van andere patiënten die reeds gescreend waren in het Centrum voor
Medische Genetica.
Exoom 1 (P11)
Voor Exoom 1 werden na analyse van het exoom 7 genen weerhouden als kandidaatgenen voor
OI: RYK, P4HA1, AKT2, TRAPPC12, SEC23B, ACAN en PLEKHM2.
Van de homozygote varianten leek RYK aanvankelijk een goede kandidaat vanwege de aard
van de mutatie (frameshift) en door zijn rol in WNT signaling pathway. De coverage van deze
variant was echter zeer laag, wat een fout in de sequenering kan betekenen. Dit samen met het
feit dat deze varianten gelegen waren in exon 1, hetgeen een moeilijk te sequeneren exon is,
wijst erop dat deze varianten waarschijnlijk vals positieven zijn. Bovendien werd ook Exoom
2 een variant gevonden in het RYK gen (zie 5.2.2). Toch kunnen we deze varianten niet negeren
vanwege de functie van dit eiwit. Verder analyse met Sanger sequencing is dan ook vereist.

69
P4HA1 is vanwege zijn rol in de collageenbiosynthese een zeer interessante kandidaat voor OI.
De variant in P4HA1 die ons het meest interesseerde was gelegen op de -1 positie, hetgeen een
belangrijke positie is voor splicing. Toch zou het, aangezien de coverage van deze variant zeer
laag is, kunnen gaan om een vals positief resultaat. Deze variant zal verder geanalyseerd worden
met Sanger sequencing.
De andere homozygote varianten, AKT2, TRAPPC12 en SEC23B, waren missense mutaties. Dit
soort mutaties geeft minder vaak aanleiding tot pathogeniciteit. De variant in SEC23B had
daarenboven een hoge populatiefrequentie (9,1%). Het is onwaarschijnlijk dat een variant met
zo’n hoge populatiefrequentie een relatief zeldzaam ziektebeeld als OI kan veroorzaken (zelfs
al gaat het om een recessieve vorm). Vanwege de rol van SEC23B in COPII vesikel transport
zal deze variant toch verder geanalyseerd worden.
De heterozygote varianten, ACAN en PLEKHM2, vinden bijval in de literatuur als
kandidaatgenen voor OI, maar een homozygote variant lijkt waarschijnlijker door de
consanguïniteit van de ouders.
Exoom 2 (P12)
De analyse van exoom 2 leverde aanvankelijk geen interessante varianten op. Na een tweede,
meer uitgebreide analyse werden volgende genen geselecteerd: RYK, COG3, EYA1, RECK,
CRTAC1, ADAMTS20 en RAB1A,. Deze genen werden voornamelijk geselecteerd op basis van
informatie uit de databanken GeneCards en OMIM. Verdere literatuurstudie omtrent deze
genen is dus nog vereist vooral er kan worden overgegaan naar de volgende stap in de analyse.
Bij de homozygote varianten zien we opnieuw RYK opduiken zoals in exoom 1. De varianten
bevinden zich ook deze keer in exon 1 en de coverage is laag. Dit wijst nogmaals in de richting
van een vals positief resultaat. Ook hier zullen we de varianten verder analyseren met Sanger
sequencing.
COG3 werd geselecteerd vanwege zijn rol als onderdeel van de normale morfologie en
localisatie van het Golgi complex. De predictie tool PolyPhen voorspelt echter een benigne
effect (SIFT en Mapp zijn niet gekend).
EYA1 codeert voor een eiwit dat een rol speelt in de ontwikkeling van het craniofaciale skelet
en in de ontwikkeling van het skelet van de romp. Ook is de PhyloP-waarde hoog, wat wijst op
een sterke conservatie van dit gen. Wel zagen we dat enkel SIFT en PolyPhen een negatief
effect voorspellen bij deze varianten, Mapp is niet gekend.

70
RECK is een MMP-regulator en speelt via MMP’s een rol in de osteogenesis en
botremodellering. SIFT, PolyPhen en Mapp voorspelde bij deze variant een negatief effect. De
PhyloP-waarde was hoog wat wijst op een sterke conservatie van dit gen.
CRTAC1 codeert voor een extracellulair matrix proteïne in kraakbeen. Enkel PolyPhen
voorspelde bij dit gen echter een negatief effect, SIFT en Mapp waren niet gekend. Bovendien
was de populatiefrequentie voor deze variant relatief hoog (8%).
ADAMTS20 speelt een rol in het transport van ER naar het Golgi-apparaat. Voor andere leden
van de ADAMTS-familie, namelijk ADAMTS-2, ADAMTS-3 en ADAMTS-14 werd reeds
aangetoond dat ze een rol spelen in de fibrilogenese van type I collageen (zie 1.1.4.6). De
predictie tools SIFT en PolyPhen voorspellen voor deze variant echter een benigne effect, enkel
Mapp geeft een negatief effect aan.
Bij de heterozygote varianten werd RAB1A geselecteerd. Heterozygote varianten zijn hier
opnieuw minder waarschijnlijk door de consanguïniteit van de ouders. Dit gen codeert voor een
GTPase dat het vesikeltransport regelt van het ER naar het Golgi-apparaat. Tegenargumenten
voor dit gen zijn de lage PhyloP-waarde en het grote verschil tussen het aantal forward reads
en het aantal reverse reads (respectievelijk 15 en 2). Dit laatste zou kunnen wijzen op een fout
tijdens de sequencing.
Exoom 3 (P13)
Voor exoom 3 selecteerden we slechts één variant, namelijk c. 1108-1G>C in TAPT1. Deze
variant was interessant vanwege zijn positie op -1. Het gen codeert voor een sterk
geconserveerd transmembranair eiwit.
Dit gen werd onlangs bevestigd als causaal gen voor OI en werd reeds geïncludeerd in OI panel
2. TAPT1 is een voorbeeld van een gen dat gevonden werd dankzij exoomsequencing. TAPT1
zou een rol spelen in transport van intracellulaire eiwitten en de organisatie van organellen zoals
het Golgi-apparaat. Verder is het ook belangrijk bij de ciliaire assemblage en signaling. Op deze
wijze kan het inwerken op de osteogenese door osteoblastdifferentiatie en
chondrocytdifferentiaties te beïnvloeden. Het werd later bevestigd met in vitro studies en
zebravisstudies. In vitro studies toonden aan dat TAPT1 gelokaliseerd was in het centrosoom
en/of in de regio van het ciliair basaal lichaam en dat defecten in TAPT1 aanleiding gaven tot
verandering in het opvouwen en de secretie, alsook een effect hebben op de morfologie van het
Golgi-apparaat en transport van het plasmamembraan naar het Golgi-apparaat. (103)

71
Sterktes en zwaktes van Whole Exome Sequencing
Whole Exome Sequencing is een efficiënte techniek om de allelen die aan de basis liggen van
zeldzame Mendeliaanse aandoeningen op te sporen. Naast de eiwit-coderende sequenties,
waarin zich de meerderheid van de causale varianten bevinden, worden ook splice acceptor en
donor sites gesequeneerd. Bovendien is deze techniek ondanks het steeds dalen van de prijs nog
steeds zo’n 5 keer goedkoper dan Whole Genome Sequencing. Ook is de ruwe data van Whole-
Exome sequencing qua grootte ongeveer één zesde van de ruwe data van Whole Genome
sequencing. Analyse van het exoom is dus minder tijdrovend. (104)
Hoewel Whole-Exome Sequencing (WES) een doeltreffend hulpmiddel is voor het
identificeren van nieuwe genen in verband met OI, heeft deze techniek ook haar beperkingen.
De belangrijkste beperking van Whole Exome sequencing is dat slechts 1% van het gehele
genoom wordt geanalyseerd. Deze techniek mist dus varianten in intronische sequenties alsook
structurele varianten zoals translocaties en inversies.
(102,104,105)(105)(105)(105)(105)(104)(104)(104)(104) Ook varianten in mitochondriaal
DNA worden gemist.
Een andere beperking is dat WES gecompliceerd wordt door genetische heterogeniteit zoals het
geval is bij OI alsook door gedupliceerde sequenties in het genoom. (105) Bij aandoeningen die
genetisch zeer heterogeen zijn, is het deel van het genoom dat bezet wordt door genen die,
wanneer zij mutaties bevatten, aanleiding geven tot ziekte (‘mutational target’) te groot. De
kans om 2 patiënten te vinden met mutaties in hetzelfde gen wordt dan ook veel kleiner. (106)
Door een tekort aan informatie over de functies van bepaalde genen is het bovendien soms
moeilijk een onderscheid te maken tussen schadelijke en benigne varianten. Varianten met deze
onbekende significantie zijn moeilijk te interpreteren. (105)
Incidental Findings
Bij WES en WGS kunnen incidental findings voorkomen. Incidental findings omvatten
bevindingen die als bijproduct van een diagnostische handeling worden bekomen, en
daarenboven niet te vermijden zijn. Een radioloog kan niet vermijden dat bij een thoracale
röntgenopname, met als doel hartfalen te evalueren, tevens een massa in de long wordt ontdekt.
(107) De snelheid waarmee NGS-technieken evolueren zorgen voor een gebrek aan
standaardisatie omtrent dergelijke zaken. Het ACMG (American College of Medical Genetics
and Genomics) richtte in 2012 een werkgroep op rond incidental findings in WGS (Whole
Genome Sequensing), om te grote verschillen in beleid omtrent incidental findings te

72
vermijden. (108) In 2013 werd door de werkgroep richtlijnen gepubliceerd, die als houvast
moesten dienen voor de klinische labo’s. In deze richtlijnen werd een extra panel van 56 genen
vooropgesteld. Er werd aangeraden dit panel bij elke indicatie voor WGS mee te screenen, en
de resultaten te rapporteren ongeachte de wil en leeftijd van de patiënt. (109)
Er ontstond echter grote verontwaardiging door dit voorstel om bij elke aanvraag van WGS het
extra panel te screenen en te rapporteren ongeacht de leeftijd en wil van de patiënt. (107,108)
Deze inbraak op de autonomie van de patiënt leidde tot een zekere uiting van protest. Het
algemene antwoord luidde dan ook: ‘Choice Matters’. (107)
Informed consent is zeker een struikelblok. De keuze omtrent het al dan niet ontvangen van
bepaalde resultaten in de klinische genetica blijkt complexer dan initieel gedacht. De data
betrokken bij klinische genetica blijft groeien. Deze info in een relevant en beperkt pakket
communiceren naar de verschillende stakeholders vormt dan ook een grote uitdaging. (108)
Een ander probleem omtrent dit extra panel was dat volgens peers de definitie van incidental
findings niet gerespecteerd werd in de richtlijnen. In de aanbevelingen wordt aangemoedigd
een bijkomende, doelbewuste analytische inspanning te leveren. Immers, indien men ervoor
kiest bepaalde genen niet te screenen, zijn er ook geen resultaten te melden. Er wordt dan ook
vanuit verschillende hoeken gepleit voor het erkennen dat het in de richtlijnen een bewust
zoeken naar secundaire bevindingen betreft. Een vorm van opportunistische screening
dus.(107)
Voor het gebruik van WGS/WES voor opportunistische screening is weinig tot geen evidentie
voorhanden. Tot op heden wordt WES slechts in specifieke klinische scenario’s toegepast,
bijvoorbeeld wanneer andere conventionele genetische tests falen in het verklaren van de
kliniek van de patiënt.
De richtlijnen echter opteren voor een opportunistische screening, wat niet noodzakelijk in een
klinische setting gebeurt. (107) Het gaat dus over een gebruik van WES in een compleet andere
populatie en setting dan deze waar we tot op dit moment vertrouwd mee zijn. Het effect van
dergelijk beleid op harde eindpunten is dan ook niet gekend. (108)
Er werd geconcludeerd dat op dit moment nog te weinig evidentie bestaat om een standard-of-
care op te stellen. Er is naast een robuuste dialoog ook nood aan empirische data betreffende de
kwaliteiten en zwaktes van WGS en WES in de algemene populatie. (107,108) Talrijke studies
zijn op dit moment aan de gang om deze data te vergaren.

73
Op dit moment hebben vele genetische labo’s hun eigen wetten omtrent het rapporteren van
secundaire bevindingen. In het centrum voor medische genetica UZ gent worden deze
gerapporteerd, maar enkel indien zij als klasse 4 of klasse 5 worden geclassificeerd. In deze
thesis werden verschillende secundaire bevindingen gevonden in exoom 1 en 2, maar geen van
hen konden als klasse 4 of 5 worden geclassificeerd.
5.3 Conclusie
De doelstelling van deze thesis was om nieuwe potentiële genen voor OI op te sporen. WES en
WGS zijn krachtige tools die steeds meer worden aangewend om naar deze genen op zoek te
gaan. Toch gaat het screenen van de OI panels hier nog steeds aan vooraf. Voor 5 van de 10
patiënten die voor deze thesis gescreend werden, werd een causale mutatie gevonden in de reeds
gekende genen voor OI. Enkel als er in de gekende genen geen causale mutatie wordt gevonden
en de kliniek zeer suggestief is voor OI, gaat men over naar WES en/of eventueel WGS. In het
kader van deze thesis zijn we erin geslaagd door middel van WES van drie patiënten een aantal
kandidaatgenen voor het OI voorop te stellen:
RYK, P4HA1, AKT2, TRAPPC12, SEC23B, ACAN, PLEKHM2, COG3, EYA1, RECK,
CRTAC1, ADAMTS20 en RAB1A.
Toch zien we dat deze krachtige hulpmiddelen ook schade kunnen berokken. De vraagstukken
omtrent bijvoorbeeld incidental findings adresseren is daarom een must. Gedeelde aandacht en
inzet is dan ook vereist om een kwaliteitsvolle implementatie van genomische geneeskunde in
de gezondheidszorg te verzekeren.
We vermelden ten slotte dat het verhaal hier niet ten einde is. Deze genen zijn potentieel
causaal, maar voor ze kunnen opgenomen worden in de OI panels moeten de varianten allereerst
bevestigd worden met Sanger sequencing. Indien ze bevestigd worden, zal men een segregatie-
onderzoek binnen de familie uitvoeren. Uiteindelijk zullen ook functionele studies verricht
worden zoals bijvoorbeeld in vitro studies en zebravis- en/of muisstudies. Op deze manier
zouden ze in de toekomst voor enkele patiënten uitsluitsel kunnen bieden.

74
6 Referenties
1. Lodish HF. Molecular cell biology. New York : W.H. Freeman and Co.; 2013.
2. Van Dijk FS, Pals G, Van Rijn RR, Nikkels PGJ, Cobben JM. Classification of
Osteogenesis Imperfecta revisited. Eur J Med Genet. 2010;53(1):1–5.
3. Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363(9418):1377–85.
4. Mienaltowski MJ, Birk DE. Structure, physiology, and biochemistry of collagens. Adv
Exp Med Biol [Internet]. 2014;802:5–29. Available from:
http://www.ncbi.nlm.nih.gov/pubmed/24443018
5. Hulmes DJS. Collagen diversity, synthesis and assembly. Collagen Struct Mech.
2008;15–47.
6. Brinckmann J. Collagens at a glance. Top Curr Chem. 2005;247:1–6.
7. Gelse K, Pöschl E, Aigner T. Collagens - Structure, function, and biosynthesis. Adv Drug
Deliv Rev. 2003;55(12):1531–46.
8. Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet [Internet].
2004;363(9418):1377–85. Available from:
http://www.sciencedirect.com/science/article/pii/S0140673604160510
9. Diegelmann RF. Collagen Metabolism. Medscape; 2001.
10. Fratzl P. Collagen: Structure and mechanics. Collagen: Structure and Mechanics. 2008.
1-506 p.
11. Van Dijk FS, Cobben JM, Kariminejad A, Maugeri A, Nikkels PGJ, Van Rijn RR, et al.
Osteogenesis imperfecta: A review with clinical examples. Molecular Syndromology.
2011. p. 1–20.
12. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: Clinical diagnosis, nomenclature
and severity assessment. Am J Med Genet Part A. 2014;164(6):1470–81.
13. Gajko-Galicka A. Mutations in type I collagen genes resulting in osteogenesis imperfecta
in humans. Acta Biochim Pol [Internet]. 2002;49(2):433–41. Available from:
http://www.ncbi.nlm.nih.gov/pubmed/12362985
14. Gelse K, Pöschl E, Aigner T. Collagens—structure, function, and biosynthesis. Adv
Drug Deliv Rev [Internet]. 2003;55(12):1531–46. Available from:
http://www.sciencedirect.com/science/article/pii/S0169409X03001820
15. Myllyharju J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix
Biol. 2003;22(1):15–24.
16. Hudson DM, Eyre DR. Collagen prolyl 3-hydroxylation: a major role for a minor post-
translational modification? Connect Tissue Res [Internet]. 2013;54(4-5):245–51.
Available from:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3995746&tool=pmcentrez
&rendertype=abstract
17. Yamauchi M, Sricholpech M. Lysine post-translational modifications of collagen.
Essays Biochem [Internet]. 2012;52:113–33. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3499978/
18. Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci.
2005;118(Pt 7):1341–53.

75
19. Terajima M, Perdivara I, Sricholpech M, Deguchi Y, Pleshko N, Tomer KB, et al.
Glycosylation and cross-linking in bone type I collagen. J Biol Chem.
2014;289(33):22636–47.
20. Brinckmann J, Notbohm H, Müller PK. Collagen Biosynthesis. Collagen:Primer in
Structure, Processing and Assembly. 2005. p. 254.
21. Ishikawa Y, Bächinger HP. A molecular ensemble in the rER for procollagen maturation.
Biochimica et Biophysica Acta - Molecular Cell Research. 2013. p. 2479–91.
22. Wilson R, Lees JF, Bulleid NJ. Protein disulfide isomerase acts as a molecular chaperone
during the assembly of procollagen. J Biol Chem. 1998;273(16):9637–43.
23. Bourhis J-M, Mariano N, Zhao Y, Harlos K, Exposito J-Y, Jones EY, et al. Structural
basis of fibrillar collagen trimerization and related genetic disorders. Nat Struct Mol Biol
[Internet]. Nature Publishing Group, a division of Macmillan Publishers Limited. All
Rights Reserved.; 2012;19(10):1031–6. Available from:
http://dx.doi.org/10.1038/nsmb.2389
24. Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci
[Internet]. 2005;118(7):1341–53. Available from:
http://jcs.biologists.org/joces/118/7/1341.full.pdf
25. Boudko SP, Engel J, Bächinger HP. The crucial role of trimerization domains in collagen
folding. International Journal of Biochemistry and Cell Biology. 2012. p. 21–32.
26. Marini JC, Blissett AR. New Genes in Bone Development: What’s New in Osteogenesis
Imperfecta. J Clin Endocrinol Metab [Internet]. Chevy Chase, MD: Endocrine Society;
2013;98(8):3095–103. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3733862/
27. van Dijk FS, Dalgleish R, Malfait F, Maugeri A, Rusinska A, Semler O, et al. Clinical
utility gene card for: osteogenesis imperfecta. Eur J Hum Genet [Internet]. Macmillan
Publishers Limited; 2013;21(6). Available from:
http://dx.doi.org/10.1038/ejhg.2012.210
28. Gajko-Galicka A. Mutations in type I collagen genes resulting in osteogenesis imperfecta
in humans. Acta Biochim Pol. 2002;49(2):433–41.
29. Valadares ER, Carneiro TB, Santos PM, Oliveira AC, Zabel B. What is new in genetics
and osteogenesis imperfecta classification? J Pediatr (Rio J) [Internet]. Sociedade
Brasileira de Pediatria; 2014;90(6):536–41. Available from:
http://www.ncbi.nlm.nih.gov/pubmed/25046257
30. Shaker JL, Albert C, Fritz J, Harris G. Recent developments in osteogenesis imperfecta
[ v1 ; ref status : indexed , http://f1000r.es/5ao ] Referee Status : 2015;4:2–11.
31. Roy Morello PWE. Osteogenesis Imperfecta. :30. Available from:
http://www.oif.org/site/DocServer/A_Chapter_on_Osteogenesis_Imperfecta_by_Roy_
Morello_and_.pdf
32. Van Dijk FS, Sillence DO. Osteogenesis imperfecta: Clinical diagnosis, nomenclature
and severity assessment. Am J Med Genet A [Internet]. Oxford, UK: BlackWell
Publishing Ltd; 2014;164(6):1470–81. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4314691/
33. Forin V. Osteogenesis imperfecta type 5 [Internet]. [cited 2016 Mar 31]. Available from:
http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=216828
34. Symoens S, Hulmes DJS, Bourhis J-M, Coucke PJ, De Paepe A, Malfait F. Type I

76
Procollagen C-Propeptide Defects: Study of Genotype–Phenotype Correlation and
Predictive Role of Crystal Structure. Hum Mutat [Internet]. 2014;35(11):1330–41.
Available from: http://dx.doi.org/10.1002/humu.22677
35. Dalgleish R. The Human Collagen Mutation Database 1998. Nucleic Acids Res.
1998;26(1):253–5.
36. Hanagata N. IFITM5 mutations and osteogenesis imperfecta. J Bone Miner Metab
[Internet]. 2015;1–9. Available from: http://dx.doi.org/10.1007/s00774-015-0667-1
37. Homan EP, Rauch F, Grafe I, Lietman C, Doll JA, Dawson B, et al. Mutations in
SERPINF1 Cause Osteogenesis Imperfecta Type VI. J Bone Miner Res [Internet].
Hoboken: Wiley Subscription Services, Inc., A Wiley Company; 2011;26:2798–803.
Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3214246/
38. Baldridge D, Schwarze U, Morello R, Lennington J, Bertin TK, Pace JM, et al. CRTAP
AND LEPRE1 MUTATIONS IN RECESSIVE OSTEOGENESIS IMPERFECTA. Hum
Mutat [Internet]. 2008;29(12):1435–42. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2671575/
39. van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PGJ, Piersma SR, Fratantoni SA, et al.
PPIB Mutations Cause Severe Osteogenesis Imperfecta. Am J Hum Genet [Internet].
Elsevier; 2009;85(4):521–7. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2756556/
40. Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, et al.
Homozygosity for a Missense Mutation in SERPINH1, which Encodes the Collagen
Chaperone Protein HSP47, Results in Severe Recessive Osteogenesis Imperfecta. Am J
Hum Genet [Internet]. Elsevier; 2010;86(3):389–98. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2833387/
41. Kelley BP, Malfait F, Bonafe L, Baldridge D, Homan E, Symoens S, et al. Mutations in
FKBP10 Cause Recessive Osteogenesis Imperfecta and Bruck Syndrome. J Bone Miner
Res [Internet]. Wiley Subscription Services, Inc., A Wiley Company; 2011;26(3):666–
72. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3179293/
42. Lapunzina P, Aglan M, Temtamy S, Caparrós-Martín JA, Valencia M, Letón R, et al.
Identification of a Frameshift Mutation in Osterix in a Patient with Recessive
Osteogenesis Imperfecta. Am J Hum Genet [Internet]. Elsevier; 2010;87(1):110–4.
Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2896769/
43. Martínez-Glez V, Valencia M, Caparrós-Martín JA, Aglan M, Temtamy S, Tenorio J, et
al. Identification of a Mutation Causing Deficient BMP1/mTLD Proteolytic Activity in
Autosomal Recessive Osteogenesis Imperfecta. Hum Mutat [Internet]. 2012;33(2):343–
50. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3725821/
44. Volodarsky M, Markus B, Cohen I, Staretz-Chacham O, Flusser H, Landau D, et al. A
Deletion Mutation in TMEM38B Associated with Autosomal Recessive Osteogenesis
Imperfecta. Hum Mutat [Internet]. 2013;34(4):582–6. Available from:
http://dx.doi.org/10.1002/humu.22274
45. Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, et al. WNT1
mutations in families affected by moderately severe and progressive recessive
osteogenesis imperfecta. Am J Hum Genet [Internet]. The American Society of Human
Genetics; 2013;92(4):590–7. Available from:
http://dx.doi.org/10.1016/j.ajhg.2013.02.009
46. Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, et al. Mutations in

77
WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013;92(4):565–74.
47. Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, et al. Signalling
mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone
formation. Nat Cell Biol [Internet]. 2009;11(10):1205–11. Available from:
http://dx.doi.org/10.1038/ncb1963
48. Symoens S, Malfait F, D’hondt S, Callewaert B, Dheedene A, Steyaert W, et al.
Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis
imperfecta in humans. Orphanet J Rare Dis [Internet]. 2013;8:154. Available from:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3850743&tool=pmcentrez
&rendertype=abstract
49. CREB3L1 Genecards [Internet]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=CREB3L1&keywords=CREB3L1
50. van Dijk FS, Zillikens MC, Micha D, Riessland M, Marcelis CLM, de Die-Smulders CE,
et al. PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med.
2013;369(16):1529–36.
51. Ha-Vinh R, Alanay Y, Bank RA, Campos-Xavier AB, Zankl A, Superti-Furga A, et al.
Phenotypic and molecular characterization of Bruck syndrome (Osteogenesis imperfecta
with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J
Med Genet. 2004;131 A(2):115–20.
52. PLOD2 - Genecards [Internet]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=PLOD2&keywords=PLOD2
53. Niyibizi C, Wang S, Mi Z, Robbins PD. Gene therapy approaches for osteogenesis
imperfecta. Gene Ther [Internet]. 2004;11(4):408–16. Available from:
http://www.ncbi.nlm.nih.gov/pubmed/14724682
54. Glorieux FH. Experience with bisphosphonates in osteogenesis imperfecta. Pediatrics.
2007;119 Suppl(March 2007):S163–5.
55. Thomas IH, DiMeglio LA. Advances in the Classification and Treatment of
Osteogenesis Imperfecta. Curr Osteoporos Rep [Internet]. 2016;1–9. Available from:
http://link.springer.com/10.1007/s11914-016-0299-y
56. Antoniazzi F, Bertoldo F, Mottes M, Valli M, Sirpresi S, Zamboni G, et al. Growth
hormone treatment in osteogenesis imperfecta with quantitative defect of type I collagen
synthesis. J Pediatr [Internet]. 1996 Sep [cited 2016 Apr 4];129(3):432–9. Available
from: http://www.sciencedirect.com/science/article/pii/S002234769670077X
57. Besio R, Forlino A. New frontiers for dominant osteogenesis imperfecta treatment:
gene/cellular therapy approaches. Cold Spring Harb Perspect Med. 2013;1.
58. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9.
Science (80- ) [Internet]. 2014;346(6213):1258096–1258096. Available from:
http://www.sciencemag.org/content/346/6213/1258096.long
59. Horwitz EM, Prockop DJ, Gordon PL, Koo WWK, Fitzpatrick LA, Neel MD, et al.
Clinical responses to bone marrow transplantation in children with severe osteogenesis
imperfecta. Blood [Internet]. 2001 Mar 1;97(5):1227–31. Available from:
http://www.bloodjournal.org/content/97/5/1227.abstract
60. Götherström C, Westgren M, Shaw SWS, Åström E, Biswas A, Byers PH, et al. Pre- and
Postnatal Transplantation of Fetal Mesenchymal Stem Cells in Osteogenesis Imperfecta:
A Two-Center Experience. Stem Cells Transl Med [Internet]. Durham, NC, USA:

78
AlphaMed Press; 2014;3(2):255–64. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3925052/
61. Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat
Biotechnol. 2000;18(February):233–4.
62. Sanger sequencing [Internet]. Available from: http://www.vce.bioninja.com.au/aos-3-
heredity/molecular-biology-technique/sequencing.html
63. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution
rates on mammalian phylogenies. Genome Res. 2010;20(1):110–21.
64. Gillespie JH. Population Genetics: A concise guide [Internet]. A Concise Guide. 2004.
214 p. Available from:
http://books.google.com/books?id=KAcAfiyHpcoC&printsec=frontcover&dq=populati
on+genetics+gillespie&hl=&cd=1&source=gbs_api\npapers3://publication/uuid/8113D
0CA-1F97-486B-84B7-86B37ACBE7DB
65. rs-nummers [Internet]. Available from: https://customercare.23andme.com/hc/en-
us/articles/202907650-What-are-all-the-rs-numbers-rsids-
66. Bhagwat M. Searching NCBI’s dbSNP database. Curr Protoc Bioinforma.
2010;(SUPP.32).
67. Nonsense mutation [Internet]. [cited 2016 Apr 3]. Available from:
http://www.nature.com/scitable/definition/nonsense-mutation-228
68. Frameshift mutation / frame-shift mutation; frameshift [Internet]. [cited 2016 Apr 3].
Available from: http://www.nature.com/scitable/definition/frameshift-mutation-frame-
shift-mutation-frameshift-203)
69. Paepe A De. Klinische genetica. Gent: Universiteit Gent; 2015.
70. Lodish BA, Zipursky SL. Mutations : Types and Causes. Molecular Cell Biology 4th
Edition [Internet]. 4th ed. 2000. p. Section 8.1 Mutations: Types and Causes. Available
from: http://www.ncbi.nlm.nih.gov/books/NBK21578/
71. RYK Gene(Protein Coding): Receptor-Like Tyrosine Kinase [Internet]. [cited 2016 Feb
27]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?id_type=entrezgene&id=6259
72. Victor A. McKusick AH. RYK RECEPTOR-LIKE TYROSINE KINASE; RYK
[Internet]. 1995. Available from: http://www.omim.org/entry/600524
73. Lu W, Yamamoto V, Ortega B, Baltimore D. Mammalian Ryk Is a Wnt Coreceptor
Required for Stimulation of Neurite Outgrowth. Cell [Internet]. 2004;119(1):97–108.
Available from: http://www.sciencedirect.com/science/article/pii/S009286740400892X
74. Ryk: A Wnt Co-receptor at the Intersection of Frizzled & Dishevelled [Internet]. [cited
2016 Feb 27]. Available from: https://www.rndsystems.com/resources/articles/ryk-wnt-
co-receptor-intersection-frizzled-dishevelled
75. Fragiadaki M, Ikeda T, Witherden A, Mason RM, Abraham D, Bou-Gharios G. High
doses of TGF-β potently suppress type I collagen via the transcription factor CUX1. Mol
Biol Cell. 2011;22:1836–44.
76. COG3 Gene (Protein Coding): Component Of Oligomeric Golgi Complex 3 [Internet].
[cited 2016 Apr 3]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=COG3
77. Burset M, Seledtsov IA, Solovyev V V. Analysis of canonical and non-canonical splice

79
sites in mammalian genomes. Nucleic Acids Res [Internet]. Oxford, UK: Oxford
University Press; 2000;28(21):4364–75. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC113136/
78. P4HA1 Gene(Protein Coding): Prolyl 4-Hydroxylase, Alpha Polypeptide I [Internet].
[cited 2016 Mar 16]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=P4HA1
79. PROCOLLAGEN-PROLINE, 2-OXOGLUTARATE-4-DIOXYGENASE, ALPHA
SUBUNIT, ISOFORM 1; P4HA1 [Internet]. Available from:
http://www.omim.org/entry/176710
80. Peng X, Xu P-Z, Chen M-L, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism,
impaired skin development, skeletal muscle atrophy, delayed bone development, and
impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev [Internet]. Cold Spring
Harbor Laboratory Press; 2003;17(11):1352–65. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC196068/
81. AKT2 Gene(Protein Coding): V-Akt Murine Thymoma Viral Oncogene Homolog 2
[Internet]. [cited 2016 Feb 28]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=AKT2
82. V-AKT MURINE THYMOMA VIRAL ONCOGENE HOMOLOG 2; AKT2 [Internet].
[cited 2016 Feb 28]. Available from: http://www.omim.org/entry/164731
83. Mukherjee A, Wilson EM, Rotwein P. Selective Signaling by Akt2 Promotes Bone
Morphogenetic Protein 2-Mediated Osteoblast Differentiation. Mol Cell Biol [Internet].
American Society for Microbiology (ASM); 2010;30(4):1018–27. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2815574/
84. TRAFFICKING PROTEIN PARTICLE COMPLEX, SUBUNIT 12; TRAPPC12
[Internet]. [cited 2016 Mar 16]. Available from:
http://www.omim.org/entry/614139?search=TRAPPC12&highlight=trappc12
85. Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, et al. TRAPP, a highly
conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion.
EMBO J [Internet]. 1998;17(9):2494–503. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1170591/
86. Scrivens PJ, Noueihed B, Shahrzad N, Hul S, Brunet S, Sacher M. C4orf41 and TTC-15
are mammalian TRAPP components with a role at an early stage in ER-to-Golgi
trafficking. Mol Biol Cell [Internet]. The American Society for Cell Biology;
2011;22(12):2083–93. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3113772/
87. Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, et al. TRAPP, a highly
conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion.
EMBO J. 1998;17(9):2494–503.
88. ACAN [Internet]. [cited 2016 Mar 16]. Available from:
https://ghr.nlm.nih.gov/gene/ACAN
89. Stattin E-L, Wiklund F, Lindblom K, Önnerfjord P, Jonsson B-A, Tegner Y, et al. A
Missense Mutation in the Aggrecan C-type Lectin Domain Disrupts Extracellular Matrix
Interactions and Causes Dominant Familial Osteochondritis Dissecans. Am J Hum Genet
[Internet]. Elsevier; 2010;86(2):126–37. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2820178/
90. Gene Family: Pleckstrin homology domain containing (PLEKH) [Internet]. [cited 2016

80
Mar 16]. Available from: http://www.genenames.org/cgi-bin/genefamilies/set/682
91. Bianchi L, Gagliardi A, Maruelli S, Besio R, Landi C, Gioia R, et al. Altered cytoskeletal
organization characterized lethal but not surviving Brtl+/− mice: insight on phenotypic
variability in osteogenesis imperfecta. Hum Mol Genet [Internet]. 2015;24(21):6118–33.
Available from: http://hmg.oxfordjournals.org/content/24/21/6118.abstract
92. Boyadjiev SA, Kim S-D, Hata A, Haldeman-Englert C, Zackai EH, Naydenov C, et al.
Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin
Genet [Internet]. Blackwell Publishing Ltd; 2011 Aug 1;80(2):169–76. Available from:
http://dx.doi.org/10.1111/j.1399-0004.2010.01550.x
93. Kim S-D, Pahuja KB, Ravazzola M, Yoon J, Boyadjiev SA, Hammamoto S, et al.
SEC23-SEC31 the Interface Plays Critical Role for Export of Procollagen from the
Endoplasmic Reticulum. J Biol Chem [Internet]. 9650 Rockville Pike, Bethesda, MD
20814, U.S.A.: American Society for Biochemistry and Molecular Biology;
2012;287(13):10134–44. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3323018/
94. Townley AK, Feng Y, Schmidt K, Carter DA, Porter R, Verkade P, et al. Efficient
coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion
and is essential for normal craniofacial development. J Cell Sci [Internet].
2008;121(18):3025–34. Available from:
http://jcs.biologists.org/content/121/18/3025.abstract
95. RAB1A Gene (Protein Coding): RAB1A, Member RAS Oncogene Family [Internet].
[cited 2016 Apr 3]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=RAB1A
96. EYA1 Gene (Protein Coding): EYA Transcriptional Coactivator And Phosphatase 1
[Internet]. [cited 2016 Apr 3]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=EYA1&keywords=EYA1
97. RECK Gene (Protein Coding): Reversion-Inducing-Cysteine-Rich Protein With Kazal
Motifs [Internet]. [cited 2016 Apr 3]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=RECK&keywords=RECK
98. Accorsi-Mendonça T, Paiva KB da S, Zambuzzi WF, Cestari TM, Lara VS, Sogayar
MC, et al. Expression of matrix metalloproteinases-2 and -9 and RECK during alveolar
bone regeneration in rat. J Mol Histol [Internet]. 2007;39(2):201–8. Available from:
http://dx.doi.org/10.1007/s10735-007-9152-z
99. CRTAC1 Gene (Protein Coding): Cartilage Acidic Protein 1 [Internet]. [cited 2016 Apr
3]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=CRTAC1&keywords=CRTAC1
100. ADAMTS20 Gene (Protein Coding): ADAM Metallopeptidase With Thrombospondin
Type 1 Motif, 20 [Internet]. [cited 2016 Apr 3]. Available from:
http://www.genecards.org/cgi-
bin/carddisp.pl?gene=ADAMTS20&keywords=ADAMTS20
101. Howell GR, Shindo M, Murray S, Gridley T, Wilson LA, Schimenti JC. Mutation of a
Ubiquitously Expressed Mouse Transmembrane Protein (Tapt1) Causes Specific
Skeletal Homeotic Transformations. Genetics [Internet]. Copyright © 2007 by the
Genetics Society of America; 2007;175(2):699–707. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1800629/
102. TAPT1 Gene(Protein Coding): Transmembrane Anterior Posterior Transformation 1

81
[Internet]. [cited 2016 Mar 16]. Available from: http://www.genecards.org/cgi-
bin/carddisp.pl?gene=TAPT1&keywords=TAPT1
103. Symoens S, Barnes AM, Gistelinck C, Malfait F, Guillemyn B, Steyaert W, et al. Genetic
Defects in TAPT1 Disrupt Ciliogenesis and Cause a Complex Lethal
Osteochondrodysplasia. Am J Hum Genet [Internet]. 2015;97(4):521–34. Available
from: http://www.sciencedirect.com/science/article/pii/S0002929715003316
104. Goh G, Choi M. Application of Whole Exome Sequencing to Identify Disease-Causing
Variants in Inherited Human Diseases. Genomics Inform [Internet]. Korea Genome
Organization; 2012 Dec 31;10(4):214–9. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3543920/
105. GENOMIC STRATEGY CONSULTING. Exome Sequencing: The Next Wave in
Identifying Rare Genes, Making Diagnoses, and Finding Effective Treatments. 2011;
106. Gilissen C, Hoischen A, Brunner HG, Veltman JA. Disease gene identification strategies
for exome sequencing. Eur J Hum Genet [Internet]. Nature Publishing Group;
2012;20(5):490–7. Available from:
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3330229/
107. Burke W, Matheny Antommaria AH, Bennett R, Botkin J, Clayton EW, Henderson GE,
et al. Recommendations for returning genomic incidental findings? We need to talk!
Genet Med. 2013;15(11):854–9.
108. Roche MI, Berg JS. Incidental Findings with Genomic Testing: Implications for Genetic
Counseling Practice. Curr Genet Med Rep [Internet]. 2015;3(4):166–76. Available from:
http://link.springer.com/10.1007/s40142-015-0075-9
109. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG
recommendations for reporting of incidental findings in clinical exome and genome
sequencing. Genet Med [Internet]. 2013;15(7):565–74. Available from:
http://www.nature.com/gim/journal/v15/n7/full/gim201373a.html\nhttp://www.nature.c
om/gim/journal/v15/n7/pdf/gim201373a.pdf\nhttp://www.ncbi.nlm.nih.gov/pubmed/23
788249
110. X-linked spondyloepiphyseal dysplasia tarda [Internet]. [cited 2016 Apr 3]. Available
from: https://ghr.nlm.nih.gov/condition/x-linked-spondyloepiphyseal-dysplasia-tarda


I
7 Bijlagen
7.1 Gebruikte materialen
Polymerase Chain Reaction (Bijlage 1)
Thermo-Fastqf® 96, non-skirted: 0,2 ml 96-well PCR plate (Thermo Scientific)
VWR International Vortex
Centrifuge Sigma (Model: 2 – 16P
Epjes
o Microcentrifuge Tubes (transparant): 1,5 ml
o Safe Seal Microcentrifuge Tubes: 0,65 ml (Sorenson BioScience, Inc)
o Thermo-Tube (transparant): 0,2 ml (Thermo Scientific)
o Thermo Scientific 8 – tube strips w/ Ultraclear attached caps 0,2 ml
Pipettips
o Pipettip voor Gilson Repetman: Brand PD – tips sterile 0,5 ml
o Grenier Bio – one 10 µl filter tips steriel
o Thermo Scientific ART tips Barrier and Non – filtered pipette tips 20 µl/100
µl/200 µl/1000 µl
Pipetten
o Gilson Repetman
o Pipet 1-10µl (Gilson)
o Pipet 0,2-2µl (Gilson)
o Pipet P20 (Gilson)
o Pipet P1000 (Gilson)
o Multichannel Thermo Scientific Finnipipette F1 1-10µl
o Multichannel Gilson 0,5 – 10µl
Seals
o Adhesive PCR Film: transparante thermo-seal
Thermal Cycler Applied Biosystems 2720
Steriele, latex- en poedervrije handschoenen (Ecoshield® )
Labojas

II
Sanger sequencing (Bijlage 2)
Thermoqf-Fast® 96, non-skirted: 0,2 ml 96-well PCR plate (Thermo Scientific)
VWR International Vortex / Vortex Genie-2
Centrifuge
o Eppendorf 5415D
o Eppendorf 5430
Epjes
o Microcentrifuge Tubes (transparant): 1,5 ml
o Safe Seal Microcentrifuge Tubes: 0,65 ml (Sorenson BioScience, Inc)
o Thermo-Tube (transparant): 0,2 ml (Thermo Scientific)
o Thermo Scientific 8 – tube strips w/ Ultraclear attached caps 0,2 ml
Pipettips
o Pipettip voor Gilson Repetman: Brand PD – tips sterile 0,5 ml
o Sapphire by Grenier Bio – one 10 µl filter tips steriel
o Thermo Scientific ART tips Barrier and Non – filtered pipette tips 100µl
Pipetten
o Gilson Repetman
o Pipet 1-10µl (Gilson)
o Pipet P1000 (Gilson)
o Multichannel Thermo Scientific Finnipipette F1 1-10µl/10 – 100 µl
o Multichannel Thermo Electron Corporation 1 – 10µl
o Multichannel Gilson 0,5 – 10µl
Seals
o Adhesive PCR Film: transparante thermo-seal
o Seal voor GSU
Thermal Cycler Applied Biosystems 2720
Magnetische plaat
Steriele, latex- en poedervrije handschoenen (Ecoshield® )
Labojas
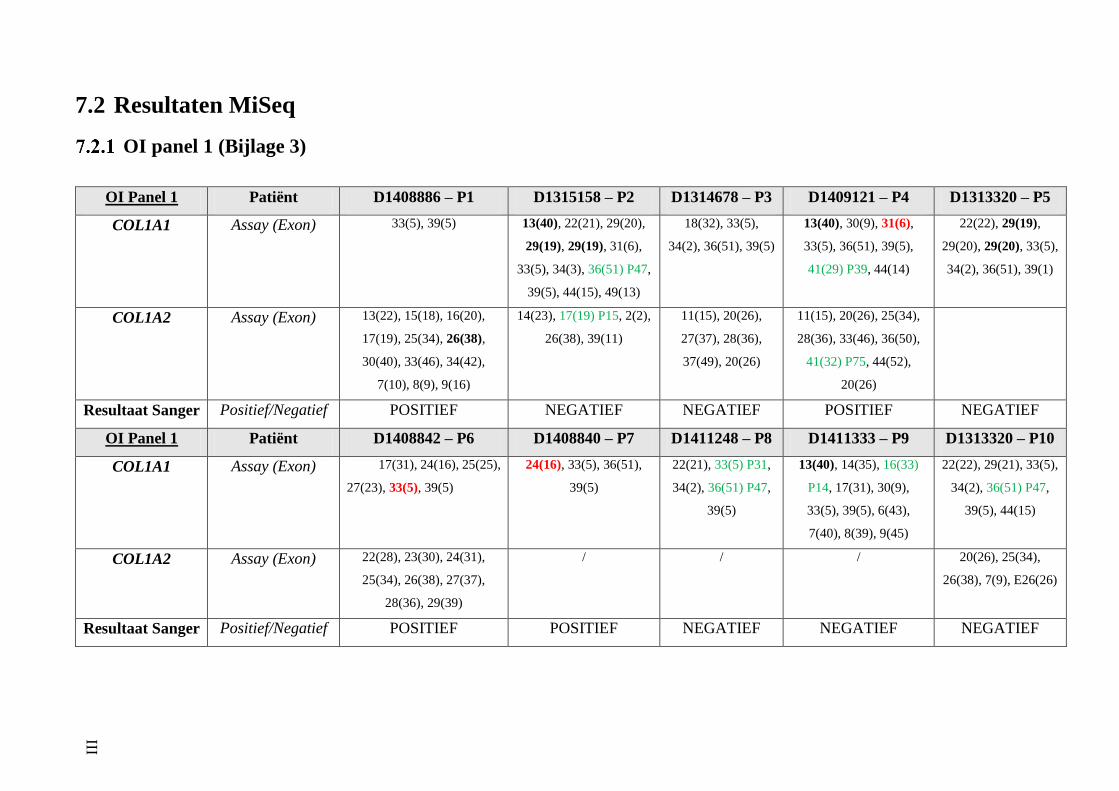
III
7.2 Resultaten MiSeq
OI panel 1 (Bijlage 3)
OI Panel 1 Patiënt D1408886 – P1 D1315158 – P2 D1314678 – P3 D1409121 – P4 D1313320 – P5
COL1A1 Assay (Exon) 33(5), 39(5) 13(40), 22(21), 29(20),
29(19), 29(19), 31(6),
33(5), 34(3), 36(51) P47,
39(5), 44(15), 49(13)
18(32), 33(5),
34(2), 36(51), 39(5)
13(40), 30(9), 31(6),
33(5), 36(51), 39(5),
41(29) P39, 44(14)
22(22), 29(19),
29(20), 29(20), 33(5),
34(2), 36(51), 39(1)
COL1A2 Assay (Exon) 13(22), 15(18), 16(20),
17(19), 25(34), 26(38),
30(40), 33(46), 34(42),
7(10), 8(9), 9(16)
14(23), 17(19) P15, 2(2),
26(38), 39(11)
11(15), 20(26),
27(37), 28(36),
37(49), 20(26)
11(15), 20(26), 25(34),
28(36), 33(46), 36(50),
41(32) P75, 44(52),
20(26)
Resultaat Sanger Positief/Negatief POSITIEF NEGATIEF NEGATIEF POSITIEF NEGATIEF
OI Panel 1 Patiënt D1408842 – P6 D1408840 – P7 D1411248 – P8 D1411333 – P9 D1313320 – P10
COL1A1 Assay (Exon) 17(31), 24(16), 25(25),
27(23), 33(5), 39(5)
24(16), 33(5), 36(51),
39(5)
22(21), 33(5) P31,
34(2), 36(51) P47,
39(5)
13(40), 14(35), 16(33)
P14, 17(31), 30(9),
33(5), 39(5), 6(43),
7(40), 8(39), 9(45)
22(22), 29(21), 33(5),
34(2), 36(51) P47,
39(5), 44(15)
COL1A2 Assay (Exon) 22(28), 23(30), 24(31),
25(34), 26(38), 27(37),
28(36), 29(39)
/ / / 20(26), 25(34),
26(38), 7(9), E26(26)
Resultaat Sanger Positief/Negatief POSITIEF POSITIEF NEGATIEF NEGATIEF NEGATIEF
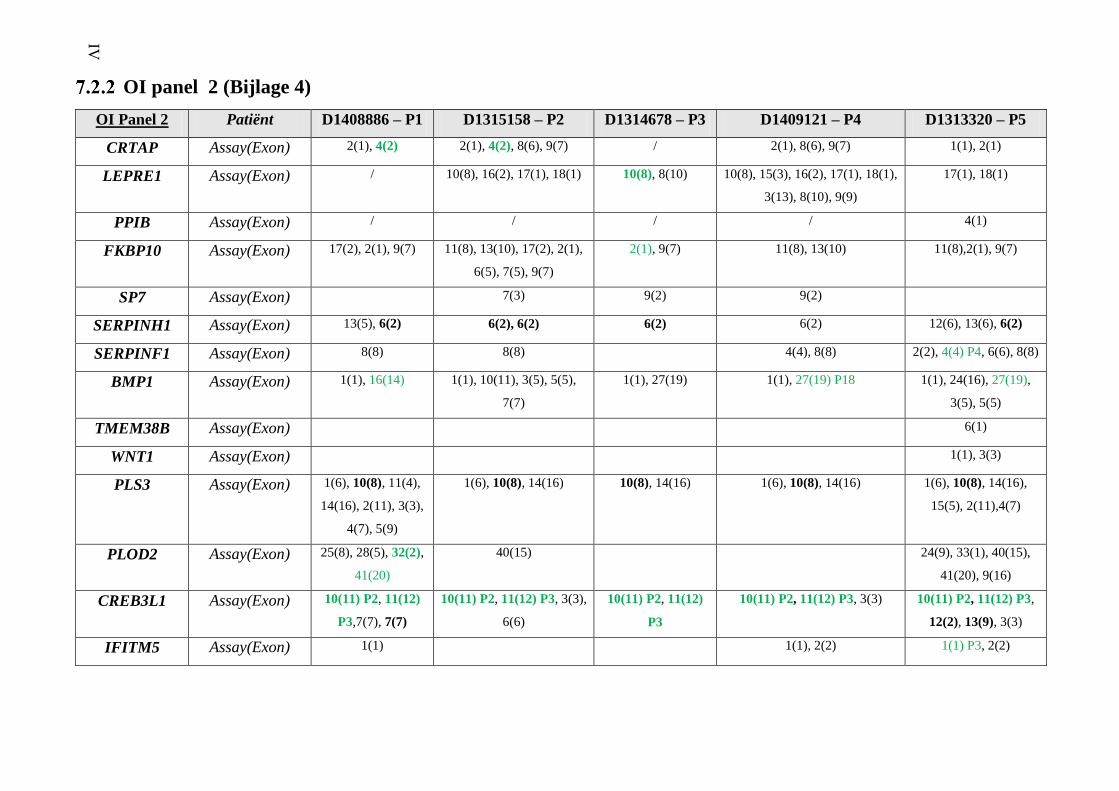
IV
OI panel 2 (Bijlage 4)
OI Panel 2 Patiënt D1408886 – P1 D1315158 – P2 D1314678 – P3 D1409121 – P4 D1313320 – P5
CRTAP Assay(Exon) 2(1), 4(2) 2(1), 4(2), 8(6), 9(7) / 2(1), 8(6), 9(7) 1(1), 2(1)
LEPRE1 Assay(Exon) / 10(8), 16(2), 17(1), 18(1) 10(8), 8(10) 10(8), 15(3), 16(2), 17(1), 18(1),
3(13), 8(10), 9(9)
17(1), 18(1)
PPIB Assay(Exon) / / / / 4(1)
FKBP10 Assay(Exon) 17(2), 2(1), 9(7) 11(8), 13(10), 17(2), 2(1),
6(5), 7(5), 9(7)
2(1), 9(7) 11(8), 13(10) 11(8),2(1), 9(7)
SP7 Assay(Exon) 7(3) 9(2) 9(2)
SERPINH1 Assay(Exon) 13(5), 6(2) 6(2), 6(2) 6(2) 6(2) 12(6), 13(6), 6(2)
SERPINF1 Assay(Exon) 8(8) 8(8) 4(4), 8(8) 2(2), 4(4) P4, 6(6), 8(8)
BMP1 Assay(Exon) 1(1), 16(14) 1(1), 10(11), 3(5), 5(5),
7(7)
1(1), 27(19) 1(1), 27(19) P18 1(1), 24(16), 27(19),
3(5), 5(5)
TMEM38B Assay(Exon) 6(1)
WNT1 Assay(Exon) 1(1), 3(3)
PLS3 Assay(Exon) 1(6), 10(8), 11(4),
14(16), 2(11), 3(3),
4(7), 5(9)
1(6), 10(8), 14(16) 10(8), 14(16) 1(6), 10(8), 14(16) 1(6), 10(8), 14(16),
15(5), 2(11),4(7)
PLOD2 Assay(Exon) 25(8), 28(5), 32(2),
41(20)
40(15) 24(9), 33(1), 40(15),
41(20), 9(16)
CREB3L1 Assay(Exon) 10(11) P2, 11(12)
P3,7(7), 7(7)
10(11) P2, 11(12) P3, 3(3),
6(6)
10(11) P2, 11(12)
P3
10(11) P2, 11(12) P3, 3(3) 10(11) P2, 11(12) P3,
12(2), 13(9), 3(3)
IFITM5 Assay(Exon) 1(1) 1(1), 2(2) 1(1) P3, 2(2)
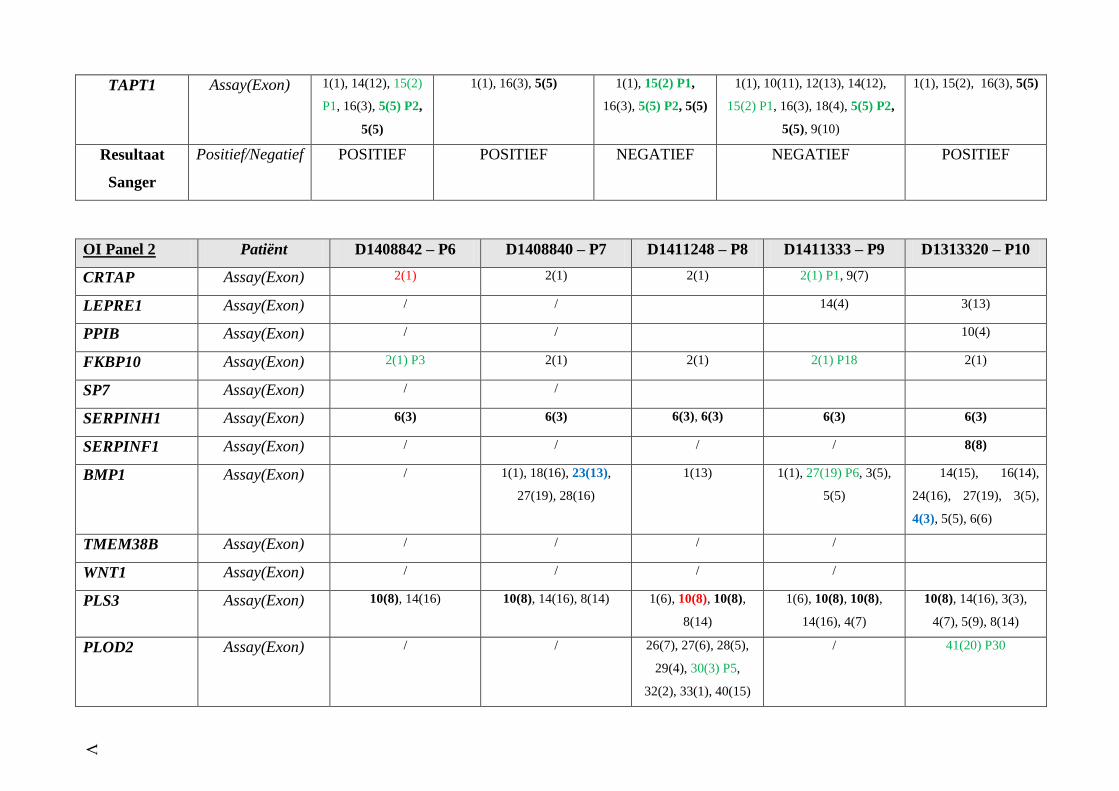
V
TAPT1 Assay(Exon) 1(1), 14(12), 15(2)
P1, 16(3), 5(5) P2,
5(5)
1(1), 16(3), 5(5) 1(1), 15(2) P1,
16(3), 5(5) P2, 5(5)
1(1), 10(11), 12(13), 14(12),
15(2) P1, 16(3), 18(4), 5(5) P2,
5(5), 9(10)
1(1), 15(2), 16(3), 5(5)
Resultaat
Sanger
Positief/Negatief POSITIEF POSITIEF NEGATIEF NEGATIEF POSITIEF
OI Panel 2 Patiënt D1408842 – P6 D1408840 – P7 D1411248 – P8 D1411333 – P9 D1313320 – P10
CRTAP Assay(Exon) 2(1) 2(1) 2(1) 2(1) P1, 9(7)
LEPRE1 Assay(Exon) / / 14(4) 3(13)
PPIB Assay(Exon) / / 10(4)
FKBP10 Assay(Exon) 2(1) P3 2(1) 2(1) 2(1) P18 2(1)
SP7 Assay(Exon) / /
SERPINH1 Assay(Exon) 6(3) 6(3) 6(3), 6(3) 6(3) 6(3)
SERPINF1 Assay(Exon) / / / / 8(8)
BMP1 Assay(Exon) / 1(1), 18(16), 23(13),
27(19), 28(16)
1(13) 1(1), 27(19) P6, 3(5),
5(5)
14(15), 16(14),
24(16), 27(19), 3(5),
4(3), 5(5), 6(6)
TMEM38B Assay(Exon) / / / /
WNT1 Assay(Exon) / / / /
PLS3 Assay(Exon) 10(8), 14(16) 10(8), 14(16), 8(14) 1(6), 10(8), 10(8),
8(14)
1(6), 10(8), 10(8),
14(16), 4(7)
10(8), 14(16), 3(3),
4(7), 5(9), 8(14)
PLOD2 Assay(Exon) / / 26(7), 27(6), 28(5),
29(4), 30(3) P5,
32(2), 33(1), 40(15)
/ 41(20) P30
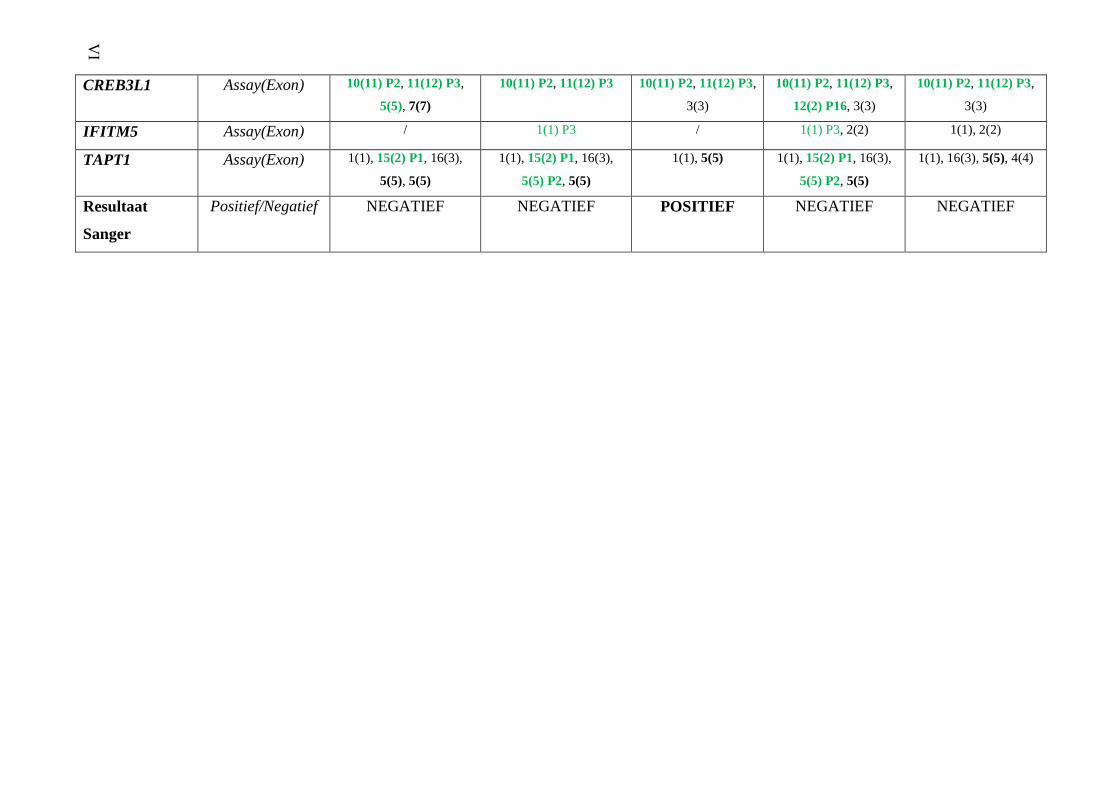
VI
CREB3L1 Assay(Exon) 10(11) P2, 11(12) P3,
5(5), 7(7)
10(11) P2, 11(12) P3 10(11) P2, 11(12) P3,
3(3)
10(11) P2, 11(12) P3,
12(2) P16, 3(3)
10(11) P2, 11(12) P3,
3(3)
IFITM5 Assay(Exon) / 1(1) P3 / 1(1) P3, 2(2) 1(1), 2(2)
TAPT1 Assay(Exon) 1(1), 15(2) P1, 16(3),
5(5), 5(5)
1(1), 15(2) P1, 16(3),
5(5) P2, 5(5)
1(1), 5(5) 1(1), 15(2) P1, 16(3),
5(5) P2, 5(5)
1(1), 16(3), 5(5), 4(4)
Resultaat
Sanger
Positief/Negatief NEGATIEF NEGATIEF POSITIEF NEGATIEF NEGATIEF

VII
7.3 Sanger Sequencing Report
Sequencing Report van de mutatie gevonden in CRTAP-4 toont een nucleotideverandering van A naar G zowel in de forward als reverse read (heterozygoot).
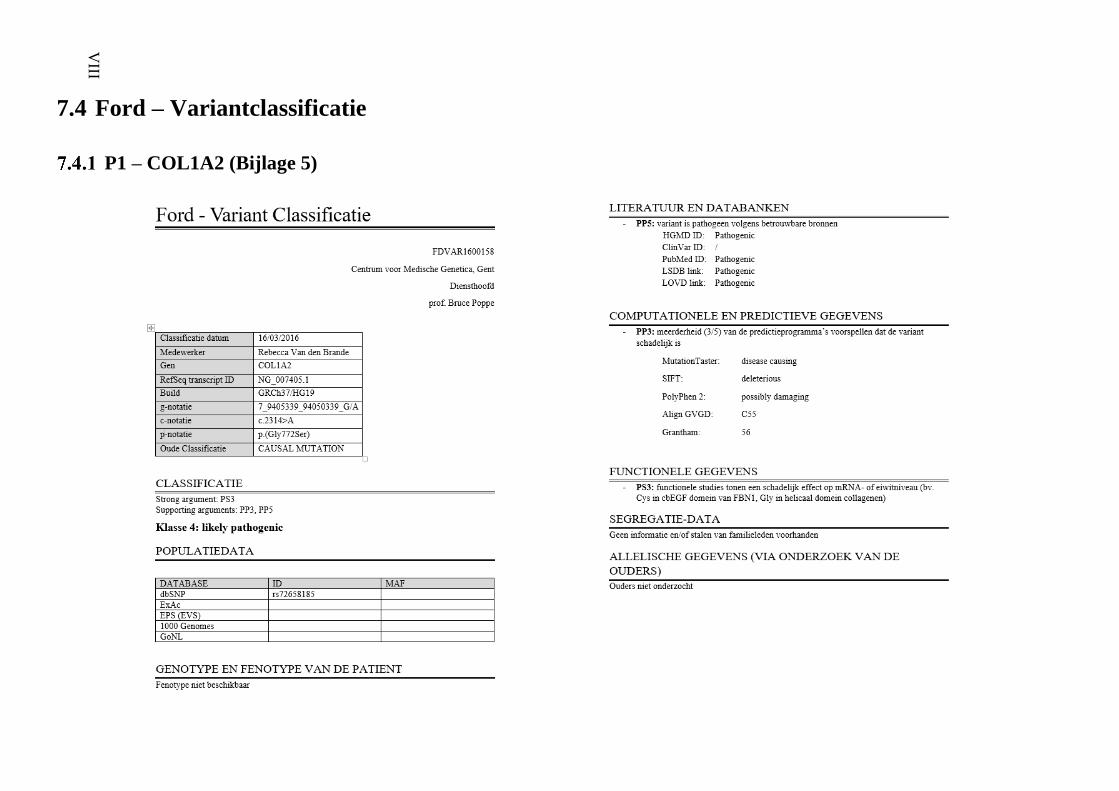
VIII
7.4 Ford – Variantclassificatie
P1 – COL1A2 (Bijlage 5)

IX
P1 en P2 – CRTAP (Bijlage 6)

X
P1 – PLOD2 (Bijlage 7)

XI
P3, P6, P7 en P9 – TAPT1 – P1 (Bijlage 8)

XII
P1, P3, P4, P7 en P9 – TAPT1 – P2 (Bijlage 9)

XIII
P4 – COL1A1 (Bijlage 10)

XIV
P6 – COL1A1 (Bijlage 11)

XV
P7 – COL1A1 (Bijlage 12)

XV
I
P5 – CREB3L1 (Bijlage 13)

XV
II
P6 – CREB3L1 (Bijlage 14)

XV
III
P7 – BMP1 (Bijlage 15)

XIX
P8 – PLS3 (Bijlage 16)
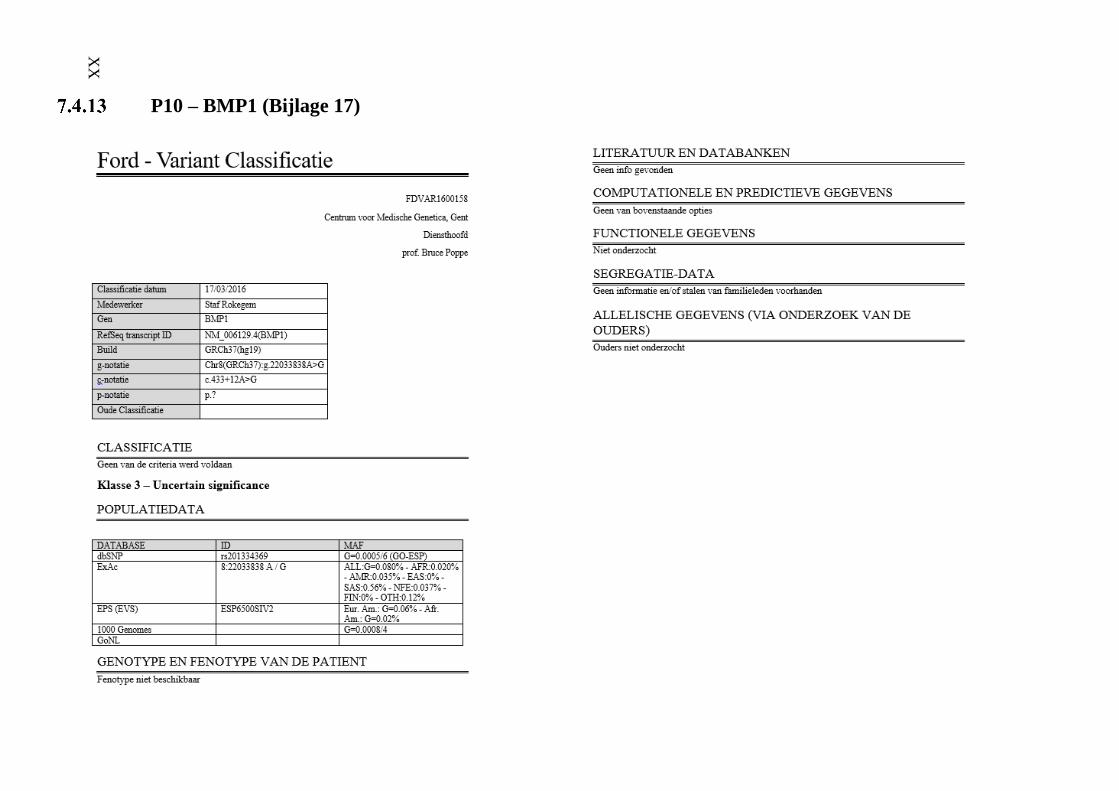
XX
P10 – BMP1 (Bijlage 17)
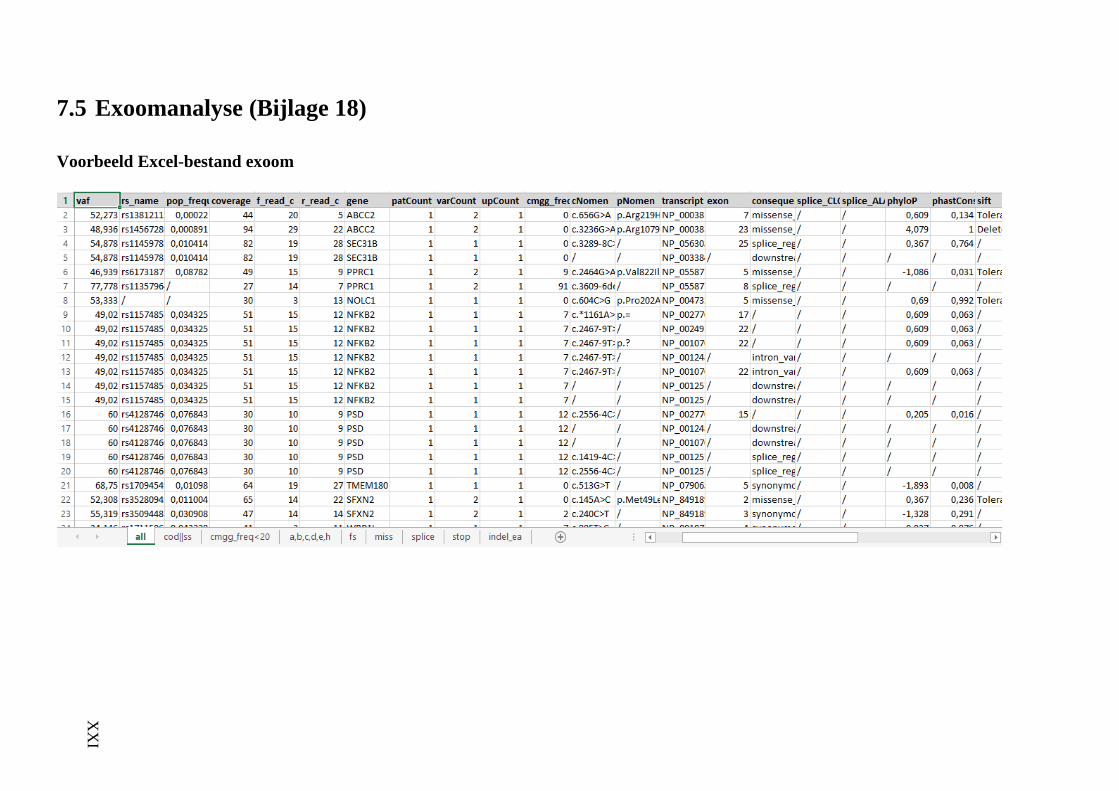
XX
I
7.5 Exoomanalyse (Bijlage 18)
Voorbeeld Excel-bestand exoom


















